
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen storage capacity and thermal stability of the CuMnO2–CeO2 composite system†
Xiubing
Huang
,
Chengsheng
Ni
,
Guixia
Zhao
and
John T. S.
Irvine
*
School of Chemistry, University of St Andrews, St Andrews, Fife, UK. E-mail: jtsi@st-andrews.ac.uk; Fax: +44 (0)1334463808; Tel: +44 (0)133463680
First published on 12th May 2015
Abstract
Fast and reversible oxygen diffusion in solid oxides depending on oxygen partial pressure at low temperatures is a promising strategy for improving the overall performance and service lifetime of many energy-related materials. However, the high energy required for the redox reaction of cations and their high thermodynamic barriers have impeded the realization of fast oxygen diffusion at low temperatures. Herein, we report enhanced oxygen diffusion and storage capacity of monoclinic crednerite CuMnO2 at a lower temperature by surface modification with CeO2. The fast and reversible oxygen uptake/release can be attributed to CeO2 that serves as a fast oxygen diffusion channel between bulk CuMnO2 and the surrounding atmospheres. Importantly, the amount of CeO2 in the CuMnO2–CeO2 composite system has a great effect on the total oxygen storage capacity and redox behaviour. Our findings could provide useful information for developing effective oxygen storage materials for wide energy-related applications.
1. Introduction
Solid-state oxygen storage materials (OSMs) have attracted considerable attention due to their wide applications in numerous oxygen-related energy and environmental fields, such as three-way catalysts for the effective removal of automobile exhaust emissions (e.g., NOx, CO, and hydrocarbons).1,2 The development of OSMs is also crucial to the success of new energy technologies, such as oxygen enrichment to improve the efficiency of chemical looping combustion.3 Ideal OSMs for practical applications should satisfy certain requirements and possess some properties, including large oxygen storage capacity (OSC), quick absorption/desorption of oxygen and their dependence on temperature and/or oxygen partial pressure, and good reversibility.The most widely investigated OSMs are based on ceria (e.g., well-known Ce1−xZrxO2+δ) due to the reversible redox reaction of Ce3+/Ce4+,2,4–8 however, their OSC is relatively small and usually achieved by the usage of reductive gas. Recently, oxides based on transition metals have attracted remarkable attention due to their excellent properties, such as their flexible oxidation states, various phase structures, possible substitutions, cationic and anionic non-stoichiometry, or lattice oxygen deficient in the framework.3,9–13 Their oxygen storage/release behaviour is generally based on the oxygen non-stoichiometry, which can be achieved by adjusting the surrounding oxygen partial pressure (i.e., oxidative air, reductive H2) or temperature.11,14 Among OSMs based on transition-metals, extensive attention has been paid to delafossite-type oxides with the general formula CuMO2 (M = trivalent cation) because of the low reductive/oxidative temperature of Cu2+/Cu+, remarkable oxygen uptake ability, wide potential applications, environmental-friendliness and abundance.15–17 However, only very limited work has been reported on the effect of trivalent cation species on the oxygen storage properties of the delafossite-type CuMO2 oxides under oxidative/reductive atmospheres.15–17 For example, Sumio Kato, et al. reported that CuMnO2 and CuFeO2 exhibited larger OSC values at lower temperature than those of CuAlO2 and CuGaO2,15 and OSC values for x = 0.1 and 0.3 in CuFe1−xAlxO2 were larger than that for x = 0 above 500 °C.17 However, the full details of the phase-transformation process under oxidative/inert atmospheres and the effect of surface-modification by other metal oxides on the oxygen diffusion, oxygen storage capacity and thermal stability of CuMO2 still remain not well-defined.
In the search for better OSMs, crednerite CuMnO2 as an alternative phase to delafossite-type CuMO2 seems to have great potential due to its changeable valence and low-temperature for oxygen uptake/release,15,18 and its wide applications, such as a hydrogen photo-evolution catalyst,19 a three-way catalyst for the removal of exhaust gases (e.g., CO, NOx),15 and hydrogen storage.20 As reported by several research groups, crednerite CuMnO2 exhibits a monoclinic structure at room temperature, which consists of edge-shared MnO6 octahedra and two-coordinated Cu+ cations at the interlayer sites, as represented in Fig. 1.15,19,21 This structure is closely related to the rhombohedral 3R delafossite structure of CuFeO2 (i.e., the Cu+ cations linearly coordinating to two O ions, and the parallel O–Cu–O chains connecting Fe3+ cations to form two-dimensional sheets of edge-shared FeO6 octahedra), but with a distortion of the MnO6 octahedron due to the Jahn–Teller effect of Mn3+.22 However, it has been reported that Cu1+xMn1−xO2 (0.08 < x < 0.12) at high temperature can exhibit a delafossite-like phase with a hexagonal structure because the thermal expansion of the structure would result in a larger lattice distortion than that from the Jahn–Teller effect.23,24
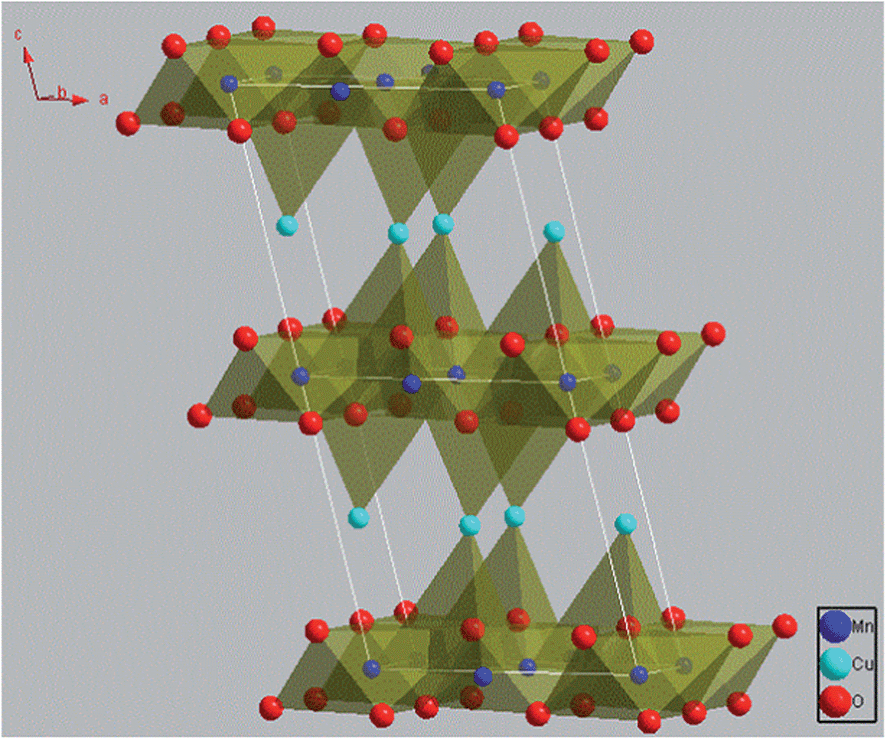 | ||
| Fig. 1 Schematic illustration of the crystal structure of CuMnO2. The illustration was drawn with Diamond 3.1d software for crystal and molecular structure visualization. | ||
It is well known that multiple valences of Mn cations can co-exist in many Mn-containing compounds, which could be beneficial to the oxygen-storage properties.25–27 The valence of Mn in CuMnO2 has been demonstrated to be +3, however the existence of Mn4+ in the nonstoichiometric Cu1+xMn1−xO2 oxides is also detected by XPS because the excess Cu atoms would occupy the octahedral site with Cu2+ while the electrical neutrality principle would result in the formation of mixed Mn3+/Mn4+.18,28 In addition, the CuxMn3−xO4 spinel, one of the oxidation products of CuMnO2, has been reported to show the coexistence of Mn3+/Mn4+, implying a possible increase in the total oxygen uptake ability under an oxidative atmosphere.27,29,30 However, pure crednerite CuMnO2 oxide is usually synthesized using the solid-state reaction at very high temperatures15,31,32 or the ion-exchange reaction with a long reaction time.32 Such a high reaction temperature or slow chemical conversion rate for reversible redox processes is impractical for many technological applications.18 Therefore, it is important to improve the oxygen diffusion ability and the overall oxygen storage capacity of crednerite CuMnO2 at low temperatures.
CeO2 has been widely investigated as a three-way catalyst or an oxygen promoter due to the fast oxygen ionic mobility between Ce4+ and Ce3+ in a reductive/oxidative atmosphere even though CeO2 could retain its fluorite structure under oxidizing and mildly reducing atmospheres.33–37 CeO2 has also been reported as an interlayer or electrolyte in the La1−xSrxCoO3−δ/CeO2 composite system to improve the oxygen ionic mobility, in which some chemical reactions can take place at the interfaces between perovskite and CeO2.38,39 Therefore, CeO2 may function as an oxygen ionic mobility channel between oxides and their surrounding atmosphere. Here, we investigated the CuMnO2–CeO2 composite system with the purpose of further optimizing the oxygen diffusion ability and overall oxygen storage capacity at low temperatures under alternating oxidative (i.e., air or O2) and inert (i.e., Ar) atmospheres. Modifying the surfaces of CuMnO2 with CeO2 may bring in both benefits of CuMnO2 and CeO2, thereafter adjusting their thermochemical properties (e.g., redox properties, non-stoichiometry, oxygen exchange constant, and formation of oxygen vacancies), further enhancing their OSC and thermal stability. Our research results indicate that modifying CuMnO2 with a small portion of CeO2 (e.g., the molar ratios of CeO2 to CuMnO2 smaller than 20%) can improve the oxygen storage capacity at low temperatures (<600 °C) in a highly reversible manner.
2. Experimental section
2.1 Sample preparation
CuMnO2–CeO2 composites were prepared by a conventional Pechini method, followed by a solid state method. In a typical process, 10 mmol of Cu(NO3)2·2.5H2O (98%) and 10 mmol of Mn(CH3COO)2·4H2O (98%) were dissolved in 100 mL of deionized H2O under continuous stirring. A certain amount (0.5, 1.0, 2.0 or 4.0 mmol) of Ce(NO3)3·6H2O (99.99%) was added into the above solution under stirring, followed by adding 20 mmol of citric acid monohydrate (99.95%) and 10 mL of ethylene glycol. After stirring for 4 h, the solvent was evaporated at 110 °C to obtain a gel. After drying, the powder was ground and pre-fired at 500 °C under static air for 1 h to obtain the precursor, then the precursor was pressed into pellets and fired at 960 °C for 12 h under flowing argon. The obtained products were referred to as CuMnO2–xCeO2, in which x is the molar ratio percent of CeO2 to CuMnO2 (i.e., x = mol of CeO2/mol of CuMnO2 × 100).2.2 Characterization
Structures of all samples were characterized by X-ray powder diffraction (XRD) on a PANalytical Empyrean Reflection Diffractometer with Cu Kα irradiation (λ = 0.15418 nm). HRTEM images and TEM elemental mapping of samples were observed on a JEM-2011 Transmission Electron Microscope (TEM) with an acceleration voltage of 200 kV equipped with an X-ray Energy Dispersive spectrometer (EDS). The morphologies of all samples were observed on a JEOL JSM-6700 Field Scanning Electron Microscope (FESEM). The oxygen uptake behaviours of these samples were measured by thermogravimetric analysis (TGA) on a NETZSCH TG 209 instrument (NETZSCH-Geraetebau GmbH, Selb, Germany) with a TASC 414/3 controller. The measurements were carried out for 50 mg specimens up to 800 °C with a heating rate of 10 °C min−1 under air or O2 with flowing rate 25 mL min−1. In addition, the reversibility of the oxygen uptake/release was carried out in the following experiments: first increasing the temperature to 600 °C under flowing O2 gas, then switching the gas from O2 to Ar with temperature from 600 to 900 °C; after cooling down to 300 °C under flowing Ar, switching the gas from Ar to O2 with temperature from 300 to 600 °C; this process was repeated between 300 and 900 °C 4 times. The thermal stability (weight and phase structural changes) of the as-prepared samples with temperature from room temperature to 1000 °C under flowing air was checked using a TGA/DTA technique carried out on a Stanton Redcroft STA-780 series thermal analyser.3. Results and discussion
XRD patterns of these as-prepared CuMnO2–xCeO2 composites obtained by post-annealing at 960 °C for 12 h under flowing Ar are shown in Fig. 2. The XRD pattern for CuMnO2 in Fig. 2a can be indexed to a pure monoclinic structure with a C2/m space group. All the peaks are in good agreement with the JCPDS card no. 50-0860 for the crednerite phase. For the CuMnO2–xCeO2 composites, they are obviously composed of crednerite CuMnO2 and fluorite CeO2, and no other impurity phases are observed. The XRD Rietveld refinements for CuMnO2 and CeO2 were carried out using GSAS software based on the monoclinic structure with the C2/m space group and cubic structure with the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group, respectively. The typical XRD patterns after final refinement for CuMnO2 and CuMnO2–40CeO2 are shown in Fig. 3, further confirming the phase composition of CuMnO2 and CeO2.21
m space group, respectively. The typical XRD patterns after final refinement for CuMnO2 and CuMnO2–40CeO2 are shown in Fig. 3, further confirming the phase composition of CuMnO2 and CeO2.21
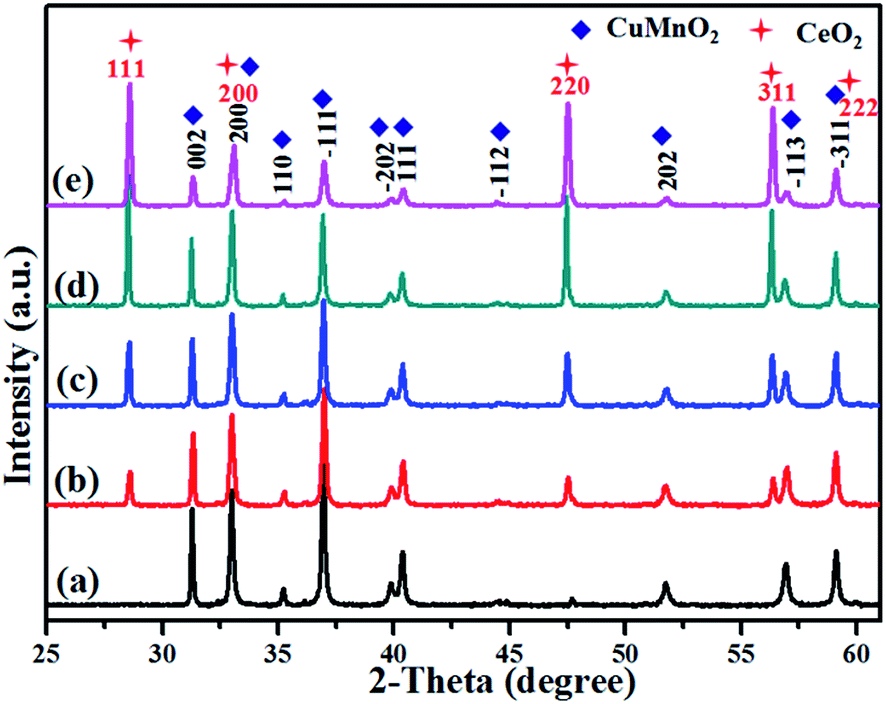 | ||
| Fig. 2 XRD patterns of the as-prepared CuMnO2–xCeO2 (i.e., post-annealed at 960 °C under flowing Ar): (a) CuMnO2, (b) CuMnO2–5CeO2, (c) CuMnO2–10CeO2, (d) CuMnO2–20CeO2, and (e) CuMnO2–40CeO2. | ||
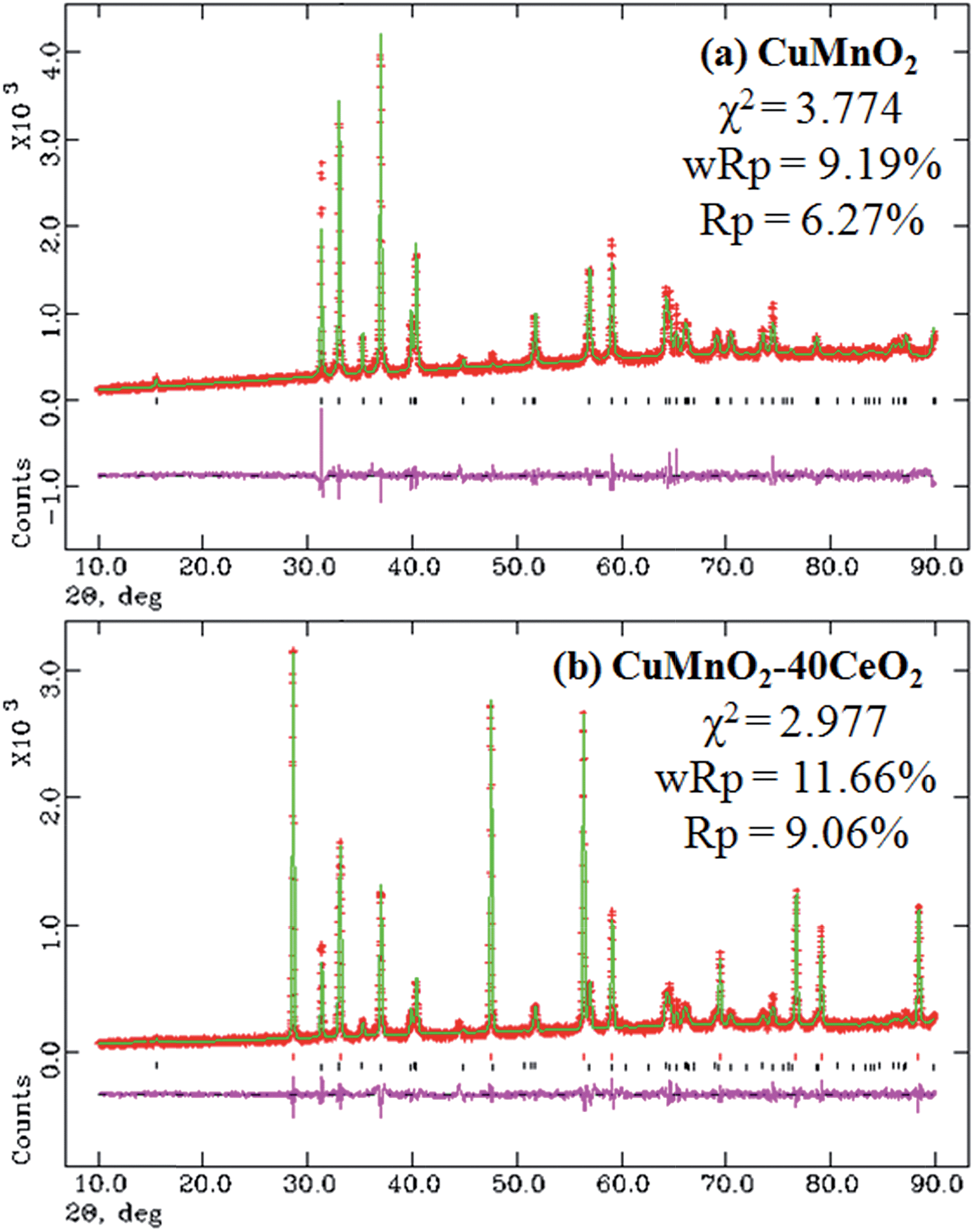 | ||
| Fig. 3 XRD patterns after final Rietveld refinements for the as-prepared products: (a) CuMnO2 and (b) CuMnO2–40CeO2. | ||
Detailed lattice parameters for pure CuMnO2 and CuMnO2–xCeO2 composites after Rietveld refinements are summarized in Table 1. There are no obvious changes in the lattice parameters a, b and c for CuMnO2 and a for CeO2 with the increasing CeO2 amount up to x = 40 for the CuMnO2–xCeO2 composite, suggesting that CuMnO2 and CeO2 exist mainly as separate phases and there may be only a little contact between the surfaces. The HRTEM images of CuMnO2 in CuMnO2–10CeO2 and CuMnO2–40CeO2 (Fig. S1†) show clear lattice fringes and these d spacings of 0.22 and 0.57 nm correspond to the (111) and (001) planes of CuMnO2, respectively. The TEM elemental mappings of Cu, Mn, O and Ce for CuMnO2–10CeO2 (Fig. S2†) and CuMnO2–40CeO2 (Fig. S3†) also indicate the individual existence of CuMnO2 and CeO2, as well as the inhomogeneous surface contact between CeO2 and CuMnO2.
| As-prepared sample | Composition | a (Å) | b (Å) | c (Å) | Cell volume (Å3) | β |
|---|---|---|---|---|---|---|
| CuMnO2 | CuMnO2 | 5.592(3) | 2.883(1) | 5.892(3) | 92.1(1) | 104.03(3) |
| CuMnO2–5CeO2 | CuMnO2 | 5.597(3) | 2.883(1) | 5.892(3) | 92.2(1) | 104.00(4) |
| CeO2 | 5.412(2) | 158.5(2) | ||||
| CuMnO2–10CeO2 | CuMnO2 | 5.593(1) | 2.883(6) | 5.892(1) | 92.19(3) | 104.06(3) |
| CeO2 | 5.412(1) | 158.5(2) | ||||
| CuMnO2–20CeO2 | CuMnO2 | 5.589(2) | 2.882(1) | 5.891(2) | 92.08(8) | 104.08(4) |
| CeO2 | 5.411(1) | 158.4(1) | ||||
| CuMnO2–40CeO2 | CuMnO2 | 5.597(3) | 2.883(1) | 5.891(3) | 92.24(9) | 104.06(7) |
| CeO2 | 5.412(2) | 158.5(3) |
The oxygen uptake behaviours of the as-prepared CuMnO2–xCeO2 composites were investigated by TGA at temperature up to 800 °C under flowing air or O2. The results shown in Fig. 4 reveal a remarkable oxygen uptake capacity of this CuMnO2–xCeO2 composite system under an oxidative atmosphere, which corresponds to the following exothermic oxidation reaction:
| CuMnO2 + 1/(6 − 2y)O2 → 1/(3 − y)CuyMn3−yO4 + (3 − 2y)/(3 − y)CuO | (1) |
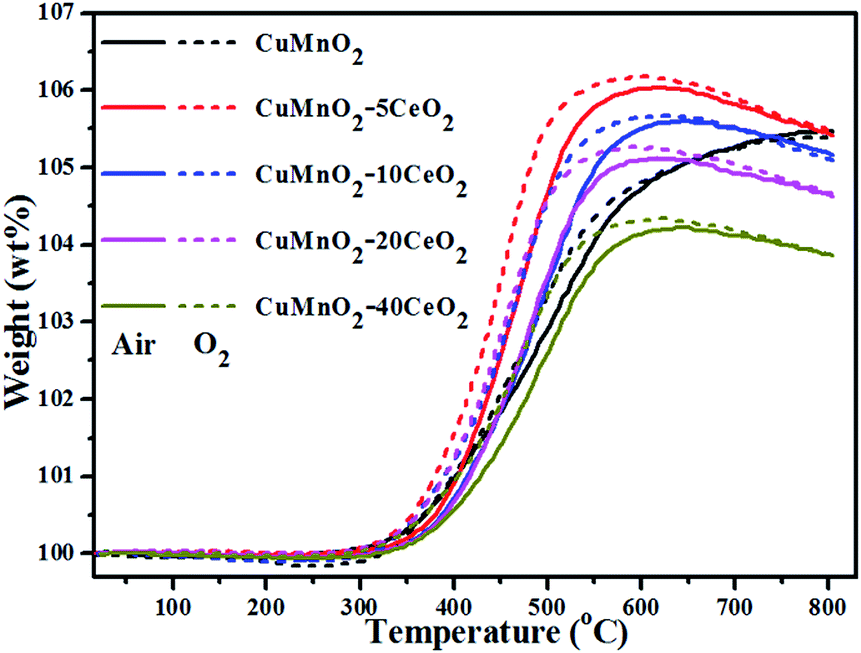 | ||
| Fig. 4 TGA curves of the as-prepared CuMnO2–xCeO2 samples (i.e., post-annealed at 960 °C under flowing Ar for 12 h) under flowing air or pure O2 with 25 mL min−1 from room temperature to 800 °C. The solid lines are the TGA curves under air and the short dashed lines are the TGA curves under pure O2. | ||
The weight for all these samples starts to increase at about 300 °C, due to the increase in the oxygen content, accompanied by the oxidation of Cu+ and/or Mn3+. Pure CuMnO2 had a continuous and smooth weight increase with increasing temperature from 300 to 800 °C, reaching the maximum value of 5.475 wt% at 800 °C under both flowing air and O2, corresponding to the formation of spinel Cu1.058Mn1.942O4. For pure CuMnO2 and CuMnO2–xCeO2 composites, the oxygen uptake rate in air is a bit slower than that in O2 and the maximum oxygen uptake amount in air is also a bit smaller than that in O2, in which CuMnO2–5CeO2 exhibits the most weight increase, reaching 6.169 wt% at 591 °C under flowing O2 and 6.031 wt% at 620 °C under flowing air, respectively, suggesting that higher oxygen partial pressure would enhance the oxygen uptake ability at lower temperatures. With the further increase of CeO2 amount in the composite, the maximum OSC decreased, which can be attributed to the offset effect of CeO2 since the weight increase is mainly caused by the oxidation of Cu+ and/or Mn3+ in CuMnO2 while CeO2 could maintain its fluorite-type structure under an inert atmosphere,40,41 as determined by the XRD results in Fig. 2. After reaching the maximum weight at around 600 °C, further increasing the temperature to 800 °C results in a slight and smooth weight decrease (<0.14 wt%) for all these CeO2-modified CuMnO2 samples, which can be attributed to the composition adjustment of spinel CuyMn3−yO4 with temperature.18 These results also indicate that CeO2-modification would favour the oxygen mobility in CuMnO2 and the formation of spinel CuyMn3−yO4 with more Mn4+ at lower temperatures. It should be noted that the oxygen uptake in CuMnO2 and CuMnO2–xCeO2 composites mainly comes from the oxidation of CuMnO2 to CuO and spinel CuyMn3−yO4. Assuming that the total weight increase only comes from the oxidation of CuMnO2, the composition of spinel CuyMn3−yO4 at the maximum weight under flowing O2 can be calculated based on eqn (1) to be Cu1.370Mn1.630O4 for the CuMnO2–5CeO2 starting mix, Cu1.317Mn1.683O4 for CuMnO2–10CeO2, Cu1.356Mn1.644O4 for CuMnO2–20CeO2 and Cu1.316Mn1.684O4 for CuMnO2–40CeO2, while the composition of spinels at the maximum weight under flowing air can be calculated to be Cu1.332Mn1.668O4 for CuMnO2–5CeO2, Cu1.296Mn1.704O4 for CuMnO2–10CeO2, Cu1.310Mn1.690O4 for CuMnO2–20CeO2 and Cu1.268Mn1.732O4 for CuMnO2–40CeO2, respectively. These calculation results suggest that higher oxygen partial pressure favours the formation of spinels with more Mn4+ from the oxidation of CuMnO2–xCeO2 composites.
XRD patterns of the oxygenated CuMnO2 and CuMnO2–xCeO2 composites after the TGA test up to 800 °C under flowing air are shown in Fig. 5. While CeO2 in these oxygenated CuMnO2–xCeO2 composites still maintained its fluorite-type cubic structure, CuMnO2 was oxidized to CuO and spinel CuyMn3−yO4 oxides, in good agreement with the TGA analysis in Fig. 4 and previous reported results.18 The lattice parameters of CeO2 and spinel CuyMn3−yO4 in the oxygenated CuMnO2 and CuMnO2–xCeO2 composites after the TGA test up to 800 °C in flowing air were refined from the XRD results in Fig. 5 using GSAS software and are summarized in Table 2. The lattice parameters of CeO2 in the oxygenated CuMnO2–xCeO2 composites are a bit smaller than those of the as-prepared CuMnO2–xCeO2 composites (as shown in Table 1), indicating the possible doping of Mn and/or Cu in the lattice of CeO2 after oxidation. In addition, the lattice parameters of spinel CuyMn3−yO4 in the oxygenated CuMnO2–xCeO2 composites are also slightly smaller than those of oxygenated pure CuMnO2, indicating the higher Cu/Mn ratios in the spinel CuyMn3−yO4 in the oxygenated CuMnO2–xCeO2 composites as the smaller ionic radii of Cu2+ than that of Mn2+ would result in a decrease of lattice parameters when copper ions replace manganese ions on tetrahedral sites.
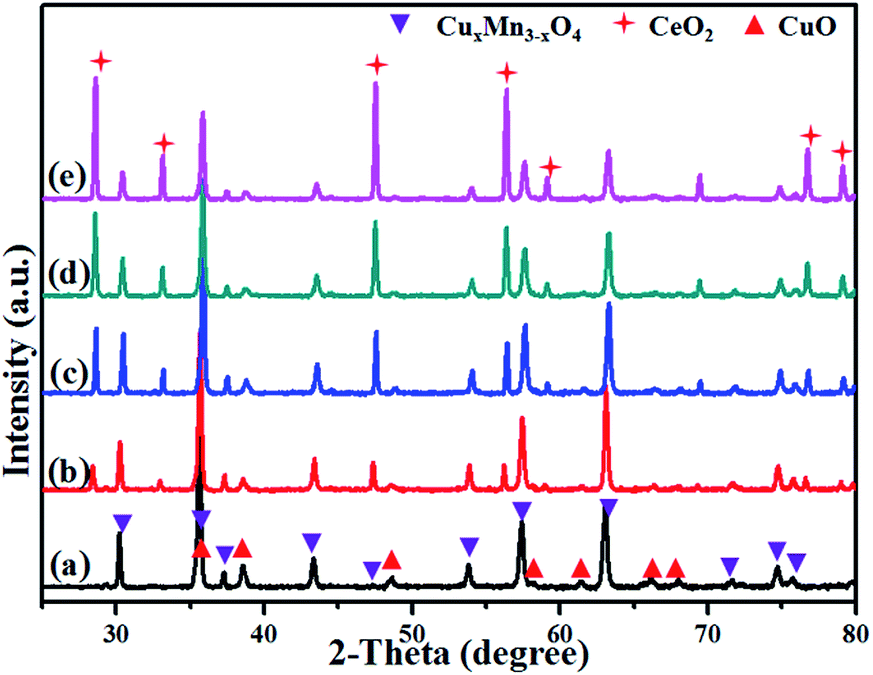 | ||
| Fig. 5 XRD patterns after the TGA test from room temperature to 800 °C under flowing air: (a) CuMnO2, (b) CuMnO2–5CeO2, (c) CuMnO2–10CeO2, (d) CuMnO2–20CeO2, and (e) CuMnO2–40CeO2. | ||
| Oxygenated CuMnO2–xCeO2 | Composition | a (Å) | Cell volume (Å3) |
|---|---|---|---|
| CuMnO2 | CuyMn3−yO4 | 8.317(2) | 575.30(7) |
| CuMnO2–5CeO2 | CuyMn3−yO4 | 8.307(2) | 573.23(4) |
| CeO2 | 5.407(8) | 158.14(7) | |
| CuMnO2–10CeO2 | CuyMn3−yO4 | 8.311(5) | 574.06(3) |
| CeO2 | 5.409(1) | 158.26(1) | |
| CuMnO2–20CeO2 | CuyMn3−yO4 | 8.309(4) | 573.64(9) |
| CeO2 | 5.410(6) | 158.39(3) | |
| CuMnO2–40CeO2 | CuyMn3−yO4 | 8.312(2) | 574.27(1) |
| CeO2 | 5.410(2) | 158.35(8) |
To further check their thermal stability with temperature under flowing air, we treated the as-prepared samples (i.e., post-annealed at 960 °C for 12 h under flowing Ar) from room temperature up to 1000 °C with heating and cooling rates of ±10 °C min−1 under flowing air, as shown in Fig. 6. As discussed in aforementioned paragraphs, the as-prepared samples can start to take up oxygen near 300 °C and reach the maximum values at near 600 °C for CuMnO2–xCeO2 composites and 800 °C for pure CuMnO2, respectively. On further heating, these samples exhibit a steep weight loss from 960 °C accompanied by an endothermic peak in the DTA curves, as shown in Fig. S4.† Such behaviour can be attributed to the transformation of spinels and CuO back into a crednerite Cu1+y′Mn1−y′O2 phase, accompanied by the release of O2.18,32 During the cooling process under flowing air from 1000 °C, there is a sharp continuous weight increase up to around 880 °C, which can be attributed to the re-oxidation of the crednerite phase to CuO and spinel oxides. The slight weight increase from 880 to 600 °C can be attributed to the composition adjustment of spinels with the cooling temperature, as determined by XRD shown in Fig. S5.† Notably, their weights at 1000 °C are higher than their pristine counterparts, suggesting that CuO and spinel CuyMn3−yO4 at high temperature under air can be converted to Cu1+y′Mn1−y′O2 rather than CuMnO2 according to eqn (2).
| 1/(3 − y)CuyMn3−yO4 + (3 − 2y)/(3 − y)CuO → αCux′Mn3−x′O4 + (2 − α)/2Cu1+y′Mn1−y′O2 + [1/(6 − 2y) − α/2]O2 | (2) |
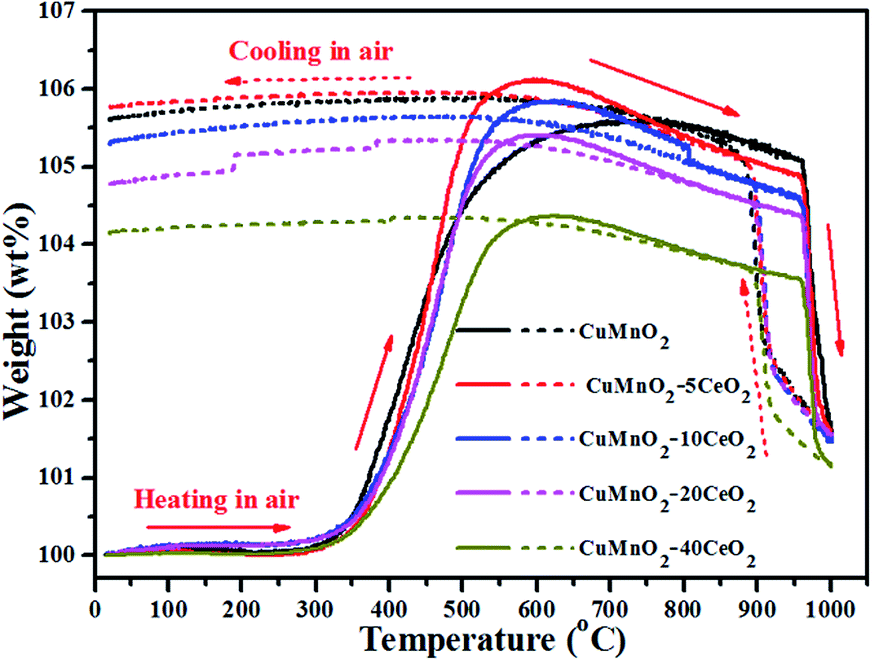 | ||
| Fig. 6 TGA curves of the as-prepared CuMnO2–xCeO2 samples (i.e., post-annealed at 960 °C for 12 h under flowing Ar) from room temperature up to 1000 °C with heating and cooling rates of 10 °C min−1 under flowing air. | ||
The TGA behaviours of the oxygenated CuMnO2–xCeO2 composites (i.e., after the TGA test up to 800 °C under flowing O2) with temperature up to 900 °C under flowing argon are displayed in Fig. 7. The results show that all these samples are stable up to around 600 °C under flowing argon. Further increasing temperature would lead to a continuous weight loss (<1.2 wt%) up to about 790 °C, in which the oxygenated CuMnO2–5CeO2 exhibited the most weight loss and the total weight loss decreased with the increase of CeO2. The small weight loss can be attributed to the removal of oxygen in spinels. At temperatures higher than 790 °C, these sharp weight losses are attributed to the further removal of oxygen to form crednerite Cu1+y′Mn1−y′O2.31 To further support this conclusion, we treated the oxygenated CuMnO2, CuMnO2–5CeO2 and CuMnO2–20CeO2 under flowing argon at 900 °C for 2 h and their XRD patterns shown in Fig. S6† indicate the reformation of the crednerite CuMnO2 structure. The total weight losses for the oxygenated CuMnO2–5CeO2, CuMnO2–10CeO2 and CuMnO2–20CeO2 are higher than that for oxygenated CuMnO2. The enhanced oxygen mobility and storage capacity of the CuMnO2–xCeO2 composite system can be attributed to the synergistic effect between CuMnO2 and CeO2, in which CeO2 can act as an oxygen transfer channel or oxygen promoter dependent on temperature and/or oxygen partial pressures. The possible doping or exsolution of Cu/Mn into or from the lattice of CeO2 on the surface depending on the temperature and oxygen partial pressure may also contribute to the enhanced performance. Therefore, with the surface modification of CeO2, CuMnO2 can take up oxygen from CeO2 rather than directly from the surrounding O2 atmosphere and vice versa, under an inert Ar atmosphere, CuO and spinel CuyMn3−yO4 can release oxygen to the CeO2 lattice and then to the surroundings, rather than directly to the surroundings.
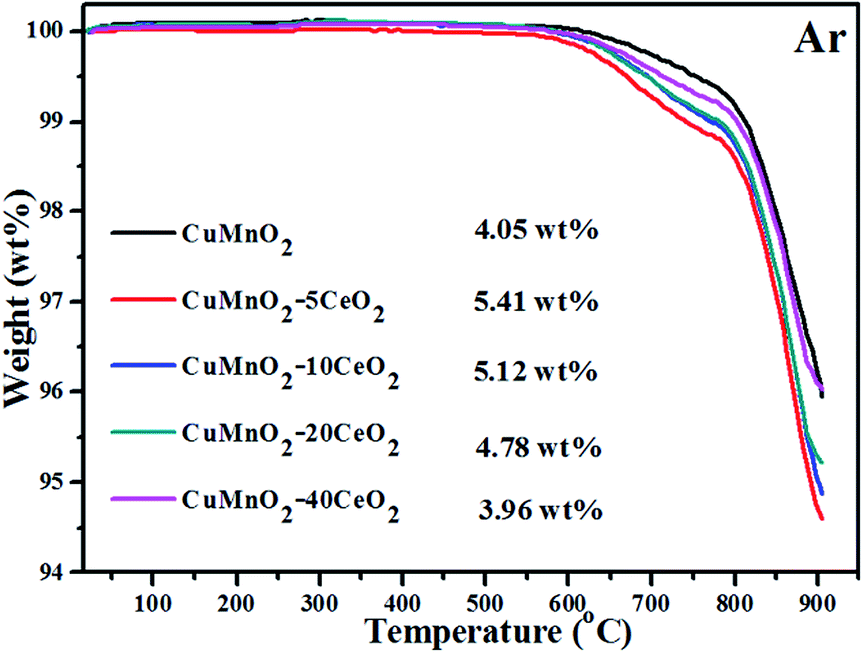 | ||
| Fig. 7 TGA curves of oxidized CuMnO2–xCeO2 samples (i.e., after the TGA test up to 800 °C under flowing O2) from room temperature to 900 °C under flowing argon with 25 mL min−1. | ||
From the TGA data in Fig. 4, it appears that these CeO2-modified CuMnO2 oxides can be oxidized from 300 °C and reach the maximum weight at around 600 °C under flowing O2, while the oxygenated samples can start to release oxygen from 600 °C under flowing argon based on the TGA results in Fig. 7. These results indicate that CeO2-modified CuMnO2 oxides can cause either oxygen uptake or release depending on the temperature and/or oxygen partial pressure. Thus, remarkable oxygen uptake/release behaviours are expected via adjusting the temperature and oxygen partial pressure. As demonstrated in Fig. 8, CuMnO2 and CeO2-modified CuMnO2 oxides can take up/release a large amount of oxygen in which CuMnO2–5CeO2 exhibits the highest OSC (ca. 6.0 wt%) and CuMnO2–20CeO2 shows the best reversibility under the alternating O2 and argon between 300 and 900 °C for four cycles, indicating that CeO2 modification would improve the oxygen storage capacity as well as the reversibility of CuMnO2. The OSC and reversibility would become worse with the increasing cycle times, as shown in Fig. S7,† but the final weight after each treatment under flowing argon during the cycles almost remains the same, indicating that the gradual weight loss may be due to the formation of spinels with less Mn4+ during the oxidation process with the increasing cycling times. As shown in Fig. S8,† after a nine cycle test under alternating O2 and argon, CuMnO2–5CeO2 still contains crednerite CuMnO2 and fluorite CeO2 phases; however, there are obvious CeO2 nanoparticles on the CuMnO2 surface, in comparison to the FESEM images in Fig. S9.† The TEM results for CuMnO2–10CeO2 after the TGA test, as shown in Fig. S10,† also indicate the appearance of CeO2 nanoparticles on the surface of CuMnO2, which resulted in a worse performance for the oxidation of CuMnO2. The TEM elemental mapping results for CuMnO2–10CeO2 in Fig. S10† suggest that some Cu is doped into the lattice of CeO2 while Mn is still in CuMnO2, which can be explained by the bigger ionic radius of Cu2+ (0.73 Å) and Cu+ (0.77 Å) than that of Mn3+ (0.58 Å) and Mn4+ (0.53 Å).
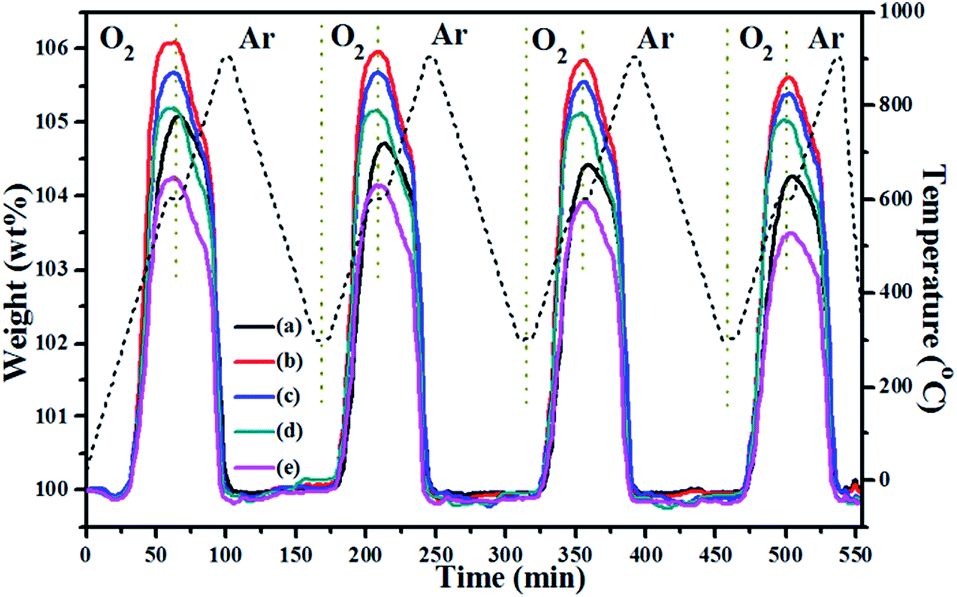 | ||
| Fig. 8 TGA curves of the as-prepared CuMnO2–xCeO2 samples (i.e., post-annealed at 960 °C for 12 h under flowing Ar) under alternating O2 and argon. The samples were first oxidized under O2 to 600 °C, then the gas was switched to argon and the temperature was increased to 900 °C. After being cooled down to 300 °C under argon, the gas was then switched back to O2 and oxidized to 600 °C. | ||
4. Conclusions
In conclusion, we found that fast and reversible oxygen mobility, and increased oxygen storage capacity of crednerite CuMnO2 could be achieved at reduced temperatures (ca. 600 °C) by surface modification with CeO2 at an amount less than 20 mol% in the CuMnO2–CeO2 composite system. The fast and reversible oxygen uptake/release and increased oxygen storage capacity at lower temperatures can be attributed to the synergistic effect of CeO2 as an oxygen diffusion channel between bulk CuMnO2 and the surrounding atmospheres, favouring the reversible formation of spinels at lower temperatures. Our findings reported here could provide a pathway for the design and development of effective oxygen storage materials for a range of energy-synergetic related applications at low temperatures.Acknowledgements
The authors gratefully thank the Engineering and Physical Sciences Research Council (EPSRC) platform grant EP/I022570/1 and EP/I022570/2 for financial support.Notes and references
- H. C. Yao and Y. F. Y. Yao, J. Catal., 1984, 86, 254–265 CrossRef CAS.
- C. E. Hori, H. Permana, K. Y. S. Ng, A. Brenner, K. More, K. M. Rahmoeller and D. Belton, Appl. Catal., B, 1998, 16, 105–117 CrossRef CAS.
- T. Motohashi, Y. Hirano, Y. Masubuchi, K. Oshima, T. Setoyama and S. Kikkawa, Chem. Mater., 2013, 25, 372–377 CrossRef CAS.
- M. Ozawa, M. Kimura and A. Isogai, J. Alloys Compd., 1993, 193, 73–75 CrossRef CAS.
- A. Gupta, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2010, 22, 5184–5198 CrossRef CAS.
- M. P. Yeste, J. C. Hernandez-Garrido, D. C. Arias, G. Blanco, J. M. Rodriguez-Izquierdo, J. M. Pintado, S. Bernal, J. A. Perez-Omil and J. J. Calvino, J. Mater. Chem. A, 2013, 1, 4836–4844 CAS.
- X. Wang, G. Lu, Y. Guo, L. Jiang, Y. Guo and C. Li, J. Mater. Sci., 2009, 44, 1294–1301 CrossRef CAS PubMed.
- Q. Dong, S. Yin, C. Guo, T. Kimura and T. Sato, RSC Adv., 2012, 2, 12770–12774 RSC.
- S. Remsen and B. Dabrowski, Chem. Mater., 2011, 23, 3818–3827 CrossRef CAS.
- O. Parkkima, H. Yamauchi and M. Karppinen, Chem. Mater., 2013, 25, 599–604 CrossRef CAS.
- S. Carter, A. Selcuk, R. J. Chater, J. Kajda, J. A. Kilner and B. C. H. Steele, Solid State Ionics, 1992, 53–56, 597–605 CrossRef CAS.
- M. Cherry, M. S. Islam and C. R. A. Catlow, J. Solid State Chem., 1995, 118, 125–132 CrossRef CAS.
- J. W. Lekse, S. Natesakhawat, D. Alfonso and C. Matranga, J. Mater. Chem. A, 2014, 2, 2397–2404 CAS.
- H. Jeen, W. S. Choi, M. D. Biegalski, C. M. Folkman, I. C. Tung, D. D. Fong, J. W. Freeland, D. Shin, H. Ohta, M. F. Chisholm and H. N. Lee, Nat. Mater., 2013, 12, 1057–1063 CrossRef CAS PubMed.
- S. Kato, R. Fujimaki, M. Ogasawara, T. Wakabayashi, Y. Nakahara and S. Nakata, Appl. Catal., B, 2009, 89, 183–188 CrossRef CAS PubMed.
- S. Kato, H. Sato, M. Ogasawara, T. Wakabayashi, Y. Nakahara and S. Nakata, Solid State Sci., 2012, 14, 177–181 CrossRef CAS PubMed.
- S. Kato, S. Suzuki, R. Kawashima, M. Ogasawara, T. Wakabayashi, Y. Nakahara and S. Nakata, J. Mater. Sci., 2013, 48, 8077–8083 CrossRef CAS.
- M. Trari, J. Töpfer, P. Dordor, J. C. Grenier, M. Pouchard and J. P. Doumerc, J. Solid State Chem., 2005, 178, 2751–2758 CrossRef CAS PubMed.
- Y. Bessekhouad, M. Trari and J. P. Doumerc, Int. J. Hydrogen Energy, 2003, 28, 43–48 CrossRef CAS.
- S. A. M. Abdel-Hameed, F. H. Margha and A. A. El-Meligi, Int. J. Energy Res., 2014, 38, 459–465 CrossRef CAS PubMed.
- Y. Bessekhouad, Y. Gabes, A. Bouguelia and M. Trari, J. Mater. Sci., 2007, 42, 6469–6476 CrossRef CAS PubMed.
- J. Topfer, M. Trari, P. Gravereau, J. P. Chaminade and J. P. Doumerc, Z. Kristallogr., 1995, 210, 184–187 CrossRef.
- F. C. M. Driessens and G. D. Rieck, Z. Anorg. Allg. Chem., 1967, 351, 48–62 CrossRef CAS PubMed.
- P. Wei, M. Bieringer, L. D. Cranswick and A. Petric, J. Mater. Sci., 2010, 45, 1056–1064 CrossRef CAS.
- J. Li, S. Xiong, X. Li and Y. Qian, Nanoscale, 2013, 5, 2045–2054 RSC.
- N. K. Radhakrishnan and A. B. Biswas, Phys. Status Solidi A, 1977, 44, 45–49 CrossRef CAS PubMed.
- D. P. Shoemaker, J. Li and R. Seshadri, J. Am. Chem. Soc., 2009, 131, 11450–11457 CrossRef CAS PubMed.
- H. Y. Chen and D. J. Hsu, J. Alloys Compd., 2014, 598, 23–26 CrossRef CAS PubMed.
- I. N. Dubrovina, V. F. Balakirev and A. V. Antonov, Inorg. Mater., 2001, 37, 76–81 CrossRef CAS.
- E. Ríos, S. Abarca, P. Daccarett, H. Nguyen Cong, D. Martel, J. F. Marco, J. R. Gancedo and J. L. Gautier, Int. J. Hydrogen Energy, 2008, 33, 4945–4954 CrossRef PubMed.
- A. P. Amrute, Z. Łodziana, C. Mondelli, F. Krumeich and J. Pérez-Ramírez, Chem. Mater., 2013, 25, 4423–4435 CrossRef CAS.
- B. Bellal, B. Hadjarab, N. Benreguia, Y. Bessekhouad and M. Trari, J. Appl. Electrochem., 2011, 41, 867–872 CrossRef CAS.
- Z. Liu, Z. Wu, X. Peng, A. Binder, S. Chai and S. Dai, J. Phys. Chem. C, 2014, 118, 27870–27877 CAS.
- L. Shi, W. Chu, F. Qu and S. Luo, Catal. Lett., 2007, 113, 59–64 CrossRef CAS.
- A. Hornés, A. B. Hungría, P. Bera, A. L. Cámara, M. Fernández-García, A. Martínez-Arias, L. Barrio, M. Estrella, G. Zhou, J. J. Fonseca, J. C. Hanson and J. A. Rodriguez, J. Am. Chem. Soc., 2009, 132, 34–35 CrossRef PubMed.
- S. D. Senanayake, D. Stacchiola and J. A. Rodriguez, Acc. Chem. Res., 2013, 46, 1702–1711 CrossRef CAS PubMed.
- I. Moog, C. Feral-Martin, M. Duttine, A. Wattiaux, C. Prestipino, S. Figueroa, J. Majimel and A. Demourgues, J. Mater. Chem. A, 2014, 2, 20402–20414 CAS.
- E. Y. Konysheva, S. M. Francis, J. T. S. Irvine, A. Rolle and R.-N. Vannier, J. Mater. Chem., 2011, 21, 15511–15520 RSC.
- E. Konysheva, R. Blackley and J. T. S. Irvine, Chem. Mater., 2010, 22, 4700–4711 CrossRef CAS.
- A. S. Ivanova, Kinet. Catal., 2009, 50, 797–815 CrossRef CAS.
- Y. Zhou and M. N. Rahaman, Acta Mater., 1997, 45, 3635–3639 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional XRD, TGA/DTA, HRTEM and FESEM images as well as TEM elemental mappings. See DOI: 10.1039/c5ta01361e |
| This journal is © The Royal Society of Chemistry 2015 |