
Ruthenium-catalyzed alkoxycarbonylation of alkenes using carbon monoxide†
Lipeng
Wu
,
Qiang
Liu
,
Ralf
Jackstell
and
Matthias
Beller
*
Leibniz-Institut für Katalyse an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
First published on 16th April 2015
Abstract
A novel protocol for ruthenium-catalyzed methoxycarbonylation reaction of alkenes is reported. Using easily available Ru3(CO)12 as catalyst in the presence of [Bmim]Cl as additive, industrial important esters are produced in yields up to 82%. Compared to traditional olefin carbonylation processes key to success is running the reaction at low carbon monoxide concentration. Notably, the ionic liquid phase which contains the catalyst can be reused for several runs.
Carbonylation reactions of alkenes represent an important basis of today's chemical industry.1 Diverse products can be produced as final products or intermediates for the production of detergents, plasticizers, lubricants, synthetic fibers, solvents and so on.2–4 In practice, most of the procedures make use of precious transition-metals, for example, rhodium complexes represent the state-of-the-art catalysts in the hydroformylation of lower aliphatic alkenes.5 Besides, in the methoxycarbonylation of olefins, palladium-based systems show the best performance (Scheme 1a and b).6–8 In order to reduce the production costs in carbonylation reactions, an alternative strategy makes use of relatively inexpensive metals as catalysts.9 Among all the noble metal catalysts, ruthenium is the least expensive one and about 20 times lower in price than rhodium and palladium.10–14
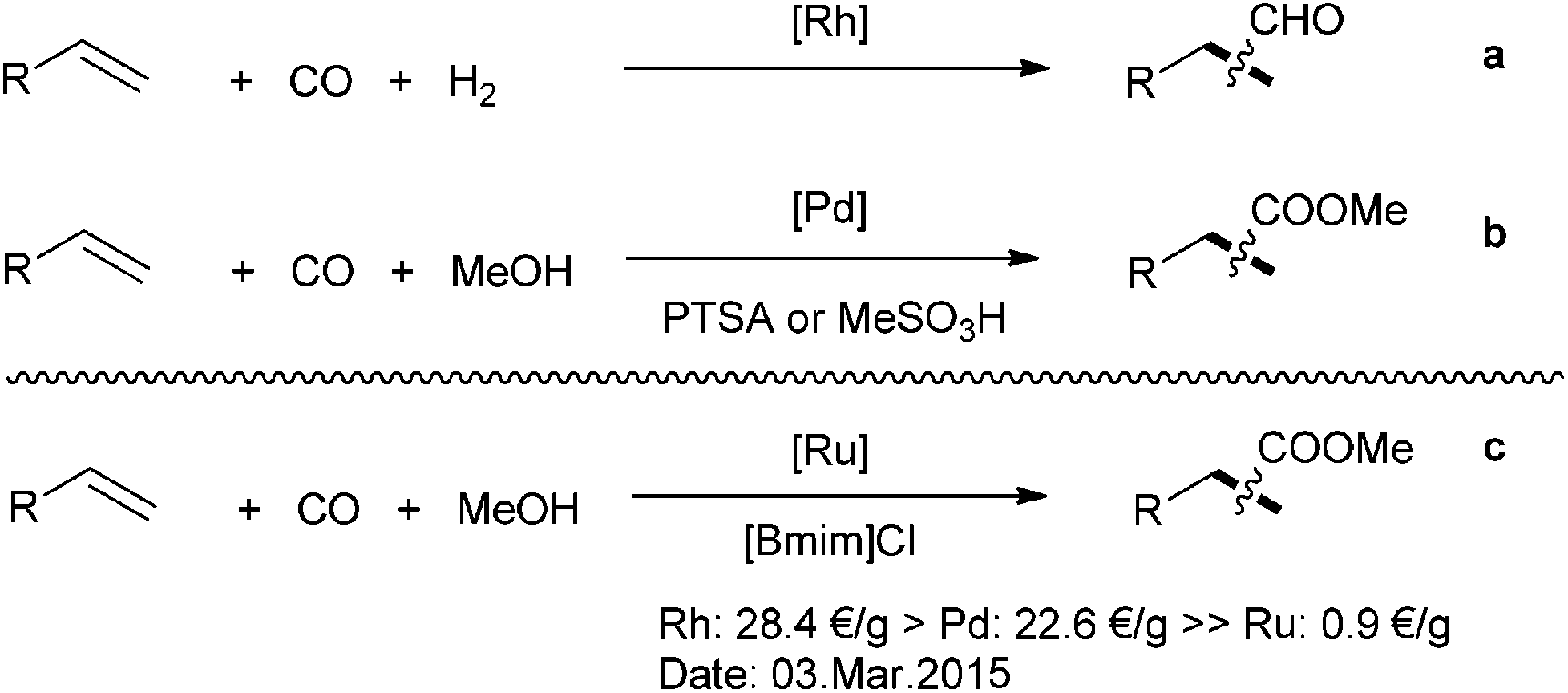 | ||
| Scheme 1 Selected examples of alkene carbonylation reactions. (PTSA = p-toluenesulfonic acid, [Bmim]Cl = 1-butyl-3-methylimidazolium chloride). | ||
Hence, it is of significant interest to study the performance of ruthenium complexes in carbonylation reactions. In this respect, recently such catalysts demonstrated interesting potential.15 For example, Nozaki and co-workers reported in 2012 the application of [Cp*Ru] complexes for hydroformylations.16 A ruthenium-catalyzed hydroformylation–acetalization reaction of olefins was presented by the group of Börner.17 Our group also developed novel ruthenium-catalyzed hydroformylation and related tandem reactions.18–20 In this latter cases, the use of special imidazole-substituted phosphine ligands was essential for achieving high yields and regioselectivites.21–23 Besides, we also developed the ruthenium-catalyzed methoxycarbonylation of alkenes with paraformaldehyde and carbon dioxide as C1 building blocks.24–27 In these works we observed that carbon monoxide is in situ generated,28,29 which allows for subsequent carbonylation.30–32 Apparently, the reaction takes place smoothly at low concentration of carbon monoxide. This finding inspired us to study ruthenium-catalyzed carbonylations of olefin at low CO pressure. To the best of our knowledge no systematic investigation has been performed before. Notably, many ruthenium-catalyzed alkoxycarbonylation reactions applying alkyl formates33–37 and alkenes have been reported, which probably make use of the same principle.38–40
Here, we present the first ruthenium-catalyzed methoxy-carbonylation reaction of alkenes with carbon monoxide (Scheme 1c). Compared to the known reactions using alkyl formates, no pre-installation of alcohol step is needed in this transformation.
As a starting point of our studies, we chose the reaction of cyclohexene (1a), carbon monoxide (2 bar), methanol (2a) in the presence of 1 mol% of Ru3(CO)12 to produce methyl cyclohexanecarboxylate (3a) at 130 °C as the model reaction. Initially, we investigated the effect of the additives. Due to the low boiling point of the substrates and methanol, we also introduced 40 bar of nitrogen to increase the pressure inside the reactor and to dilute the carbon monoxide. Selected results are summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additives | Additive amount [mol%] | Conversionb [%] | Yieldb [%] | |
| 3a | 3a′ | ||||
| a Reaction conditions: cyclohexene 2.0 mmol, Ru3(CO)12 1 mol%, CO 2 bar, N2 40 bar, methanol 2 mL, 130 °C, 48 h. b Data was determined by GC analysis using 0.4 mL isooctane as internal standard. c Ru3(CO)12 0.5 mol%. d Ru(methylallyl)2(COD) 3 mol%. e [RuCl2(p-cymene)]2 1.5 mol%. f 110 °C. g 40 bar CO. | |||||
| 1 | — | — | — | — | — |
| 2 | PTSA | 4 | 51 | 2 | 48 |
| 3 | AcOH | 4 | 0 | — | — |
| 4 | [Bmim]Cl | 10 | 27 | 9 | — |
| 5 | [Bmim]Cl | 100 | 49 | 41 | — |
| 6 | [Bmim]Cl | 200 | 76 | 70 | — |
| 7c | [Bmim]Cl | 200 | 33 | 31 | — |
| 8 | LiCl | 200 | 70 | 61 | 4 |
| 9d | [Bmim]Cl | 200 | 47 | 24 | — |
| 10e | [Bmim]Cl | 200 | 22 | 3 | 8 |
| 11f | [Bmim]Cl | 200 | — | — | — |
| 12g | [Bmim]Cl | 200 | — | — | — |

Cyclohexene was not consumed at all without any additives (Table 1, entry 1). In the presence of PTSA, though 51% of conversion was achieved, most of the cyclohexene was reduced to cyclohexane (Table 1, entry 2). In the presence of acetic acid as an example of a weaker acid, again there was no conversion (Table 1, entry 3). The fact that Cl− is pivotal in the methoxycarbonylation reaction using carbon dioxide and paraformaldehyde promoted us to add 10 mol% of [Bmim]Cl in the reaction solution. Here, in fact 9% of the desired ester 3a was produced (Table 1, entry 4). It is proposed that the presence of Cl− is crucial for the formation of [Ru3(Cl)(CO)12−n]− (n = 1–3) species.41 Apparently, the strong anti-effect of Cl− facilitate the CO dissociation and alkene association. As expected the amount of [Bmim]Cl is relevant. Increasing [Bmim]Cl from 10 to 200 mol% enhanced the product yield gradually from 9 to 70% (Table 1, entries 5–6). Other Cl− sources like LiCl gave slightly inferior results (Table 1, entry 8). Reducing the catalyst loading led to lower yield (31%) (Table 1, entry 7). Interestingly, other ruthenium pre-catalysts, e.g. Ru(methylallyl)2(COD) and [RuCl2(p-cymene)]2 also failed to promote this transformation (Table 1, entries 9–10). A minimum reaction temperature of 130 °C proved crucial since at 110 °C there was no conversion of the cyclohexene observed (Table 1, entry 11). In order to show clearly the effect of the carbon monoxide concentration, the model reaction was performed at higher pressure, too. As expected using 40 bar of carbon monoxide, no reaction at all occurred (Table 1, entry 12).
Next, the optimized conditions were applied to various alkenes (Scheme 2). Apart from cyclohexene, other cyclic olefins, e.g. cyclopentene gave the ester 3b in good yield (66%) as well. Olefins with larger rings like cyclooctene reacted not well and the corresponding carboxylic acid ester 3c was only obtained in low yield (23%). On the other hand, norbornene reacted well to deliver 3d in 65% yield. The reaction of 3,3-dimethylbut-1-ene gave highly selectively the corresponding terminal ester 3e in good yield (82%). Linear aliphatic alkenes such as allylbenzene and allylcyclohexane worked similar to give the corresponding esters 3f and 3g in 72% and 75% yield, respectively. Regioselectivities around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were observed with all the linear aliphatic alkenes. Starting from 1-octene we obtained 59% of ester 3h. The lower regioselectivity is explained by fast isomerization of 1-octene in the presence of ruthenium carbonyl complexes. With 2-octene only 29% of ester 3h was produced with somewhat lower regioselectivity. The slower isomerization of 2-octene to 1-octene takes place too, which gives the normal isomer via the following carbonylation process. However, the product yield can be improved to 48% using higher reaction temperature, though the regioselectivity retained the same.
:1 were observed with all the linear aliphatic alkenes. Starting from 1-octene we obtained 59% of ester 3h. The lower regioselectivity is explained by fast isomerization of 1-octene in the presence of ruthenium carbonyl complexes. With 2-octene only 29% of ester 3h was produced with somewhat lower regioselectivity. The slower isomerization of 2-octene to 1-octene takes place too, which gives the normal isomer via the following carbonylation process. However, the product yield can be improved to 48% using higher reaction temperature, though the regioselectivity retained the same.
 | ||
| Scheme 2 Ruthenium-catalyzed methoxycarbonylation: variation of alkenes. (Reaction conditions: alkenes 2.0 mmol, Ru3(CO)12 1 mol%, CO 2 bar, N2 40 bar, methanol 2 mL, 130 °C, 48 h; yield and regioselectivity were determined by GC analysis using 0.4 mL isooctane as internal standard, number in parameter are isolated yield; only linear products are shown). | ||
Then, applying cyclohexene 1a as starting material, the scope of diverse alcohols 2 was studied as well (Scheme 3). The present catalyst system transformed both lower and higher primary aliphatic alcohols into the corresponding esters in good yields (3a, 4a–c). The reaction yields were slightly decreased with the extension of the carbon chain. Aliphatic alcohols with aryl group worked well and yielded esters 4d–e in 68% and 70%, respectively. More complex alcohol with furyl-group was also tolerated in this transformation. Besides, secondary alcohol-isopropanol was successfully used to produce 4g in 61% yield.
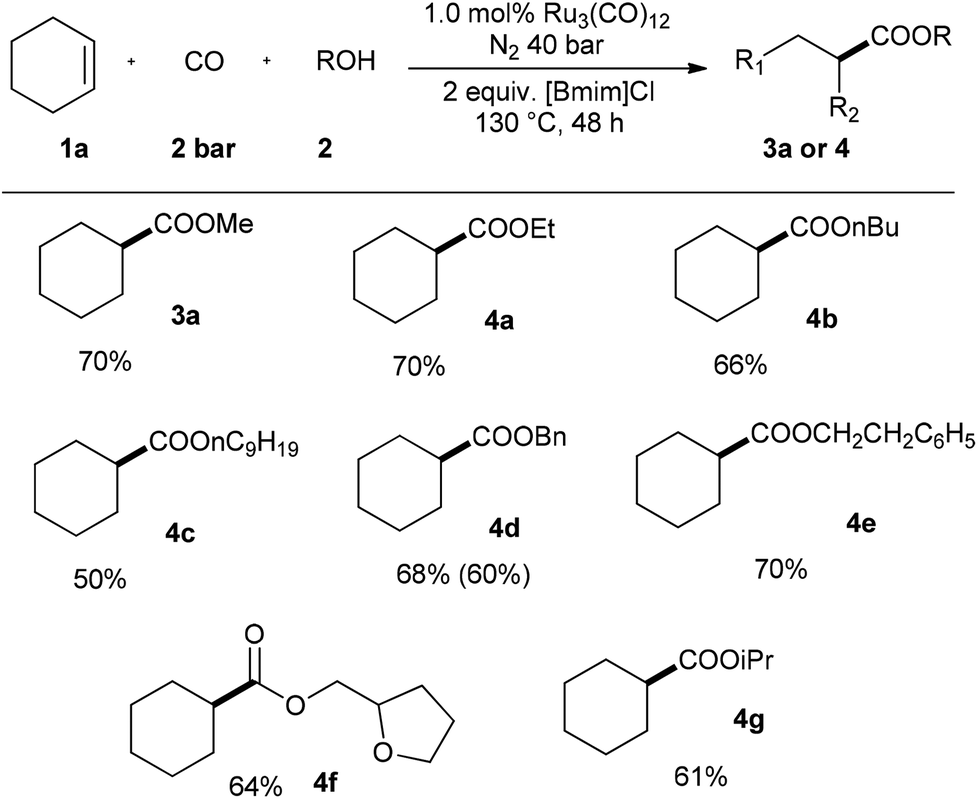 | ||
| Scheme 3 Ruthenium-catalyzed alkoxycarbonylation: variation of alcohols. (Reaction conditions: cyclohexene 2.0 mmol, Ru3(CO)12 1 mol%, CO 2 bar, N2 40 bar, alcohols 2 mL, 130 °C, 48 h; yield were determined by GC analysis using 0.4 mL isooctane as internal standard, number in parameter are isolated yield). | ||
Finally, in order to illustrate the stability of the catalyst system in this transformation, recycling tests were carried out via adding a new portion of alkene, gas and methanol after each run (Table 2). To our delight, the catalyst was recyclable although slightly lower efficiency was observed in the next cycle. Notably, the ionic phase containing catalyst is air-stable. Hence, even after exposure of the reaction solution to air for 5 days, the isolated ionic phase was still active for this transformation and gave the ester in 55% yield.
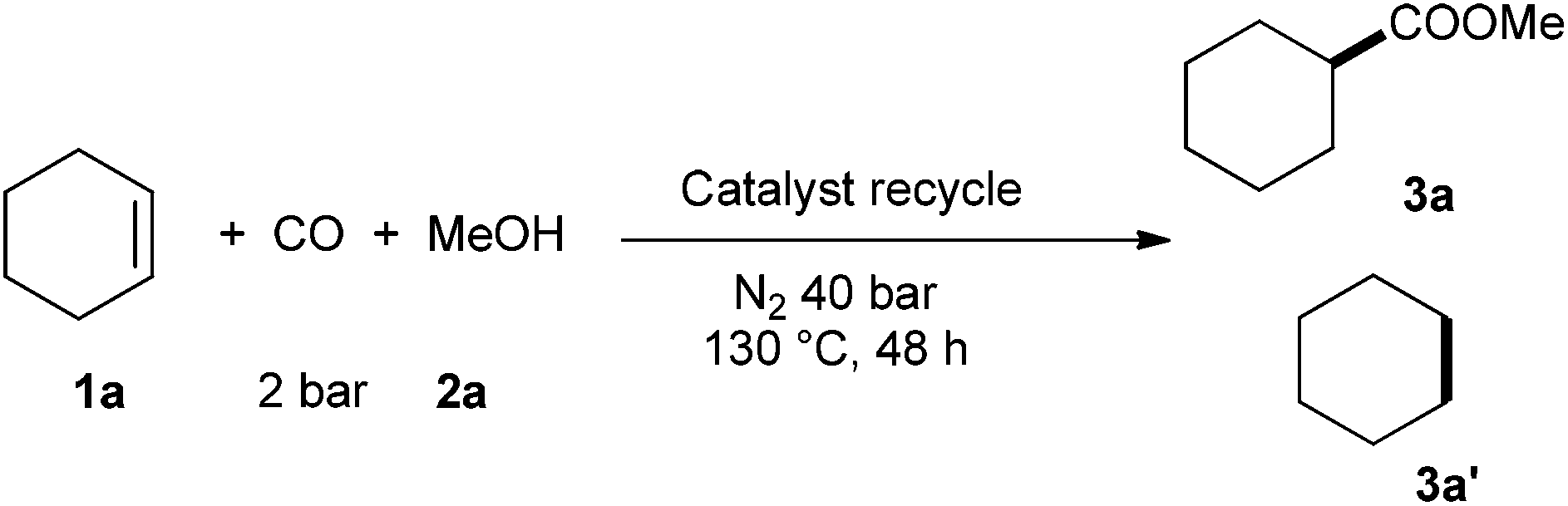
Conclusions
Using easily available Ru3(CO)12 as pre-catalyst in the presence of [Bmim]Cl as additive the hydroesterification of alkenes with alcohols proceeds smoothly at low carbon monoxide concentration. This process constitutes the first example of a ruthenium-catalyzed olefin alkoxycarbonylation using simply carbon monoxide as carbonyl source. Industrial relevant esters are produced in medium to high yields up to 82% at relatively low temperature. Notably, the ionic liquid phase which contains the catalyst can be reused for several runs, and was stable even after 5 days exposed to air.We are grateful to the funding support from the State of Mecklenburg-Vorpommern, BMBF, Alexander von Humboldt Foundation (grants for Q.L.) and the analytic department in Likat.
Notes and references
- W. Bertleff, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed.
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- E. V. Gusevskaya, J. Jiménez-Pinto and A. Börner, ChemCatChem, 2014, 6, 363–363 CrossRef PubMed.
- P. W. N. M. Van Leeuwen and C. Claver, Rhodium Catalyzed Hydroformylation, Springer, Berlin Heidelberg, 2008 Search PubMed.
- C. Jimenez Rodriguez, D. F. Foster, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2004, 1720–1721 RSC.
- I. del Río, C. Claver and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 2001, 2001, 2719–2738 CrossRef.
- C. Godard, A. Ruiz and C. Claver, Helv. Chim. Acta, 2006, 89, 1610–1622 CrossRef CAS PubMed.
- J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- L. M. Geary, B. W. Glasspoole, M. M. Kim and M. J. Krische, J. Am. Chem. Soc., 2013, 135, 3796–3799 CrossRef CAS PubMed.
- T. Smejkal, H. Han, B. Breit and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 10366–10367 CrossRef CAS PubMed.
- S. Gülak, L. Wu, Q. Liu, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 7320–7323 CrossRef PubMed.
- A. Behr, U. Kanne and W. Keim, J. Mol. Catal., 1986, 35, 19–28 CrossRef CAS.
- A. Tlili, J. Schranck, J. Pospech, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 6293–6297 CrossRef CAS PubMed.
- K. Takahashi, M. Yamashita, Y. Tanaka and K. Nozaki, Angew. Chem., Int. Ed., 2012, 51, 4383–4387 CrossRef CAS PubMed.
- J. Norinder, C. Rodrigues and A. Börner, J. Mol. Catal. A: Chem., 2014, 391, 139–143 CrossRef CAS PubMed.
- I. Fleischer, L. Wu, I. Profir, R. Jackstell, R. Franke and M. Beller, Chem. – Eur. J., 2013, 19, 10589–10594 CrossRef CAS PubMed.
- L. Wu, I. Fleischer, R. Jackstell, I. Profir, R. Franke and M. Beller, J. Am. Chem. Soc., 2013, 135, 14306–14312 CrossRef CAS PubMed.
- L. Wu, I. Fleischer, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2013, 135, 3989–3996 CrossRef CAS PubMed.
- T. Schulz, C. Torborg, B. Schaffner, J. Huang, A. Zapf, R. Kadyrov, A. Borner and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 918–921 CrossRef CAS PubMed.
- D. B. Grotjahn, C. R. Larsen, J. L. Gustafson, R. Nair and A. Sharma, J. Am. Chem. Soc., 2007, 129, 9592–9593 CrossRef CAS PubMed.
- D. B. Grotjahn, C. D. Incarvito and A. L. Rheingold, Angew. Chem., Int. Ed., 2001, 40, 3884–3887 CrossRef CAS.
- L. Wu, Q. Liu, I. Fleischer, R. Jackstell and M. Beller, Nat. Commun., 2014, 5 CAS.
- T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 CrossRef CAS PubMed.
- L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310–6320 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, ChemCatChem, 2014, 6, 2805–2809 CrossRef CAS PubMed.
- P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061–6071 CrossRef CAS PubMed.
- C. Brancour, T. Fukuyama, Y. Mukai, T. Skrydstrup and I. Ryu, Org. Lett., 2013, 15, 2794–2797 CrossRef CAS PubMed.
- K. Tsuchiya, J.-D. Huang and K.-I. Tominaga, ACS Catal., 2013, 3, 2865–2868 CrossRef CAS.
- K.-i. Tominaga and Y. Sasaki, J. Mol. Catal. A: Chem., 2004, 220, 159–165 CrossRef CAS PubMed.
- Q. Liu, L. Wu, I. Fleischer, D. Selent, R. Franke, R. Jackstell and M. Beller, Chem. – Eur. J., 2014, 20, 6809–6809 CrossRef CAS PubMed.
- N. Lugan, G. Lavigne, J. M. Soulie, S. Fabre, P. Kalck, J. Y. Saillard and J. F. Halet, Organometallics, 1995, 14, 1712–1731 CrossRef CAS.
- S. Fabre, P. Kalck and G. Lavigne, Angew. Chem., Int. Ed. Engl., 1997, 36, 1092–1095 CrossRef CAS PubMed.
- H. Konishi, T. Ueda, T. Muto and K. Manabe, Org. Lett., 2012, 14, 4722–4725 CrossRef CAS PubMed.
- S. Ko, Y. Na and S. Chang, J. Am. Chem. Soc., 2002, 124, 750–751 CrossRef CAS PubMed.
- N. Armanino, M. Lafrance and E. M. Carreira, Org. Lett., 2013, 16, 572–575 CrossRef PubMed.
- Y. Katafuchi, T. Fujihara, T. Iwai, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2011, 353, 475–482 CrossRef CAS PubMed.
- T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 CrossRef CAS PubMed.
- I. Fleischer, R. Jennerjahn, D. Cozzula, R. Jackstell, R. Franke and M. Beller, ChemSusChem, 2013, 6, 417–420 CrossRef CAS PubMed.
- G. Lavigne, Eur. J. Inorg. Chem., 1999, 1999, 917–930 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure and esters characterization data. See DOI: 10.1039/c5qo00071h |
| This journal is © the Partner Organisations 2015 |