
Cu-catalyzed aerobic oxidative amidation of aryl alkyl ketones with azoles to afford tertiary amides via selective C–C bond cleavage†
Wen
Ding
a and
Qiuling
Song
*ab
aInstitute of Next Generation Matter Transformation, College of Chemical Engineering at Huaqiao University, 668 Jimei Boulevard, Xiamen, Fujian 361021, P. R. China. E-mail: qsong@hqu.edu.cn
bBeijing National Laboratory for Molecular Sciences, Beijing 100190, China
First published on 5th May 2015
Abstract
Chemoselective cleavage of the C(CO)–C(alkyl) bond in aryl ketones leading to azole amides is disclosed with a broad substrate scope. Aryl ketones with a variety of long-chain alkyl groups have been demonstrated to be active substrates and mechanism studies suggested that molecular oxygen serves both as an oxidant and a reactant in this strategy.
Selective cleavage of carbon–carbon bonds is a long standing theme and a holy grail in synthetic organic chemistry and has attracted great attention over the past few decades.1 Among carbon–carbon bonds, selective cleavage of the C–C single bond is more challenging due to the following reasons: (1) C–C single bonds are more stable, thus are inert to most reaction conditions,2 (2) C–C single bonds are less polar,3 (3) C–C single bonds have weaker coordination to metal catalysts, making them difficult to be activated,4 (4) C–C single bonds widely exist in one molecule, making the selective cleavage difficult.5 Currently, there are three strategies to cleave the C–C single bonds: (1) design special substrates, such as employing strained structure scaffolds like three- and four-membered rings, to lower the C–C single bond stability;6 (2) prefunctionalize substrates to increase C–C single bond reactivities, such as preinstalling some activating groups adjacent to the C–C single bonds,7 and (3) develop suitable catalytic systems to activate C–C single bonds by decreasing their activation energy.8 Although great advances have been achieved in cleavage of C–C single bonds, the harsh reaction conditions, expensive and toxic transition-metal catalysts, stoichiometric oxidants (especially peroxides) and limited substrate scope make it highly desirable to develop more efficient and sustainable protocols to cleave unstrained C–C single bonds.
Aryl ketones are among the ubiquitous structural motifs in organic compounds and the direct manipulation of ketones is one of the most versatile transformations in organic synthesis.9 In very recent times, a few elegant examples of aerobic oxidative C(CO)–C(alkyl) bond cleavage of ketones have been reported.10–12 Despite significant progress in the area of aerobic oxidative C(CO)–C(alkyl) bond cleavage in aryl ketones, the direct aerobic oxidative tertiary amide formation through C(CO)–C(alkyl) single bond cleavage with molecular oxygen as the oxidant has not been reported to date. Meanwhile, aryl ketones with a wide variety of long-chain groups are either inert or have not been reported in the above reported (CO)–C(alkyl) single bond cleavage reactions yet; therefore, activation of such aryl ketones under dioxygen is a big challenge and a fascinating area for the synthetic community.
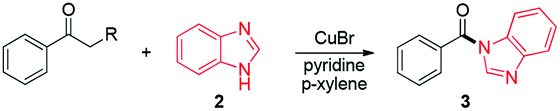
Azoles, particularly benzimidazoles, are prevalent scaffolds in a broad spectrum of bioactive natural products and pharmaceutically active molecules.13 Therefore, it is highly desirable to develop novel and practical methods for the construction of functionalized azoles. Traditionally, acylation of azoles was performed either with free carboxylic acids or with acyl chloride.14 In the former process, preactivation of the free carboxylic acids is always required with stoichiometric activating reagents, and in the latter reaction corrosive byproducts are often generated during the reaction. Recently, a metal-free N-acylation of azole was reported from aldehydes and azoles.15 However, an excess amount of peroxides was required for the success of the transformation. Molecular oxygen is an ideal oxidant owing to its well-known sustainable properties.16 Acylation of azoles via aerobic oxidative C(CO)–C single bond cleavage with aryl ketones has not been reported thus far. As part of our ongoing interest in copper catalyzed aerobic oxidative C–C bond activation reaction,17 herein we would like to report a copper-catalyzed aerobic oxidative amidation between aryl alkyl ketones and azoles. Azole tertiary amides were found to be formed with molecular oxygen as the sole terminal oxidant in good yields with a broad substrate scope. Notably, numerous aryl ketones with a wide variety of long-chain groups have been demonstrated to be active substrates in this novel strategy, thus greatly broadening the substrate scope (Scheme 1).
 | ||
| Scheme 1 Azole amide formation from various starting substrates. | ||
At the outset, acetophenone (1) and benzimidazole (2) were selected as substrates in model reactions. As we can see, copper salts demonstrated good activities in this novel transformation (for details see the ESI†) and 76% of the desired product was obtained with the superb CuBr with pyridine (3 equiv.) at 130 °C in 1 mL of p-xylene under O2 in a sealed tube (Table 1, entry 2). When the solvent was changed to DMSO, DMF or toluene, the yield of the desired product was dramatically decreased to 33% with toluene (entry 3) and no desired product was detected with DMSO and DMF.18 Ligand screening indicated that pyridine is the superior choice to 2-phenyl-pyridine, 2-methyl-pyridine, 2-amino-pyridine, Et3N and DMEDA.18 When the temperature was decreased to 120 °C and 110 °C, the yields of the desired product 3 were dropped to 22% and 4% correspondingly (entries 6–7). Copper salt, pyridine and dioxygen were all essential for this transformation, which have been indicated by entries 8–11: without CuBr, no desired product was ever detected (entry 8); without pyridine, the yield of the desired product was reduced to 33% with other unknown compounds generated (entry 9); when the reaction was conducted under an N2 atmosphere or in air, only a trace or a small amount of the product was detected (entries 10 and 11). Furthermore, ligand and catalyst loading and fine tuning (entries 12–15) revealed that the optimal reaction conditions for 3 were acetophenone 1 (0.25 mmol), benzimidazole 2 (0.5 mmol), CuBr (10 mol%), pyridine (3 equivalents), p-xylene (1 mL) at 130 °C under oxygen (entry 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Atmosphere | Ligand (equiv.) | Temperature | Solvent | Yield of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a Reaction conditions: acetophenone (1, 0.25 mmol), benzoimidazole (2, 0.5 mmol), Cu salt (10 mol%), ligand, solvent (1 mL), 16 h, temp. 130 °C, corresponding atmosphere. b GC yield. c Isolated yield. d Acetophenone (1, 0.25 mmol), benzoimidazole (2, 0.25 mmol). | ||||||
| 1 | CuCl (10) | O2 | Pyridine (3) | 130 °C | p-Xylene | 16 |
| 2 | CuBr (10) | O2 | Pyridine (3) | 130 °C | p-Xylene | 76c |
| 3 | CuBr (10) | O2 | Pyridine (3) | 130 °C | Toluene | 38 |
| 4 | CuBr (10) | O2 | 2-Methyl-py (3) | 130 °C | p-Xylene | 33 |
| 5 | CuBr (10) | O2 | Pyridine (0.5) | 130 °C | p-Xylene | Trace |
| 6 | CuBr (10) | O2 | Pyridine (3) | 120 °C | p-Xylene | 22 |
| 7 | CuBr (10) | O2 | Pyridine (3) | 110 °C | p-Xylene | 4 |
| 8 | — | O2 | Pyridine (3) | 130 °C | p-Xylene | No product |
| 9 | CuBr (10) | O2 | — | 130 °C | p-Xylene | 33 |
| 10 | CuBr (10) | N2 | Pyridine (3) | 130 °C | p-Xylene | No product |
| 11 | CuBr (10) | Air | Pyridine (3) | 130 °C | p-Xylene | 11 |
| 12 | CuBr (10) | O2 | Pyridine (2) | 130 °C | p-Xylene | 74 |
| 13 | CuBr (10) | O2 | Pyridine (4) | 130 °C | p-Xylene | 64 |
| 14 | CuBr (20) | O2 | Pyridine (3) | 130 °C | p-Xylene | 4 |
| 15d | CuBr (10) | O2 | Pyridine (3) | 130 °C | p-Xylene | 39c |

To explore the scope of this transformation, a variety of acetophenones and azoles were applied under the optimal conditions. As shown in Scheme 2, acetophenone derivatives with H, alkyl substituents and halo substituents at the para position performed well, giving the desired products in moderate to good yields (3–12) and among them, halo substituted desired products provided the feasibility for further structural manipulation (10–12). Electron-donating groups like methoxy, methylthiol and bis-benzyloxyl substituents on the aromatic rings also indicated good reactivity (13–15). Polyphenylene substrates, such as 1-acetonaphthone, 2-acetonaphthone, 4-phenylacetophenone and 4-phenylethynylacetophenone, were all tolerated under the standard conditions to afford the corresponding desired products in good yields (16–19). Moreover, other aromatic rings, such as 3-thiophene methyl ketone, were compatible in this reaction as well (20). Bis-substituted benzimidazole was also found to be tolerated for this transformation to give the desired product in 68% yield (21). Intrigued by the above results, we further investigated the azole scope by extending benzimidazoles to other heterocycles. 3-Phenyl pyrazole and a variety of indazoles were found to show good reactivities in this transformation (22–29). Notably, indazoles could react very well with acetophenones which bear strong electron-withdrawing groups on the aromatic rings to afford the corresponding desired products in good to excellent yields (25–27). Meanwhile, heteroaromatic methyl ketones, for example benzofuranylethanones reacted well with indazoles to generate the desired product 28 in 52% yield. All the above results have demonstrated that a very broad scope of (hetero)aryl methyl ketones and azoles are suitable substrates for the copper-catalyzed aerobic oxidative C(CO)–C single bond cleavage reaction, which infers that our method might be an efficient and practical method to convert readily available acetophenones to acyl synthons for a tertiary amide synthesis.
 | ||
| Scheme 2 Substrate scope for the formation of azole amides from aryl methyl ketones and azoles. | ||
With the broad substrate scope of acetophenone derivatives established for this transformation, we were fascinated about the possibility of aryl ketones with different long-chain substituents under the standard conditions (Table 2). Delightfully, in addition to various aryl alkyl ketones (30, 31 and 32), which have been demonstrated to be active substrates by Jiao and co-workers in their primary amide formation with NaN3,11a sterically hindered compounds 33 (isopropyl) and 34 (benzyl) were also compatible substrates in this transformation, albeit the yields were a little bit lower than the former ones. Most attention should be paid to compound 33 since a similar compound was inert in ester formation from aryl ketones,11b which implies that the mechanism of this reaction is quite different from the precedent reports.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ketones | Yield of the product (%) | Entry | Ketones | Yield of the product (%) |
| a Reaction conditions: aryl ketone (0.25 mmol), benzoimidazole (0.5 mmol), CuBr (10 mmol%), pyridine (0.75 mmol), p-xylene (1 mL), 130 °C, 16 h, O2 in a sealed tube. b Isolated yield based on aryl ketones. ND = not detected. | |||||
| 1 |

|
68 | 7 |
|
77 |
| 2 |

|
71 | 8 |

|
46 |
| 3 |

|
74 | 9 |
|
47 |
| 4 |
|
37 | 10 |
|
49 |
| 5 |
|
59 | 11 |

|
ND |
| 6 |

|
55 | 12 |

|
0 |
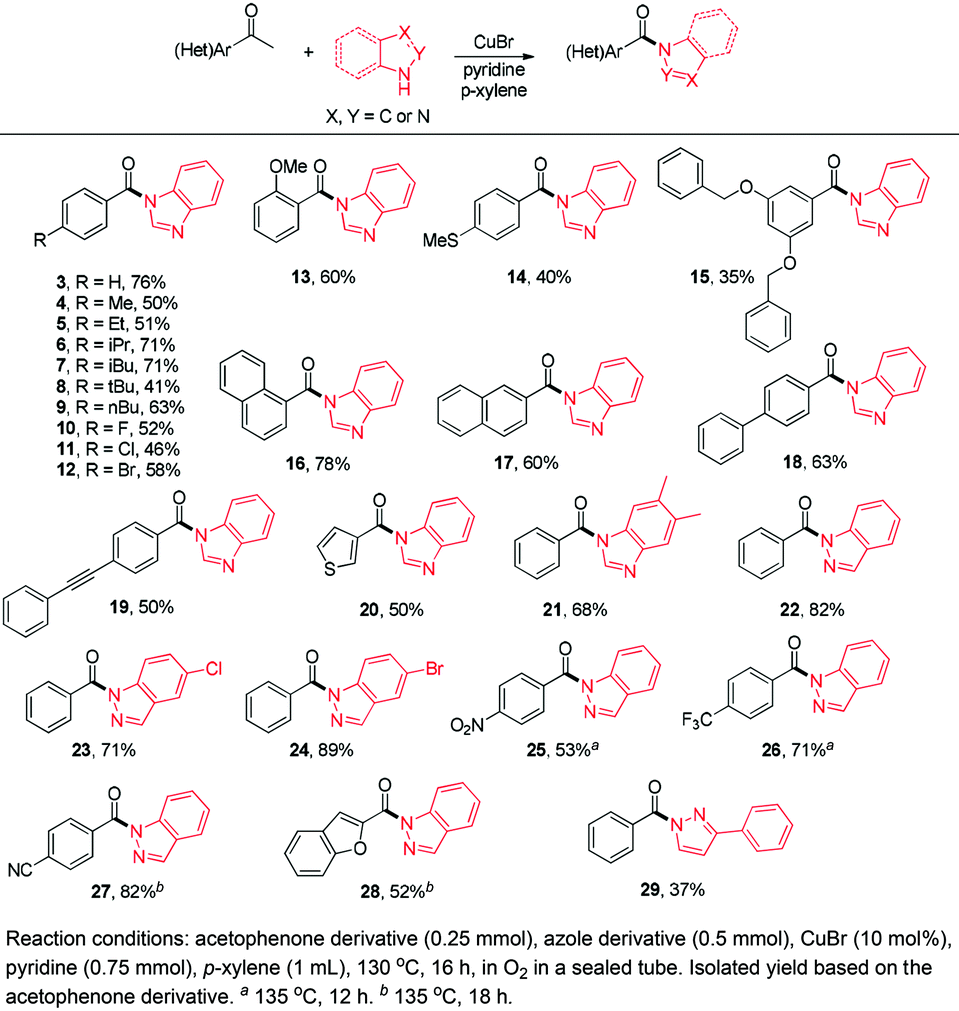
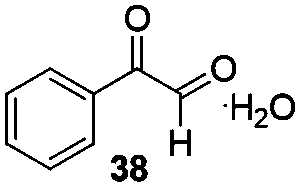
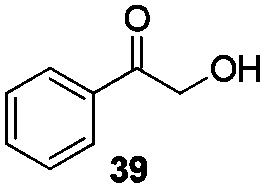
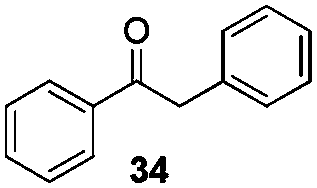
Gratifyingly, 1,3-dicarbonyl compounds, such as ethyl 3-oxo-3-phenylpropanoate (35) and 1,3-diphenylpropane-1,3-dione (36), exhibit good reactivities under the standard conditions as well. Among the above aryl ketones, compounds 33–36 were all first reported active substrates in such Cu-catalyzed aerobic oxidative C(CO)–C single bond cleavage reaction, which greatly broadens the substrate scope. Meanwhile, 2-oxo-2-phenylacetic acid (37), 2-oxo-2-phenylacetaldehyde monohydrate (38) and 2-hydroxy-1-phenylethanone (39) were all applied under the standard conditions and moderate yields of the desired products were obtained (see a detailed explanation on these three compounds in Scheme 2 for control experiments). Interestingly, diethyl β-keto-phosphonate (40) and benzaldehyde (41) are inert in this transformation and no cleavage of the C(CO)–C(alkyl) single bond was ever detected.
In order to gain insight into the reaction mechanism of the novel C(CO)–C single cleavage reaction, several control experiments were carried out. As shown in Scheme 3, the reactions were almost totally inhibited by radical scavengers 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and BHT (2,6-di-tert-butyl-4-methylphenol), indicating that a radical pathway might be involved in this reaction (eqn (1)). No desired product 3 was obtained from benzaldehyde (41) and benzimidazole (2), suggesting that benzaldehyde (41) is not the intermediate for this reaction (eqn (2)). The reactions of 2-hydroxy-1-phenylethanone (39), 2-oxo-2-phenylacetaldehyde monohydrate (38), and 2-oxo-2-phenylacetic acid (37) with benzimidazole (2) under standard conditions were also investigated and the desired products were afforded in 49%, 47% and 46% yields respectively (eqn (3), (4) and (5)); these results demonstrated that they might be the intermediates during the reaction transformation (yet compared to 76% of 3 in Table 1, entry 4, the yields were still quite lower). If only acetophenone was exposed under the standard conditions without a benzimidazole, no major product was detected (eqn (6)).
 | ||
| Scheme 3 Control experiments under standard conditions. | ||
As shown in Table 1, oxygen was crucial for the success of this transformation (Table 1, entries 10 and 11). To understand the originality of the oxygen in the amide product, the reaction was conducted first under 18O2. Surprisingly, ca. 40% of 3 was labeled with 18O (Scheme 4, eqn (7)). Furthermore, isotope labeled control experiments demonstrated that the amide product 3 could only slightly undergo oxygen exchange with water (eqn (10)). Moreover, when the reaction was conducted in the presence of 5.0 equiv. of H218O under the standard conditions (with 16O2), product 3 was obtained only in 43% yield with 28% 18O labeled; at the same time, 18% of benzoic acid was generated (eqn (8)) with a great amount of benzimidazole remaining, which indicated that water plays a deleterious effect on the reaction and slows down the entire process (competitive reaction vs. azole). Yet when the reaction was performed under 18O2 with 5.0 equiv. of H2O, 45% 18O labeled product 3 was detected (eqn (9)) and the total yield of 3 was only 39% with 19% of benzoic acid formed. When acetophenone (1) was exposed under the optimal conditions with 5.0 equiv. of H218O, it was totally decomposed and no major product was detected (eqn (11)); if the reaction was shortened to 6 h, only the starting material remained and no oxygen scrambling was detected. All the above reactions reveal that the oxygen atom of the amide product partially originated from molecular oxygen in this novel transformation, which is consistent with the 18O incorporation in the byproduct benzoic acid (eqn (8) and (9)) and consistent with previous reports11b as well.
 | ||
| Scheme 4 Isotope labeled control experiments. | ||
It is noteworthy that when compound 34 was subjected to the standard conditions (Table 2, entry 5), in addition to the desired product 3 (59% yield), benzaldehyde (41) and benzil were obtained in 17% and 35% yield respectively (eqn (12)), which suggested that oxidation on the methylene group adjacent to the carbonyl group in aryl ketones might occur before the nucleophilic addition of ketones with azoles, and benzaldehyde (41) might be a byproduct in this transformation.
 | (12) |
On the basis of the above results and the reported literature,11,19 a tentative mechanism is given below (Scheme 5): in path a, the methylene group adjacent to the carbonyl group was oxidized before the nucleophilic addition of ketones with azoles in the presence of Cu salt and molecular oxygen20via a single electron transfer (SET) process, generating superoxide intermediate A. Then, the nucleophilic attack of the carbonyl group with azoles in intermediate A occurred, leading to intermediate B. Subsequently, the SET reduction and protonation of intermediate B generates hydroperoxide intermediate C,21 in which the cleavage of the C–C bond occurred during the rearrangement of intermediate C, affording an amide along with aldehyde D.22 In this pathway, the carbonyl group in the desired product 3 stays intact without the O atom incorporation; in path b, the nucleophilic addition of ketones with azoles took place before the aerobic oxidation reaction, affording intermediate E. Dehydration of intermediate E generates enamine F. Subsequently, dioxetane intermediate G was formed23 from enamine F in a Cu(II)/oxygen system in the presence of pyridine.24 Eventually, the C−C bond and O−O bond cleavage of dioxetane intermediate G affords amide 3 and the aldehyde byproduct D.23
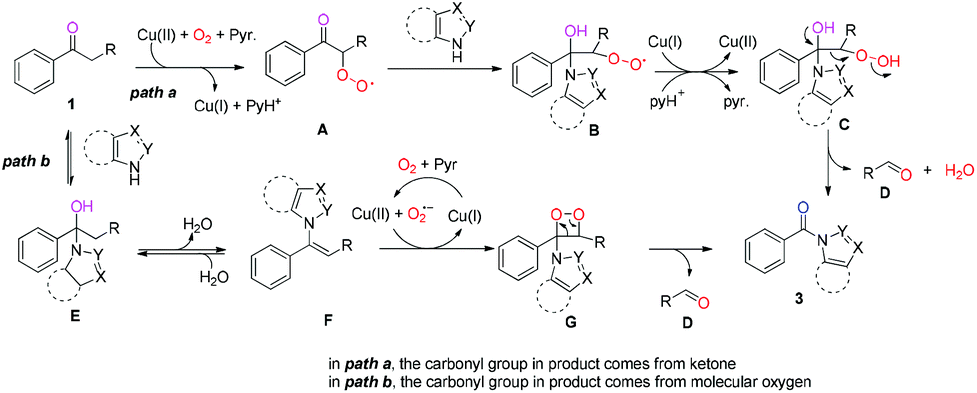 | ||
| Scheme 5 Proposed mechanism. | ||
Conclusions
In summary, a novel Cu-catalyzed aerobic oxidative amidation via C(CO)–C single bond cleavage of aryl ketones with various azoles has been demonstrated. Azole amides were obtained in moderate to good yields with a very broad substrate scope. This reaction has many advantages like cheap copper salt as the catalyst, oxygen as the sole terminal oxidant, cheap and readily available starting materials and a wide substrate scope. Meanwhile, aryl ketones with a variety of long-chain groups have been demonstrated to be active substrates in this novel strategy, and most of them are inert or have not been reported in the previously reported (CO)–C(alkyl) single bond cleavage reactions yet. Therefore, our strategy greatly widens the aryl ketone substrate scope in C(CO)–C single bond cleavage reaction and shows a potential application in organic synthesis. Further investigation on new C–C single bond cleavage is under way in our laboratory.Acknowledgements
Financial support from the National Science Foundation of China (21202049), the Recruitment Program of Global Experts (1000 Talents Plan), the Fujian Hundred Talents Program, and the Program for Innovative Research Team of Huaqiao University is gratefully acknowledged.Notes and references
- (a) A. D. Meijere, Chem. Rev., 2003, 103, 931 CrossRef; (b) K. C. Bishop III, Chem. Rev., 1976, 76, 461 CrossRef; (c) R. H. Rabtree, Chem. Rev., 1985, 85, 245 CrossRef; (d) L. J. Gooßen, N. Rodríguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed; (e) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef; (f) Carbocyclic Three- and Four-Membered Ring Compounds, Houben-Weyl methods in Organic Chemistry, ed. A. de Meijere, Thieme, Stuttgart, 1996, vol. E17a–c Search PubMed; (g) C.-H. Jun, Chem. Soc. Rev., 2004, 33, 610 RSC.
- (a) P. Steenwinkel, R. A. Gossage and G. Koten, Chem. – Eur. J., 1998, 4, 759 CrossRef CAS; (b) M. Murakami and Y. Ito, Topics in Organometallic Chemistry, ed. S. Murai, Springer, Berlin, Germany, 1999, pp. 97–129 Search PubMed.
- C.-Q. Sun, H.-L. Bai, B.-K. Tay, S. Li and E.-Y. Jiang, J. Phys. Chem. B, 2003, 107, 7544 CrossRef CAS.
- B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870 CrossRef.
- M. Murakami, T. Itahashi and Y. Ito, J. Am. Chem. Soc., 2002, 124, 13976 CrossRef CAS PubMed.
- (a) M. Murakami, H. Amii, K. Shigeto and Y. Ito, J. Am. Chem. Soc., 1996, 118, 8285 CrossRef CAS; (b) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 1999, 121, 11010 CrossRef CAS; (c) P. A. Wender, A. G. Correa, Y. Sato and R. Sun, J. Am. Chem. Soc., 2000, 122, 7815 CrossRef CAS; (d) S. C. Bart and P. J. Chirik, J. Am. Chem. Soc., 2003, 125, 886 CrossRef CAS PubMed; (e) T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340 CrossRef CAS PubMed.
- (a) C.-H. Jun and H. Lee, J. Am. Chem. Soc., 1999, 121, 880 CrossRef CAS; (b) A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412 CrossRef CAS PubMed; (c) H. Wakui, S. Kawasaki, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2004, 126, 8658 CrossRef CAS PubMed; (d) H. Li, W. Li, W. Liu, Z. He and Z. Li, Angew. Chem., Int. Ed., 2011, 50, 2975 CrossRef CAS PubMed; (e) M. Arisawa, M. Kuwajima, F. Toriyama, G. Li and M. Yamaguchi, Org. Lett., 2012, 14, 3804 CrossRef CAS PubMed; (f) S. S. Stahl, Science, 2005, 309, 1824 CrossRef CAS PubMed.
- (a) Y. Kuninobu, T. Uesugi, A. Kawata and K. Takai, Angew. Chem., Int. Ed., 2011, 50, 10406 CrossRef CAS PubMed; (b) M. A. Halcrow, F. Urbanos and B. Chaudret, Organometallics, 1993, 12, 955 CrossRef CAS; (c) I. Omae, Chem. Rev., 1979, 79, 287 CrossRef CAS.
- Modern Carbonyl Chemistry, ed. J. Otera, John Wiley & Sons, Weiheim, 2000 Search PubMed.
- L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303 CrossRef CAS PubMed.
- (a) C. Tang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 6528 CrossRef CAS PubMed; (b) X. Huang, X. Li, M. Zou, S. Song, C. Tang, Y. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS PubMed.
- P. Subramanian, S. Indu and K. P. Kaliappan, Org. Lett., 2014, 16, 6212 CrossRef CAS PubMed.
- (a) D. Spinks, H. B. Ong, C. P. Mpamhanga, E. J. Shanks, D. A. Robinson, I. T. Collie, K. D. Read, J. A. Frearson, P. G. Wyatt, R. Brenk, A. H. Fairlamb and I. H. Gilbert, ChemMedChem, 2011, 6, 302 CrossRef CAS PubMed; (b) S. K. Gupta, S. S. Pancholi, M. K. Gupta, D. Agrawal and M. P. Khinchi, J. Pharm. Sci. Res., 2010, 2, 228 CAS; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (d) M. L. McKee and S. M. Kerwin, Bioorg. Med. Chem., 2008, 16, 1775 CrossRef CAS PubMed; (e) K. Bahrami, M. M. Khodaei and F. Naali, J. Org. Chem., 2008, 73, 6835 CrossRef CAS PubMed.
- B. Tan, N. Toda and C. F. Barbas, Angew. Chem., Int. Ed., 2012, 51, 12538 CrossRef CAS PubMed.
- (a) J. Zhao, P. Li, C. Xia and F. Li, Chem. Commun., 2014, 50, 4751 RSC; (b) L. Yu, M. Wang and L. Wang, Tetrahedron, 2014, 70, 5391 CrossRef CAS PubMed.
- (a) M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227 CrossRef CAS PubMed; (b) W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736 CrossRef CAS PubMed; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (d) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (e) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed; (f) T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692 CrossRef CAS PubMed; (g) R. Lin, F. Chen and N. Jiao, Org. Lett., 2012, 14, 4158 CrossRef CAS PubMed; (h) C. Zhang, Z. Xu, T. Shen, G. Wu, L. Zhang and N. Jiao, Org. Lett., 2012, 14, 2362 CrossRef CAS PubMed.
- (a) Q. Song, Q. Feng and M. Zhou, Org. Lett., 2013, 15, 5990 CrossRef CAS PubMed; (b) Q. Song, Q. Feng and K. Yang, Org. Lett., 2014, 16, 624 CrossRef CAS PubMed; (c) Q. Feng and Q. Song, Adv. Synth. Catal., 2014, 356, 1649 CrossRef PubMed; (d) Q. Feng and Q. Song, Adv. Synth. Catal., 2014, 356, 2445 CrossRef CAS PubMed; (e) Q. Feng and Q. Song, J. Org. Chem., 2014, 79, 1867 CrossRef CAS PubMed; (f) M. Zhou and Q. Song, Synthesis, 2014, 1853 Search PubMed; (g) X. Xu, W. Ding, Y. Lin and Q. Song, Org. Lett., 2015, 17, 516 CrossRef CAS PubMed.
- Refer to the ESI† for a complete listing of the reaction conditions that were screened.
- (a) H. Liu, C. Dong, Z. Zhang, P. Wu and X. Jiang, Angew. Chem., Int. Ed., 2012, 51, 12570 CrossRef CAS PubMed; (b) L. Zhang, X. Bi, X. Guan, X. Li, Q. Liu, B.-D. Barry and P. Liao, Angew. Chem., Int. Ed., 2013, 52, 11303 CrossRef CAS PubMed; (c) Ref. 10, 11 .
- M. Gomez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef CAS PubMed.
- A. Pinter, A. Sud, D. Sureshkumar and M. Klussmann, Angew. Chem., Int. Ed., 2010, 49, 5004 CrossRef CAS PubMed.
- (a) J. Mecinovic, R. B. Hamed and C. J. Schofield, Angew. Chem., Int. Ed., 2009, 48, 2796 CrossRef CAS PubMed; (b) S. Chiba, L. Zhang, G. Y. Ang and B. W.-Q. Hui, Org. Lett., 2010, 12, 205 CrossRef PubMed.
- (a) M. Tokunaga, Y. Shirogane, H. Aoyama, Y. Obora and Y. Tsuji, J. Organomet. Chem., 2005, 690, 5378 CrossRef CAS PubMed; (b) A. Wang and H. Jiang, J. Am. Chem. Soc., 2008, 130, 5030 CrossRef CAS PubMed.
- (a) S. Borah, M. S. Melvin, N. Lindquist and R. A. Manderville, J. Am. Chem. Soc., 1998, 120, 4557 CrossRef CAS; (b) F.-T. Du and J.-X. Ji, Chem. Sci., 2012, 3, 460 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00101c |
| This journal is © the Partner Organisations 2015 |