
Application of online microdialysis coupled with liquid chromatography-tandem mass spectrometry method in assessing neuroprotective effect of Rhizoma coptidis on diabetic rats
Qianqian
Wang
ab,
Jing
Zhang
c,
Zifeng
Pi
a,
Zhong
Zheng
a,
Junpeng
Xing
a,
Fengrui
Song
a,
Shu Liu
*a and
Zhiqiang
Liu
*a
aNational Center for Mass Spectrometry in Changchun & Key Laboratory for Traditional Chinese Medicine Chemistry and Mass Spectrometry of Jilin Province & Chemical Biology Laboratory, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun 130022, Jilin, China. E-mail: liuzq@ciac.jl.cn; Fax: +86 0431 85262236; Tel: +86 0431 85262236
bUniversity of Chinese Academy of Sciences, Beijing 100039, China
cSchool of Pharmaceutical Science, Jilin University, Changchun 130021, China
First published on 12th September 2014
Abstract
Monitoring the in vivo dynamics of neurochemicals is very important for studies on learning and memory impairment. However, drawbacks in conventional sampling and off-line analysis methods require the employment of a more satisfying system. We present an online microdialysis coupled with liquid chromatography-tandem mass spectrometry method (LC-MS/MS) to determine eight neurochemicals simultaneously, including glutamate, aspartic acid, γ-aminobutyric acid, serine, taurine and acetylcholine, as well as dopamine and serotonin. A self-assembled automated injector with a high-pressure-resisting six-port valve was utilized to control the switch of the two states between loading and injection, which potentially simplified the sample preparation procedures and avoided the restrictions offered by small sample volume. Good linearity (R2 > 0.997) was observed in each analyte dynamic range. The inter-day and intra-day precisions (RSD) were <15%, and the accuracy (RE) was from −13.84% to 11.16%. The overall matrix effect was within an acceptable range, from 87.79% to 111.71%. Morris water maze (MWM) test was performed to evaluate the differences of cognitive abilities among normal control, diabetes mellitus (DM) model and Rhizoma coptidis (R. coptidis)-treated DM group rats. Moreover, the developed method was applied for the further exploration of the R. coptidis efficacies on the eight analytes changes in the hippocampus of diabetic rats with mild cognitive impairment. It was found that an improvement in neurochemicals imbalance was observed in diabetic rats after treatment with R. coptidis. In conclusion, the developed online analysis method is selective, sensitive and accurate, which is suitable for in vivo neurochemicals monitoring and could be extended to pharmacology studies.
1. Introduction
A biological sample providing more biochemical event information of interest and containing less matrix interference is of crucial importance in the fields of pathology and pharmacology. In conventional sampling methods, the collections of blood, tissue or excrement are the main sources for biological analysis. Series of complicated sample preparation processes are usually required, for instance, homogenate, protein precipitation1 and liquid–liquid extraction,2 which are tedious and time-consuming. However, microdialysis (MD) is a membrane-based sampling technique, which has been widely used in the sampling of extracellular components and other research.3,4 While implanting a probe into blood or a tissue of interest, molecules in the extracellular space may be carried out by the dialysate according to their concentration gradients. In the entire process, there is negligible loss of body fluids and minor tissue damage to animals. For making the probe through a miniature semipermeable membrane, which could exclude large molecules such as proteins and lipids, the dialysate is protein- and cell-free without the need for further complex sample preparations. Furthermore, continuous in vivo monitoring of target substances at multiple sites simultaneously is realized by this technique.5 Compared with the traditional sampling, the number of animals needed is dramatically reduced.A key to the application of microdialysis is the analysis of sample fractions. Temporal resolution of sampling is related to the dialysate volume. The relative recoveries of probes for each analyte are mainly influenced by the factors of the diffusion coefficient of the compound, the length and diameter of the probe, time interval, temperature and perfusion flow rate.6 In order to achieve better temporal resolution and higher relative in vivo recovery, sample fractions are typically collected in 10–30 min intervals at low flow rates of 1.0–3.0 μL min−1.7–9 For the property of small volume and low analytes concentration in the dialysate, a highly sensitive and selective detecting technique is necessary. Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) is a powerful tool for determining trace compounds in complex biological matrices. Prior to molecular detection, the HPLC technique can separate target chemicals efficiently. Then, analytes are detected in the most selective and sensitive mode of multiple reaction monitoring (MRM), which could effectively lead to fewer false-negative and false-positive results. Some studies have reported the off-line analysis of dialysate by LC-MS/MS method.8,9 By following the MD sampling strategy, sample preparation steps have been simplified considerably. However, there are still some drawbacks in the off-line MD-based analytical systems. Unexpected contamination or degradation of the analytes may occur during the storage and transfer steps. Moreover, temporal resolution has to be compromised sometimes to reach the limitation of the sample volume for injection. Therefore, determining an appropriate interface to realize the combination of MD with LC-MS/MS technique for online in vivo monitoring is critical. Relevant online MD coupled with HPLC methodss have been developed in some studies.10–13 However, the interfaces utilized are typically compatible with the HPLC system and not the UPLC system, which would be unsuitable for a more rapid and improved temporal resolution analysis.
Neurochemicals, including monoamine transmitters, acetylcholine (Ach) and neuroactive amino acids, which participate in a broad range of physiological processes, play a significant role in neural communication. The alteration in the activities of these compounds may result in a variety of neuronal dysfunction and neurological disorders such as Alzheimer's disease,14 anxiety and depression.15 As a result, measuring concentrations of neurochemicals in the extracellular space of the central nervous system (CNS) dynamically helps us to improve our understanding of the relationship between neurotransmission and the behavioral, cognitive, and pathological states of an organism. Diabetes mellitus (DM) is a prevalent disease among the middle-aged and elderly population, and is often associated with severe complications due to the acute hypo- and hyper-glycemia.16,17 Research has confirmed that type 2 DM has a strong relationship with degenerative and functional disorders of the CNS.18 The new term ‘diabetes-associated cognitive decline’ (DACD) was proposed by Mijnhout et al.19 to strengthen awareness of this disease and to promote research into this disorder. Rhizoma coptidis (R. coptidis) is a classic traditional Chinese medicine. In recent decades, R. coptidis has been widely used for the treatment of diabetes20 and Alzheimer's disease.21 Some research results show that alkaloids extracted from R. coptidis have the effect of reducing blood sugar,22 inhibiting aldose reductase23 and anti-Alzheimer activities.24 However, there was little research reported on the effects of R. coptidis on neurochemicals changes in diabetic rats.
In our study, a simple and sensitive online MD-based LC-MS/MS analytical system was constructed to determine eight neurochemicals in vivo and applied for exploring the effects of R. coptidis on the eight analytes changes in diabetic rat hippocampus. Compared with off-line analysis, our proposed method is simple, rapid and avoids the restrictions offered by small sample volume, which is valuable to neuroscience and pharmacology studies.
2. Material and methods
2.1 Chemicals and reagents
Glutamate (Glu), aspartic acid (Asp), γ-aminobutyric acid (GABA), serine (Ser), taurine (Tau), dopamine (DA), serotonin (5-HT), acetylcholine (Ach), heptafluoro-butanoic acid (HFBA) and streptozotocin (STZ) were purchased from Sigma-Aldrich (USA). HPLC-grade methanol was purchased from Fisher Scientific (USA). HPLC-grade formic acid and acetic acid were obtained from Tedia (USA). Water was obtained from Millipore Milli-Q Ultra-pure water purification system (Belgium). Artificial cerebrospinal fluid (aCSF), consisting of 1.2 mM CaCl2; 2.0 mM Na2HPO4·12H2O; 1.0 mM MgCl2; 2.7 mM KCl; and 145 mM NaCl adjusted to a pH of 7.4, was prepared as perfusate for microdialysis. R. coptidis was purchased from Jilin Dispensary, China, and had been identified in our previous study.252.2 Preparation of R. coptidis extract
R. coptidis (250 g) was immersed with 2500 mL water for 20 min, followed by refluxing extraction for 45 min. The solution was filtered through four sheets of gauze. Residues were extracted again under the same conditions for 30 min. After filtration, the combined solution was concentrated with rotary evaporation to yield an extract (about 55.23 g). The dried powder was stored at 4 °C before use.2.3 Instruments
A Waters Acquity-Ultra Performance LC system (Waters, USA) coupled with a Xevo TQ MS spectrometer with an electrospray ion source (ESI) (Waters, Manchester, UK) was used. Chromatographic separations were performed with a Venusil ASB C18 HPLC column (4.6 mm × 250 mm, 5 μm) from Agela Technologies. MD systems consisted of a CMA 402 syringe pump (CMA Microdialysis AB, Sweden), a self-assembled online injector with a 20 μL sample loop and a CMA 120 system for freely moving animals (CMA Microdialysis AB, Sweden). MAB 6 microdialysis probes (4 mm long, 15 kD cut-off) were purchased from Microbiotech AB (Stockholm, Sweden). A WMT-100 Morris water maze analysis system was purchased from Chengdu Taimeng Technology Co. LTD (Chengdu, China).2.4 Animal experiment
A probe trial was performed 24 hours later after conducting the place navigation task, which reflected the extent of memory consolidation after learning. The platform was removed from water, and each rat was placed in the water facing the wall at a zone directly opposite to the platform quadrant. The times of crossing the platform quadrant and the percentage of swimming track within 120 s in the former platform quadrant compared to that in all quadrants were recorded and calculated.
2.5 Tandem mass spectrometric and chromatographic conditions
MS analysis was operated using an electrospray ionization source (ESI) in the positive mode. The optimized parameters included the following: a capillary voltage of 2.5 kV; source temperature, 150 °C; desolvation temperature, 350 °C; desolvation gas flow, 800 L h−1; and cone gas flow 50 L h−1. Nitrogen was used as the cone and desolvation gas, and argon was used as the collision gas. The MRM transitions for eight analytes as well as their respective cone voltages and collision energies are listed in Table 1. To ensure the sensitivity of detection, the analytes were divided into two groups according to their retention times, and only one group of analytes was monitored simultaneously during each running period.| Analytes | Cone voltage (V) | Quantitation transition (m/z) | Confirmation transition (m/z) | RTs (min) |
|---|---|---|---|---|
| Collisiona (eV) | Collisiona (eV) | |||
| a Collision energy (eV) is given in brackets. | ||||
| Glu | 20 | 148.03 > 83.99 (16) | 148.03 > 102.01 (10) | 6.60 |
| Asp | 22 | 134.08 > 73.92 (14) | 134.08 > 87.97 (10) | 6.07 |
| GABA | 20 | 104.02 > 87.05 (10) | 104.02 > 68.97 (14) | 5.72 |
| Ser | 19 | 105.99 > 59.99 (10) | 105.99 > 87.94 (14) | 5.78 |
| Tau | 26 | 126.03 > 108.08 (10) | 126.03 > 44.04 (14) | 6.09 |
| Ach | 18 | 146.01 > 87.03 (14) | 146.01 > 43.06 (22) | 9.99 |
| DA | 18 | 154.18 > 136.97 (10) | 154.18 > 90.96 (20) | 10.47 |
| 5-HT | 15 | 177.02 > 160 (12) | 177.02 > 132 (20) | 14.89 |
Mobile phase A was acidified water containing 0.12% formic acid (pH 2.64), and mobile phase B was 33% methanol containing 0.06% formic acid aqueous (pH 2.78). The solutions were degassed before use. A gradient profile was used in the chromatographic separation: 0–3.5 min, 0% B; 3.5–4 min, 0–22% B; 4–8 min, 22–24% B; 8–8.5 min, 24–100% B; 8.5–15.5 min, 100% B; 15.5–16 min, 100–0% B; and 16–19.5 min, 0% B. The total run time was 19.5 min with a flow rate of 500 μL min−1 and the injected volume was 20 μL. To avoid contaminating the ion source by the salts in aCSF, the first 3 min of effluent was directed into a waste bottle.
2.6 Method validation
A stock solution containing each analyte was prepared with HPLC water at a concentration of 1.0 mM and stored at −80 °C. The stock solution was diluted with a solution of 0.12% formic acid/a-CSF (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to prepare a series of working standard solutions for calibration and quality control (QC) samples. Seven-point calibration curves for eight analytes were constructed by plotting the peak areas of the quantification MRM transitions against the theoretical concentrations. Each point was obtained as the average of three replicates with an injection volume of 20 μL.
:1, v/v) to prepare a series of working standard solutions for calibration and quality control (QC) samples. Seven-point calibration curves for eight analytes were constructed by plotting the peak areas of the quantification MRM transitions against the theoretical concentrations. Each point was obtained as the average of three replicates with an injection volume of 20 μL.
Three QC concentrations (n = 5 for each concentration level) were analyzed to assess the intra- and inter-day accuracy and precision on three separate days. The relative error (RE) of each QC point served as the measure of accuracy. Precision was evaluated by the relative standard deviation (RSD) of the determinations per QC concentration. The limit of detection (LOD) and the limit of quantification (LOQ) were studied based on the signal-to-noise ratios of 3:1 and 10:1, respectively.
The post-extraction spike method29 was adopted in our study to evaluate the matrix effect. Endogenous concentrations of the eight analytes in the dialysate were analyzed according to the method we developed, and then the same dialysate spiked at three concentration levels of QC standard solutions was checked. The peak areas obtained in neat solution standards were depicted as A, the corresponding peak areas for standards spiked into dialysate as B, and peak areas for analytes in non-spiked dialysate as C. The matrix effect (ME) values were calculated as follows:
| ME (%) = (B − C)/A × 100 |
2.7 Data processing
HPLC-MS/MS data acquisitions were performed with the MassLynx V4.1 software and processed for the calibration and for the quantification of the analytes using Targetlynx V4.1 software. Statistical analysis was managed with SPSS 18.0 version software, and p < 0.05 was considered significant.3. Results and discussion
3.1 Interface between MD probe and HPLC-MS/MS
Online MD coupled with the HPLC system has received increased attention because of the remarkable merits mentioned in the introduction. However, the significant limitation is that the analysis time must be shorter than the sampling interval, if not, the valuable sample would be lost. This puts forward higher requirements for efficiency in analysis. The particle size of packing materials in HPLC columns results in limited speed and peak capacity.30 When multiple compounds need to be separated in a single run, HPLC may not fit for a sufficiently rapid analysis. The UPLC system dramatically increased efficiency, resolution and sensitivity while retaining the practicality and principles of HPLC. We present a self-assembled online injector with a high pressure-resisting six-port valve to be the interface between the MD probe and HPLC-MS/MS, as shown in Fig. 1, which could satisfy different pressure demands by HPLC and UPLC columns. Port 1 was used to sample from the outlet of the MD probe. A 20 μL sample loop connected port 3 and port 6 for loading dialysate. Combined solvents were pumped into port 4, and port 5 was connected with the chromatographic column. The functions of injection and loading were controlled through switching the valve automatically. In the schematic, solid lines represented loading states that lasted for 20 min and dashed lines signified 10 s injection states. Sample loops with different volumes could be fixed to match HPLC or UPLC columns; therefore, more data points would be achieved on the basis of shorter analysis time and improved temporal resolution. | ||
| Fig. 1 Diagram of the online microdialysis-LC-MS/MS method. | ||
3.2 HPLC-MS/MS method development
A standard mixture (amino acids 10 μM, Ach, DA, 5-HT 1.0 μM) was used to optimize the LC-MS/MS method. Amino acids are molecules with a structure that contains both basic (such as amine) and acidic (such as carboxylic acid) functional groups. Therefore, the positive and negative ionization modes were both tested to determine the most abundant or the most specific transition for each amino acid in the MRM mode. The best spectrometry performances for most analytes were achieved in the positive ESI mode. Capillary voltage, gas flow and desolvation temperature were optimized for each chemical. Because the variation of these three parameters did not lead to a significant effect on the signal intensity, compromised values for such arguments were chosen and used in the further investigation. Cone voltages (CVs) for each precursor ion and collision energies (CE) for each fragment ion were optimized by the Intellistart function, and the results are listed in Table 1.Based on the previous work in our lab for the separation of underivatized amino acids in dry blood spots,31 some conditions were improved in this study. For glutamic acid and aspartic acid, we found that adding formic acid to achieve an appropriate pH value of the mobile phase resulted in a more satisfactory peak shape compared to acetic acid. Therefore, 0.12% formic acid was finally selected as the aqueous phase. In order to achieve better separation degree, we investigated the effect of heptafluoro-butanoic acid at 1 mM in the method development process. However, the separation effect did not improve significantly, and remarkable ion suppressions for GABA and Tau were observed. At the same time, different gradients, source temperatures between 120–150 °C and flow rates ranging from 0.3 to 0.8 mL min−1 were studied, and a flow rate of 0.5 mL min−1 was found to be the optimum flow rate to attain an acceptable peak shape and high sensitivity for target analytes with a total run time of 19.5 min. To reduce the influence of non-volatile salts in microdialysis samples, the gradient elution with an initial mobile phase containing a high proportion of water at a flow rate of 0.5 mL min−1 was used to rapidly elute the salt. In addition, the first 3 min of effluent were directed into a waste bottle through a diversion valve to prevent salts from entering the ion source.
3.3 Method validation
A sensitive LC-MS/MS coupled with online MD method was validated to simultaneously quantify Glu, Asp, GABA, Ser, Tau, Ach, DA and 5-HT concentrations from the in vivo dialysate. Applying the optimized tandem mass spectrometry and chromatography conditions, we observed a series of parameters such as linearity, limits of detection (LODs), limits of quantification (LOQs), accuracy, precision and stability to characterize the method developed.The dynamic range, correlation coefficients, LODs and LOQs are shown in Table 2. The correlation coefficients are 0.997–0.999, which indicate that the method developed had excellent linearity. The sensitivity was evaluated by determining the LODs and LOQs of the analytes. From our observation, the LODs ranged from 0.005 nM (Ach) to 100 nM (Tau) and the LOQs from 0.02 nM to 250 nM. Although Tau exhibited lower sensitivity in the positive mode, the LOD and LOQ were still suitable enough for quantification in rat brain dialysate. In previous studies, capillary electrophoresis coupled to light-induced fluorescence (CE-LIF), liquid chromatography (LC) coupled to fluorescence detection or electrochemical detection (FLD or ECD) techniques were used for the multi-analyte detections of amino acids and monoamines. For example, preceding studies reported that the LODs of Glu and Asp were 70 nM and 94 nM,32 respectively, which are higher than those obtained in our work. LODs measured for DA and 5-HT were 0.2–2.5 nM,33,34 whereas the lower values of 0.025 nM and 0.075 nM were obtained in the present study. In other words, the LODs and LOQs were improved, and better sensitivity was achieved in the proposed method.
| Analytes | Dynamic range (μM) | Slope (mean ± SD) | Y-intercept (mean ± SD) | R 2 | LODs (nM) | LOQs (nM) |
|---|---|---|---|---|---|---|
| Glu | 0.5–10 | 13.11 ± 0.69 | 2099.6 ± 237.67 | 0.9994 | 9 | 50 |
| Asp | 0.1–5 | 16.74 ± 0.67 | 2043 ± 400.85 | 0.9984 | 35 | 80 |
| GABA | 0.1–2.5 | 4.64 ± 0.099 | 241.27 ± 42.02 | 0.9986 | 12 | 48 |
| Tau | 0.5–10 | 0.76 ± 0.081 | 332.7 ± 97.09 | 0.9974 | 100 | 250 |
| Ser | 0.5–10 | 7.49 ± 0.40 | 2451.8 ± 198.27 | 0.9994 | 25 | 75 |
| Ach | 1.0 × 10−4 to 1.0 × 10−2 | 785.58 ± 51.47 | 1004.7 ± 127.79 | 0.9995 | 0.005 | 0.02 |
| DA | 1.0 × 10−4 to 1.0 × 10−2 | 356.48 ± 49.13 | −0.54 ± 14.49 | 0.9981 | 0.01 | 0.025 |
| 5-HT | 1.0 × 10−4 to 1.0 × 10−2 | 296.85 ± 17.94 | −9.32 ± 13.10 | 0.9996 | 0.02 | 0.075 |
The results of accuracy and precision at three QC levels are presented in Table 3. The inter-day and intra-day accuracies are in the range of −13.84–7.63% and −11.56–11.16%, respectively. The inter-day and intra-day precision, expressed as the relative standard deviation (RSD), are lower than 15% for each analyte. These results are in good agreement with the FDA guideline on bioanalytical method validation.35 Matrix effect data are also shown in Table 3, and the matrix effect of all analytes is ranged from 87.79% to 111.71%, which demonstrates that there was no significant matrix interference in our method.
| AAs | Added | Intra-day (n = 5) | Inter-day (n = 3) | Matrix effect (%) (n = 5) | ||||
|---|---|---|---|---|---|---|---|---|
| Observed | Precision (RSD, %) | Accuracy (RE, %) | Observed | Precision (RSD, %) | Accuracy (RE, %) | |||
| a The concentration level is μM. b The concentration level is nM. | ||||||||
| Glua | 0.5 | 0.50 ± 0.01 | 2.33 | 0.11 | 0.52 ± 0.03 | 4.45 | 4.58 | 93.99 ± 3.07 |
| 1.25 | 1.17 ± 0.03 | 1.68 | −6.10 | 1.22 ± 0.07 | 5.31 | −2.61 | 92.96 ± 4.12 | |
| 10 | 10.03 ± 0.46 | 4.01 | 0.17 | 10.87 ± 0.94 | 9.41 | 9.74 | 96.89 ± 4.83 | |
| Aspa | 0.2 | 0.18 ± 0.01 | 6.32 | −9.99 | 0.22 ± 0.03 | 11.76 | 11.16 | 87.79 ± 5.98 |
| 0.75 | 0.71 ± 0.03 | 2.93 | −7.09 | 0.73 ± 0.05 | 7.72 | −2.69 | 93.89 ± 4.72 | |
| 2.5 | 2.51 ± 0.09 | 3.78 | 0.21 | 2.75 ± 0.36 | 11.60 | 9.50 | 107.84 ± 4.25 | |
| GABAa | 0.1 | 0.1 ± 0.01 | 8.18 | −6.83 | 0.09 ± 0.01 | 9.11 | −3.40 | 93.84 ± 3.81 |
| 0.5 | 0.53 ± 0.02 | 2.81 | 7.63 | 0.54 ± 0.01 | 2.34 | 8.76 | 96.77 ± 1.98 | |
| 2.5 | 2.52 ± 0.06 | 2.44 | 0.35 | 2.46 ± 0.09 | 3.54 | −0.61 | 95.81 ± 3.32 | |
| Taua | 0.5 | 0.45 ± 0.04 | 9.39 | −9.03 | 0.49 ± 0.05 | 9.75 | −3.74 | 95.43 ± 8.26 |
| 1.25 | 1.16 ± 0.08 | 6.72 | −8.22 | 1.27 ± 0.14 | 11.92 | 1.50 | 97.21 ± 5.79 | |
| 10 | 10.07 ± 0.41 | 3.00 | 0.64 | 10.96 ± 0.73 | 6.39 | 8.32 | 101.90 ± 6.49 | |
| Sera | 0.75 | 0.67 ± 0.02 | 4.59 | −13.84 | 0.70 ± 0.04 | 5.96 | −11.56 | 103.85 ± 5.81 |
| 2.5 | 2.49 ± 0.06 | 2.46 | −0.04 | 2.43 ± 0.11 | 5.43 | −1.75 | 111.71 ± 3.08 | |
| 10 | 9.87 ± 0.21 | 1.96 | −0.29 | 10.35 ± 0.42 | 3.35 | 2.59 | 93.17 ± 3.08 | |
| Achb | 0.5 | 0.45 ± 0.03 | 7.48 | −2.47 | 0.44 ± 0.05 | 9.72 | −5.39 | 104.87 ± 7.26 |
| 2.0 | 2.06 ± 0.11 | 4.92 | 3.44 | 2.12 ± 0.16 | 5.36 | 5.49 | 102.63 ± 4.57 | |
| 7.5 | 7.42 ± 0.20 | 2.85 | −1.74 | 7.14 ± 0.26 | 3.58 | −4.66 | 98.73 ± 4.69 | |
| DAb | 0.5 | 0.48 ± 0.02 | 5.62 | −3.96 | 0.47 ± 0.05 | 8.71 | −4.18 | 96.25 ± 5.78 |
| 2.0 | 2.03 ± 0.08 | 3.49 | 1.57 | 1.96 ± 0.07 | 3.30 | −1.34 | 107.84 ± 6.09 | |
| 7.5 | 7.31 ± 0.36 | 4.29 | −2.15 | 7.08 ± 0.43 | 5.60 | −5.22 | 105.43 ± 3.52 | |
| 5-HTb | 0.5 | 0.48 ± 0.04 | 7.69 | −4.35 | 0.46 ± 0.03 | 6.51 | −7.26 | 104.26 ± 6.81 |
| 1.5 | 1.54 ± 0.09 | 6.37 | 2.48 | 1.59 ± 0.15 | 9.14 | 5.79 | 97.61 ± 3.74 | |
| 5.0 | 4.82 ± 0.16 | 2.83 | −3.61 | 5.14 ± 0.31 | 5.75 | 3.42 | 95.64 ± 5.24 | |
The stability was tested by reanalyzing standard solutions under different temperatures and storage conditions. The percentage of each analyte retained in the solution was expressed as the index of stability. The data suggested that the stock solution was stable at least for a month at −80 °C. The stability of QCs was evaluated by comparing the peak areas of solutions that were freshly prepared with those that were kept in the dark at 4 °C for two weeks. Under this condition, there was no significant decrease in signal intensity. Since the microdialysate samples were analysed online, freeze/thaw stability was not studied.
3.4 Performance of Morris water maze test
The Morris water maze test was carried out to assess cognitive abilities after blood glucose checks in three groups. Blood glucose values were higher in both groups of diabetic mice compared with the controls, DM model group with 26.83 ± 2.71 mmol L−1 glucose, R. coptidis-DM group with 21.36 ± 2.64 mmol L−1 and the controls with 9.47 ± 0.89 mmol L−1 glucose, respectively. However, R. coptidis-DM group rats exhibited more mildly elevated blood sugar than DM model rats over the course of 10 weeks of feeding with R. coptidis extract after the induction of diabetes. Thus, treating with R. coptidis extract appeared to reduce the elevated blood glucose in the diabetic mouse model. Observed from the data of the Morris water maze test, the mean ELT decreased during the trials in all groups (Fig. 2A). Diabetic rats exhibited markedly prolonged ELT compared to control group rats (p < 0.01), indicating an impairment in learning ability under high blood glucose. ELT in the control and R. coptidis-DM groups became progressively shorter in a day-dependent manner. The poor performance was improved by treatment with R. coptidis extract as evidenced by significantly decreased ELT from the third day of the trails (72.27 ± 6.41 s) compared to diabetic rats (87.72 ± 6.64 s) (p < 0.01).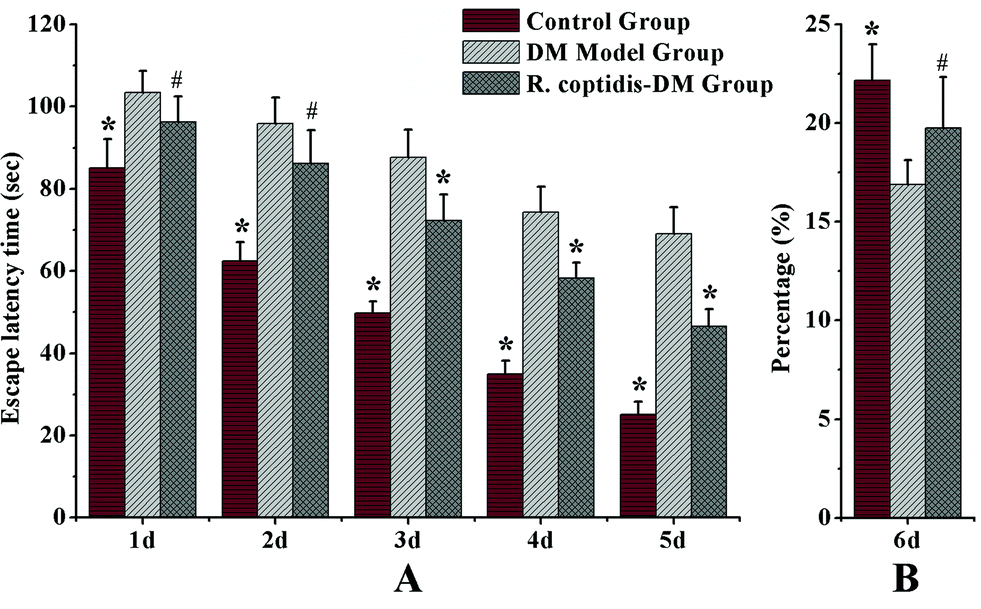 | ||
| Fig. 2 (A) Escape latency time (ELT) in Morris water maze test from one to five days. (B) Percentage of swimming track within 120 s in target quadrant compared to that in all quadrants. Data presented as mean ± SD (n = 7–8 per group). # p < 0.05, * p < 0.01 vs. DM model group. | ||
The probe trial studies how well the animals had learned and consolidated the platform location during the five days of training trails, which specifically correlates with learning impairment. The data from the probe trial is depicted in Fig. 2B. Diabetic rats displayed a worse learned bias navigating towards the target quadrant, which previously contained the platform. The percentage of swimming tracks in target quadrants (16.90 ± 1.21) was significantly lower than that of the control rats (22.17 ± 1.82) (p < 0.01). The rats treated with R. coptidis displayed improved learning bias, as evidenced by the higher percentage of swimming tracks in the former platform quadrant (19.75 ± 2.57) than diabetic animals (p < 0.05). The water maze test showed the protective effect of R. coptidis extract on cognitive abilities of diabetic rats from the perspective of behavior. To further explore the protective mechanism, we determined eight neurochemicals in the hippocampus.
3.5 Changes in neurochemicals levels
Although as the protagonists of information transmission, few studies on the relationship between neurochemicals and mild cognitive impairment, including diabetes-induced learning/memory deficiencies, were conducted. Our developed and validated MD-based LC-MS/MS online detection method directly, successionally and dynamically evaluated the extracellular levels of eight neurochemicals in the hippocampus region of freely moving male rats. The MRM transitions in the positive mode are presented in Fig. 3. The in vitro recovery was tested according to the literature,36 and it was found to be 36.78% for Glu, 27.12% for Asp, 35.08% for GABA, 26.37% for Ser, 29.86% for Tau, 36.96% for Ach, 33.88% for DA and 28.72% for 5-HT. The concentration of analytes in the brain dialysate was calibrated by the recovery, and the related analytical results were depicted in Fig. 4. Compared with the control rats, significant increase was observed in six analytes, including Glu, Asp, GABA, Ser, DA and 5-HT, in the hippocampus of brains in diabetic rats. However, Tau and Ach levels significantly decreased in diabetic rats. Glu, Asp and GABA were major amino acid neurotransmitters in the mammalian CNS. In particular, glutamate, which has a close relationship with long-term potentiation functions,37 was considered as a two-edged sword for its neurotoxicity when there was exorbitant ambient glutamate. Note that Ser and Tau were essential neuromodulators. Previous studies reported that taurine supplementation reduced the oxidative stress in nerves and prevented the impairment of calcium handling in sensory neurons of STZ-treated diabetic rats.38 As the neurotransmitters are closely related with cognitive functions such as learning and memory, DA, 5-HT and Ach have been focused on numerous neurodegeneration diseases.39–41 During 10 weeks of treatment with R. coptidis, the imbalance of eight neurochemical levels were significantly attenuated compared to DM rats. Associated with the results of MWM test, we found some correlations between neurochemicals and MWM trial outcomes. The greater memory impairments accompanied with the higher hippocampal excitatory amino acids and monoamine concentrations and the lower hippocampal Tau and Ach concentrations were observed. Although the sample size was small, we think that it at least indicated a tendency between the changed neurochemical levels and cognitive decline. In addition, similar levels of neurochemicals were reported in rat brain samples. For instance, the concentrations of Glu, Asp and GABA obtained in our study were in accordance with the values found by Fan et al. using electrochemical detection.42 The values of DA were similar with the published concentrations.43,44 Compared with nonspecific methods such as electrochemical detection or fluorescence detection,45 the analyte can be identified by both its retention time and molecular weight in the LC-MS/MS method, which provides a specific and direct measurement. Furthermore, an additional dimension of structurally specific filtering for individual analytes is achieved by the MRM scan function. As a result, the signal-to-noise ratio of an ion chromatographic peak is significantly improved. On the whole, the LC-MS/MS technique provides a direct and structurally specific measurement for individual analytes with high sensitivity and minimal baseline drift. | ||
| Fig. 3 Extracted ion chromatograms of eight analytes in control rat's hippocampus. | ||
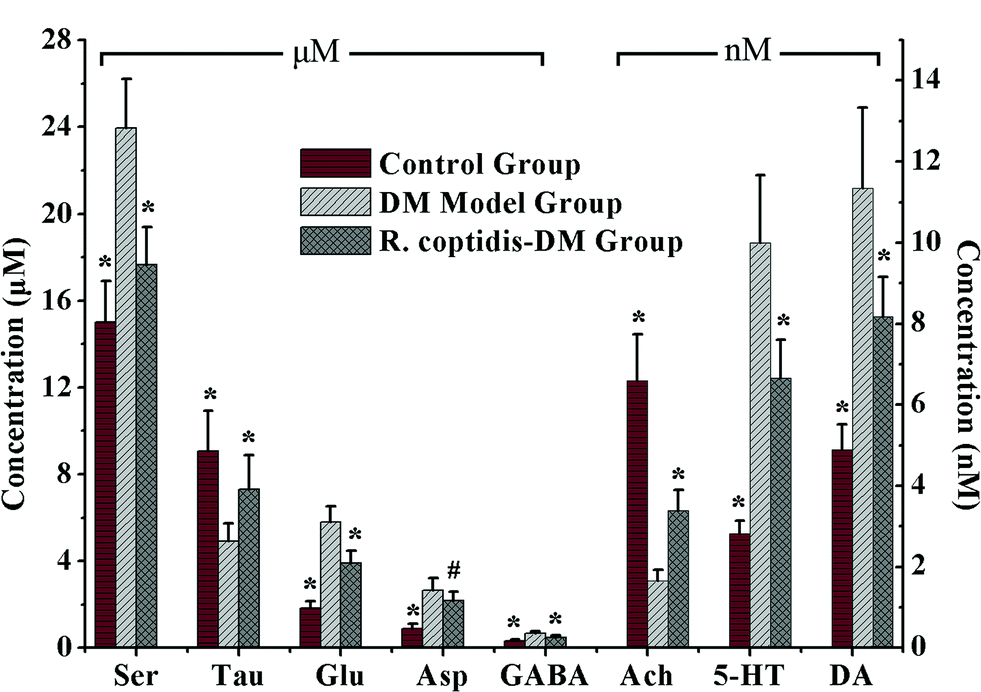 | ||
| Fig. 4 Levels of eight neurochemicals in rat hippocampus among three groups (n = 7–8 per group). Data presented as mean ± SD. # p < 0.05, * p < 0.01 vs. DM model group. | ||
4. Conclusion
An online MD coupled with LC-MS/MS method that employed a high-pressure-resisting automated injector as the interface was developed. In addition, eliminating additional sample pretreatment procedures, the online detection system also avoids the restrictions of small sample volume and acquires immediate feedback, which is conducive for making this method widely applicable for monitoring the in vivo dynamics of chemicals in real time. As a demonstration, eight neurochemicals in rat hippocampus were determined simultaneously among control, DM and R. coptidis-DM group rats. Treatment with R. coptidis significantly reversed diabetes-induced neurochemicals changes and memory impairment. Therefore, our proposed rapid, stable and sensitive method is valuable for the studies of neuroscience and pharmacology.Acknowledgements
The authors are grateful to the Innovation Method Fund of China (Grant no. 2012IM030600) and the National Natural Science Foundation of China (Grant no. 81373952 and 81473537) for the financial support for this work.References
- X. M. Chen, S. S. Zhang, J. S. Ma, S. P. Hu, S. H. Shi and Z. Xiang, Anal. Methods, 2014, 6, 1197–1202 RSC.
- C. W. N. Damen, H. Rosing, J. H. M. Schellens and J. H. Beijnen, Anal. Bioanal. Chem., 2009, 394, 1171–1182 CrossRef CAS PubMed.
- E. P. Corcoles, S. Deeba, G. B. Hanna, P. Paraskeva, M. G. Boutelle and A. Darzi, Anal. Methods, 2011, 3, 2010–2016 RSC.
- S. Kaul, M. D. Faiman and C. E. Lunte, Anal. Methods, 2011, 3, 1514–1520 RSC.
- J. Li, V. von Pfoestl, D. Zaldivar, X. Zhang, N. Logothetis and A. Rauch, Anal. Bioanal. Chem., 2012, 402, 2545–2554 CrossRef CAS PubMed.
- H. Benveniste, J. Neurochem., 1989, 52, 1667–1679 CrossRef CAS PubMed.
- P. L. Tsai and T. H. Tsai, J. Chromatogr. A, 2002, 961, 125–130 CrossRef CAS.
- C. Wang, S. Li, Y. J. Tang, S. W. Wang, Y. L. Zhang, G. R. Fan, L. Q. Li and Y. Zhang, J. Pharm. Biomed. Anal., 2012, 64, 1–7 Search PubMed.
- X. Zhou, J. Qiao, W. Yin, L. Zhu and H. F. Kung, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2011, 879, 3041–3046 CrossRef CAS PubMed.
- G. W. Cheng, H. L. Wu and Y. L. Huang, Talanta, 2009, 79, 1071–1075 CrossRef CAS PubMed.
- D. Wang, C. S. Liu, Y. X. Liu, Z. L. An, P. F. Li, L. H. Liu and J. H. Hu, Chin. J. Anal. Chem., 2012, 40, 164–168 CAS.
- P. S. H. Wong, K. Yoshioka, F. Xie and P. T. Kissinger, Rapid Commun. Mass Spectrom., 1999, 13, 407–411 CrossRef CAS.
- G. Damsma, D. P. Boisvert, L. A. Mudrick, D. Wenkstern and H. C. Fibiger, J. Neurochem., 1990, 54, 801–808 CrossRef CAS PubMed.
- M. Haris, K. Nath, K. Cai, A. Singh, R. Crescenzi, F. Kogan and R. Reddy, NMR Biomed., 2013, 26, 386–391 CrossRef CAS PubMed.
- W. Krzysciak, Acta Biochim. Pol., 2011, 58, 461–466 CAS.
- R. C. Turner, R. R. Holman, C. A. Cull, I. M. Stratton, D. R. Matthews and V. Frighi, Lancet, 1998, 352, 837–853 CrossRef.
- V. R. Kondepati and H. M. Heise, Anal. Bioanal. Chem., 2007, 388, 545–563 CrossRef CAS PubMed.
- B. d. Almeida-Pititto, C. d. M. Almada Filho and M. S. Cendoroglo, Arq. Bras. Endocrinol. Metabol., 2008, 52, 1076–1083 Search PubMed.
- G. S. Mijnhout, P. Scheltens, M. Diamant, G. J. Biessels, A. M. Wessels, S. Simsek and R. J. Heine, Diabetologia, 2006, 49, 1447–1448 CrossRef CAS PubMed.
- G. Chen, F. Lu, L. Xu, H. Dong, P. Yi, F. Wang and X. Zou, Phytomedicine, 2013, 20, 780–786 CrossRef CAS PubMed.
- H. J. Hong, P. Y. Chen, T. C. Shih, C. Y. Ou, M. D. Jhuo, Y. Y. Huang and J. G. Chung, Mol. Med. Rep., 2012, 5, 142–147 CAS.
- X. Tang, J. Tang, Q. Zhang, W. Dong, Y. Xiong, B. Li and G. L. Xu, Chin. J. Clin. Pharmacol. Ther., 2010, 15, 967–971 Search PubMed.
- H. A. Jung, N. Y. Yoon, H. J. Bae, B.-S. Min and J. S. Choi, Arch. Pharmacal Res., 2008, 31, 1405–1412 CrossRef CAS PubMed.
- H. A. Jung, B.-S. Min, T. Yokozawa, J. H. Lee, Y. S. Kim and J. S. Choi, Biol. Pharm. Bull., 2009, 32, 1433–1438 CAS.
- Q. Q. Du, X. Zhang, F. R. Song, Z. Q. Liu and S. Y. Liu, Chem. J. Chin. Univ., 2010, 31, 1332–1336 CAS.
- S. H. Yin, Journal of Practical Diabetology, 2008, 4, 58–59 Search PubMed.
- J. Wang, Z. M. Yuan, H. W. Kong, Y. Li, X. Lu and G. W. Xu, Chin. J. Chromatogr., 2012, 30, 8–13 CrossRef CAS.
- G. Paxinos and C. Watson, The rat brain in stereotaxic coordinates – preface, Elsevier Acdemic Press Inc, San Diego, 4th edn, 1998 Search PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS.
- M. E. Swartz, LCGC North Am., 2005, 8–14 CAS.
- C. Y. Wang, H. B. Zhu, W. Y. Zhang, F. R. Song, Z. Q. Liu and S. Y. Liu, Amino Acids, 2013, 44, 661–671 CrossRef CAS PubMed.
- L. Denoroy, S. Parrot, L. Renaud, B. Renaud and L. Zimmer, J. Chromatogr. A, 2008, 1205, 144–149 CrossRef CAS PubMed.
- T. Yoshitake, J. Kehr, S. Yoshitake, K. Fujino, H. Nohta and M. Yamaguchi, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 807, 177–183 CrossRef CAS PubMed.
- H. Xu, D. Wang, W. Zhang, W. Zhu, K. Yamamoto and L. Jin, Anal. Chim. Acta, 2006, 577, 207–213 CrossRef CAS PubMed.
- US Department of Health and Human Services, FDA, Center for Drug Evaluation and Research, 2001.
- C. Huang, K. C. Chen, C. F. Chen and T. H. Tsai, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 716, 251–255 CrossRef CAS.
- A. Malgaroli and R. W. Tsien, Nature, 1992, 357, 134–139 CrossRef CAS PubMed.
- F. Li, I. G. Obrosova, O. Abatan, D. Tian, D. Larkin, E. L. Stuenkel and M. J. Stevens, Am. J. Physiol.: Endocrinol. Metab., 2005, 288, E29–E36 CrossRef CAS PubMed.
- Y. Xu, J. Yan, P. Zhou, J. Li, H. Gao, Y. Xia and Q. Wang, Prog. Neurobiol., 2012, 97, 1–13 CrossRef CAS PubMed.
- A. van Waarde, N. K. Ramakrishnan, A. A. Rybczynska, P. H. Elsinga, K. Ishiwata, I. M. Nijholt and R. A. Dierckx, Behav. Brain Res., 2011, 221, 543–554 CrossRef CAS PubMed.
- J. M. Alisky, Med. Hypotheses, 2006, 67, 556–560 CrossRef CAS PubMed.
- Y. Fan, J. Zhang, X. L. Sun, L. Gao, X. N. Zeng, J. H. Ding and G. Hu, J. Neurosci. Res., 2005, 82, 458–464 CrossRef CAS PubMed.
- L. H. Parsons and J. B. Justice, J. Neurochem., 1992, 58, 212–218 CrossRef CAS PubMed.
- M. E. Hows, L. Lacroix, C. Heidbreder, A. J. Organ and A. J. Shah, J. Neurosci. Methods, 2004, 138, 123–132 CrossRef CAS PubMed.
- J. P. Danaceau, E. E. Chambers and K. J. Fountain, Bioanalysis, 2012, 4, 783–794 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |