
Glycosylation with N-acetyl glycosamine donors using catalytic iron(III) triflate: from microwave batch chemistry to a scalable continuous-flow process†‡
Amandine
Xolin
a,
Arnaud
Stévenin
a,
Mathieu
Pucheault
b,
Stéphanie
Norsikian
a,
François-Didier
Boyer
*ac and
Jean-Marie
Beau
*ad
aCentre de Recherche de Gif, Institut de Chimie des Substances Naturelles, CNRS, 1 avenue de la Terrasse, F-91198 Gif-sur-Yvette, France. E-mail: francois-didier.boyer@cnrs.fr; jean-marie.beau@cnrs.fr
bInstitut des Sciences Moléculaires, UMR 5255 Bâtiment A11, CNRS-Université de Bordeaux 1, F-33405 Talence, France
cInstitut Jean-Pierre Bourgin, UMR1318 INRA-AgroParisTech RD10, F-78126 Versailles, France. E-mail: francois-didier.boyer@versailles.inra.fr
dUniversité Paris-Sud and CNRS, Laboratoire de Synthèse de Biomolécules, Institut de Chimie Moléculaire et des Matériaux, F-91405 Orsay, France. E-mail: jean-marie.beau@u-psud.fr
First published on 11th August 2014
Abstract
Efficient and highly selective glycosylation reactions of peracetylated β-D-N-acetyl gluco- and galactosamine are described using catalytic iron(III) triflate under microwave conditions or in a continuous flow process. Simple β-glycosides and β-(1 → 6), β-(1 → 2) and β-(1 → 3) linked disaccharides bearing various protecting groups were obtained in high yields. Insights into the glycosylation mechanism are discussed.
Introduction
Numerous natural glycoconjugates (oligomeric structures and small molecules)1,2 contain N-acetylated-D-glucosamine residues connected through a 1,2-trans linkage. They are implicated in important biological systems, such as structural polysaccharides (chitin), circulating signaling molecules (chitooligosaccharides3 and lipo-chitooligosaccharides4,5), tumor markers (sialyl-Lewis X), anticoagulants (heparin), glycoproteins (multi-antennary complex type N-glycans)6 or as an essential part of small molecules for various bioactivities.7 In all cases these structures are difficult to obtain from natural sources. The main challenge of these syntheses is the glycosidic linkage formation through glycosylation, one of the most studied reactions in organic synthesis,8 especially in the case of N-acetyl D-glucosamine.Under glycosylation conditions, the 2-acetamido group in sugar donors bearing various leaving groups at C-1 forms a rather stable 1,2-O,N-oxazoline, which must be opened by acceptors under appropriate conditions to give the trans glycoside.
Numerous β-selective glycosylation methods8,9 have been developed using elaborate glucosamine donors possessing temporary participating groups10–12 of the 2-amino function such as the well-known phthaloyl,13,14 trichloroethoxycarbonyl,15 trichloro- and trifluoroacetyl (TCA and TFA) groups,16,17 and more recently, the N-acetyl-2,3-oxazolidinone group.18,19 The appropriate leaving groups at C-1 are the trichloroacetimidate,20 phosphite,21 or thio groups.22 The reactions generally proceed at low temperature with high yields but require separate steps for the introduction of the protecting groups and the post-coupling conversion to the 2-acetamido substituent found in natural products. To date, these methods have been most commonly used for the synthesis of glycoconjugates bearing an N-acetylated-D-glucosamine residue.
Glycosylation with glycosyl acetate donors23 is a straightforward alternative to the above methods using donors bearing complex leaving groups at the anomeric position.9 It involves a direct acid-catalyzed exchange of the anomeric oxygen to provide the glycosidic acetal. Recently, stoichiometric cupric salts (CuCl2, CuBr2),24 30 mol% Yb(OTf)3,25 15 mol% rare earth metal triflates [Sc(OTf)3, Sm(OTf)3, La(OTf)3, Dy(OTf)3, Nd(OTf)3],26,27 H2SO4-silica under microwave conditions,28 and TsOH29 were used as promoters in the synthesis of glycosides of N-acetyl D-glucosamine (GlcNAc), directly or via the isolated 1,2-O,N-oxazolines. Activation using FeCl3 was also previously described for anomeric ester donors incorporating a C-2 amide functionality (N-acetyl, N-phthaloyl, N-chloroacetyl glycosyl acetate donors)30,31via the oxazolinium cations. It was also reported for other donors having a C-2 ester participatory group32–34 that react via the 1,2-acyloxonium ion. It involved a large excess of both FeCl3 and glycosyl donors producing, in the case of fluorogenic and serine acceptors, rather the α-anomer under anomerization conditions.35
Mild conditions using triflates of rare earth metals were previously reported.9,26,27 Iron36–39 has a number of advantages over other metals typically used in catalysis since it is cheap, non-toxic, environmentally friendly and abundant. In carbohydrate chemistry,40 iron(III) triflate has only been utilized in a few instances: oxidative C–C bond cleavage,41 thioglycosylation of peracetylated glycosides42 and type I Ferrier rearrangement of 2,4,6-tri-O-acetyl-D-glucal.43 Over the past few years, our laboratory has developed several step-saving options that have significantly shortened the synthetic route to bioactive glycoconjugates.44–49 Along these lines, we recently communicated50 the glycosylation of the stable and commercially available glucosaminyl donor 1β using, as the activator, catalytic amounts of stable and non-hygroscopic Fe(OTf)3·6.2DMSO.51 We present here a full account of this glycosylation: the catalysis design, the scope and limitations of the method, the scale-up using flow chemistry and some mechanistic elements.
Results and discussion
Catalytic system design
For optimization conditions of the glycosylation, glucose derivative 352 was selected as a test sugar acceptor and the results are presented in Table 1. The glycosylation reaction of donor 1β, prepared by acetylation of 1,3,4,6-tetra-O-acetyl-2-amino-2-deoxy-β-D-glucopyranose hydrochloride,53 was very slow at r.t. (entries 1 and 2, Table 1, 12–38%) and required heating under refluxing conditions for 84 hours in CH2Cl2 to furnish 4 in good yields with both Fe(OTf)3·6.2DMSO and Fe(OTf)3 (entries 5 and 6, Table 1, 86–87%). The same range of yields was also obtained using microwave irradiation at 110 °C for 45 min (entries 7 and 8, Table 1, 89–93%). At r.t., the catalyst Fe(OTf)3·6.2DMSO was less efficient than Fe(OTf)3 (entries 1 and 2, Table 1) and addition of 2,4,6-tri-tert-butylpyrimidine (TTBP) blocked the reaction (entries 3 and 4, Table 1). This did not occur under microwave irradiation at 110 °C (entries 7 and 8, Table 1). In previous experiments,50 we established that glycosylation of oxazoline 2 produced, similarly, the β-1,6 disaccharide using the Fe(OTf)3 solvate (15 mol%) under microwave irradiation and that no reaction occurred with the more stable donor 1α (Fig. 1).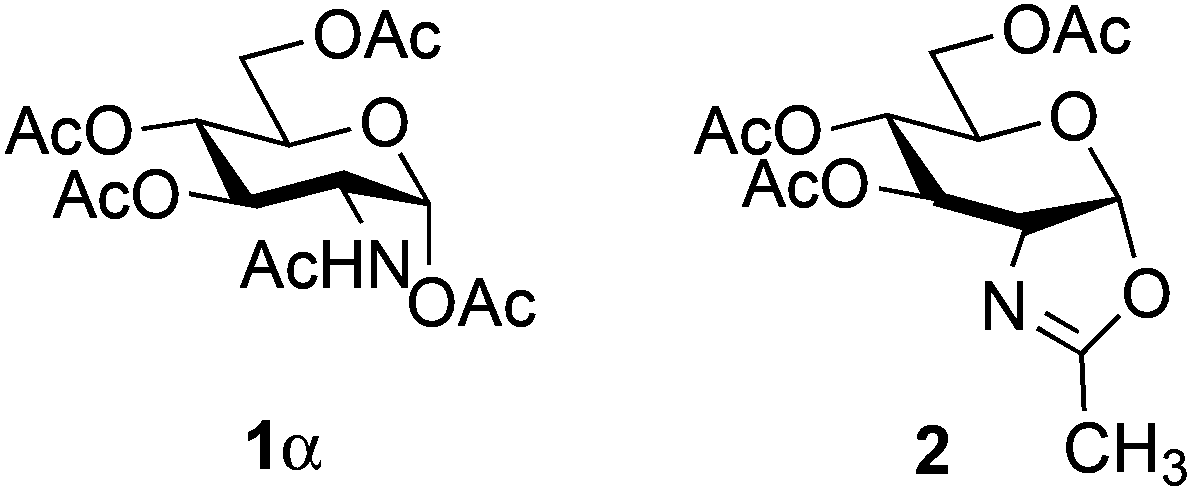 | ||
| Fig. 1 Glucosaminyl donor 1α and oxazoline 2. | ||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (15 mol%) | TTBPa | Temperature, time | Yieldb (product 4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a TTBP = 2,4,6-tri-tert-butylpyrimidine. b Yield after silica gel chromatography. c Microwave irradiation (Anton Paar device). d No reaction. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Fe(OTf)3·6.2DMSO | — | r.t., 96 h | 12% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Fe(OTf)3 | — | r.t., 96 h | 38% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Fe(OTf)3·6.2DMSO | 2 equiv. | r.t., 96 h | nrd | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Fe(OTf)3 | 2 equiv. | r.t., 96 h | nrd | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Fe(OTf)3·6.2DMSO | — | Reflux, 84 h | 87% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Fe(OTf)3 | — | Reflux, 84 h | 86% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Fe(OTf)3·6.2DMSO | 2 equiv. | 110 °Cc, 45 min | 89% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Fe(OTf)3 | 2 equiv. | 110 °Cc, 45 min | 93% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
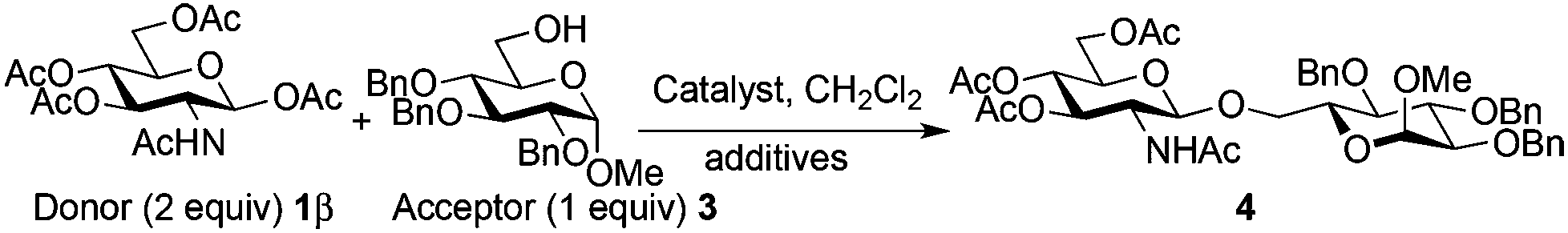
Table 2 shows that Fe(OTf)3·6.2DMSO or Fe(OTf)3 was superior to other Fe(III) salts (FeI3, FeCl3, Fe(NTf2)3·6.2DMSO) (entries 1, 4 vs. 3, 5, 13, Table 2, 90–92% vs. 31–59%), Sc(OTf)3 (entry 2, Table 2, 62% in our hands),26 and acidic conditions (TfOH) (entries 6 and 7, Table 2, 47–70%). The addition of TTBP (2 equiv.) (entries 7 and 8, Table 1 and entry 8, Table 2, 89–98%) optimized the procedure. Using another base such as 2,6-lutidine with Fe(OTf)3·6.2DMSO was inefficient to carry out the transformation. It is interesting to note that in dichloromethane, the Fe(OTf)3 solvate was not soluble at the onset of the reaction while the complex became soluble in the final medium. The dissolving of the Fe(III) salts occurred in acetonitrile but the yield of glycosylation decreased (entries 9 vs. 8, Table 2, 43 vs. 89–98%). The use of a mixture of CH2Cl2–CH3CN (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) or CHCl3–CH3CN (7:3) provided a soluble mixture all along the reaction course with only a slight decrease in the glycosylation yield (entries 10, 11 vs. 8, Table 2, 76–80 vs. 89–98%). This enabled the development of the reaction using a millifluidic device (see below). Interestingly, under our optimized conditions, Bi(OTf)3, an alternative cheap, non-toxic, environmentally friendly and abundant metal complex54–56 already described for the glycosylation of sialyl acetates,57 proved to be as effective as Fe(OTf)3·6.2DMSO (entries 12 vs. 8, Table 2, 88 vs. 89–98%).
:3) or CHCl3–CH3CN (7:3) provided a soluble mixture all along the reaction course with only a slight decrease in the glycosylation yield (entries 10, 11 vs. 8, Table 2, 76–80 vs. 89–98%). This enabled the development of the reaction using a millifluidic device (see below). Interestingly, under our optimized conditions, Bi(OTf)3, an alternative cheap, non-toxic, environmentally friendly and abundant metal complex54–56 already described for the glycosylation of sialyl acetates,57 proved to be as effective as Fe(OTf)3·6.2DMSO (entries 12 vs. 8, Table 2, 88 vs. 89–98%).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | TTBP (equiv.) | Solvent | Time | Yield %c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Under microwave irradiation at 80 °C (CEM Discover). b Under microwave irradiation at 110 °C (Anton Paar device). c Yield after silica gel chromatography. d The yield varies with the device used (CEM device 80 °C or Anton Paar device 110 °C). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Fe(OTf)3·6.2DMSOa (15) | — | CH2Cl2 | 30 min | 92% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Sc(OTf)3a (15) | — | CH2Cl2 | 180 min | 62% (75%)26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Fe/I2a (20) | — | CH2Cl2 | 60 min | 59% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Fe(OTf)3a (15) | — | CH2Cl2 | 30 min | 90% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | FeCl3a (15) | — | CH2Cl2 | 30 min | 31% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | TfOHa 0.45 equiv. | — | CH2Cl2 | 30 min | 47% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | TfOHa 0.45 equiv. | 2 | CH2Cl2 | 30 min | 70% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Fe(OTf)3·6.2DMSOaorb (15) | 2 | CH2Cl2 | 45 min | 89–98%d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Fe(OTf)3·6.2DMSOb (15) | 2 | CH3CN | 45 min | 43% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Fe(OTf)3·6.2DMSOb (15) | 2 | CH2Cl2–CH3CN 7:3 |
45 min | 80% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | Fe(OTf)3·6.2DMSOb (15) | 2 | CHCl3–CH3CN 7:3 |
45 min | 76% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | Bi(OTf)3a (15) | 2 | CH2Cl2 | 45 min | 88% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | Fe(NTf2)3·6.2DMSOa (15) | — | CH2Cl2 | 30 min | 51% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
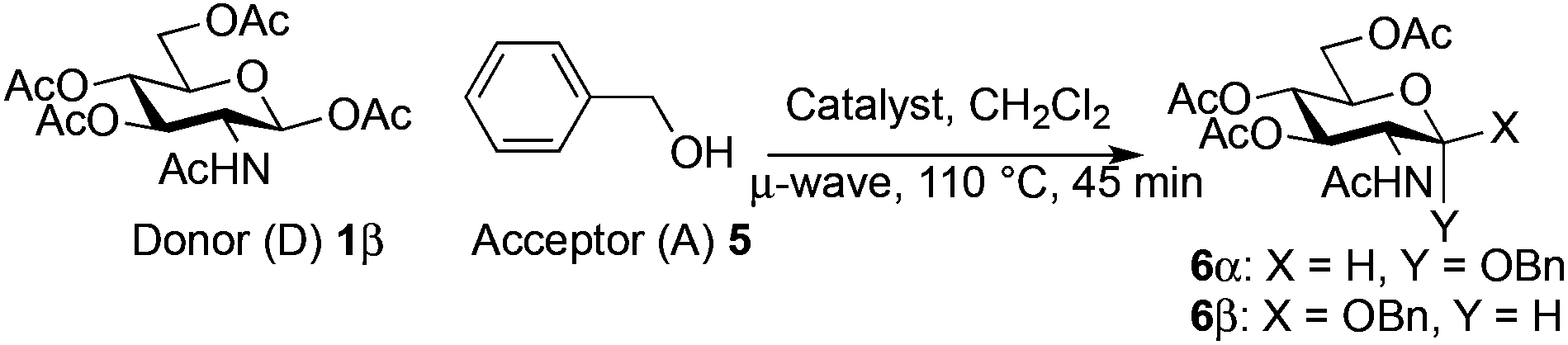
Using an excess of the reactive benzyl alcohol acceptor with the commercially available Fe(OTf)3 without TTBP (entry 5, Table 3), a large amount of α-anomer 635 was produced (α/β, 3:7). This was also observed but to a lesser extent with Fe(OTf)3·6.2DMSO (entry 7, Table 3, α/β 1:9). Proceeding with an excess of the donor (2 equiv.) and/or adding TTBP with the catalyst Fe(OTf)3 or Fe(OTf)3·6.2DMSO prevented this α-anomerization to occur26 (entries 1–4 and 6, Table 3).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | D:A (equiv.) |
Catalyst (15 mol%) | TTBP (equiv.) | Yield %b (ratio α/β)c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Under microwave irradiation with an Anton Paar device. b Yield after silica gel chromatography. c Ratio determined by 1H NMR. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 2:1 |
Fe(OTf)3·6.2DMSOa | 2 | 95% (<5:95) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 2:1 |
Fe(OTf)3·6.2DMSOa | — | 96% (<5:95) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 2:1 |
Fe(OTf)3a | 2 | 95% (<5:95) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 2:1 |
Fe(OTf)3a | — | 89% (5:95) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 1:2 |
Fe(OTf)3a | — | 79% (30:70) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 1:2 |
Fe(OTf)3a | 2 | 97% (<5:95) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 1:2 |
Fe(OTf)3·6.2DMSOa | — | 77% (10:90) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Iron triflate-catalyzed glycosylation under microwave irradiation
The scope of the β-glycosylation was evaluated with different acceptors using glycosyl donor 1β (Table 4). Our conditions led to an efficient glycosylation with highly reactive acceptors BnOH (entries 1, 3 and 6, Table 3, 95–97%) and 4-ClBnOH (entry 1, Table 4, 95%). Glycosylation of 2-chloroacetic acid provided a poor yield of the α/β-anomeric esters 10 (entry 2, 21%, α/β, 4/1, Table 4). The use of TTBP enabled the glycosylation of silylated or benzylidene acceptors (compounds 11, 13, 15 and 17) without degradation (entries 3–6, Table 4) with the recovered acceptor. For instance, the β(1 → 3) linked disaccharide 14 was obtained in 74% yield from donor 1β (entry 4, Table 4). This method could also be applied to the efficient formation of β(1 → 2) linked disaccharides 16 and 18 (entries 5 and 6, Table 4, 61–53%) with almost quantitative recovery of the acceptor. The reaction was tested in the synthesis of a β-1,4-glycosidic linkage between two D-glucopyranosyl units (donor 1β and acceptors of the glucose and glucosamine series 19, 21, 23, entries 7–10, Table 4). Very moderate yields were obtained (20–26%) (entries 7, 9 and 10, Table 4), with however a quantitative recovery of the acceptor (entry 7, Table 4). In the glucosaminyl series with a phthaloyl group at the C2 position, a 3-O-acetyl (compound 21) or 3-O-benzyl group (compound 23) (entries 9 and 10, Table 4) furnished the same amount of β-1,4-disaccharide 22 or 24 (23–25%). The N-acetyl-2,3-oxazolidinone58 acceptor 25 or the 1,6-anhydro acceptor 2759 (entries 11 and 12, Table 4), developed to enhance the nucleophilicity of the hydroxyl group at the C4 position, gave only traces of the glycosylation product 26 or no glycosylation. This could be due to the degradation of these acceptors or products in the reaction mixture. However, optimization by proceeding at higher concentration (0.65 M in acceptor) led to a slight increase in the yield of glycoside 20 to 37% (entry 8, Table 4).| Entry | Acceptor (A) | Product | D:A |
[A] (M) | Time | Yield %a | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Yield after silica gel chromatography. b 110 °C (Anton Paar device). c 70–80 °C (CEM device); for details see ESI. d 20 mol%, Fe(OTf)3; no TTBP. e Yield based on the recovered acceptor. f Donor was recovered as a mixture of anomers (1α/1β = 1/1; 24% combined yield). g No TTBP. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
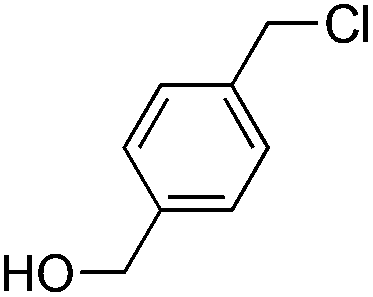
|
7 |
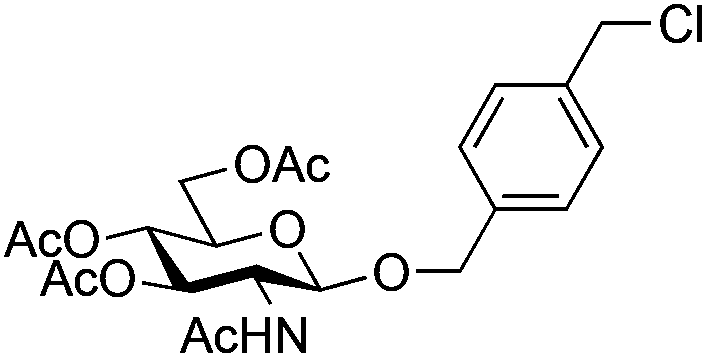
|
8 | 2:1 |
0.065 | 30 min | 95%b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |
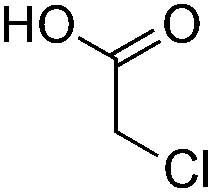
|
9 |
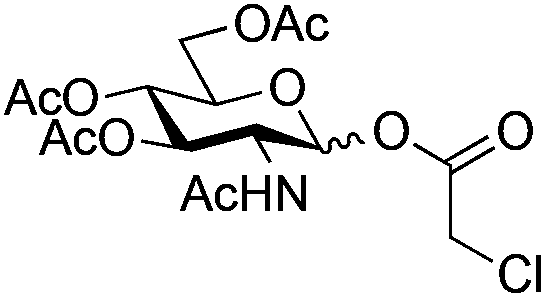
|
10 | 2.5:1 |
0.065 | 45 min | 21% (α/β 8:2)c,d |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |
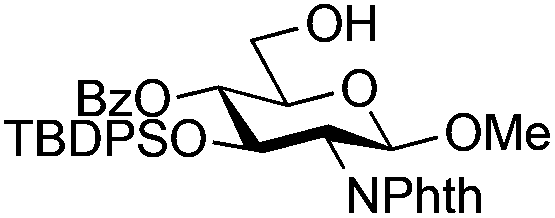
|
11 |
|
12 | 2:1 |
0.065 | 45 min | 76%c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
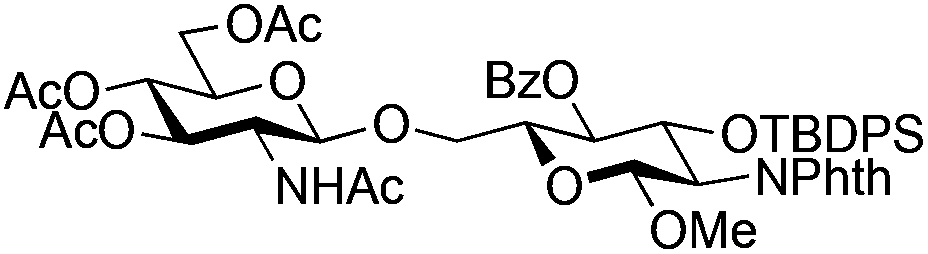
| 4 |
|
13 |
|
14 | 2:1 |
0.065 | 45 min | 74%b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |
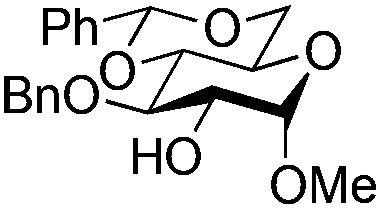
|
15 |
|
16 | 2:1 |
0.065 | 1 h | 61%b (90%)e | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
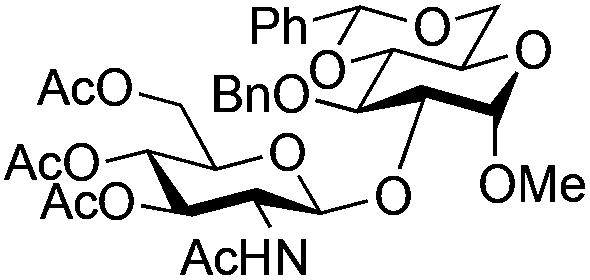
| 6 |
|
17 |
|
18 | 2:1 |
0.065 | 1 h | 53%b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 |
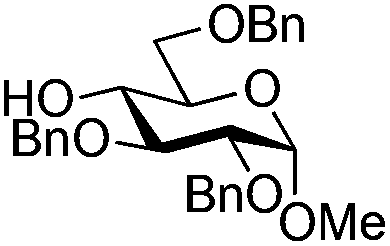
|
19 |
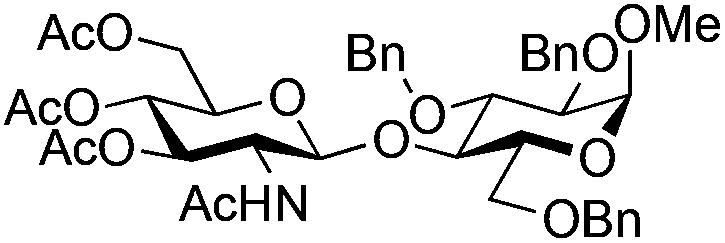
|
20 | 2:1 |
0.065 | 3 h | 20–26%b,c,f (>95%)e | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 19 | 20 | 2:1 |
0.65 | 3 h | 37%b (75%)e | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 |
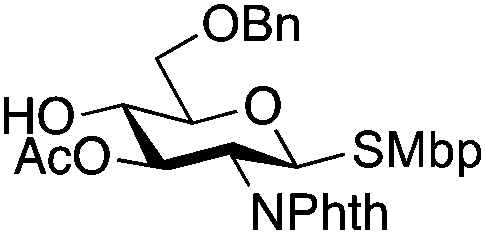
|
21 |
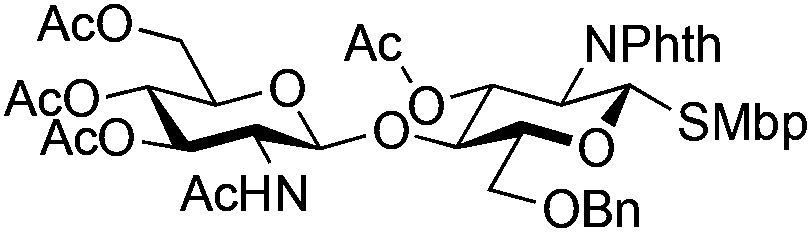
|
22 | 5:1 |
0.065 | 11 h | 23%c,g | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 |
|
23 |
|
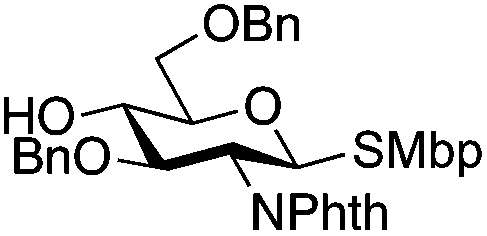
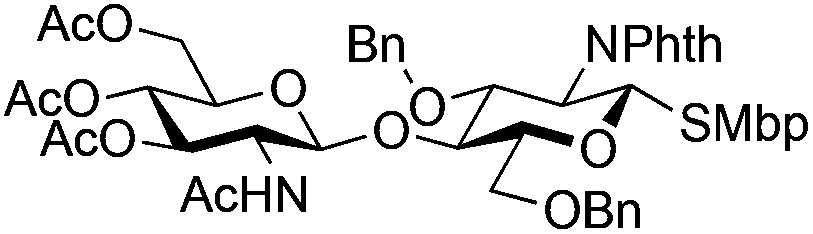
24 | 1:2 |
0.065 | 3 h | 25%c,g | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 |
|
25 |
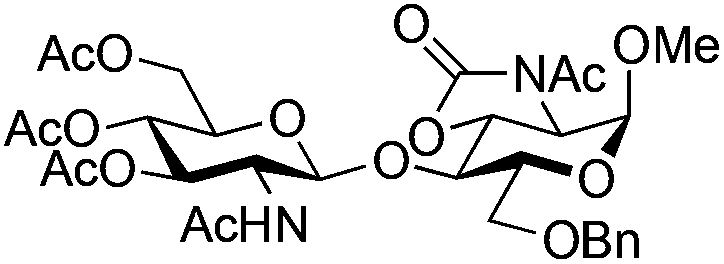
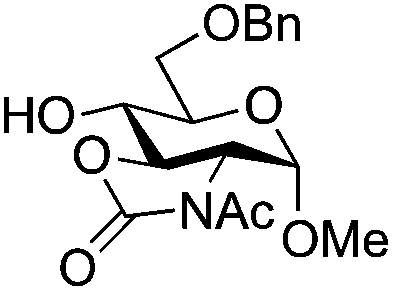
|
26 | 2:1 |
0.065 | 3 h | 7%c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 |
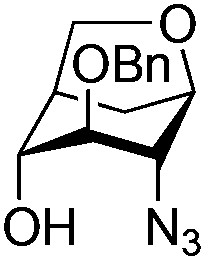
|
27 |
|
28 | 2:1 |
0.065 | 45 min | 0%c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
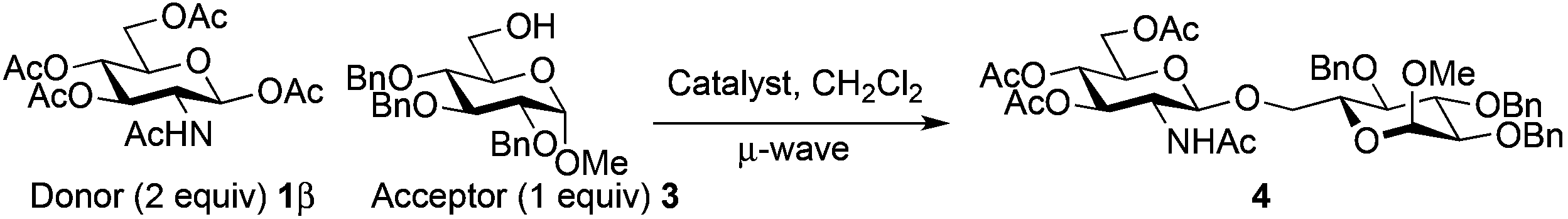
The glycosylation scope was then evaluated with various N-acetyl-D-glucosamine donors (10, 2, 29, 32) in the formation of β-1,6 and β-1,4-glycosidic linkages between two D-glucopyranosyl units using glycosyl acceptors 3, 19 and 21 (Table 5). Compared with donor 1β, the replacement of the anomeric acetate group by a chloroacetate group or the acetates at the 3,4,6-positions by benzyl groups had no significant effect on the glycosylation (entries 1–3, Table 5). However, the benzylidene donor 32 failed to give the expected β-(1 → 6) linked disaccharide (entry 4, Table 5). This result is in accordance with the stereoelectronic effect of the 4,6-O-benzylidene acetals of pyranosides stabilizing the C–O bond at the anomeric center.60 Oxazoline 2 furnished the β-(1 → 4) linked disaccharide 20 in poor yield (13%) (entry 5, Table 5). Glycosylation with the commercially available N-acetyl D-galactosamine donor 33 gave results similar to those of the N-acetyl D-glucosamine donor 1β in the formation of β-1,6; β-1,3; β-1,2 and β-1,4-glycosidic linkages (95–26%, entries 6–10, Table 5, versus (89–98%)–(20–26%), entry 8, Table 2 and entries 4–7, Table 4) with a quantitative recovery of acceptors. Under our harsh reaction conditions (microwave irradiation at 80–110 °C), variations of the oxygen protecting group at the 1, 3, 4 and 6 positions in donor or acceptor had no effect on the disaccharide yield and the course of the reaction. With our device for microwave irradiation, the iron triflate-catalyzed glycosylation scale-up was limited to the use of a 30 mL reactor versus a 10 mL reactor. This change induced a slight decrease in the yield (77% vs. 89% for 4, and 89% vs. 95% for 6β) probably as a consequence of the impaired heat transfer.
| Entry | Donor | Acceptor | Product (yield %)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Yield after silica gel chromatography. 15 mol% of Fe(OTf)3·6.2DMSO and TTBP (2 equiv.) in CH2Cl2. b 45–180 min, 80 °C, CEM Discover®; for details see ESI. c No reaction. d 30–180 min, 110 °C, Anton Paar Monowave 300®; for details see ESI. e Yield based on the recovered acceptor. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
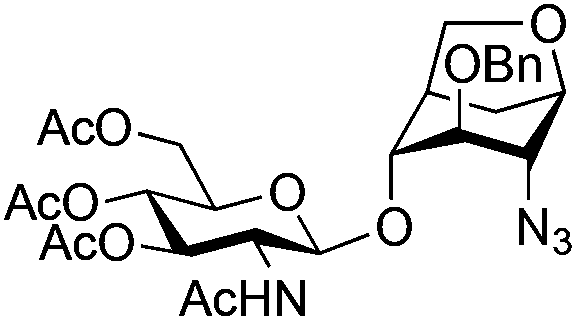
| 1 | 10 | 3 | 4 (73%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 29 (α/β 1/2) | 3 | 30 (86%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
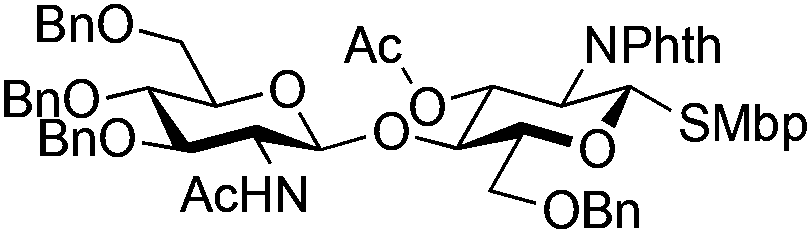
| 3 | 29 | 21 | 31 (14%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
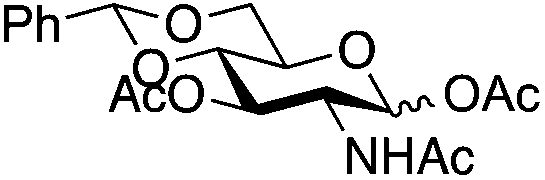
| 4 | 32 (α/β 1/1) | 3 | nrb,c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 2 | 19 | 20 (13%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
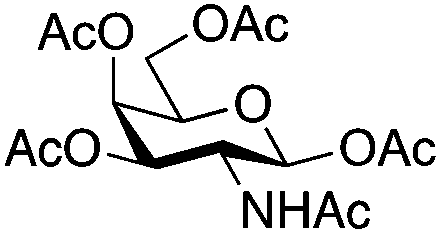
| 6 | 33 | 3 | 34 (95%)d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
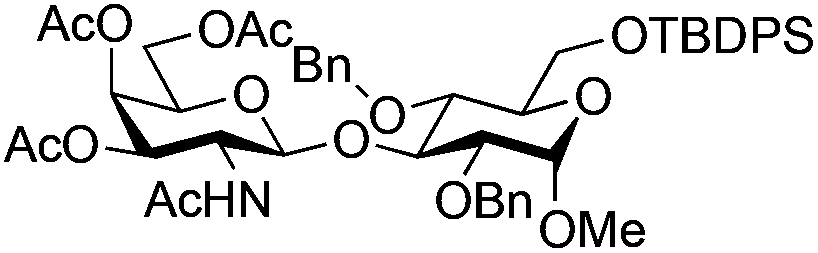
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 33 | 13 | 35 (75%)d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
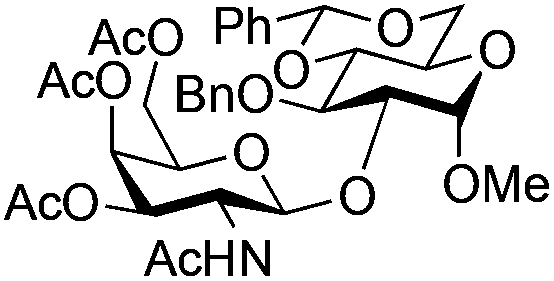
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 33 | 15 | 36 (63%)d (>95%)e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
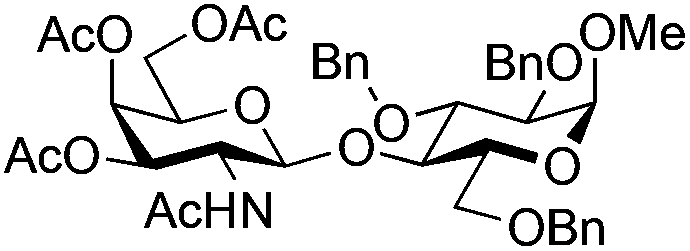
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 33 | 17 | 37 (55%)d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
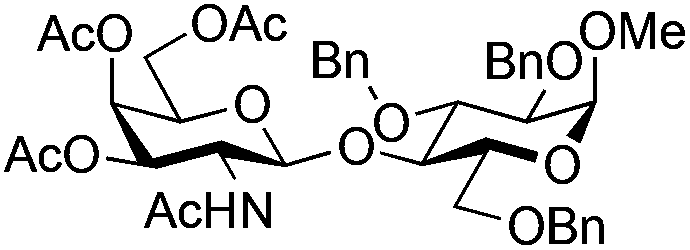
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
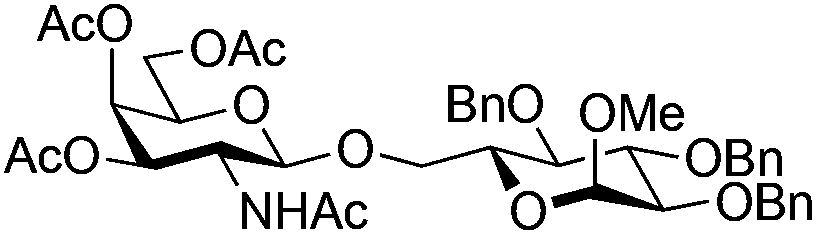
| 10 | 33 | 19 | 38 (26%)d (>95%)e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Flow chemistry
The above limitation can be overcome by transposing the reaction in flow chemistry.61,62 It has been demonstrated that micro- or minifluidic flow devices fitted with a backpressure regulator mimic high temperatures and pressures attainable in a sealed-vessel microwave chemistry batch experiment. Flow chemistry has already been used for glycosylation with success.63–65 The major limitation was the low solubility of donor 1β that required the use of a mixture of solvents (CH2Cl2–acetonitrile or CHCl3–acetonitrile) which induced a yield decrease under microwave irradiation (76–80% vs. 89–98%, entries 10 and 11 vs. 8, Table 2). In this study a Vapourtec R4-Unit was used as a millifluidic system. This system suppressed the tendency to block and does not limit the flow capacity observed with micro reactors when preparing substantial amounts of the product.66 The formation of disaccharides 4, 20 and benzyl glycosides 6β and 8 was studied using donor 1β and acceptor 3, 5, 7 or 19.The use of TTBP dramatically slowed down the process and decreased the yield of the reaction (entries 1 vs. 2, Table 6, 25 vs. 62%). A slight decrease of the yield was also observed with a decrease of the loading of Fe(OTf)3·6.2DMSO (entries 3 vs. 2, Table 6, 51 vs. 62%). The optimized temperature of the reactor was 110 °C (entries 2 vs. 4, Table 6, 62 vs. 44%) and higher temperatures increased degradation. A higher pressure (33 vs. 25 bar) associated with a longer residence time (70 vs. 45 min) and a more concentrated reaction mixture in the acceptor (0.15 M) with an excess of donor 1β gave a 78% yield of 4 with high recovery of the unreacted acceptor. This was also obtained with the commercially available Fe(OTf)3 which provided a 75% yield of 4 (entry 8, Table 6). The same yield range was obtained for benzyl glycosides 6β (77%, entry 9, Table 6) and 8 (75%, entry 12, Table 6). An excess of benzyl alcohol 5 (2 equiv./1β) decreased the yield of 6β (62%, entry 10, Table 6) without the formation of 6α as observed under microwave heating. A residence time of only 30 min, more practical for a g-scale production, allowed to maintain an acceptable yield of 6β (73%, entry 11, Table 6) as well with the Fe(OTf)3 catalyst (77%). Our conditions were ineffective for the formation of the β-1,4-glycosidic linkage (<10%, entry 13, Table 6).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Acceptor (equiv.) | Catalyst (mol%) | TTBP | 1β: equiv.; concentration | Pressure | Temperature | Residence time | Product (yield %)a | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Yield after silica gel chromatography. b Yield based on the recovered acceptor. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 3 (1) | Fe(OTf)3·6.2DMSO (15) | 2 equiv. | 2; 0.1 M | 25 bar | 110 °C | 45 min | 4 (25%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 3 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.1 M | 25 bar | 110 °C | 45 min | 4 (62%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 3 (1) | Fe(OTf)3·6.2DMSO (10) | — | 2; 0.1 M | 25 bar | 110 °C | 45 min | 4 (51%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 3 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.1 M | 25 bar | 100 °C | 45 min | 4 (44%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 3 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.1 M | 33 bar | 110 °C | 45 min | 4 (70%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 3 (1) | Fe(OTf)3·6.2DMSO (15) | — | 1; 0.1 M | 33 bar | 110 °C | 45 min | 4 (45%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 3 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.15 M | 33 bar | 110 °C | 70 min | 4 (74–78%) (86%)b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 3 (1) | Fe(OTf)3 (15) | — | 2; 0.15 M | 33 bar | 110 °C | 70 min | 4 (75%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 5 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.15 M | 33 bar | 110 °C | 45 min | 6β (77%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 5 (2) | Fe(OTf)3·6.2DMSO (15) | — | 1; 0.15 M | 33 bar | 110 °C | 45 min | 6β (62%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 5 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.15 M | 33 bar | 110 °C | 30 min | 6β (73%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 7 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.15 M | 33 bar | 110 °C | 45 min | 8 (75%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 19 (1) | Fe(OTf)3·6.2DMSO (15) | — | 2; 0.15 M | 33 bar | 110 °C | 70 min | 20 (<10%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
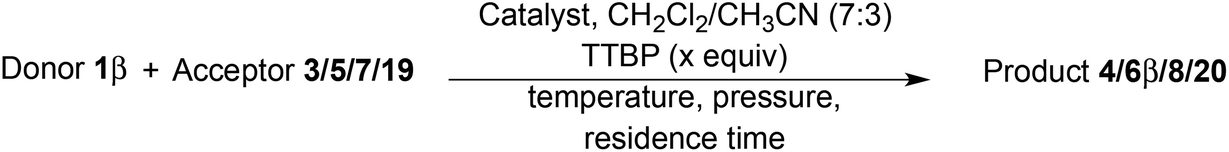
Extending the glycosylation reaction to a continuous flow process without further changes proceeded with good yields (75–78%) using chloroform instead of dichloromethane. Due to its high volatility, dichloromethane was not suitable with our flow chemistry device for long injection times. This procedure delivered 2 g (2.52 mmol) of disaccharide 4 with a 50 mL injected volume (Fig. 2).
 | ||
| Fig. 2 Flow system used for glycosylation reactions under optimized conditions. | ||
Mechanistic studies
Starting from GlcNAc glycosyl donors for the one-step synthesis of β-D-GlcNAc glycopyranosides, oxazolinium ion C (Scheme 1) is expected to be the intermediate, justifying the high β-stereoselectivity.1,9 However, the glycosylation results with the less nucleophilic 4-OH acceptor 19 and oxazoline 2 compared to β-acetate 1 (13% yield, entry 5, Table 5vs. 20–26% yield, entry 7, Table 4) were different. This suggested that the reaction may not proceed via this intermediate and another route to the glycoside may operate. To study this possibility, glycosylation with iron(III) triflate was examined by modulating the electronic and/or the steric properties of the N-substituent in D-glucosaminyl donors 39–47. This was done by choosing the glycosylation of primary alcohol 3 under the optimized conditions (entry 1, Table 7) as a reference glycosylation reaction. Similar to 1β, formyl amide 39 and tolyl amide 40 (entries 3 and 4, Table 7) provided the expected glycosides 48 and 49, while carbamate 41, trichloroacetamide 42, trifluoroacetamide 43, phthalimide 46 or pivaloyl amide 45 (entries 6, 7, 9, 13 and 11, Table 7) were completely ineffective or significantly less effective (chloroacetamide 44, entry 10, Table 7). In the case of the diacetamide 47, one acetyl group was transferred to the acceptor providing 58, without detecting the formation of the disaccharide (entries 14, Table 7). These negative results should be compared with 2-deoxy-2-trichloroacetamido17 and 2-deoxy-2-trifluoroacetamido10,67,68 derivatives equipped with a good leaving group at the anomeric carbon (e.g., trichloroacetimidate). When activated with appropriate promoters (e.g., Me3SiOTf), they are known to be good glycosyl donors through the formation of the oxazolinium ion intermediate.17 In the absence of nucleophile, oxazolines 2, 56 and 57 (entries 2, 8 and 12, Table 7) were not detected except for oxazoline 55 from the tolylamide 40 (entry 5, Table 7). These experiments suggest that glycosylation would proceed through an alternative intermediate and not necessarily through the oxazolinium ion. Glycosylation may require a pre-complexation of the catalyst by a suitable amide group such as the acetamide present in 1β (see A, Scheme 1), the tolyl amide in 40 or the formyl amide in 39 before the activation of the anomeric acetate occurs. Effective amide pre-complexation of Fe(OTf)3·6.2DMSO may be partially or totally prevented for electronic reasons (NHTCA, donor 42; NHTFA, donor 43; NHAcCl, donor 44) or steric grounds (NHPiv, donor 45; NPhth, donor 46), thus preventing glycoside formation as experimentally observed. Alpha–ionic pair B from 1β, instead of oxazolinium ion C, would then encourage the glycosylation to take place from the β face by shielding the α face.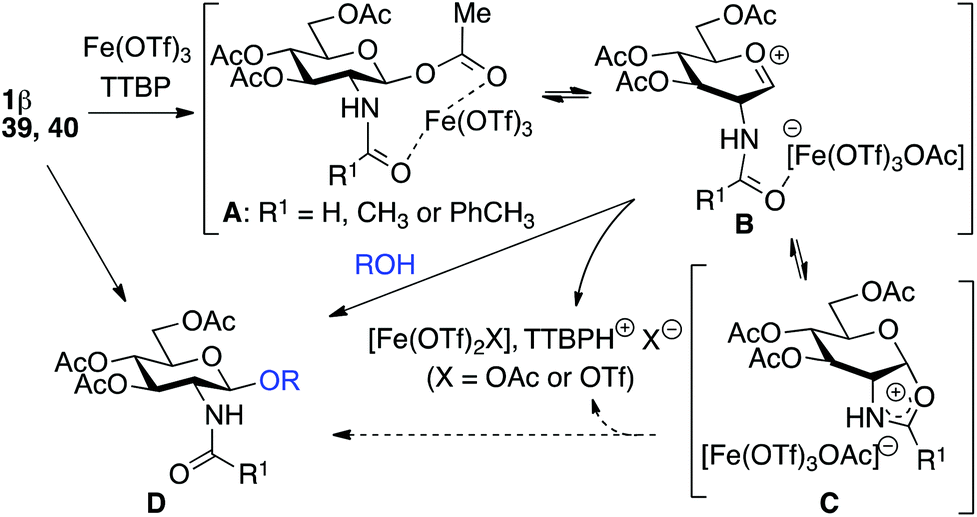 | ||
| Scheme 1 Possible mechanism for the iron triflate-catalyzed glycosylation. | ||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Product (yield %)a | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Yield after silica gel chromatography. b Reaction performed in the presence of TTBP (2 equiv.) with 15 mol% of Fe(OTf)3·6.2DMSO in CH2Cl2/CEM device 80 °C, 45–60 min. c 1β/1α ratio of 4/1. d No reaction. e Inseparable mixture with the donor, conversion determined by 1H NMR. f Traces detected by UPLC-MS/DAD. g Reaction performed in the presence of TTBP (2 equiv.)/Anton Paar device 110 °C, 45 min. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1β | 3 | 4 (98%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1β | None | 1α–1β (100%)c | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 39 R1 = R2 = H | 3 | 48 (50%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 40 R1 = PhCH3, R2 = H | 3 | 49 (82%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 40 R1 = PhCH3, R2 = H | None | 55 (62%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 41 R1 = OCH2Ph, R2 = H | 3 | nrb,d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 42 R1 = CCl3, R2 = H | 3 | 50 (<5%)b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 42 R1 = CCl3, R2 = H | None | nrb,d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 43 R1 = CF3, R2 = H | 3 | nrb,d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 44 R1 = CH2Cl, R2 = H | 3 | 51 (30%)b,e | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 45 R1 = C(CH3)3, R2 = H | 3 | 52 (<5%)b,f | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 45 R1 = C(CH3)3, R2 = H | None | nrb,d | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 46 R1 = R2 = Phth | 3 | 53 (<5%)f,g | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 47 R1 = CH3, R2 = COCH3 | 3 |
5869 (40%)b |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
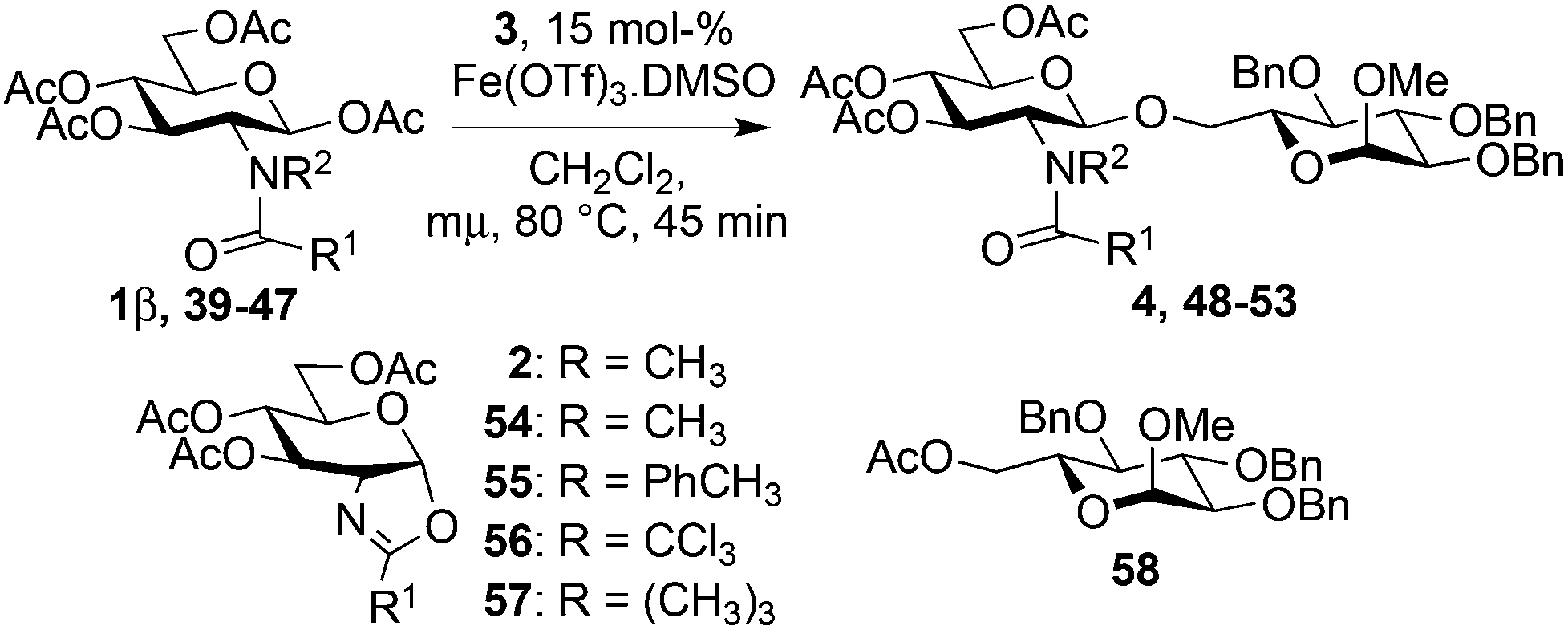
It is noteworthy that the reaction scale-up in the preparation of disaccharide 4 allowed the isolation of a small amount of oxazoline 2, suggesting a partial contribution of the oxazolinium ion C in the formation of the glycoside. A possible equilibrium between B and C could be envisioned depending on the nature of the group R1 favoring one or the other.
Conclusion
This novel catalytic glycosylation using peracetylated β-GlcNAc 1β and β-GalNAc 33 as glycosyl donors with Fe(III) triflate and TTBP is effective in the direct synthesis of β-GlcNAc and β-GalNAc glycosides but has not yet been efficient using less nucleophilic sugar acceptors. Our results suggest a possible mechanism which proceeds mostly by intermediates not involving the unique oxazolinium ion. We have demonstrated that the Fe(III) triflate glycosylation conducted under microwave irradiation is amenable to flow chemistry without requiring the presence of TTBP.Experimental section
Typical procedure for microwave-assisted glycosylation
The donor 1β (50 mg, 0.128 mmol, 2 equiv.), TTBP (32 mg, 0.129 mmol, 2 equiv.) and Fe(OTf)3·6.2DMSO (10 mg, 0.010 mmol, 15 mol%) were added to the acceptor 3 (30 mg, 0.065 mmol, 1 equiv.) in an oven-dried, argon-purged microwave vial equipped with a magnetic stirring bar. Everything was flushed under argon and dry CH2Cl2 (1 mL) was added. After sealing the vial, the reaction mixture was heated to 110 °C under microwave irradiation for 45 min (1 minute ramp time from room temperature to 110 °C and 45 min hold time at 110 °C, stirring set at 800 rpm). The reaction mixture was diluted with CH2Cl2 (20 mL) and washed with a saturated aqueous solution of NaHCO3 (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel (heptane–EtOAc 5:5 to 0:1) to afford the pure product 4 (46 mg, 89%, white amorphous solid).
Typical procedure for glycosylations under continuous flow conditions
The donor 1β (2.92 g, 7.50 mmol, 2 equiv.) and Fe(OTf)3·6.2DMSO (556 mg, 0.56 mmol, 15 mol%) were added to the acceptor 3 (1.74 g, 3.75 mmol, 1 equiv.) in an oven-dried, argon purged vial equipped with a magnetic stirring bar. A dry mixture of chloroform–acetonitrile 7:3 (50 mL) was added and the reaction mixture was stirred and sonicated for a few minutes (until complete homogenisation). After setting up and drying the whole flow system with dry chloroform–acetonitrile 7:3, the pump was primed and the reaction mixture (contained in an argon overpressured vial) is pumped into two 10 mL stainless steel reactors in series, heated at 110 °C with a flow rate of 0.286 mL min−1 (corresponding to a residence time of 70 min). The system pressure, controlled with a back pressure regulator, was fixed at 33 bars and the reaction mixture was finally collected into a single receptor. The reaction mixture was diluted with dichloromethane (250 mL) and washed with a saturated aqueous solution of NaHCO3 (100 mL). The aqueous layer was extracted with CH2Cl2 (4 × 100 mL) and the combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The crude product was purified by flash chromatography on silica gel (heptane–EtOAc 5:5 to 0:1) to afford the pure product 4 (2.00 g, 78%, white amorphous solid).
Acknowledgements
The authors thank R. Beau for her comments on the manuscript. We are grateful to the Institut de Chimie des Substances Naturelles (ICSN), the Institut Universitaire de France (IUF) for the financial support of this study. The CHARM3AT Labex program is also acknowledged for its support.Notes and references
-
P. Sinaÿ, B. Ernst and G. Hart, Carbohydrates in Chemistry and Biology, Wiley VCH, 2000 Search PubMed
.
-
A. Varki, J. D. Esko, R. D. Cummings, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of Glycobiology, Cold Spring Harbour Laboratory Press, Cold Spring Harbor, NY, 2nd edn, 2009 Search PubMed
- N. Shibuya and E. Minami, Physiol. Mol. Plant Pathol., 2001, 59, 223–233 CrossRef CAS
- J. Dénarié, F. Debellé and J. C. Promé, Annu. Rev. Biochem., 1996, 65, 503–535 CrossRef PubMed
- F. Maillet, V. Poinsot, O. André, V. Puech-Pagès, A. Haouy, M. Gueunier, L. Cromer, D. Giraudet, D. Formey, A. Niebel, E. A. Martinez, H. Driguez, G. Bécard and J. Dénarié, Nature, 2011, 469, 58–63 CrossRef CAS PubMed
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G. J. Boons, Science, 2013, 341, 379–383 CrossRef CAS PubMed
- M. E. Jung and P. Koch, Org. Lett., 2011, 13, 3710–3713 CrossRef CAS PubMed
- X. M. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed
-
A. V. Demchenko, Handbook of Chemical Glycosylation, Wiley-VCH, 2008 Search PubMed
- A. F. G. Bongat and A. V. Demchenko, Carbohydr. Res., 2007, 342, 374–406 CrossRef CAS PubMed
- R. Enugala, L. C. Carvalho, M. J. Dias Pires and M. M. Marques, Chem. – Asian J., 2012, 7, 2482–2501 CrossRef CAS PubMed
- Y. Yang and B. Yu, Tetrahedron, 2014, 70, 1023–1046 CrossRef CAS PubMed
- R. U. Lemieux, T. Takeda and B. Chung, Abstr. Pap. Am. Chem. Soc., 1976, 6–6 Search PubMed
- J. Banoub, P. Boullanger and D. Lafont, Chem. Rev., 1992, 92, 1167–1195 CrossRef CAS
- W. Dullenkopf, J. C. Castro-Palomino, L. Manzoni and R. R. Schmidt, Carbohydr. Res., 1996, 296, 135–147 CrossRef CAS
- M. L. Wolfrom and H. B. Bhat, J. Org. Chem., 1967, 32, 1821–1823 CrossRef CAS
- G. Blatter, J.-M. Beau and J.-C. Jacquinet, Carbohydr. Res., 1994, 260, 189–202 CrossRef CAS
- Y. Geng, L.-H. Zhang and X.-S. Ye, Chem. Commun., 2008, 597–599 RSC
- J. D. M. Olsson, L. Eriksson, M. Lahmann and S. Oscarson, J. Org. Chem., 2008, 73, 7181–7188 CrossRef CAS PubMed
- M. R. E. Aly, E.-S. I. Ibrahim, E. S. H. El Ashry and R. R. Schmidt, Carbohydr. Res., 2001, 331, 129–142 CrossRef CAS
- R. Arihara, S. Nakamura and S. Hashimoto, Angew. Chem., Int. Ed., 2005, 44, 2245–2249 CrossRef CAS PubMed
- S. Yamago, T. Yamada, T. Maruyama and J. Yoshida, Angew. Chem., Int. Ed., 2004, 43, 2145–2148 CrossRef CAS PubMed
- B. Helferich and E. Schmitz-Hillebrecht, Ber. Dtsch. Chem. Ges., 1933, 66, 378–383 CrossRef PubMed
- V. Wittmann and D. Lennartz, Eur. J. Org. Chem., 2002, 1363–1367 CrossRef CAS
- C. F. Crasto and G. B. Jones, Tetrahedron Lett., 2004, 45, 4891–4894 CrossRef CAS PubMed
- H. Christensen, M. S. Christiansen, J. Petersen and H. H. Jensen, Org. Biomol. Chem., 2008, 6, 3276–3283 CAS
- J. Krag, M. S. Christiansen, J. G. Petersen and H. H. Jensen, Carbohydr. Res., 2010, 345, 872–879 CrossRef CAS PubMed
- S. Mandal, N. Sharma and B. Mukhopadhyay, Synlett, 2009, 3111–3114 CAS
- Y. Cai, C.-C. Ling and D. R. Bundle, Org. Lett., 2005, 7, 4021–4024 CrossRef CAS PubMed
- M. Kiso and L. Anderson, Carbohydr. Res., 1985, 136, 309–323 CrossRef CAS
- F. Dasgupta and L. Anderson, Carbohydr. Res., 1990, 202, 239–255 CrossRef CAS
- S. K. Chatterjee and P. Nuhn, Chem. Commun., 1998, 1729–1730 RSC
- S. Koto, M. Hirooka, T. Tashiro, M. Sakashita, M. Hatachi, T. Kono, M. Shimizu, N. Yoshida, S. Kurasawa, N. Sakuma, S. Sawazaki, A. Takeuchi, N. Shoya and E. Nakamura, Carbohydr. Res., 2004, 339, 2415–2424 CrossRef CAS PubMed
- J. Seibel, L. Hillringhaus and R. Moraru, Carbohydr. Res., 2005, 340, 507–511 CrossRef CAS PubMed
- G. H. Wei, X. Lv and Y. Du, Carbohydr. Res., 2008, 343, 3096–3099 CrossRef CAS PubMed
- B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS PubMed
- D. D. Diaz, P. O. Miranda, J. I. Padron and V. S. Martin, Curr. Org. Chem., 2006, 10, 457–476 CrossRef
- C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed
- E. B. Bauer, Curr. Org. Chem., 2008, 12, 1341–1369 CrossRef CAS
-
J.-M. Beau, Y. Bourdreux, F.-D. Boyer, S. Norsikian, D. Urban, G. Doisneau, B. Vauzeilles, A. Gouasmat, A. Lemétais, A. Mathieu, J.-F. Soulé, A. Stévenin and A. Xolin, in Carbohydrate Chemistry, The Royal Society of Chemistry, 2014, vol. 40, pp. 118–139 Search PubMed
- S. Ichikawa, I. Tomita, A. Hosaka and T. Sato, Bull. Chem. Soc. Jpn., 1988, 61, 513–520 CrossRef CAS
- S. S. Weng, Tetrahedron Lett., 2009, 50, 6414–6417 CrossRef CAS PubMed
- P. Chen and S. Wang, Tetrahedron, 2012, 68, 5356–5362 CrossRef CAS PubMed
- N. Grenouillat, B. Vauzeilles, J. J. Bono, E. Samain and J.-M. Beau, Angew. Chem., Int. Ed., 2004, 43, 4644–4646 CrossRef CAS PubMed
- A. Français, D. Urban and J.-M. Beau, Angew. Chem., Int. Ed., 2007, 46, 8662–8665 CrossRef PubMed
- Y. Bourdreux, A. Lemétais, D. Urban and J.-M. Beau, Chem. Commun., 2011, 47, 2646–2648 RSC
- J.-F. Soulé, A. Mathieu, S. Norsikian and J.-M. Beau, Org. Lett., 2010, 12, 5322–5325 CrossRef PubMed
- S. Zameo, B. Vauzeilles and J.-M. Beau, Angew. Chem., Int. Ed., 2005, 44, 965–969 CrossRef CAS PubMed
- A. Malapelle, Z. Abdallah, G. Doisneau and J.-M. Beau, Angew. Chem., Int. Ed., 2006, 45, 6016–6020 CrossRef CAS PubMed
- A. Stévenin, F.-D. Boyer and J.-M. Beau, Eur. J. Org. Chem., 2012, 1699–1702 CrossRef PubMed
- S. Antoniotti and E. Duňach, Chem. Commun., 2008, 993–995 RSC
- T. Ishikawa, Y. Shimizu, T. Kudoh and S. Saito, Org. Lett., 2003, 5, 3879–3882 CrossRef CAS PubMed
- H. Myszka, D. Bednarczyk, M. Najder and W. Kaca, Carbohydr. Res., 2003, 338, 133–141 CrossRef CAS
- H. Gaspard-Iloughmane and C. Le Roux, Eur. J. Org. Chem., 2004, 2517–2532 CrossRef CAS PubMed
- V. Mandadapu, F. Wu and A. I. Day, Org. Lett., 2014, 16, 1275–1277 CrossRef CAS PubMed
- J. R. Desmurs, M. Labrouillère, C. Le Roux, H. Gaspard, A. Laporterie and J. Dubac, Tetrahedron Lett., 1997, 38, 8871–8874 CrossRef CAS
- K. Ikeda, Y. Torisawa, T. Nishi, J. Minamikawa, K. Tanaka and M. Sato, Bioorg. Med. Chem., 2003, 11, 3073–3076 CrossRef CAS
- D. Crich and A. U. Vinod, J. Org. Chem., 2005, 70, 1291–1296 CrossRef CAS PubMed
- D. Tailler, J.-C. Jacquinet and J.-M. Beau, J. Chem. Soc., Chem. Commun., 1994, 1827–1828 RSC
- H. H. Jensen, L. U. Nordstrom and M. Bols, J. Am. Chem. Soc., 2004, 126, 9205–9213 CrossRef CAS PubMed
- T. N. Glasnov and C. O. Kappe, Chem. – Eur. J., 2011, 17, 11956–11968 CrossRef CAS PubMed
- K. S. Elvira, X. Casadevall i Solvas, R. C. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905–915 CrossRef CAS PubMed
- D. M. Ratner, E. R. Murphy, M. Jhunjhunwala, D. A. Snyder, K. F. Jensen and P. H. Seeberger, Chem. Commun., 2005, 578–580 CAS
- F. R. Carrel, K. Geyer, J. D. C. Codee and P. H. Seeberger, Org. Lett., 2007, 9, 2285–2288 CrossRef CAS PubMed
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed
- J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC
- L. G. Weaver, Y. Singh, J. T. Blanchfield and P. L. Burn, Carbohydr. Res., 2013, 371, 68–76 CrossRef CAS PubMed
- D. J. Silva, H. Wang, N. M. Allanson, R. K. Jain and M. J. Sofia, J. Org. Chem., 1999, 64, 5926–5929 CrossRef CAS
- M. Giordano, A. Iadonisi and A. Pastore, Eur. J. Org. Chem., 2013, 3137–3147 CrossRef CAS PubMed
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Preparation, 1H and 13C NMR spectra for novel compounds. See DOI: 10.1039/c4qo00183d |
| This journal is © the Partner Organisations 2014 |