
N-heterocyclic carbene catalysed oxidative esterification of aliphatic aldehydes†‡
Ramesh C.
Samanta
and
Armido
Studer
*
Organisch-Chemisches Institut and NRW Graduate School of Chemistry, Westfalische Wilhelms-Universitat, Corrensstraße 40, 48149, Munster, Germany. E-mail: studer@uni-muenster.de; Fax: (+49) 251 83-36523
First published on 22nd July 2014
Abstract
N-heterocyclic carbene-catalyzed oxidative esterification of various aliphatic aldehydes is reported. A commercially available bisquinone is used as an external oxidant and rubidium carbonate is best suited as base to run these processes. With the novel method various aliphatic esters are directly obtained in good to excellent yields from the corresponding aldehydes and alcohols.
Esters are ubiquitous functional groups in organic chemistry due to their wide abundance and because they are easily converted to other valuable functional groups.1 Esters are generally prepared via stoichiometric activation of a carboxylic acid to an intermediate active acyl donor with subsequent acyl transfer to an alcohol.
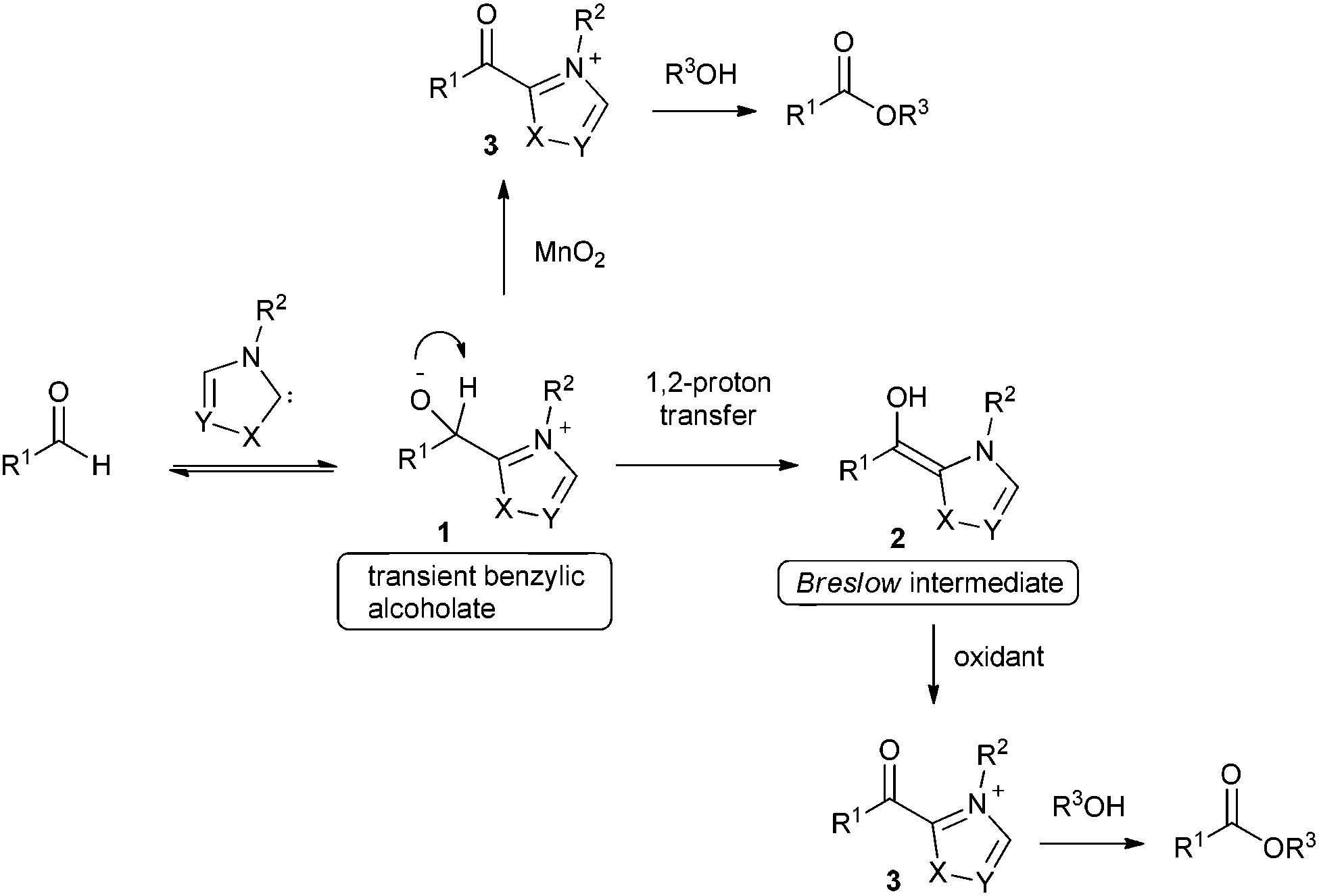
As an alternative, great progress has been made on oxidative esterification of aldehydes using N-heterocyclic carbene catalysis over the last few years.2 The proposed mechanism for most of these transformations involves addition of a carbene catalyst to the aldehyde resulting in formation of intermediate 1 followed by subsequent proton transfer to give the corresponding Breslow intermediate 2 (Scheme 1). Electron-rich enol 2 is then oxidized to acyl azolium ion 3 either by employing internal oxidants3 or via stoichiometric external oxidants e.g. nitrobenzene,4 MnO2,5 TEMPO,6 diphenoquinone,7 azobenzene,8 riboflavin, phenazine9 and a catalytic metal oxidant in combination with molecular oxygen as cheap terminal oxidant.10 Moreover, oxidative esterification was also achieved by anodic electrochemical oxidation11 of the Breslow intermediate or by using atmospheric oxygen as oxidant.5h,12
 | ||
| Scheme 1 Oxidative NHC-catalysed esterification of aldehydes. | ||
In contrast to most other oxidants reported which act on the Breslow intermediate, MnO2 was suggested to oxidize the transient benzyl alcoholate of type 1 (see Scheme 1). However, as documented by many examples, oxidative esterification via the Breslow intermediate seems to be more valuable and the diphenoquinone 7 (see Table 1) turned out to be an ideal stoichiometric reagent to oxidize intermediate 2 to an acyl azolium ion 3, which readily reacts with various alcohols to the corresponding esters. Therefore, this approach was also pursued for the current work. Although the reported 7-mediated NHC catalysed aldehyde oxidation has high potential in terms of functional group tolerance, the process works efficiently only for oxidative esterification of enals, alkynals, aromatic and heteroaromatic aldehydes. Aliphatic aldehydes turned out to be not good substrates for that transformation. Herein we present an efficient method for oxidative esterification of aliphatic aldehydes.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Precatalyst (eq.) | Base (eq.) | Temperature | Time | Yield (%) |
| 1 | 4 (0.20) | DBU (1.0) | rt | 72 h | 70 |
| 2 | 4 (0.20) | DBU (1.0)/catechol (0.20) | rt | 46 h | 58 |
| 3 | 4 (0.20) | DBU (1.0) | 60 °C | 30 h | 68 |
| 4 | 4 (0.20) | 5 (1.0) | rt | 72 h | 65 |
| 5 | 4 (0.20) | 6 (1.0) | 60 °C | 26 h | 44 |
| 6 | 4 (0.20) | K2CO3 (1.0) | rt | 40 h | 47 |
| 7 | 4 (0.20) | Cs2CO3 (1.0) | rt | 24 h | Traces |
| 8 | 4 (0.20) | Rb2CO3 (1.0) | rt | 40 h | 86 |
| 9 | 4 (0.20) | Rb2CO3 (0.50) | rt | 20 h | 88 |
| 10 | 4 (0.20) | Rb2CO3 (1.5) | rt | 12 h | 88 |
| 11 | 4 (0.20) | Rb2CO3 (2.0) | rt | 6 h | 88 |
| 12 | 4 (0.20) | Rb2CO3 (2.5) | rt | 6 h | 89 |
| 13 | 4 (0.10) | Rb2CO3 (2.0) | rt | 26 h | 77 |
| 14 | 4 (0.050) | Rb2CO3 (2.0) | rt | 48 h | 60 |
| 15 | 4 (0.050) | Rb2CO3 (2.0) | 60 °C | 22 h | 75 |
| 16 | 4 (0.10) | Rb2CO3 (2.0) | 60 °C | 16 h | 84 |
| 17 | 4 (0.075) | Rb2CO3 (2.0) | 60 °C | 20 h | 82 |
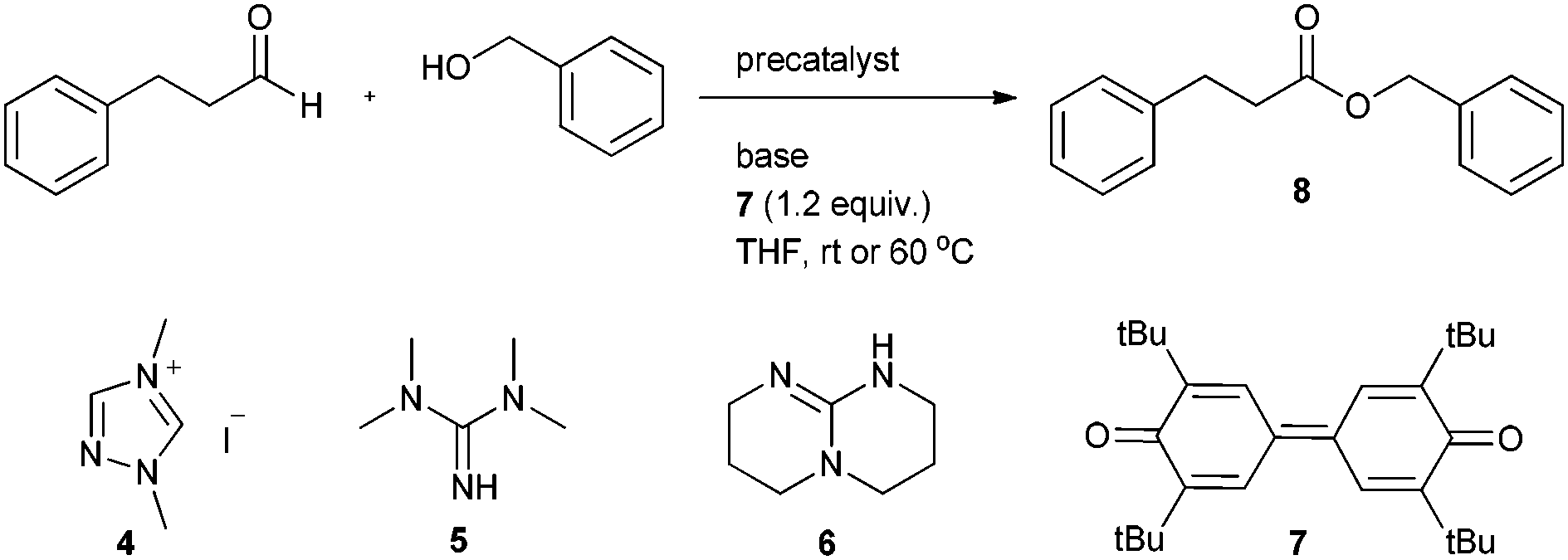
Initial attempts to esterify aliphatic aldehydes were performed with 3-phenyl propionaldehyde (1.0 equiv.) as substrate which was reacted with benzyl alcohol (1.5 equiv.) in the presence of 1,3-dimethyl triazolium iodide (4) as the precatalyst (0.20 equiv.). DBU was added as base (1.0 equiv.) and diphenoquinone 7 (1.2 equiv.) was chosen as oxidant. Initial reaction was conducted in THF (0.1 M) at room temperature. We noted very sluggish oxidation and the targeted benzyl 3-phenylpropionate (8) was obtained in 70% isolated yield after 72 h reaction time (Table 1, entry 1). To accelerate oxidative esterification we chose catechol as an additive (0.20 equiv.)13 under otherwise identical conditions and obtained 8 in 58% yield after 46 h (Table 1, entry 2). Prolonging reaction time did not lead to a better result, clearly revealing that catechol has no beneficial effect on the process. Reaction at 60 °C in the absence of any additive provided benzyl ester 8 in 68% yield within 30 h (entry 3).
We next tested several bases in the oxidative esterification. Reaction with 1,1,3,3-tetramethylguanidine (5) at room temperature delivered 8 in 65% yield after 72 h (entry 4). The use of 2,3,4,6,7,8-hexahydro-1H-pyrimido[1,2-α]pyrimidine (6) did not provide a better result (entry 5). We also tested inorganic bases and found that K2CO3 at room temperature afforded 47% of the desired ester after 40 h reaction time (entry 6) and with Cs2CO3 only a trace amount of 8 was formed besides other side products (entry 7). However, upon switching to Rb2CO3, yield substantially increased and 8 was isolated in 86% yield after 40 h (entry 8). Yield was not affected upon varying the amount of added base (0.5 to 2.5 equivalents); however, we noted that the concentration of base has an effect on the reaction rate. Fastest transformation was achieved upon using 2 equivalents of Rb2CO3 (entries 9–12). Lowering of the catalyst loading led to a lower yield along with longer reaction time (entries 13, 14). Upon increasing reaction temperature to 60 °C, an acceptable yield was also achieved at 5 mol% catalyst loading (entries 15–17). In terms of catalyst loading and reaction efficiency we regarded the conditions used in entry 17 as ideal (precatalyst 4 (0.075 equiv.), Rb2CO3 (2.0 equiv.) and diphenoquinone 7 (1.2 equiv.) in THF (0.1 M) at 60 °C for 20 h).
These investigations revealed that rubidium carbonate is the most appropriate base for this reaction, whereas potassium carbonate and cesium carbonate did not give a satisfactory yield. The size of the metal ion increases from potassium to cesium and also the basicity increases in the same direction. Cesium carbonate is obviously too basic leading to several side reactions (aldolization). We currently assume that the inorganic base plays an important role on the proton transfer in converting the carbene aldehyde adduct 1 into the Breslow intermediate 2. We believe that rubidium carbonate having a moderate basicity and a medium size cation catalyses the proton transfer for Breslow intermediate formation in a two-step sequence as suggested in Scheme 2.
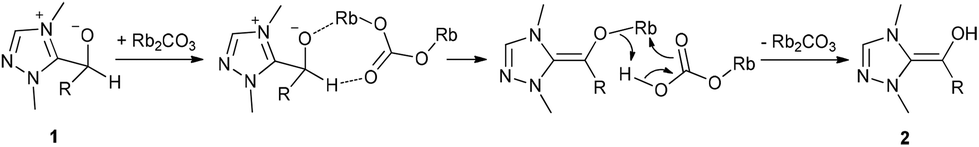 | ||
| Scheme 2 Potential mode of activation of Rb2CO3 for the proton transfer in going from 1 to 2. | ||
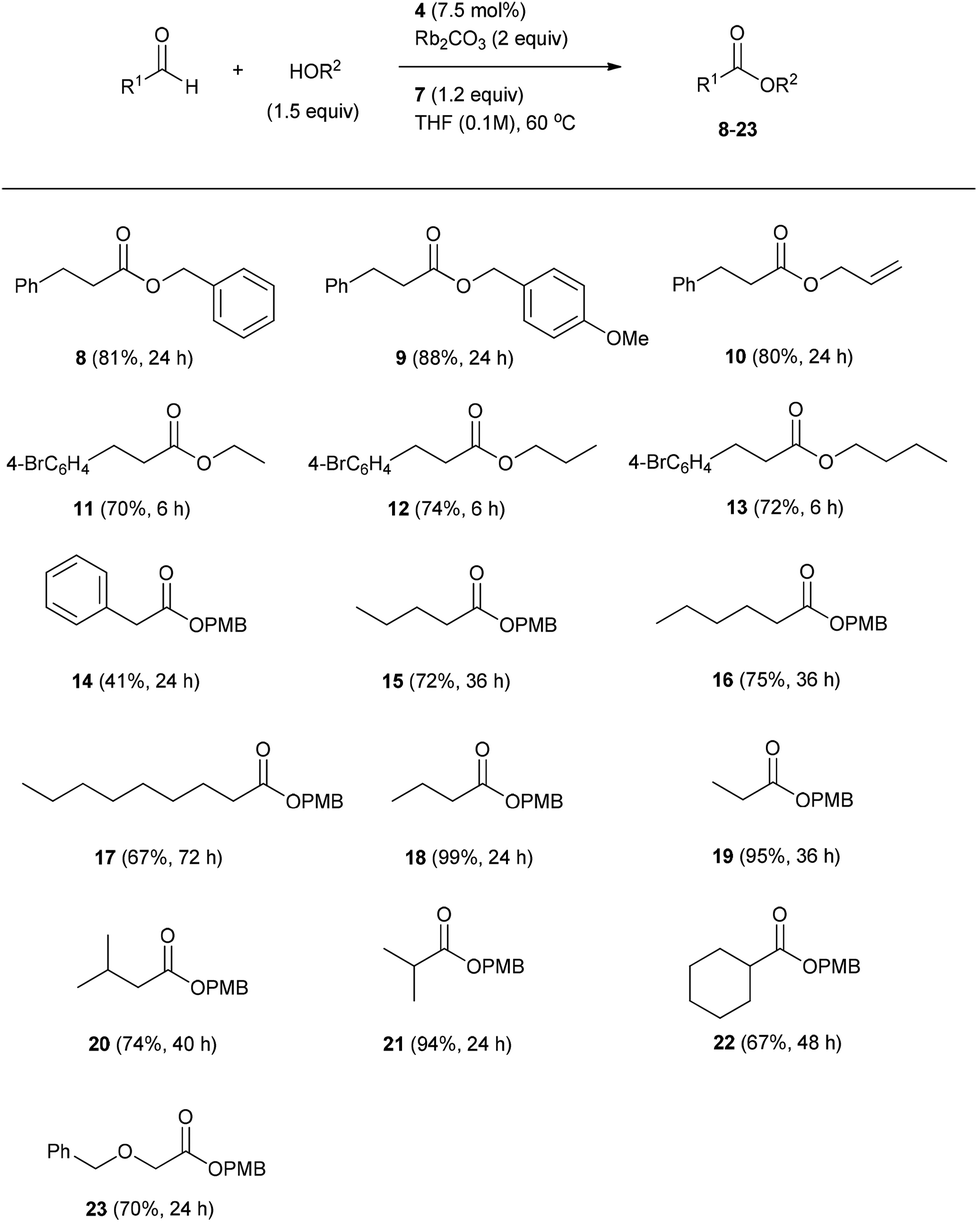
Under optimized conditions (Table 1, entry 17), the effect of the alcohol component on the oxidative esterification of 3-phenyl propanal was studied. Benzyl alcohol delivered the corresponding ester 8 in 81% yield within 24 h (Scheme 3). A similar result was achieved with allyl alcohol (see 10) and para-methoxy benzyl alcohol even provided a slightly better yield (9, 88%).
 | ||
| Scheme 3 Substrate scope. | ||
Aliphatic alcohols were tested in combination with 3-para-bromophenyl propanal as aldehyde component. The bromo-derivative was chosen in order to avoid volatility problems which might occur during isolation of the corresponding aliphatic esters. We found ethanol, propanol and butanol to react efficiently to the corresponding esters 11–13 which were isolated in 70 to 74% yield.
We continued the studies by varying the aldehyde component. To this end, different aliphatic aldehydes were reacted with 4-methoxy benzyl alcohol (PMBOH) to provide the corresponding esters 14–23. In case of 2-phenyl acetaldehyde, ester 14 was isolated in only 41% yield. The lower yield as compared to the other examples is likely due to the high acidity of the α-protons in the starting aldehyde and also in the intermediate acyl azolium ion derived from 2-phenyl acetaldehyde. This might lead to enolate formation and in turn to unwanted side reactions. For linear aldehydes the yield of the corresponding esters decreased with increasing chain length. Aldehydes with small alkyl chains such as butyraldehyde and propanal were esterified with excellent yields (18, 99%; 19, 95%). A larger scale experiment (1 mmol) revealed a slightly lower yield in the formation of ester 18 (60%). We were pleased to find that also α- and β-branched aliphatic aldehydes could be transformed in good to excellent yields to the corresponding esters (3-methylbutanal: 20, 74%, isobutyraldehyde: 21, 94%, cyclohexanecarbaldehyde: 22, 67%). Furthermore, an α-reducible aldehyde provided the corresponding ester 23 in good yield (70%) without undergoing any internal redox chemistry.
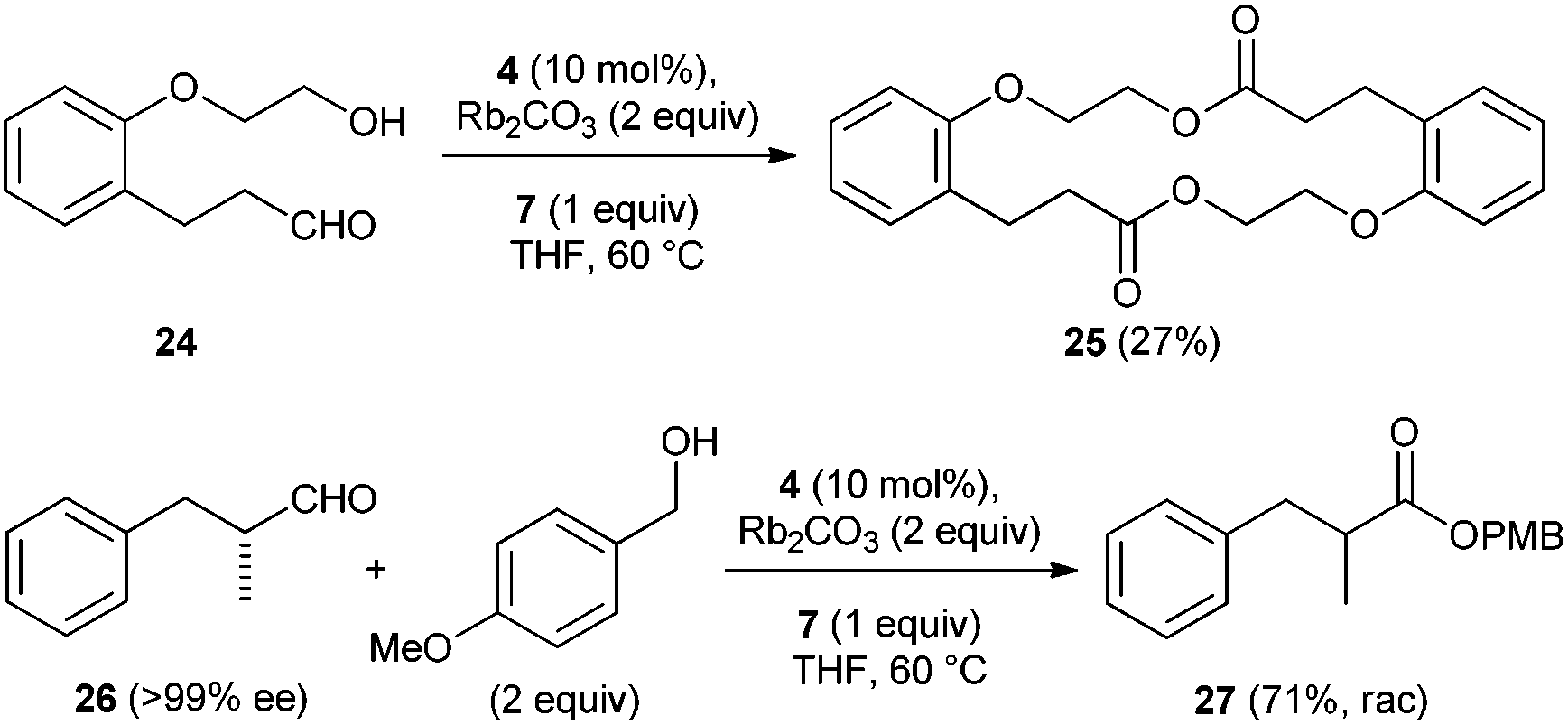
We also tested the intramolecular variant of this reaction as an approach towards the preparation of macrolactones. We chose compound 24 as model substrate for intramolecular lactonization under oxidative esterification conditions. The 18-membered-ring macrolactone 25 was isolated in 27% yield and the smaller 9-membered congener was not identified in the crude reaction mixture (Scheme 4). Besides 25, other side products which could not be isolated were also formed. Finally, we tested whether oxidative esterification occurs stereospecifically under optimized conditions. To this end, aldehyde 26 (>99% ee) was transformed to ester 27 which was isolated in 71% yield, unfortunately as racemic mixture.
 | ||
| Scheme 4 Macrolactonization and experiment to check stereospecificity of the oxidative esterification. | ||
In summary, we have developed an efficient method for oxidative esterification of enolizable aliphatic aldehydes. Notable, existing protocols for NHC-catalyzed oxidative esterification of aldehydes do not work well for aliphatic aldehydes. Rubidium carbonate was found to be an ideal base which likely assists 1,2-proton transfer in going from the carbene aldehyde adduct 1 to the Breslow intermediate 2. Various aliphatic aldehydes were esterified in good to excellent yields. For aliphatic linear aldehydes, the length of the alkyl chain influences reactivity. Smaller aldehydes such as propanal and butanal were oxidized in near quantitative yields whereas congeners with longer alkyl chains delivered lower but still good yields (around 70%). Moreover, α- and β-branched aliphatic aldehydes were successfully oxidatively esterified using the novel method and an initial experiment showed that the process has potential to be used for macrolactonization.
Acknowledgements
We thank the NRW Graduate School of Chemistry and the University of Münster for continuous support. We also thank Srikrishna Bera for conducting two experiments during revision of this paper and Martina Prekel for HPLC analysis.Notes and references
-
(a)
J. Otera, Esterification: Methods, Reactions and Applications, Wiley, New York, 2003 CrossRef
; (b) R. C. Larock, Comprehensive Organic Transformations, VCH, New York, 1989 Search PubMed
-
(a) C. E. I. Knappke, A. Imami and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 937 CrossRef CAS
-
(a) K. Y.-K. Chow and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 8126 CrossRef CAS PubMed
-
(a) A. Miyashita, Y. Suzuki, M. Kobayashi, N. Kuriyama and T. Higashino, Heterocycles, 1996, 43, 509 CrossRef CAS PubMed
-
(a) B. E. Maki and K. A. Scheidt, Org. Lett., 2008, 10, 4331 CrossRef CAS PubMed
-
(a) J. Guin, S. De Sarkar, S. Grimme and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 8727 CrossRef CAS PubMed
-
(a) S. De Sarkar, S. Grimme and A. Studer, J. Am. Chem. Soc., 2010, 132, 1190 CrossRef CAS PubMed
-
(a) H. Inoue and K. Higashiura, J. Chem. Soc., Chem. Commun., 1980, 549 RSC
-
(a) S. W. Tam, L. Jimenez and F. Diederich, J. Am. Chem. Soc., 1992, 114, 1503 CrossRef CAS
-
(a) R. S. Reddy, J. N. Rosa, L. F. Veiros, S. Caddick and P. M. P. Gois, Org. Biomol. Chem., 2011, 9, 3126 RSC
- E. E. Finney, K. A. Ogawa and A. J. Boydston, J. Am. Chem. Soc., 2012, 134, 12374 CrossRef CAS PubMed
-
(a) E. G. Delany, C.-L. Fagan, S. Gundala, A. Mari, T. Broja, K. Zeitler and S. J. Connon, Chem. Commun., 2013, 49, 6510 RSC
- D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402 CrossRef CAS PubMed
Footnotes |
| † This paper is dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00164h |
| This journal is © the Partner Organisations 2014 |