Visible-light photocatalytic oxidation of 1,3-dicarbonyl compounds and carbon–carbon bond formation†‡
Marion
Daniel
ab,
Louis
Fensterbank
*ab,
Jean-Philippe
Goddard
*abc and
Cyril
Ollivier
*ab
aSorbonne Universités UPMC Univ Paris 06, UMR 8232, Institut Parisien de Chimie Moléculaire, 4 place Jussieu, C. 229, F-75005 Paris, France. E-mail: louis.fensterbank@upmc.fr; jean-philippe.goddard@uha.fr; cyril.ollivier@upmc.fr; Fax: +33(0)44277360
bCNRS, UMR 8232, Institut Parisien de Chimie Moléculaire, 4 place Jussieu, C. 229, F-75005 Paris, France
cLaboratoire de Chimie Organique et Bioorganique EA 4566, Université de Haute-Alsace, Ecole Nationale Supérieure de Chimie de Mulhouse, 3 rue Alfred Werner, F-68093 Mulhouse Cedex, France
First published on 30th April 2014
Abstract
Visible-light photocatalytic oxidation of 1,3-dicarbonyl compounds, using Ru(bpy)3Cl2 as a photocatalyst and combining molecular oxygen and triphenylcarbenium as a new sacrificial acceptor, has been developed for carbon–carbon bond formation giving access to dimerization, allylation and polycyclization products.
Oxidative radical transformations involving 1,3-dicarbonyl compounds have been extensively developed in both inter- and intramolecular manifolds, participating in original and relevant cascade reactions. Several transition metal salts based inter alia on manganese(III),1,2 cerium(IV)3 or iron(III)4 have proved to be effective in promoting such radical processes. This fruitful chemistry remains a field of great interest with still many challenges ahead. As stoichiometric amounts of metal reagents and elevated temperatures are often required for complete conversion, developing new catalytic versions and milder conditions is of paramount importance today in the growing context of sustainable chemistry.
Recently, visible-light photoredox catalysis has emerged as a powerful tool in radical chemistry. As such, it represents a valuable alternative to generate radicals by single electron transfer reactions from an appropriate photocatalyst that absorbs light in the visible region, which can be an organic dye5 or a polypyridine complex such as the well-known Ru(bpy)3Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 or Ir(ppy)3.7 Since the pioneering studies of Kellogg8 and Deronzier,9 important contributions have been reported for the development of both photoreduction and photooxidation for synthetic purposes.10–12 Notably, α-oxyamination of a series of 1,3-dicarbonyl compounds has been undertaken successively by Tan13 and Akita14 using either Rose Bengal or Ir(ppy)2(dtbbpy)PF6 as a photocatalyst and TEMPO playing both roles of an oxidative quencher and a radical trapper (Scheme 1). But these conditions have remained limited to carbon–oxygen bond formation. Obviously, the presence of TEMPO limits other types of functionalization considering its high ability to trap intermediate carbon radicals. In this context, we aim to elaborate new photocatalytic oxidation conditions of 1,3-dicarbonyl derivatives to perform carbon–carbon bond forming reactions (Scheme 1).
6 or Ir(ppy)3.7 Since the pioneering studies of Kellogg8 and Deronzier,9 important contributions have been reported for the development of both photoreduction and photooxidation for synthetic purposes.10–12 Notably, α-oxyamination of a series of 1,3-dicarbonyl compounds has been undertaken successively by Tan13 and Akita14 using either Rose Bengal or Ir(ppy)2(dtbbpy)PF6 as a photocatalyst and TEMPO playing both roles of an oxidative quencher and a radical trapper (Scheme 1). But these conditions have remained limited to carbon–oxygen bond formation. Obviously, the presence of TEMPO limits other types of functionalization considering its high ability to trap intermediate carbon radicals. In this context, we aim to elaborate new photocatalytic oxidation conditions of 1,3-dicarbonyl derivatives to perform carbon–carbon bond forming reactions (Scheme 1).
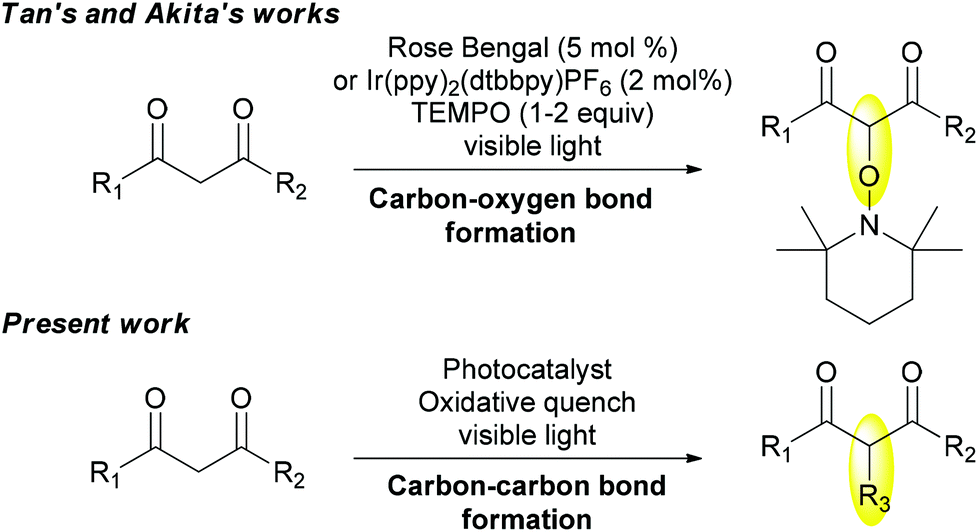 | ||
| Scheme 1 Photooxidation of 1,3-dicarbonyl compounds. | ||
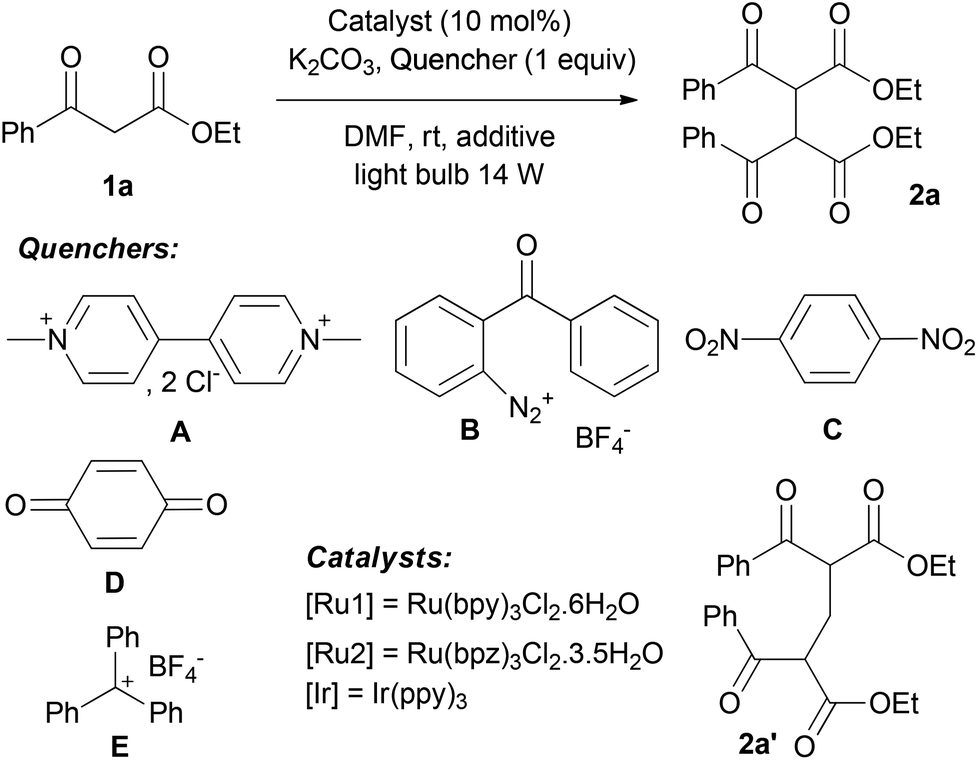
We first decided to investigate the dimerization of ketoester 1a (Table 1).15 The numbers of parameters had to be fixed in order to access an efficient procedure for the formation of the ketoester radical. Optimization has been pursued under basic conditions (K2CO3, 1.2 equiv.) to favour the formation of the corresponding enolate and then facilitate mild oxidative processes of such species. We first tested the usual Ru(bpy)3Cl2 ([Ru1], E° Ru(III)/Ru(II) = 1.29 V vs. SCE) with a catalytic loading of 10 mol% in DMF under visible light irradiation (fluorescent bulb, 14 W) and examined molecular oxygen as a possible oxidative quencher, known to be very efficient for this purpose (Table 1, entry 1).16 In an open flask manner, dimer 2a was formed in 56% yield, as a 1:1 mixture of meso/DL diastereomers. Other sacrificial acceptors have been investigated instead of oxygen in order to avoid potential side reactions of oxygen spin trapping. Methyl viologen A did not afford the dimer 2a but the methylene-bridged compound 2a′ in 32% yield (Table 1, entry 2). Such types of compounds have already been observed under similar photocatalytic conditions with benzylamine derivatives as methylene donors.17 1,4-Benzoquinone D and 1,4-dinitrobenzene C appeared to be inefficient for this transformation (Table 1, entries 3 and 4). 2-Benzoylbenzenediazonium tetrafluoroborate B was introduced by Deronzier as an active oxidative quencher of the ruthenium photocatalyst.9 Under our conditions, it proved to be suitable for the oxidation of 1a leading to 2a in 79% yield, but presumably contaminated by ca. 10% of diazoic coupling by-product resulting from addition of the ketoester enolate to the diazonium salt (Table 1, entry 5). To our delight, commercially available triphenylcarbenium tetrafluoroborate E (E° = 0.54 vs. NHE)18 gave pure 2a in 41% yield in only 24 h (Table 1, entry 6). To the best of our knowledge, this compound has never been used before as a sacrificial acceptor for this purpose.19 Moreover, a synergic effect was observed between oxygen and the triphenylcarbenium when E was used in an open flask with 10 mol% of [Ru1]. Indeed, quantitative formation of 2a was reached in 24 h. With these optimized conditions in hand, we tried to decrease the amount of the catalyst to 5 mol% (Table 1, entry 8). We observed a longer reaction time still with a high yield for the formation of 2a. The influence of the photocatalyst has been investigated. In spite of a lower redox potential, Ir(ppy)3 (E° Ir(IV)/Ir(III) = 0.77 V vs. SCE) afforded 2a in 89% yield (Table 1, entry 9) while a stronger oxidant such as Ru(bpz)3Cl2 (E° Ru(III)/Ru(II) = 1.86 V vs. SCE) appeared to be less active (Table 1, entry 10). As reported by Tan,13 organic dyes appeared to be interesting organocatalysts for the photooxidation of 1,3-dicarbonyl compounds. Thus, Rose Bengal (E° = 1.09 V vs. SCE) was investigated but only 31% of 2a was isolated in extended reaction time (Table 1, entry 11). Control experiments confirmed that light, catalyst and base are essential and a simultaneous action is needed. Finally, we accessed optimized conditions in DMF involving Ru(bpy)3Cl2 as a photocatalyst (10 mol%) upon visible light irradiation (light bulb, 14 W) with oxygen and triphenylcarbenium tetrafluoroborate E (1 equiv.) as sacrificial acceptors under basic conditions (1.2 equiv. of K2CO3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Acceptor/quencher | Additive | Time | Yield (%) |
| a 2a was obtained as an inseparable mixture with the diazoic coupling by-product. b 5 mol% of catalyst was used. c The reaction was carried out in the dark. d The reaction was conducted without K2CO3. e The reaction was done in MeCN. | |||||
| 1 | [Ru1] | Air | — | 48 h | 56 |
| 2 | [Ru1] | A | — | 24 h | 32 (2a′) |
| 3 | [Ru1] | C | — | 48 h | SM |
| 4 | [Ru1] | D | — | 48 h | 4 |
| 5 | [Ru1] | B | — | 36 h | 79a |
| 6 | [Ru1] | E | — | 24 h | 41 |
| 7 | [Ru1] | E | Air | 24 h | 100 |
| 8b | [Ru1] | E | Air | 48 h | 85 |
| 9 | [Ir] | E | Air | 24 h | 89 |
| 10 | [Ru2] | E | Air | 24 h | 54 |
| 11b,e | Rose Bengal | — | — | 62 h | 31 |
| 12c | [Ru1] | E | Air | 60 h | SM |
| 13 | — | E | Air | 60 h | SM |
| 14 | — | — | Air | 48 h | SM |
| 15d | [Ru1] | E | Air | 60 h | SM |
Then, we extended the substrate scope to diversely substituted 1,3-dicarbonyl derivatives (Scheme 2). As mentioned above, ketoester 1a dimerized in quantitative yield under the optimized conditions. When the phenyl ring was substituted by a para-methyl group 1b, the corresponding dimer 2b was obtained in 85% yield while in the presence of a methyl group in meta position, 1c gave 2c in a lower 43% yield. A more electron-donating substituent like a methoxy group did not impact the efficiency of the dimerization and 2d was obtained in 80% yield. A slight decrease of the yield was observed with a para-fluoro substituent; 2e was isolated in 70% yield. A total degradation was observed with substrate 1f, certainly due to the presence of a nitro group. Such nitro-substituted aromatic carbonyl derivatives are known to be good oxidative quenchers of photoactive [Ru(bpy)3]2+ resulting in their SET reduction.20 Interestingly, 1,3-diketone 1g was oxidized to generate the corresponding dimer 2g. Unfortunately, the purification of 2g did not allow us to obtain an analytically pure product.
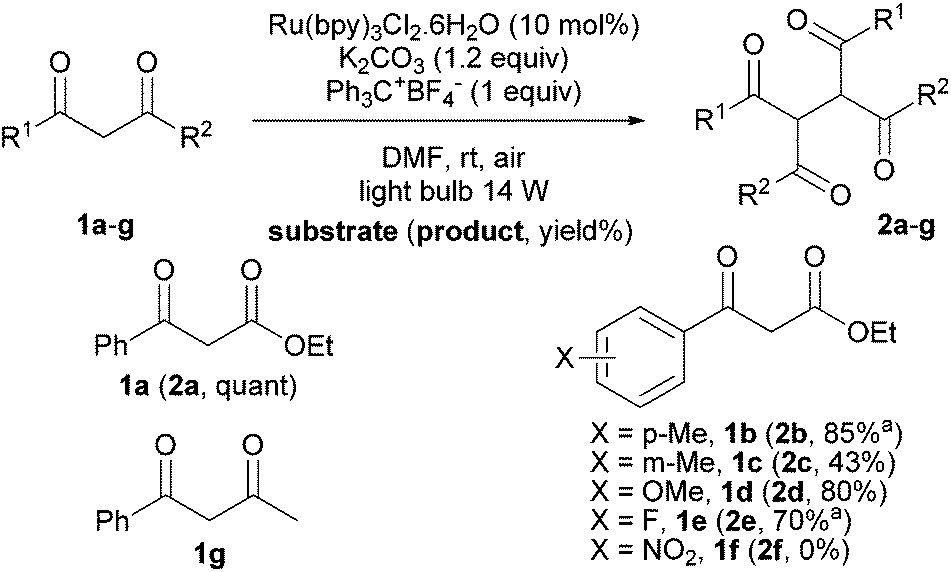 | ||
| Scheme 2 Dimerization of 1,3-dicarbonyl compounds. aContaminated by ca. 10% (estimated by 1H NMR) of a byproduct which may result from overoxidation of the corresponding dimer. | ||
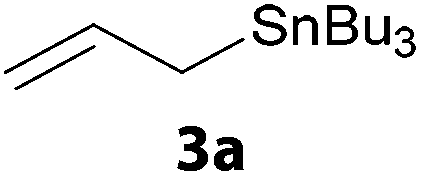
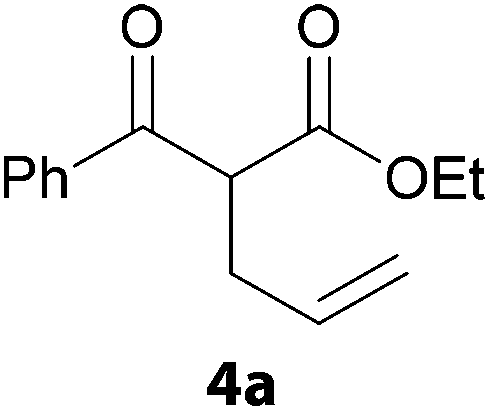
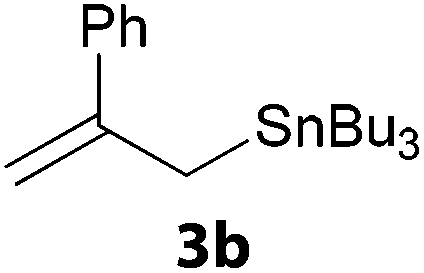
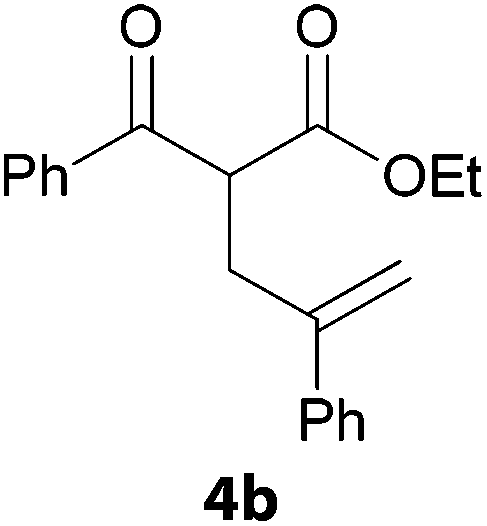
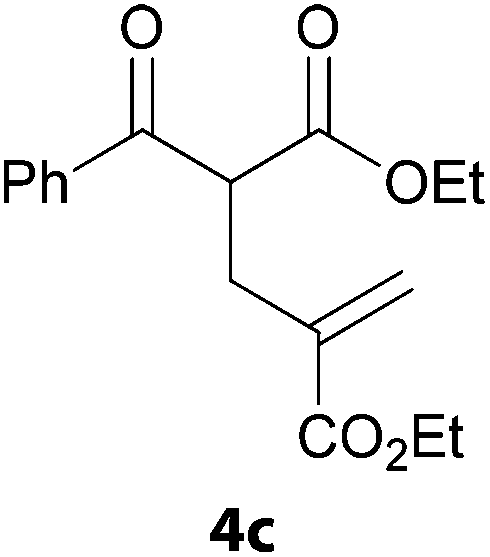
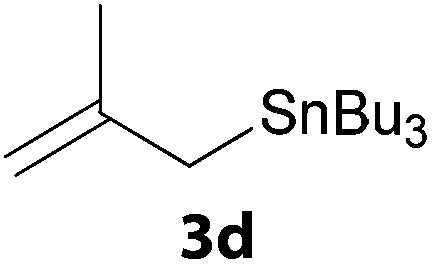
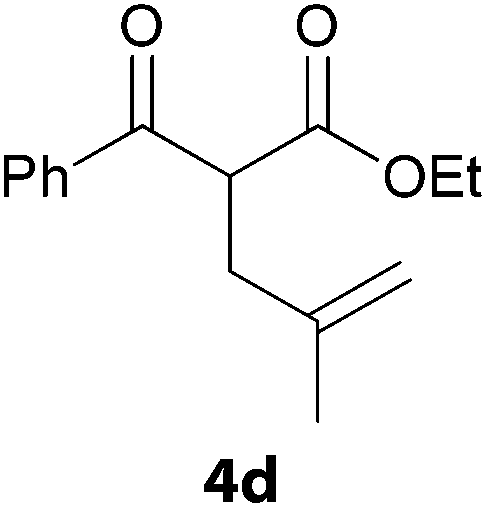
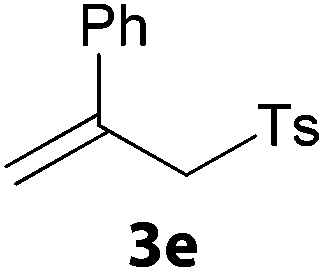
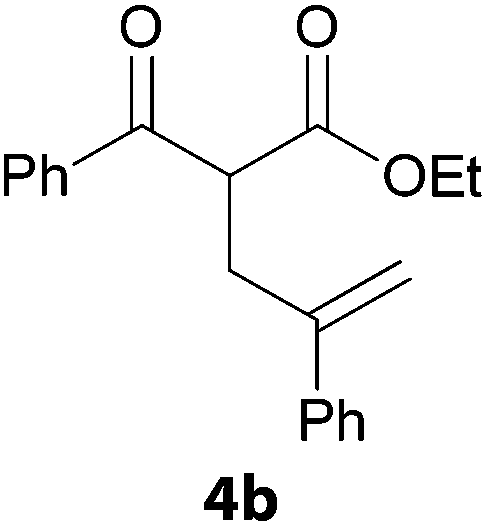
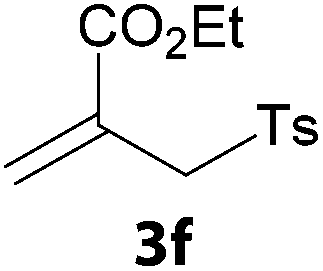
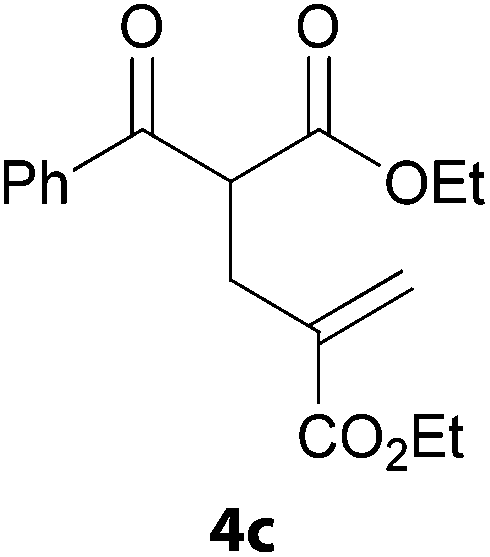
In a second part, we applied these conditions to allylation reactions. Experiments were accomplished on ethyl benzoylacetate 1a by addition of ten equivalents of variously substituted allylstannanes to the previous mixture (Table 2). Thus, reaction of allyltributyltin 3a with 1a gave the desired product 4a but in low yield. Better results up to 72% were achieved with α-phenyl, -ester and -methyl substituted reagents 3b–d. These results partly confirm the radical character of the process.21,22 The same transformation carried out with the corresponding tin free allylsulfones proceeded in 30% and 62% starting from 3e and 3f, respectively.12 In contrast, the sole dimer was observed with allyl p-tolyl sulfone or in mixture with side-products for its α-methylated counterpart.
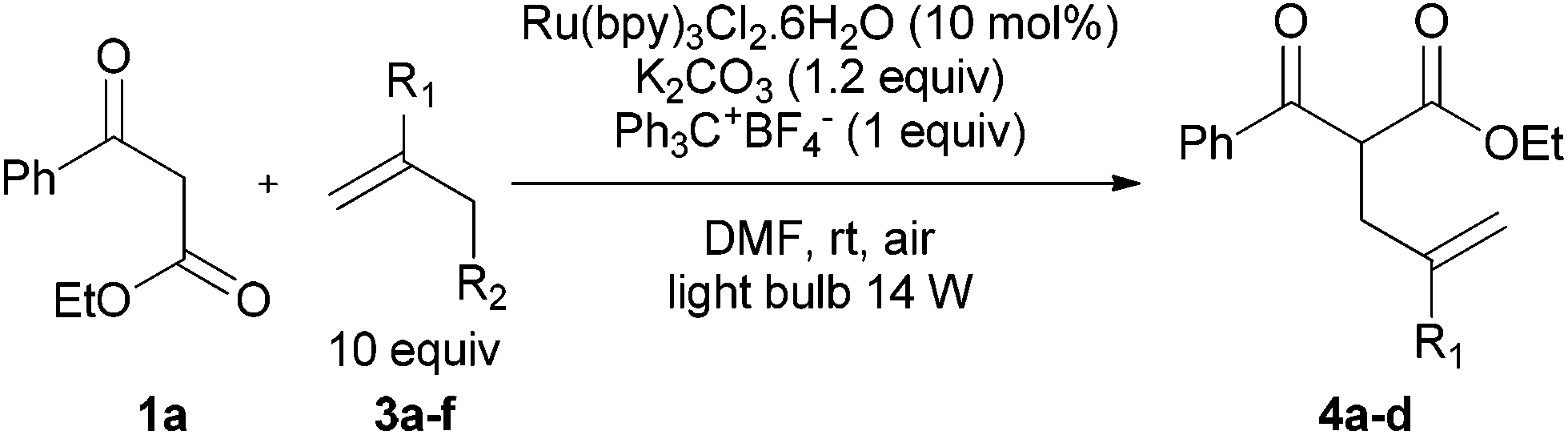
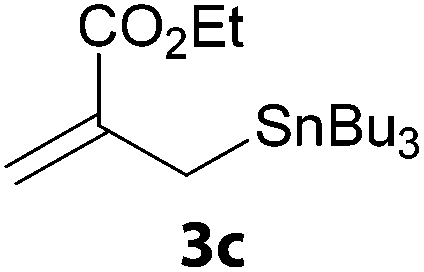
We finally turned our attention to intramolecular cascade processes for accessing polycyclic products (Scheme 3). Monoelectronic oxidation of two unsaturated phenyldiketones 5, previously reported by Snider15a,b and 7 in the presence of over-stoichiometric amounts of Mn(OAc)3, has been realized under photocatalytic conditions. Upon treatment with the Ru(bpy)3Cl2/Ph3CBF4/O2 oxidative system, less than 5% of tricyclic product 6 was formed. Addition of another equivalent of Ph3CBF4 allowed us to obtain a better 29% yield of 6. A possible explanation is that another equivalent is necessary for recovering the phenyl ring by oxidative aromatization of the transient cyclohexadienyl radical F2.23 Adding more equivalents of triphenylcarbenium did not improve the yield. The same reactivity was observed with diketone 7, and the tricyclic adduct 8 was formed in 31% yield in one step. Although the yields are still low, these two transformations are the first examples of radical cascade cyclization of 1,3-diketones promoted by visible-light photooxidation catalysis.
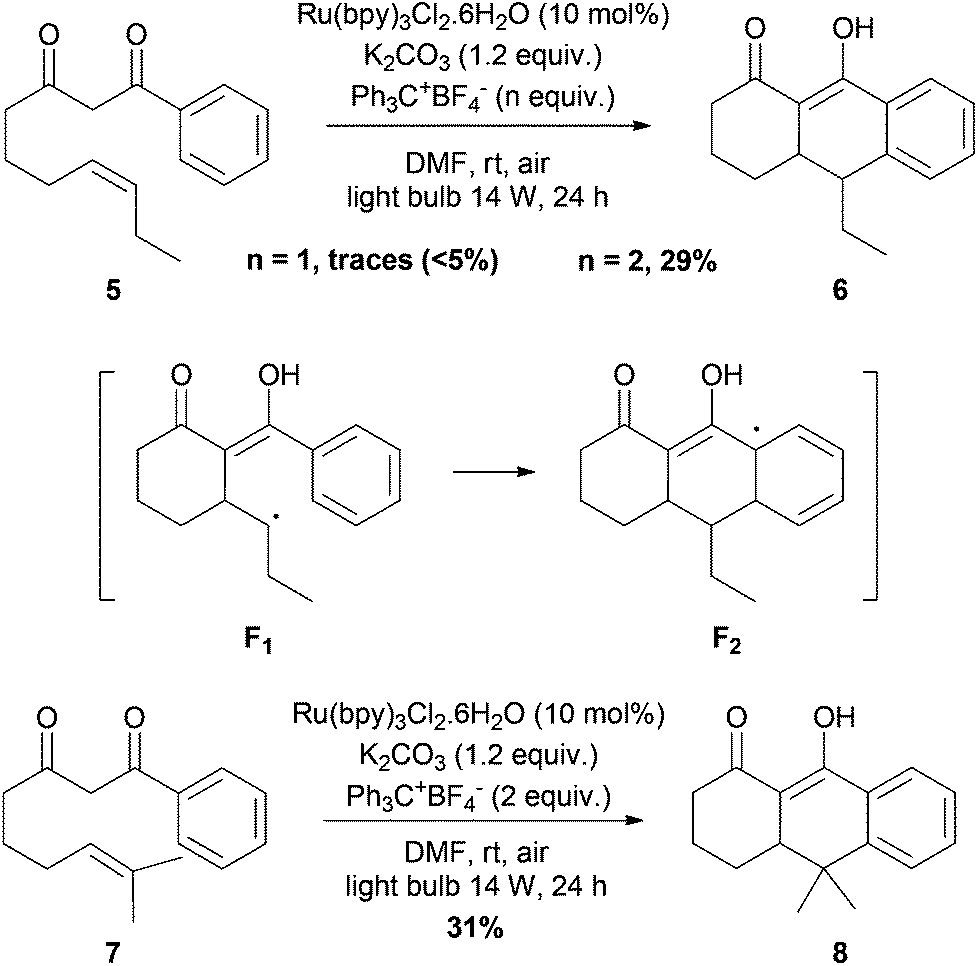 | ||
| Scheme 3 Intramolecular cascade reactions with 1,3-diketones 5 and 7. | ||
In summary, we have developed a new photocatalytic process to promote monoelectronic oxidation of 1,3-dicarbonyl compounds using Ru(bpy)3Cl2 as a photocatalyst and visible-light as a mild source of energy. The system triphenylcarbenium tetrafluoroborate/oxygen has been successfully developed as a new sacrificial acceptor. Under basic conditions, we were able to perform inter- as well as intramolecular carbon–carbon bond formation with ketoesters and diketones. Mechanistic studies should be performed to rationalize the role of each component and extend these new conditions to other synthetic transformations.
Notes and references
-
(a) G. G. Melikyan, Org. React., 1997, 49, 427 CAS
; (b) A. S. Demir and M. Emrullahoglu, Curr. Org. Synth., 2007, 4, 223 CrossRef
-
(a) B. B. Snider, Chem. Rev., 1996, 96, 339 CrossRef CAS PubMed
-
(a) V. Nair, J. Mathew and J. Prabhakaran, Chem. Soc. Rev., 1997, 26, 127 RSC
- U. Jahn, Top. Curr. Chem., 2012, 320, 191 CrossRef CAS
-
(a) M. A. Miranda and H. García, Chem. Rev., 1994, 94, 1063 CrossRef CAS
-
(a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS
-
(a) M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970 CrossRef CAS PubMed
-
(a) D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 19, 1255 CrossRef
-
(a) H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517 CrossRef CAS
- For recent reviews, see ref. 5 and
(a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785 CrossRef CAS PubMed
- The renewal of interest for photoredox catalysis in synthesis was inspired by the following studies:
(a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 77, 322 Search PubMed
- For our contribution in this field, see:
(a) M. H. Larraufie, R. Pellet, L. Fensterbank, J. P. Goddard, E. Lacôte, M. Malacria and C. Ollivier, Angew. Chem., Int. Ed., 2011, 123, 4555 CrossRef
- H. Liu, W. Feng, C. W. Kee, Y. Zhao, D. Leow, Y. Pan and C. H. Tan, Green Chem., 2010, 12, 953 RSC
- T. Koike, Y. Yasu and M. Akita, Chem. Lett., 2012, 41, 999 CrossRef CAS
-
(a) B. B. Snider, J. J. Patricia and S. A. Kates, J. Org. Chem., 1988, 53, 2137 CrossRef CAS
- Q. G. Mulazzani, H. Sun, M. Z. Hoffman, W. E. Ford and M. A. J. Rodgers, J. Phys. Chem., 1994, 98, 1145 CrossRef CAS
- W.-J. Yoo, A. Tanoue and S. Kobayashi, Chem – Asian J., 2012, 7, 2764 CrossRef CAS PubMed
- E. M. Arnett, R. A. Flowers II, R. T. Ludwig, A. E. Meekhof and S. A. Walek, J. Phys. Org. Chem., 1997, 10, 499 CrossRef CAS
- For a recent application of triphenylcarbenium salts as oxidants, see: Z. Xie, L. Liu, W. Chen, H. Zheng, Q. Xu, H. Yuan and H. Lou, Angew. Chem., Int. Ed., 2014, 53, 3904 CrossRef CAS PubMed
- H.-B. Kim, N. Kitamura, Y. Kawanishi and S. Tazuke, J. Phys. Chem., 1989, 93, 5757 CrossRef CAS
-
I. J. Rosenstein, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, Weinheim, 2001, vol. 1, p. 50 Search PubMed
- M. Pirtsch, S. Paria, T. Matsuno, H. Isobe and O. Reiser, Chem. – Eur. J., 2012, 18, 7336 CrossRef CAS PubMed
- For each type of transformation, we hypothesize two possible catalytic redox cycles in which: (1) the Ph3CBF4/oxygen system acts as an oxidative quenching agent for the photoactivated Ru(bpy)32+ to generate the transient Ru(bpy)33+ that in turn oxidizes the potassium enolate to generate the corresponding radical; (2) the enolate is involved in a reductive quenching cycle providing the desired radical and on its part, the Ph3CBF4/oxygen system should regenerate the photocatalyst by oxidation. Then, the formed radical undergoes either inter- (dimerization or allylation) or intramolecular transformations following classical homolytic steps.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00071d |
| This journal is © the Partner Organisations 2014 |