DOI:
10.1039/D3CE00846K
(Paper)
CrystEngComm, 2024,
26, 268-276
The design of a series of cocrystals featuring dicarboxylic acid with a modified isoniazid derivative†
Received
25th August 2023
, Accepted 6th December 2023
First published on 6th December 2023
Abstract
A series of cocrystals featuring an isoniazid derivative was synthesized and characterized. Isoniazid was derivatised using acetone to make N′-(propan-2-ylidene)pyridine-4-carbohydrazide (izact). An attempt to form seven new cocrystals was made, which involved cocrystalizing izact an aliphatic dicarboxylic acid of varying chain lengths ranging from malonic acid (C3) to sebacic acid (C10), excluding succinic acid (C4) since this cocrystal was reported before. Out of these, only five cocrystals were successfully synthesized; since an attempt to form a cocrystal with malonic acid (C3) and glutaric acid (C5) failed, leaving the successful cocrystals to contain dicarboxylic acids ranging from adipic acid (C6) to sebacic acid (C10). These izact–dicarboxylic acid cocrystals were then compared to their isoniazid counterparts, as well as that with the previously reported izact–C4 cocrystal. This comparison showed that the cocrystals of izact with C4, C6, C8 were isostructural, while the other cocrystals reported here showed unique aspects that differentiated them from their counterparts. Single-crystal X-ray diffraction (SC-XRD) was used to determine the crystal structures, while powder X-ray diffraction (PXRD) was used to determine the bulk phase purity. Differential scanning calorimetry was used to determine any additional phases but only melting was observed.
Introduction
Crystal engineering is the ability to design and control the structure of solid state materials using an understanding and manipulation of intermolecular interactions present in that solid state system.1,2 One method for controlling the intermolecular interactions in a crystal structure is through the design and synthesis of cocrystals and molecular salts. A cocrystal can be defined as a single-phase crystalline solid composed of at least two different neutral compounds in a stoichiometric ratio,3,4 with further classifications depending on if the coformers are ionic or neutral, or contains solvate molecules and/or water.4 The development of cocrystals of active pharmaceutical ingredients (API's) is used as a means to alter the physicochemical, mechanical, and pharmacokinetic properties of those API's.5 The approach to designing cocrystals is to examine the structure of the API and identify parts of the structure involved with forming particular intermolecular interactions, such as π⋯π stacking, hydrogen bonding, van der Waals forces etc.6 One potential source for searching for coformers is the Cambridge Structural Database (CSD).7 Although the CSD is a good source for determining potential coformers, it is possible that it may not contain many examples of suitable coformers for a particular API, or the cocrystals reported may contain a diverse range of coformers that do not share much structural similarities with each other to note any strong or particular trends in the intermolecular interactions across these structures. Therefore, it is vital to undertake research using structurally similar coformers, where any trends can give insight in how the API can form cocrystals.
The aliphatic dicarboxylic acids are an interesting class of organic materials and have been studied extensively.8,9 The aliphatic dicarboxylic acids have a general formula of HOOC–CnH2n–COOH, where the dicarboxylic acids of interest for this work ranged from malonic acid (C3) to sebacic acid (C10), with the abbreviations used in this work refers to the total number of carbons present. The full list of names of these dicarboxylic acids is given in Table 1, while the structures of these dicarboxylic acids is given in Scheme 1. The most prominent feature of this series is the odd–even effect of the carbon chain length, where properties such as the melting point and solubility alternates between the even-chain and odd-chain members.9 It has also been observed that succinic acid (C4) and the odd-chain dicarboxylic acids exhibits crystal polymorphism, existing in at least two forms, labelled α and β.8–10 In all forms the dicarboxylic acids form a centrosymmetric carboxylic acid hydrogen-bonded dimer. Other significant observations between the even- and odd-chained members is that even-chain members are offset along their length while odd-chained members are uniformly aligned, and that odd-chained members exhibit twisted molecular conformations when compared to their even-chained members.9 The “twisting” feature of odd-chain dicarboxylic acids is to avoid O⋯O repulsion between two neighboring chains, while for even-chained members this “twisting” is avoided by being offset.11 Overall, the dicarboxylic acids exhibits a trend in properties and packing that alternates between even- and odd-chained members.
Table 1 List of dicarboxylic acids used for this work, along with the abbreviations and masses used for this work
| Dicarboxylic name |
Abbreviation |
Mass used/mg |
| Malonic acid |
C3 |
38 |
| Glutaric acid |
C5 |
48 |
| Adipic acid |
C6 |
53 |
| Pimelic acid |
C7 |
58 |
| Suberic acid |
C8 |
40 |
| Azelaic acid |
C9 |
69 |
| Sebacic acid |
C10 |
74 |
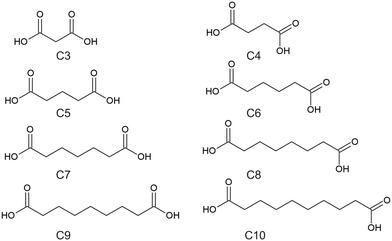 |
| | Scheme 1 Structures of the dicarboxylic acids of interest to this work. | |
Isoniazid (inh) is an anti-bacterial used to treat Mycobacterium tuberculosis in combination with other drugs.12 Inh has received considerable attention by solid-state chemists, as several numbers of cocrystals/molecular salts of its unmodified13,14 and modified forms15,16 have been reported in the literature. In particular, several cocrystals with inh and different aliphatic dicarboxylic acids have been reported.17–20 It should be noted that a cocrystal containing isoniazid and azelaic acid (C9) has not been reported, despite several attempts to synthesize said cocrystal.20 There is a trend with the inh cocrystal with dicarboxylic acids where the stoichiometric ratio in the asymmetric unit is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inh:dicarboxylic acid for odd-chain dicarboxylic acids and 2:1 inh:dicarboxylic acid for even-chain dicarboxylic acids.15,17 The hydrogen bonding observed in cocrystals of inh with dicarboxylic acids are diverse, with some of these hydrogen bonding motifs presented in Scheme 2. All cocrystals of inh with the dicarboxylic acids form the pyridine⋯carboxylic acid hydrogen bond motif. However, in some of these cocrystals only one of the carboxylic acid groups forms this hydrogen bond motif. For example, in the cocrystal of inh + C7 motif 5 (Scheme 2) forms.19 When comparing the even-chained and odd-chained members, there is very little that represents a pattern. For example, among the even-chained members, hydrogen bond motif 4 (Scheme 2) is observed among cocrystals with inh and C4, C8 and C10. In this case, both polymorphic forms of inh + C6 does not form this hydrogen bond motif, instead favouring the formation hydrogen bond motif 3 (Scheme 2) instead. Apart from the stoichiometric ratios between the odd-chained and even-chained members, there is no other clear pattern between the cocrystals of inh with these dicarboxylic acids, which is in contrast in the trends observed in the crystal structures of the pure dicarboxylic acids. This also appears in contrast to many other reported works where a series of cocrystals containing varying chain lengths of dicarboxylic acids with some target molecule (API) usually resulted in a series with some trend in packing or properties being observed.21–24 There is also no clear reason why the inh + C9 cocrystal does not form, despite several different attempts.20
:1 inh:dicarboxylic acid for odd-chain dicarboxylic acids and 2:1 inh:dicarboxylic acid for even-chain dicarboxylic acids.15,17 The hydrogen bonding observed in cocrystals of inh with dicarboxylic acids are diverse, with some of these hydrogen bonding motifs presented in Scheme 2. All cocrystals of inh with the dicarboxylic acids form the pyridine⋯carboxylic acid hydrogen bond motif. However, in some of these cocrystals only one of the carboxylic acid groups forms this hydrogen bond motif. For example, in the cocrystal of inh + C7 motif 5 (Scheme 2) forms.19 When comparing the even-chained and odd-chained members, there is very little that represents a pattern. For example, among the even-chained members, hydrogen bond motif 4 (Scheme 2) is observed among cocrystals with inh and C4, C8 and C10. In this case, both polymorphic forms of inh + C6 does not form this hydrogen bond motif, instead favouring the formation hydrogen bond motif 3 (Scheme 2) instead. Apart from the stoichiometric ratios between the odd-chained and even-chained members, there is no other clear pattern between the cocrystals of inh with these dicarboxylic acids, which is in contrast in the trends observed in the crystal structures of the pure dicarboxylic acids. This also appears in contrast to many other reported works where a series of cocrystals containing varying chain lengths of dicarboxylic acids with some target molecule (API) usually resulted in a series with some trend in packing or properties being observed.21–24 There is also no clear reason why the inh + C9 cocrystal does not form, despite several different attempts.20
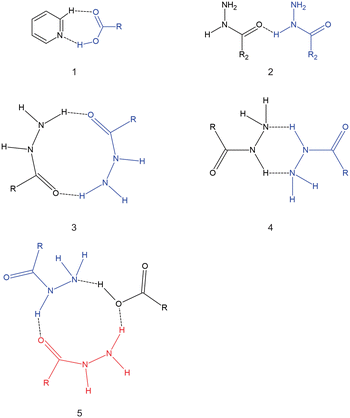 |
| | Scheme 2 Hydrogen bonding motifs observed for cocrystals of inh with dicarboxylic acids. | |
As such, the question that arises is: does this diversity arise due to the variability of the hydrogen bonding of the hydrazine group or does the chain length have a significant effect on the overall packing of the crystal structure? This work aims to provide some clues by synthesizing and characterizing a similar series of cocrystals by using a modified form of inh. The modifier of choice is acetone, due to its relatively simplicity and its ability to react with inh very easily, producing N′-(propan-2-ylidene)pyridine-4-carbohydrazide (izact). The result of the modification is that the number of hydrogen bonding pairs that arises from the hydrazine group is reduced. Therefore, for this work, it is expected that the resulting izact–dicarboxylic acid structures to be more uniform in terms of its packing and the intermolecular interactions observed. Also, it is expected that the stoichiometric ratio between izact and the respective odd-chain dicarboxylic acid coformer would change to 2:1 izact:dicarboxylic acid coformer due to the change of the hydrazine group. In this work we present the cocrystals of izact with dicarboxylic acids ranging from C6 to C10. We also report our attempt to form cocrystals of izact + C3 and izact + C5 but failing to do so.
Experimental
All materials and solvents were purchased from Sigma-Aldrich, and used as is without further purification.
2.1 Synthesis
Izact was synthesized using a previously described method.16 500 mg of inh and 1 mL of acetone were dissolved in 5 mL of absolute ethanol in a small vial. This vial was closed using a lid and stirred for 24 hours at room temperature. After which, the lid was replaced with a lid with a hole in it and the solution was then left to evaporate until crystals of izact were left behind. These vials were stored in the dark in a cupboard. In order to synthesize the cocrystals, a stoichiometric ratio of izact and the respective dicarboxylic acid in a 2:1 molar ratio was dissolved in 5 mL of absolute ethanol and stirred using a stirrer bar until all powder was dissolved. This vial was placed in a dark cupboard and closed with a lid with a hole in it to allow solvent to evaporate. After several days of letting the solvent evaporate, a few crystals were obtained.
Single-crystal X-ray diffraction (SC-XRD)
Intensity data for the cocrystals presented in this work were collected on a Bruker D8 Venture Bio PHOTON III 28 pixel array area detector (208 × 128 mm2) diffractometer with a Mo Kα (λ = 0.71073 Å) IμS DIAMOND source (50 kV, 1.4 mA). The collection method involved ω- and φ-scans, and 1536 × 1024 bit data frames. The unit cell and full data set were collected using APEX4; SAINT was used to integrate the data, and SADABS was used to make empirical absorption corrections and scale the data and the program SADABS was used to make empirical absorption corrections.25 Space group assignments were made using XPREP on all compounds.25 In all cases, the structures were solved in the WinGX Suite of programs26 by using intrinsic phasing with ShelXT27 and refined using ShelXL refinement package using least squares minimization.27 All non-hydrogen atoms were refined anisotropically. Thereafter, all hydrogen atoms attached to N or O atoms were located in the difference Fourier map and their coordinates and isotropic thermal displacement parameters were refined freely. Diagrams and publication material were generated using ORTEP-3 (ref. 28) and Mercury.29
Powder X-ray diffraction (PXRD)
Powder X-ray diffraction data for all compounds were measured at 293 K on a Bruker D2 Phaser diffractometer which employs a sealed tube Co Kα1 X-ray source (λ = 1.78896 Å), operating at 30 kV and 10 mA, and LynxEye PSD detector in Bragg–Brentano geometry. Powder patterns for each sample is presented in the ESI,† where the experimentally obtained data is compared to the calculated patterns obtained from the SC-XRD data. It should be noted that the experimental patterns were collected at room temperature while the calculated patterns were obtained from data collected from crystals at 173 K. As such, this temperature difference would yield a significant change in the positions of the peak.
Thermal analysis (DSC)
Differential scanning calorimetry data were collected using a Mettler Toledo DSC 3 with aluminium pans under nitrogen gas (flow rate = 10 mL min−1). Exothermic events were shown as peaks. Samples were heated and cooled to determine melting points as well as any additional phase transitions. The temperature and energy calibrations were performed using pure indium (purity 99.99%, m.p. 156.6 °C, heat of fusion: 28.45 J g−1) and pure zinc (purity 99.99%, m.p. 479.5 °C, heat of fusion: 107.5 J g−1). Samples were heated to 250 °C from 25 °C before being cooled back down at 25 °C at a heating or cooling rate of 10 °C min−1.
Fourier transform infrared spectroscopy
FTIR spectra was collected using the Bruker Alpha II model equipped with the Eco-ATR sampling module. Background noise was subtracted and small amount of the sample of interest was placed onto the ATR crystal. Spectra were measured from 600 to 4000 cm−1 range, resolution 4 cm−1 with 24 scans per sample. Spectra were collected using the ATR-FTIR in a room with air conditioning set at 22 °C. After spectral acquisition the ATR crystal was cleaned using isopropanol. Spectra are presented in the ESI,† along with some assignments of important functional groups.
Cambridge Structural Database analysis
The CSD (version 5.44)7 was used to analyse and compare the cocrystals prepared here with the relevant literature references.
Results and discussion
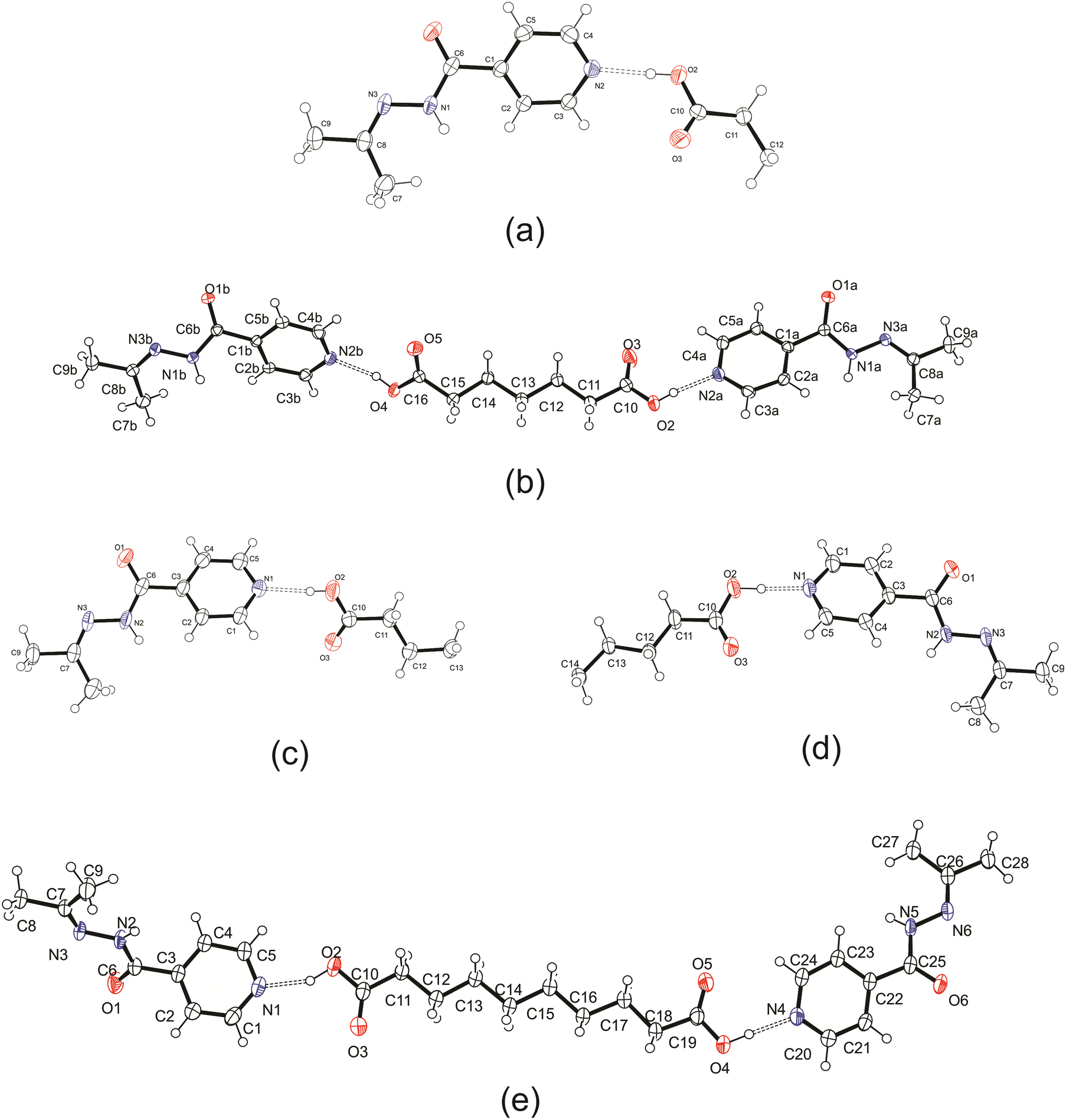
Five cocrystals containing a dicarboxylic acid and an izact molecule were obtained. Each crystal structure is described below and some comparison is given to its inh counterpart. It should be noted that we attempted to obtain cocrystals featuring both C3 and C5, but were unsuccessful in doing so. In our attempt to form a cocrystal with izact + C3 using both solvent evaporation and mechanochemical methods at varying stoichiometric ratios (izact:C3 as 1:1, 2:1 and 1:2), we obtained an oil each time. In our observations during grinding using both a ball mill and a mortar and pestle, the solid material would start transforming into a wax-like substance after 2–3 minutes of grinding. We also attempted to synthesize a cocrystal containing izact + C5 using varying stoichiometric ratios (izact:C5 as 1:1, 2:1 and 1:2) and using both solution-based and mechanochemical methods, which yielded no results. It should be noted that izact is not suitable for thermal-based methods as heating tends to allow the izact to undergo hydrolysis (water from atmosphere) to reform inh. It should also be noted that for the cocrystal system of izact + C8, we did not let the solution to evaporate slowly as it would lead to the formation of an oil. Instead the solvent was removed using a rotary evaporator to allow small crystals to form quickly. The ORTEP diagrams of the crystal structures are presented in Fig. 1 below, while the experimental details are listed in the ESI,† in Table S1.
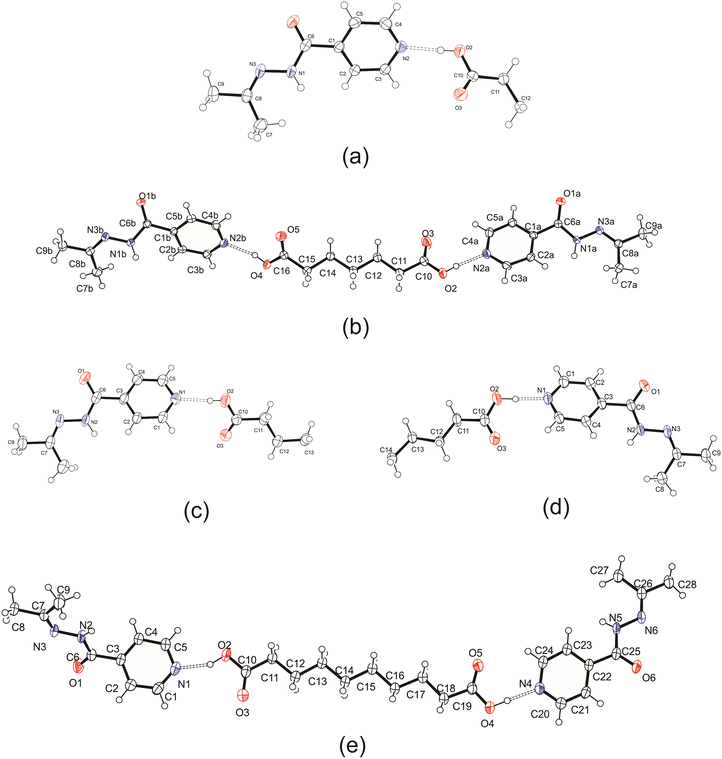 |
| | Fig. 1 ORTEP diagram for the crystal structures presented in this work, (a) izact + C6, (b) izact + C7, (c) izact + C8, (d) izact + C9, and (e) izact + C10. Ellipsoids were calculated at 50% probability level. | |
Crystal structure of izact + C6
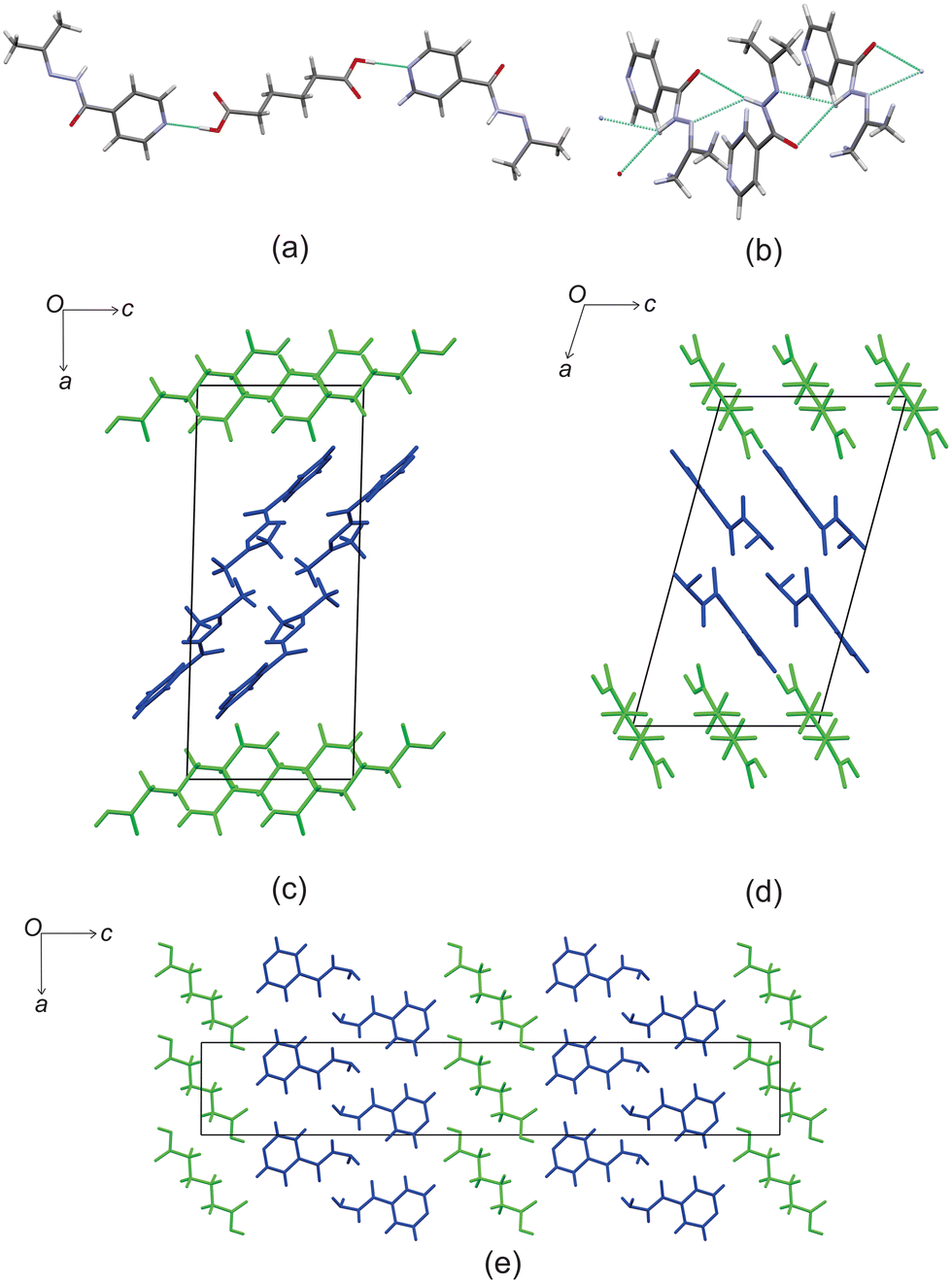
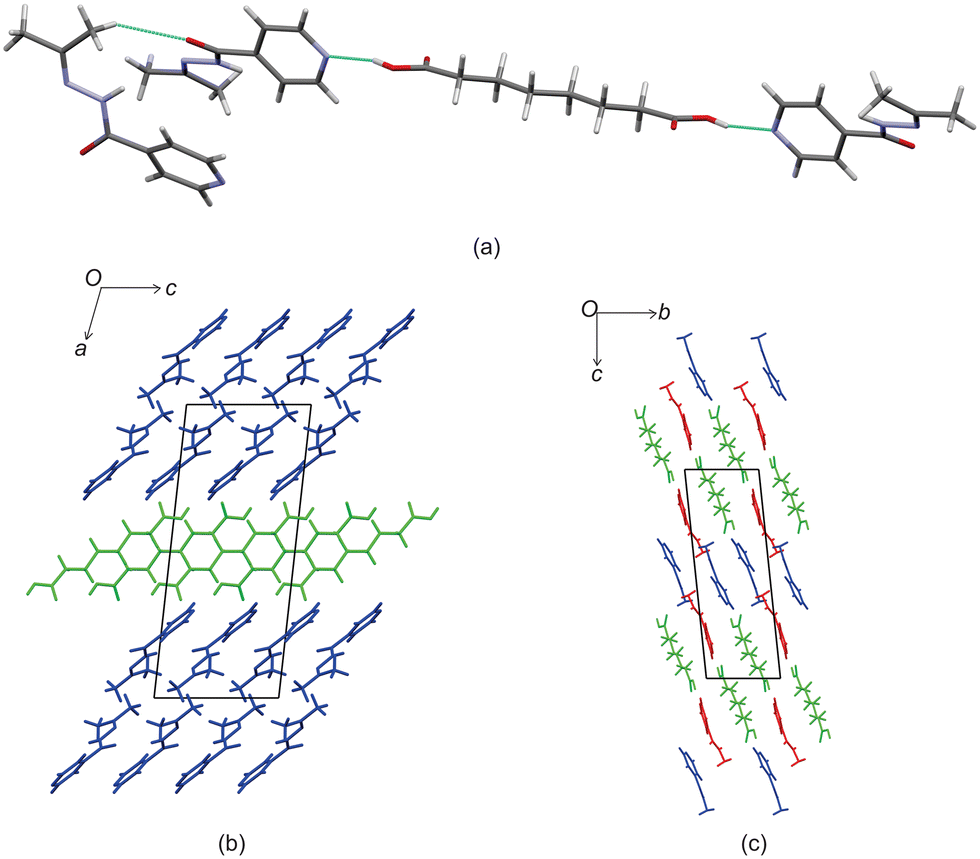
Izact formed a cocrystal with C6, forming colourless blocks. The asymmetric unit consists of one molecule of izact and half a molecule of C6, with the C6 molecule sitting on an inversion site. The izact + C6 cocrystal crystalizes in the P21/c space group. Like the cocrystal of inh + C6, a carboxylic acid⋯pyridine hydrogen bond motif formed between the dicarboxylic acid and pyridine group of izact (Fig. 2a). The dicarboxylic acid features no other strong hydrogen bonding. The izact molecule forms a bifurcated hydrogen bond between the nitrogen of the imine group and the oxygen of the amide group with the hydrogen of the amide group from a neighbouring izact molecule, which ultimately forms a chain hydrogen bond motif C11(4) (Fig. 2b). The packing of izact + C6 may be described as rows of C6 molecules separated by two rows of izact molecules. Within this structure, the methyl groups from the izact molecules would face each other, forming a hydrophobic layer within the crystal system, which is followed by a hydrophilic layer where the strong hydrogen bonding is present. Inh + C6 is a polymorphic cocrystal system with two forms20 that both crystalise in the P21/c space group, just like that of izact + C6. In terms of packing, izact + C6 is very similar to the packing of both forms of inh + C6, where there is alternating layers of inh molecules with C6 molecules. However, the inh molecules form strong hydrogen bonds together, whereas izact molecules interact with each other using fewer strong hydrogen bonds and the weaker van der Waals forces formed between the methyl groups.
 |
| | Fig. 2 The crystal structure of izact + C6 showing (a) the hydrogen bonding between izact and C6 (b) the hydrogen bonding between neighbouring izact molecules, (c) the packing of izact + C6 (with izact in blue and C6 in green), (d) the packing of inh + C6 form II (with inh in blue and C6 in green, Refcode FADGUO01) and (e) the packing of inh + C6 form I (with inh in blue and C6 in green, Refcode FADGUO). | |
Crystal structure of izact + C7
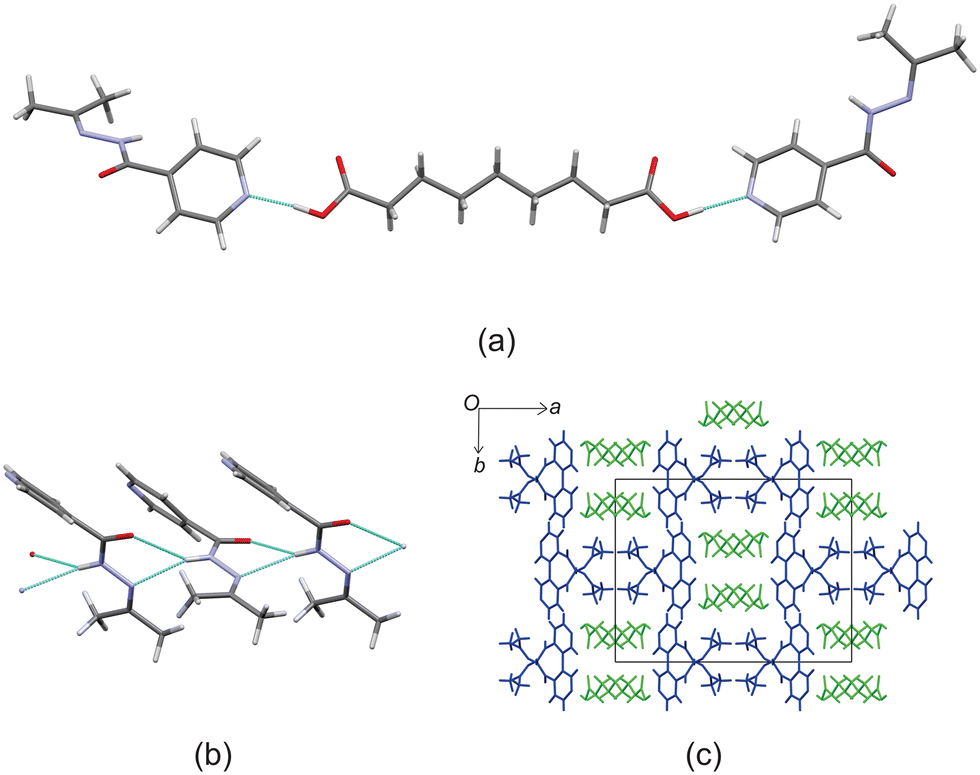
Izact formed a cocrystal with C7, which formed as colourless plates. The asymmetric unit consists of two molecules of izact and one molecule of C7. This cocrystal crystalizes in the C2/c space group. Like its inh + C7 cocrystal counterpart, the cocrystal of izact + C7 features the same carboxylic acid⋯carboxylic acid synthon. The other strong hydrogen bond that forms is one between the hydrogen atom of the amide group with the oxygen of the amide group from a neighbouring izact molecule (N1–H1A–O1A), which ultimately forms a chain hydrogen bond motif C11(4) (Fig. 3a). In this case, the imine does not form a hydrogen with this hydrogen. The packing of izact + C7 is made up of rows alternating between two molecules of izact and one molecule of C7 (Fig. 3b). In comparison, the cocrystal of inh + C7 (Refcode: FADHAV),19 the packing of inh + C7 is almost similar to the packing of izact + C7, where rows of molecules alternating between inh and C7 form (Fig. 3c). In addition, the cocrystal of inh + C7 exists in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Fig. 3c) while the izact + C7 exits in the C2/c space group. This is probably a result of the symmetry that arises from the carboxylic acid⋯pyridine synthon that forms on both ends of C7 as opposed to the only one that forms in inh + C7.
space group (Fig. 3c) while the izact + C7 exits in the C2/c space group. This is probably a result of the symmetry that arises from the carboxylic acid⋯pyridine synthon that forms on both ends of C7 as opposed to the only one that forms in inh + C7.
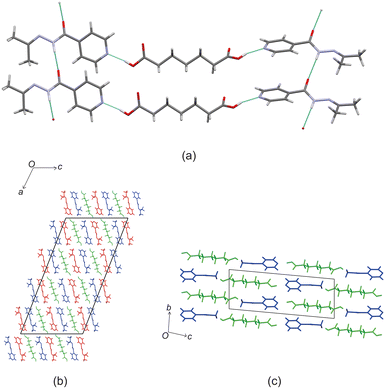 |
| | Fig. 3 The crystal structure of izact + C7 showing (a) the hydrogen bonding. (b) The packing (with izact in red and blue and C7 in green) and (c) the packing of inh + C7 (with C7 in green and inh in blue, Refcode: FADHAV). | |
Crystal structure of izact + C8
Izact formed a cocrystal with C8, which formed as colourless plates. The asymmetric unit consists of one molecule of izact and half a molecule of C8. This cocrystal crystalizes in the P21/c space group. The only strong hydrogen bonds that form is the carboxylic acid⋯pyridine. As such, the amide group of izact does not form any strong hydrogen bonds (Fig. 4a), in contrast to the other cocrystals featuring izact that is presented in this work. The only other hydrogen bond that has been observed using the short contact option of the Mercury software was a weak C–H bond with the oxygen (C8–H8⋯O1) (Fig. 4a). The methyl groups of izact point towards each other as a result of the van der Waals interactions that formed between them. Packing of izact + C8 is made up of rows of C8 molecules separated by rows of two molecules of izact (Fig. 4b). Here, the methyl groups izact molecules face each other due to the van der Waals interactions between the methyl group. This causes the C8 molecules to occupy the middle of the unit cell, as observed in Fig. 4b. In comparison to the cocrystal of inh + C8 (Refcode: SETRUG),19 inh + C8 exists in the P space group. As opposed to making a clear pattern of alternating rows like izact + C8, the packing of inh + C8 was orientated to favour maximizing the hydrazine⋯hydrazine interactions (synthon 4, Scheme 2) as well as the carboxylic acid⋯hydrazine interaction. As a result, the C8 molecules in inh + C8 pack in a more zigzag pattern than that of its izact counterpart (Fig. 4c).
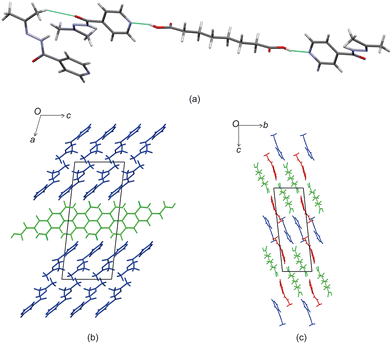 |
| | Fig. 4 The crystal structure of izact + C8 showing (a) the hydrogen bonding, (b) the packing (izact in blue, C8 in green) and (c) the packing of inh + C8 (inh in green and red, C8 in blue, Refcode: SETRUG). | |
Crystal structure of izact + C9
Izact forms a cocrystal with C9, which formed as colourless needles. The asymmetric unit consists of one molecule of izact and half a molecule of C9. This cocrystal crystalizes in the C2/c space group. Like the other cocrystals presented here, the cocrystal of izact + C9 features the carboxylic acid⋯pyridine synthon. In addition to the carboxylic acid–pyridine hydrogen bond sets (Fig. 5a), izact + C9 also features a bifurcated hydrogen bond formed between the nitrogen of the imine group and the oxygen of the amide group with the hydrogen of the amide group from a neighbouring izact molecule, which ultimately forms a chain hydrogen bond motif C11(4) (Fig. 5b). Packing consists of rows of izact molecules followed by rows of C9, with the alkyl groups of izact pointing towards the C9 molecule, which differentiates it from the previous cocrystals which had the alkyl groups pointing at each other (Fig. 5c). It should be noted that the cocrystal of izact + C9 is rather unique as, at the time at which this work is being reported, it did not have an inh + C9 cocrystal counterpart.
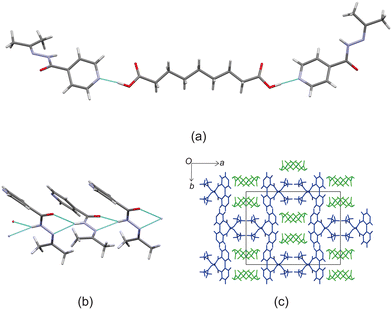 |
| | Fig. 5 The crystal structure of izact + C9 showing (a) the hydrogen bonding between neighbouring izact molecules, (b) the hydrogen bonding between izact and C9 and (c) the packing (with izact in blue and C9 in green). | |
Crystal structure of izact + C10
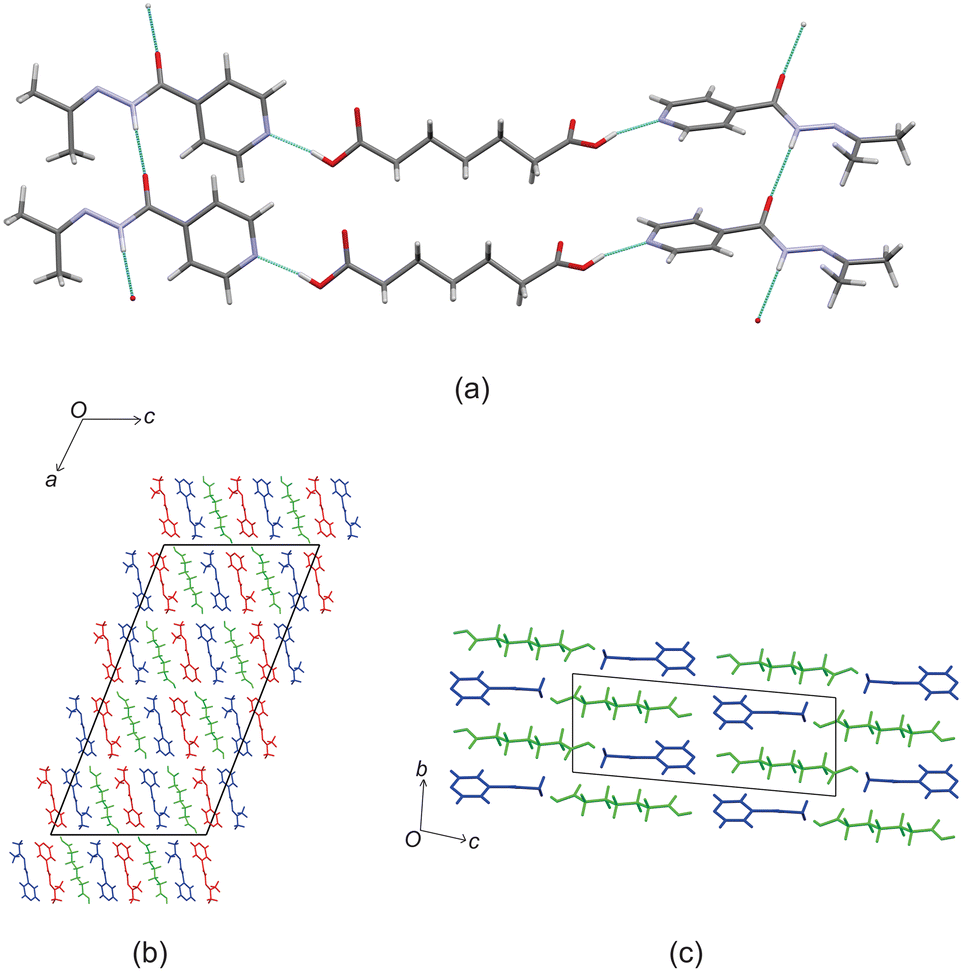
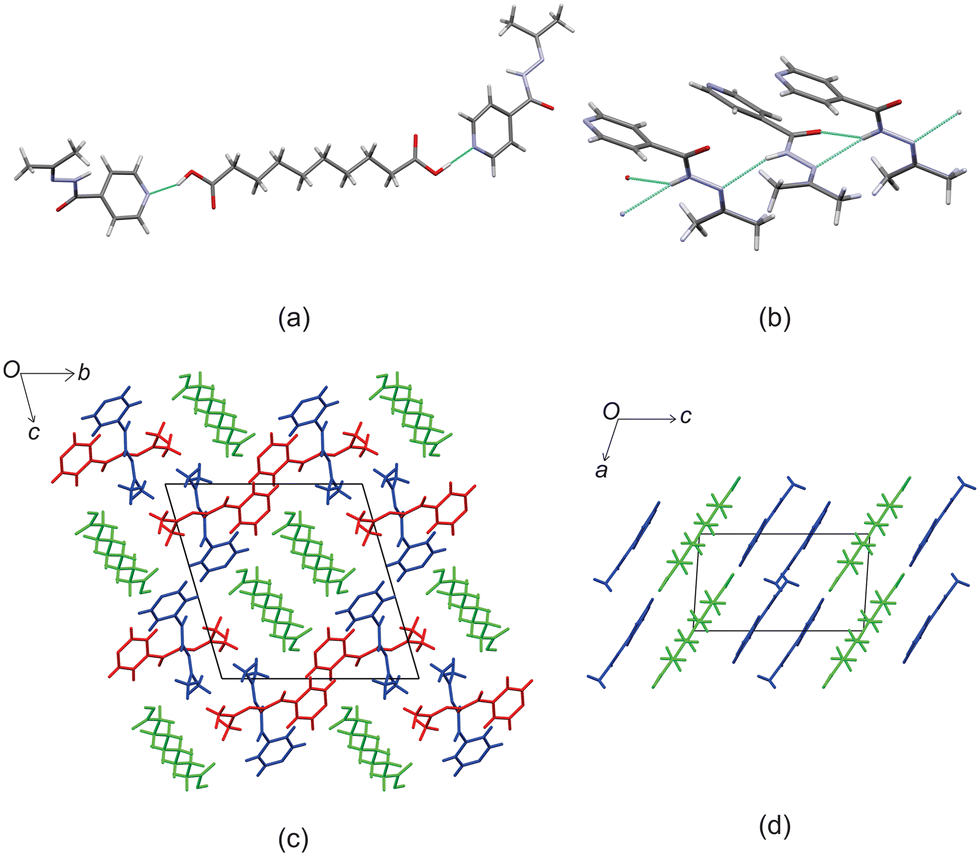
Izact formed a cocrystal with C10, crystalizing as colourless plates. The cocrystal of izact + C10 crystalizes in the P space group. The cocrystal of izact + C10 also features the carboxylic acid⋯pyridine synthon (Fig. 6a). Izact molecules also interact with each other via the amide group. A chain hydrogen bond motif formed when a single hydrogen bond formed between the hydrogen of the amide to the oxygen of a neighbouring amide, which is then then followed by a bifurcated hydrogen bond formed between the nitrogen of the imine and the oxygen of the amide to hydrogen of the amide from a neighbouring izact molecule. This results in an alternating pattern between a single hydrogen bond followed by a bifurcated hydrogen bond, as observed in Fig. 6b. Packing of izact + C6 represents channels formed by izact molecules occupied with molecules of C10 (Fig. 6c). In comparison, inh + C10 and izact + C10 also crystalizes in the P space group, but instead seems to form sheets as opposed to channels (Fig. 6d).
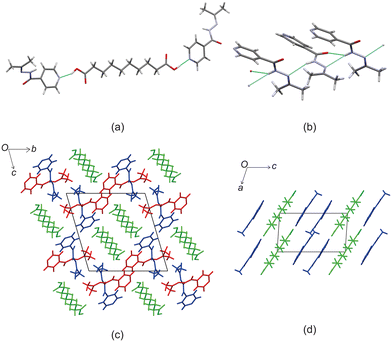 |
| | Fig. 6 The crystal structure of izact + C10 showing (a) the carboxylic acid⋯pyridine synthon formed between izact and C10, (b) the hydrogen bonding between izact molecules showing alteration between single and bifurcated hydrogen bonds, (c) the packing of inh + C10 (with izact as the red and blue molecules and C10 as the green molecules) and (d) the packing of inh + C10 showing the sheet packing (with inh in blue and C10 in green, Refcode: SETROA). | |
Discussion
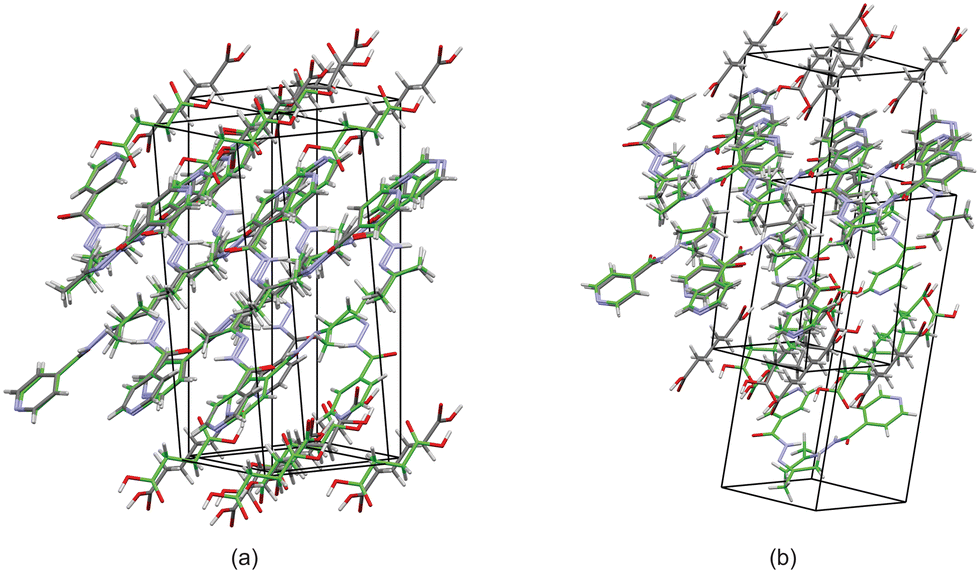
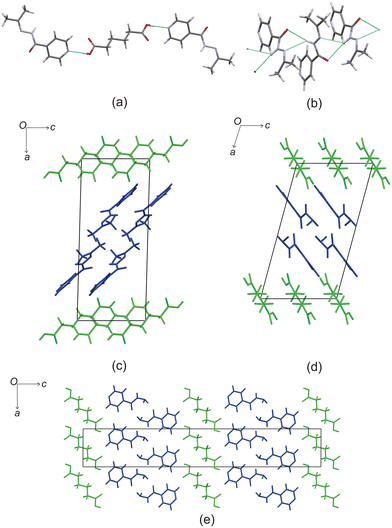
Based on these results, there is a few trends that can be observed in this series. When comparing the packing of izact + C4 (Refcode LATKEY), izact + C6 and izact + C8, the packing is isostructural. This was confirmed using the crystal structure packing similarity tool of Mercury, and can be observed in Fig. 7. However, the packing of izact + C10 is drastically different to that of izact + C4, izact + C6 and izact + C8, indicating a break in this trend. It is most likely the chain length of the C10 molecule is the cause of this deviation. Among the cocrystals of izact with odd-chained members, it is difficult to compare izact + C7 with izact + C9 in isolation. The only other consistency between the different cocrystals presented here is the formation of a 2:1 izact:dicarboxylic acid molecular ratio, as well as the fact the both carboxylic groups are involved with the carboxylic acid⋯pyridine synthon. One of the major reasons for this is that in the cocrystals containing inh and an odd-chain dicarboxylic acid is that one end of the molecule will be involved with hydrogen bonding with the pyridine ring of inh while the other end will form a hydrogen bond (which differs between the cocrystals of inh and the different dicarboxylic acids) with the hydrazine group of another inh molecule. This hydrogen bond between the dicarboxylic acid and the hydrazine group of inh cannot form readily in cocrystals of izact due to the bulky methyl groups present. Although this leaves the amide group of the izact molecule to interact with the amide group other izact molecules, the degree at which this occurs differs between the different cocrystal. This can range from being non-existent (izact + C8) to one which alternates between a single and bifurcated hydrogen bonds (izact + C10). However, when comparing the structures of izact + C9 and izact + C10 with the rest of the cocrystals reported here, it seems there is a transition that occurs. Going from izact + C6 to izact + C8 the packing mostly consists of alternating rows, while izact + C9 and izact + C10 seems to form channels formed by the izact molecules where the dicarboxylic acid occupies, as observed in Fig. 5c and 6c. As such, this definitely indicates that the chain length does play a significant effect on the packing of these cocrystals.
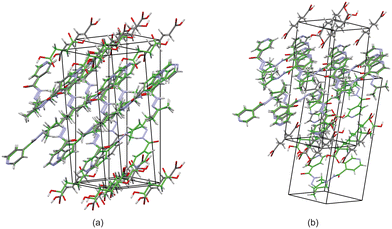 |
| | Fig. 7 The overlay of (a) izact + C4 (in green) with izact + C6 (in standard colours) and (b) the overlay of izact + C8 with izact + C6. | |
Thermal analysis (DSC)
DSC scans were collected to determine the melting points of the cocrystals, as well as to determine the presence of any other phase changes. All thermal data including the onset and enthalpies is available in Table 2, including data of the melting point of izact, dicarboxylic acid and inh–dicarboxylic acid cocrystals for comparison. The DSC curves were plotted and are available in the ESI.† All cocrystals showed no other phase changes prior to melting, indicating there are no potential polymorphic forms upon heating. Upon cooling, there were no further phase changes observed in each of the cocrystals. When opening the DSC pans after each completed run, a dark, glass-like substance remained, which indicated that the cocrystals decomposed during melting. Comparing the izact cocrystals presented here with both the melting points of the dicarboxylic acids and inh–dicarboxylic acid cocrystals, the melting points of most of the izact cocrystals are much lower overall. This decrease was expected as the decrease in the number of strong hydrogen-bonding pairs present between the two sets of structures would yield to an overall weaker collection of intermolecular bonding present. It should also be noted that there does not seem to be any particular trend between the melting points of these cocrystals.
Table 2 Thermodynamic data
| Carboxylic acid |
C4 |
C6 |
C7 |
C8 |
C9 |
C10 |
| Melting point of acid/°C (izact melting point: 160 °C (ref. 17)) |
184 |
152 |
103–105 |
140–144 |
109 |
131–134 |
| Melting point of inh cocrystal counterpart |
143–144 (ref. 19) |
138–140 (ref. 19) |
129–130 (ref. 19) |
111–113 (ref. 15) |
— |
112–114 (ref. 15) |
| Onset of melting of izact cocrystal/°C |
126.6 (ref. 17) |
99.3 |
85.7 |
108.4 |
117.7 |
106.6 |
| Peak of melting of izact cocrystal/°C |
— |
104.1 |
88.47 |
111.4 |
122.2 |
116.6 |
| Enthalpy of melting of izact cocrystal/J g−1 |
31.8 (ref. 17) |
176.3 |
109.0 |
97.7 |
133.7 |
63.68 |
Conclusions
Five new cocrystals containing izact and a dicarboxylic acid were synthesized and characterized. The dicarboxylic acids ranged from C6 to C10, excluding C3, C4 and C5, as the cocrystal of izact + C4 has already been reported and we could not obtain cocrystals of izact + C3 and izact + C5. All cocrystals presented here have a stoichiometric ratio of 2:1 izact:dicarboxylic acid in the asymmetric unit. The cocrystals containing the even-chained members of C4, C6 and C8 respectively were isostructural, sharing a similar packing. This trend was broken at the cocrystal of izact + C10. The odd-chained members did not share much similarity in their packing. The cocrystals of izact + C9 and izact + C10 share some similarities in their packing, where channels were made by the izact molecules where the respective dicarboxylic acid sits. In this series the chain length does play a significant role in the packing of these cocrystals, as there seems to be a significant shift in the packing between the cocrystals with a dicarboxylic acid chain length of eight or smaller to those with a chain length greater than eight. Also as expected, the melting points of the izact cocrystal is much lower than their inh counterparts due to the reduced number of strong hydrogen bonding pairs. As such, despite the dicarboxylic acids having an observable trend in terms of the crystal packing and its properties between its odd-chained and even-chained members, it seems that such trends does not necessarily cross over into that of its cocrystals.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This material is based upon work supported financially by the University of the Witwatersrand and the Molecular Sciences Institute. The National Research Foundation National Equipment Programme is thanked for financing the purchase of the dual wavelength hybrid diamond anode X-ray diffractometer (Bruker D8 Venture equipped with Mo and Cu X-ray sources) under NEP Grant No 129920. The DSI/NRF Centre of Excellence in Strong Materials is thanked for use of the Bruker D2-phaser.
Notes and references
- A. Mukherjee, Building upon supramolecular synthons: Some aspects of crystal engineering, Cryst. Growth Des., 2015, 15, 3076–3085 CrossRef CAS.
- G. R. Desiraju, Crystal engineering: From molecule to crystal, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. K. Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Polymorphs, Salts, and Cocrystals: What’s in a Name?, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- E. Grothe, H. Meekes, E. Vlieg, J. H. Ter Horst and R. De Gelder, Solvates, Salts, and Cocrystals: A Proposal for a Feasible Classification System, Cryst. Growth Des., 2016, 16, 3237–3243 CrossRef CAS.
- P. S. Panzade and G. R. Shendarkar, Pharmaceutical cocrystal: a game changing approach for the administration of old drugs in new crystalline form, Drug Dev. Ind. Pharm., 2020, 46, 1559–1568 CrossRef CAS PubMed.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Pharmaceutical cocrystals: An overview, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- R. Srinivasa Gopalan, P. Kumaradhas, G. U. Kulkarni and C. N. R. Rao, An experimental charge density study of aliphatic dicarboxylic acids, J. Mol. Struct., 2000, 521, 97–106 CrossRef CAS.
- V. R. Thalladi, M. Nüsse and R. Boese, The melting point alternation in α,ω-alkanedicarboxylic acids, J. Am. Chem. Soc., 2000, 122, 9227–9236 CrossRef CAS.
- A. Burger, J. O. Henck and M. N. Dünser, On the Polymorphism of Dicarboxylic Acids: I Pimelic Acid, Microchim. Acta, 1996, 122, 247–257 CrossRef CAS.
- H. Zhang, C. Xie, Z. Liu, J. Gong, Y. Bao, M. Zhang, H. Hao, B. Hou and Q. X. Yin, Identification and molecular understanding of the odd-even effect of dicarboxylic acids aqueous solubility, Ind. Eng. Chem. Res., 2013, 52, 18458–18465 CrossRef CAS.
- J. F. Murray, D. E. Schraufnagel and P. C. Hopewell, Treatment of tuberculosis: A historical perspective, Ann. Am. Thorac. Soc., 2015, 12, 1749–1759 CrossRef PubMed.
- B. Swapna, D. Maddileti and A. Nangia, Cocrystals of the tuberculosis drug isoniazid: Polymorphism, isostructurality, and stability, Cryst. Growth Des., 2014, 14, 5991–6005 CrossRef CAS.
- L. F. Diniz, M. S. Souza, P. S. Carvalho, C. C. P. da Silva, R. F. D'Vries and J. Ellena, Novel Isoniazid cocrystals with aromatic carboxylic acids: Crystal engineering, spectroscopy and thermochemical investigations, J. Mol. Struct., 2018, 1153, 58–68 CrossRef CAS.
- I. Sarcevica, L. Orola, M. V. Veidis, A. Podjava and S. Belyakov, Crystal and molecular structure and stability of isoniazid cocrystals with selected carboxylic acids, Cryst. Growth Des., 2013, 13, 1082–1090 CrossRef CAS.
- A. Lemmerer, Covalent assistance to supramolecular synthesis: Modifying the drug functionality of the antituberculosis API isoniazid in situ during co-crystallization with GRAS and API compounds, CrystEngComm, 2012, 14, 2465–2478 RSC.
- A. Lemmerer, J. Bernstein and V. Kahlenberg, Covalent assistance in supramolecular synthesis: In situ modification and masking of the hydrogen bonding functionality of the supramolecular reagent isoniazid in co-crystals, CrystEngComm, 2011, 13, 5692–5708 RSC.
- S. Cherukuvada and A. Nangia, Fast dissolving eutectic compositions of two anti-tubercular drugs, CrystEngComm, 2012, 14, 2579–2588 RSC.
- A. Lemmerer, J. Bernstein and V. Kahlenberg, One-pot covalent and supramolecular synthesis of pharmaceutical co-crystals using the API isoniazid: A potential supramolecular reagent, CrystEngComm, 2010, 12, 2856–2864 RSC.
- I. Sarceviča, A. Kons and L. Orola, Isoniazid cocrystallisation with dicarboxylic acids: Vapochemical, mechanochemical and thermal methods, CrystEngComm, 2016, 18, 1625–1635 RSC.
- C. B. Aakeröy, S. Forbes and J. Desper, Using cocrystals to systematically modulate aqueous solubility and melting behavior of an anticancer drug, J. Am. Chem. Soc., 2009, 131, 17048–17049 CrossRef PubMed.
- C. B. Aakeröy, S. Forbes and J. Desper, Altering physical properties of pharmaceutical co-crystals in a systematic manner, CrystEngComm, 2014, 16, 5870–5877 RSC.
- S. Tothadi and A. Phadkule, Does stoichiometry matter? Cocrystals of aliphatic dicarboxylic acids with isonicotinamide: odd-even alternation in melting points, CrystEngComm, 2019, 21, 2481–2484 RSC.
- H. F. Su, L. Xue, Y. H. Li, S. C. Lin, Y. M. Wen, R. Bin Huang, S. Y. Xie and L. S. Zheng, Probing hydrogen bond energies by mass spectrometry, J. Am. Chem. Soc., 2013, 135, 6122–6129 CrossRef CAS PubMed.
-
Bruker, APEX4 version 2021.4-1 data collection software which includes SAINT version 8.40B, SADABS-2016/2 and XPREP version 2014/2, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- L. J. Farrugia, WinGX suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, ORTEP-3 for windows - A version of ORTEP-III with a graphical user interface (GUI), J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, Mercury: Visualization and analysis of crystal structures, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Andreas
Lemmerer
and
Andreas
Lemmerer