
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe concentration-dependent effect of hydrocortisone on the structure of model lung surfactant monolayer by using an in silico approach†
Mohammad Zohurul Islam‡
 *a,
Sheikh I. Hossainb,
E. Deplazesba,
Zhen Luoa and
Suvash C. Saha*a
*a,
Sheikh I. Hossainb,
E. Deplazesba,
Zhen Luoa and
Suvash C. Saha*a
aSchool of Mechanical and Mechatronic Engineering, University of Technology Sydney, 15 Broadway, Ultimo 2007, NSW, Australia. E-mail: Suvash.Saha@uts.edu.au
bSchool of Life Sciences, University of Technology Sydney, 15 Broadway, Ultimo 2007, NSW, Australia. E-mail: Evelyne.Deplazes@uts.edu.au
First published on 22nd November 2022
Abstract
Understanding the adsorption mechanism of corticosteroids in the lung surfactant requires the knowledge of corticosteroid molecular interactions with lung surfactant monolayer (LSM). We employed coarse-grained molecular dynamics simulation to explore the action of hydrocortisone on an LSM comprised of a phospholipid, cholesterol and surfactant protein. The structural and dynamical morphology of the lung surfactant monolayer at different surface tensions were investigated to assess the monolayer compressibility. The simulations were also conducted at the two extreme ends of breathing cycles: exhalation (0 mN m−1 surface tension) and inhalation (20 mN m−1 surface tension). The impact of surface tension and hydrocortisone concentration on the monolayer compressibility and stability are significant, resulting the monolayer expansion at higher surface tension. However, at low surface tension, the highly compressed monolayer induces monolayer instability in the presence of the drug due to the accumulation of surfactant protein and drug. The constant area per lipid simulation results demonstrate that the surface pressure-area isotherms show a decrease in area-per-lipid with increased drug concentration. The drug-induced expansion causes considerable instability in the monolayer after a specific drug concentration is attained at inhalation breathing condition, whereas, for exhalation breathing, the monolayer gets more compressed, causing the LSM to collapse. The monolayer collapse occurs for inhalation due to the higher drug concentration, whereas for exhalation due to the accumulation of surfactant proteins and drugs. The findings from this study will aid in enhancing the knowledge of molecular interactions of corticosteroid drugs with lung surfactants to treat respiratory diseases.
1 Introduction
Hydrocortisone is a glucocorticoid that is used as an anti-inflammatory and immunosuppressive drug to treat a range of inflammatory and allergy conditions,1,2 including many pulmonary illnesses such as asthma, chronic obstructive pulmonary disease, influenza and bronchitis,3 and bronchopulmonary dysplasia in infants.4–6 A recent study conducted by Petersen et al.,7 also showed that corticosteroids, including hydrocortisone, reduce the mechanical ventilation required by patients with severe acute respiratory syndrome caused by SARS-COV-2. Hydrocortisone might be as useful as dexamethasone to treat covid-19.8Normal breathing and lung function rely on the activity of lung surfactants, which are secreted and synthesised from alveolar type II cells, and create a bilayer-associated monolayer at the alveolar air–water interface9–11 known as the lung surfactant monolayer (LSM). The functions of LSM include supporting the mechanical work of breathing by dropping the surface tension and preventing alveolar collapse at exhalation when surface tension reaches values close to zero.12–14 The LSM serves as the first line of defense against potential inhaled pathogens/particles entering the lung, and of course, the same barrier also protects again many inhaled entities.
All these functions, interfacial surface area regulation and compressibility of the lung surfactant are highly dependent on the lung surfactant composition, surfactant molecules organisation at the air–liquid interface in the alveoli.15 Lack of surfactant or any imbalance of surfactant can cause reduced lung surfactant activity. In mammalian lung surfactants, phospholipids are the most abundant component comprising approximately 85% by weight.16 Of the phospholipids, the zwitterionic phosphatidylcholine (PC) lipids are the most abundant species amounting to ∼70% w/w. More than 50% w/w of total PC lipids is di-saturated 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), and the remaining species are either mono or di-unsaturated phospholipids such as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC).17,18 The next common phospholipids, account for ∼10% w/w, are negatively charged phosphatidylglycerol (PG) lipids such as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) or 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG).10,15,19 In addition to phospholipids, the LSM also contains nearly ∼8% w/w cholesterol (CHOL).15 Besides lipids, the LSM contains ∼8% w/w surfactant protein.20,21 They are divided into two categories: hydrophilic proteins (SP-A and SP-D) that are found in the aqueous subphase of the lipid bilayer, and hydrophobic proteins (SP-B and SP-C) that are anchored in the membrane involving the surface-tension regulating activity of the LSM.22,23
During regular breathing, the LSM is continuously compressed and expanded. The surface tension lies between ≈ 0 mN m−1 near the end of exhalation, at maximal compression, and between 20–25 mN m−1 during inhalation, at maximal expansion of the monolayer.12,24 As a result of these compression and expansion properties, the LSM passes through different phases; the liquid-condensed (LC) phase, a transitional phase (LC + LE), in which the LC and liquid-expanded phase (LE) coexist and the liquid-expanded phase (LE) of the monolayer.25,26 The structure and, thus, phase behaviour of the LSM is strongly related to the packing and movement of lipids in the monolayer. The phase behaviour of the monolayer can be expressed in a function of the surface pressure and surface tension according to the equation π = γa–w − γeq, where, π is the pressure, γa–w (∼72 mN m−1 at 310 K) is the surface tension of the pure water at the air–water interface, and γeq is the surface tension of the monolayer.27–29 Monolayer phase transition from LC phase to LE phase induced from the high surface pressure to low surface pressure, respectively. Due to the complexity of the LSM and changing the structure of LSM during breathing cycles, understanding the interaction of inhaled drug such as corticosteroid drugs is not trivial. As each phase shows different lipid packing, compressibility and surface tension, the interaction of drug with LSM is not only dependent on the drug concentration but also on the LSM phase. Nevertheless, elucidating these interactions is important to understand how corticosteroid drugs are adsorbed into the LSM.
Understanding the interactions of lung surfactants with corticosteroids is also essential for improving drug delivery to treat pulmonary illnesses. The lack and dysfunction of lung surfactants is linked to severe respiratory diseases.30 In some cases, treatment includes exogenous surfactants, which might include the addition of corticosteroids.31 Furthermore, the delivery of corticosteroid to the alveoli and their incorporation into endogenous lung surfactants might be improved by incorporating the drug into exogenous surfactants.32 While the main therapeutic target of corticosteroids is receptors, inhaled drugs can reach the LSM, where they alter the structure and dynamics of the monolayer with detrimental effects on normal lung function. Thus, exogenous lung surfactants might improve corticosteroid delivery and reduce side effects of inhaled glucocorticoids. The molecular interactions between the drug and surfactants are critical to understand how corticosteroid drugs can be incorporated into exogenous surfactants.
The interaction of the corticosteroids such as beclomethasone, budesonide and fluticasone with exogenous lung surfactant was investigated by Cimato et al.,.33 They explored the concentration-dependent effect of these three corticosteroids as well as cholesterol on surfactant fluidity and compressibility using pulsating bubble surfactometer and polarised microscopy experiments. In the absence of corticosteroids or cholesterol, the monolayer formed by exogenous lung surfactants showed film compressibility of 60%. This was reduced to less than 40% in the presence of the drugs or cholesterol. The study also shows that at low compressibility, the LSM can decrease surface tension to less than 2 mN m−1, which is close to the 0 mN m−1 required for normal breathing. Similar surface tension lowering capabilities were also reported by other biophysical studies for corticosteroid containing natural and exogenous surfactant.34,35 This experimental study also demonstrates that the drug does not destabilise the surfactant monolayer at lower drug concentration. Cimato et al.,36 conducted an experimental study to investigate the corticosteroid-associated lung surfactant formulation to advance the spreading mechanism of corticosteroids. In their study, the authors explored corticosteroid budesonide interaction with an exogenous pulmonary surfactant to advance drug delivery to the alveoli. In a study by Wang et al.,35 combined the Langmuir balance and AFM experiments to investigate the corticosteroid interaction with commercially available natural surfactant (Infasurf) and two corticosteroids (budesonide and beclomethasone propionate) drugs. The characteristic isotherms of Infasurf at 0.1, 1, and 10% budesonide and beclomethasone propionate were compared to investigate the corticosteroid effects on the activity of the monolayer. Budesonide and beclomethasone propionate do not significantly interfere with the Infasurf monolayer stability up to corticosteroid concentrations of ≤1% w/w for budesonide and ≤10% w/w for beclomethasone propionate. Once these concentrations were exceeded, the corticosteroids induce instability of the monolayer due to monolayer fluidisation. Thus, Infasurf loaded with drugs below the above-mentioned critical concentration of drug could be useful to measure the spreading of corticosteroids to the lung surfactant correctly at the alveolar air–water surface.
Dos Santos et al.,37 performed an in vitro experimental study using the monolayer composition (POPC-DOPC-CHOL) to investigate how corticosteroid drug budesonide affects monolayer biophysics including monolayer fluidity and surface pressure in the presence of budesonide and budesonide mixed cyclodextrin oligosaccharides complex. The study demonstrates that cholesterol changes lipid packing and lipid desorption with the help of the corticosteroid drug budesonide. Budesonide increased membrane fluidity and permeability, according to their findings. It is also found that budesonide induced the disruption of lipid monolayer in cholesterol-enriched raft-like domains that change the lipid packing in the monolayer. In addition to the above-mentioned studies, a wide range of wet-lab methods have also been investigated to study the structure and physicochemical characteristics of the LSM, including Langmuir trough,38–43 atomic force microscopy (AFM),35 electron resonance spectroscopy33 and captive bubble surfactometer experiments.44 These techniques can be used to study the interaction of the LSM with steroids45–47 and non-steroid drugs,29,48,49 but they do not provide proper molecular-level mechanistic interaction between drugs and LSM. To gain better insight into molecular level mechanistic interaction of corticosteroid drugs and surfactant monolayer, it is preferable to use coarse-grained lung surfactant models because coarse-grained simulations provide precise insights into molecular level interaction.
Computer simulation is one of the techniques to study the structural and dynamical properties of LSM. This method has been used to investigate the stability and biophysical characteristics of lung surfactant monolayers and bilayers. The interaction of the corticosteroid drug prednisolone with a simple model LSM was investigated by Estrada-López et al.,50 using coarse-grained (CG) MD simulations at surface tension 0, 10 and 20 mN m−1. Results revealed that the film collapses at low surface tensions (0 mN m−1). In a recent study, the CG MD simulation was used to investigate the effect of mometasone on an LSM model comprised of DPPC-POPC-POPG-CHOL and surfactant protein (SP-B and SP-C) containing monolayer.45 The results revealed that mometasone alters the structure of the LSM as a function of drug concentration, and causes monolayer to collapse, which is influenced by surface tension and surfactant protein. The accumulation of drug molecules limits the drug capacity to spread into the surfactant monolayer at high drug concentration, resulting monolayer collapse. The findings from these studies highlight the complexity of these systems and specify the impact of corticosteroid is facilitated by specific interactions between the drug and surfactants. The drug-induced changes in surfactant properties are concentration-dependent and rely on lipid composition, surface tension or alternatively surface pressure.
However, molecular interactions with corticosteroids from such experiments are little known, and more research into lung surfactant stability and mechanical properties is needed to figure out how to distribute corticosteroid drugs into the targeted region of the alveoli by avoiding lung surfactant damage. Drug interaction with lung surfactants has contributed to the development of more efficient and less intrusive ways for a therapeutic dose, controllable corticosteroid release, and off-targeting drug deposition on LSM.51
In this study, the CG MD simulation is used to investigate the minimum and maximum compressibility of the LSM for the drug-free and drug-containing monolayer by constant surface tension simulation ranging from 0 to 50 mN m−1. The mechanistic changes of the LSM at inhalation (20 mN m−1) and exhalation (0 mN m−1) breathing conditions for different drug concentration is also investigated. The broader ranges of surface tensions beyond the exhalation and inhalation surface tension are used to explore the extremum area per lipid (APL) values (lowest and highest APL) of the LSM and their phase analysis. To analyse phase behaviour of the LSM and monolayer-to-multilayer transformation of the LSM at LC and LE phases, the fixed APL simulation were carried out at APL values ranging from 0.47 to 0.61 nm2. Finally, to isolate the effect of hydrocortisone concentration on breathing dynamics, the drug-containing systems were simulated at exhalation breathing (0 mN m−1) and inhalation breathing (20 mN m−1). For all the underlying LSM model used in this study is composed of DPPC-POPC-POG-CHOL (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10:10) and surfactant protein (SP-B-SP-C, 1:1) to mimic the fraction of surfactant components observed in human lung surfactants.19 This amount of surfactant molecules ensures the trustworthy composition of LSM in mol%15–21 that, equivalent to the number of surfactant molecules, 65% DPPC, 21% POPC, 5% POPG, 5% CHOL and 4% surfactant proteins (SP-B and SP-C) by mass.
:20:10:10) and surfactant protein (SP-B-SP-C, 1:1) to mimic the fraction of surfactant components observed in human lung surfactants.19 This amount of surfactant molecules ensures the trustworthy composition of LSM in mol%15–21 that, equivalent to the number of surfactant molecules, 65% DPPC, 21% POPC, 5% POPG, 5% CHOL and 4% surfactant proteins (SP-B and SP-C) by mass.
2 Methodology
2.1 Simulation parameter and simulation model setup
The monolayer was built by using two symmetric lipid-protein monolayers of dimensions 25.0 × 25.0 × 60.0 nm3 that were put into a water box of dimensions 25.0 × 25.0 × 12.0 nm3. Monolayers were spaced sufficiently far apart to avoid the interaction between them (Fig. 1a). Each monolayer is comprised of 2032 phospholipids and cholesterol molecules at a molar ratio DPPC-POPC-POPG-CHOL (60:20:10:10) with surfactant protein B and C (SP-B and SP-C, 1:1). Sixteen (16) monolayer-associated surfactant proteins (8 SP-B and 8 SP-C) were incorporated into the monolayer. The number of water beads was ∼50000. The monolayers were built by using the python script insane.py52 according to the protocol stated by Hossain et al.,.53 The CG parameters for DPPC, POPC, POPG, CHOL and water were obtained standard MARTINI force field.54–58 The CG structure of DPPC, POPC, POPG, CHOL, SP-B, SP-C and hydrocortisone are represented in Fig. S1,† 1b and c. The structure of the surfactant proteins (SP-B and SP-C) were based on the all-atom structure of SP-B from the protein data bank (PDB ID: 1DFW59) and SP-C (PDB ID: 1SPF60). For SP-B1–25 sequence, FPIPLPYCWLCRALIKRIQAMIPKG was chosen.61–64 For SP-C, the sequence LRIPCCPVNLKRLLVVVVVVVLVVVVIVGALLMGL was used that contains 35-residues containing the 23 hydrophobic residues showing activities in LSM surface.65,66 The net charge of SP-B1–25 and SP-C model are +4 and +3.66 An ionic concentration of 150 mM NaCl was incorporated into the LSM system due to negatively charged phospholipid (POPG) and positively charged surfactant proteins to confirm that the system was electrically neutral (Table S2†). The python script matinize.py was used to convert the protein structures from all-atoms to CG. The CG topology of hydrocortisone was described based on other sterols/steroids previously reported50,67 and using standard MARTINI beads.58 The detailed description of the topology and bead types of hydrocortisone are found in the ESI section (S1.1, Table S1,† Fig. 1b and c). The hydrocortisone parameter was validated by calculating the octanol–water partition coefficient from the free energy required to move the drug molecule from an octanol phase into the water phase. The free energy was achieved from a potential mean force (PMF) calculation using standard umbrella sampling simulations. The detailed procedure of these PMF measurement is described in the ESI (S1.2).†
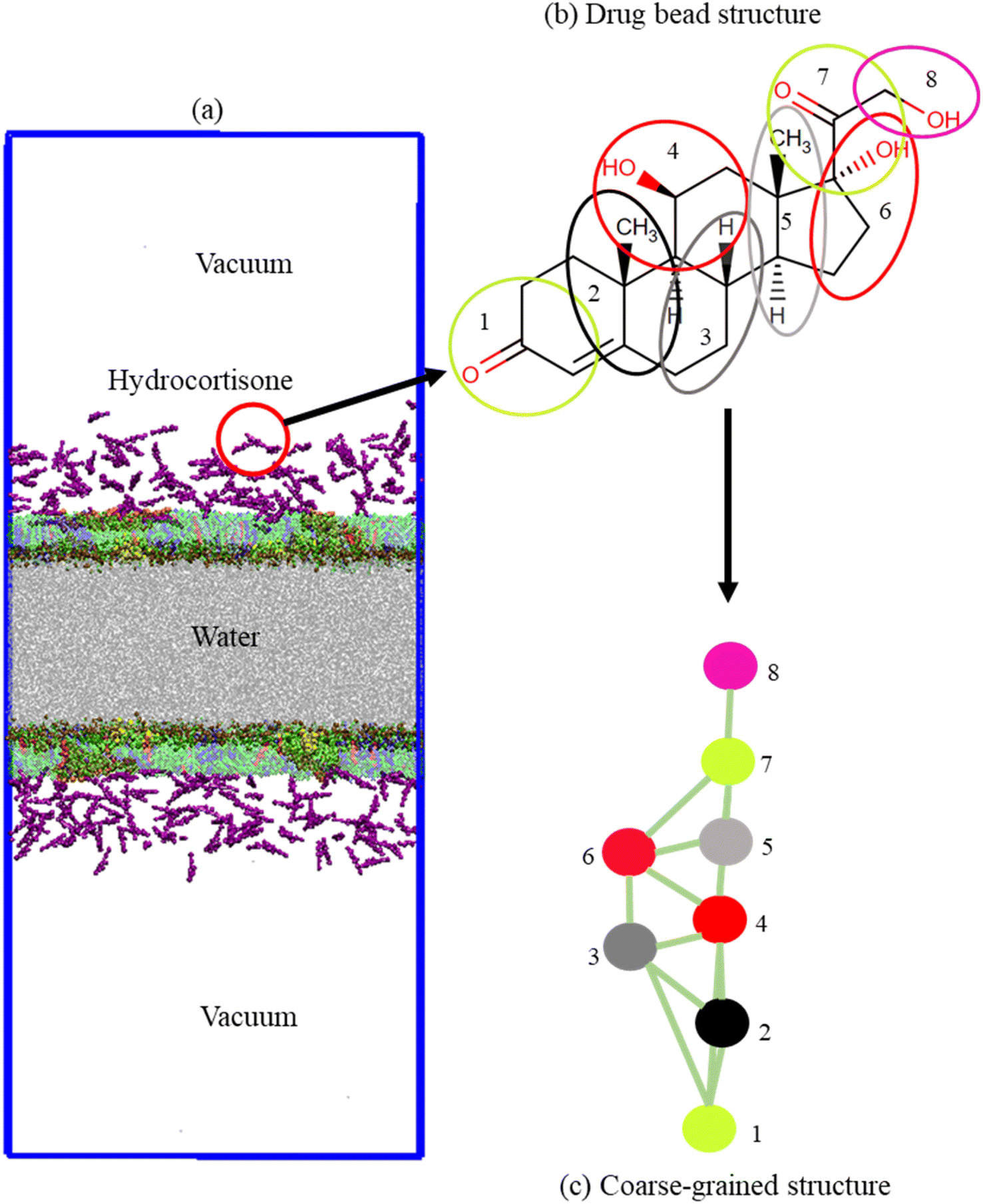 | ||
| Fig. 1 (a) Schematic representation of the system composed of surfactant components in the presence of 2.84% w/w hydrocortisone molecules. The system consists of two surfactant monolayers detached by a 12 nm water box, and 21 nm vacuum on each side of the layer. The monolayer is comprised of DPPC, POPC, POPG and cholesterol with surfactant proteins B and C (SP-B and SP-C). The system components are indicated by DPPC (green), POPC (blue), POPG (cyan), cholesterol (red) and hydrocortisone (purple), SP-B1–25 (yellow), SP-C (orange) and water (silver). (b) Chemical structure of hydrocortisone and (c) coarse-grained bead representation of hydrocortisone. | ||
2.2 Molecular dynamics simulation
:POPC:POPG:CHOL:SP-B1–25:SP-C (60:20:10:10:1:1) was simulated at NPγT ensemble using surface tensions ranging from 0 to 50 mN m−1 with an interval of 10 mN m−1 to reproduce highly compressed (0 mN m−1) to highly expanded (50 mN m−1) monolayers (system I). Monolayers with eight different APL values of 0.47, 0.49, 0.51, 0.53, 0.55, 0.57, 0.59 and 0.61 nm2 were extracted from previously built reference systems. These monolayers were inserted into a simulation box with dimensions 24.90 × 24.90 nm2, 24.49 × 24.49 nm2, 24.10 × 24.10 nm2, 23.64 × 23.64 nm2, 23.21 × 23.21 nm2, 22.77 × 22.77 nm2 and 22.32 × 22.32 nm2, respectively to ensure APL values remain at the target APL values during NVT simulations. Each of these eight systems (system II) was equilibrated first 500 ns and then 2 μs final simulation in the NVT ensemble.
:POPC:POPG:CHOL (60:20:10:10) and surfactant proteins (SP-B1–25 and SP-C, 1:1) in the presence of hydrocortisone (0 to ∼10.5% w/w). The systems are simulated at NPγT and NVT ensemble. Nhydrocortisone refers the number of hydrocortisone molecule per leaflet of the monolayer. Each of all systems were simulated at least two repeated runs
| System | Nhydrocortisone/monolayer | Concentration (% w/w) |
|---|---|---|
| NPγT simulation at surface tension: γ = 0, 10, 20, 30, 40 and 50 mN m−1 (system I) | ||
| I | 0 | 0% |
| 120 | 5.52% | |
|
||
| NVT simulation at fixed APL: 0.47, 0.49, 0.51, 0.53, 0.55, 0.57, 0.59 and 0.61 nm2 (system II) | ||
| II | 0 | 0% |
| 10 | 0.49% | |
| 30 | 1.44% | |
| 60 | 2.84% | |
| 120 | 5.52% | |
| 240 | 10.46% | |
|
||
| NPγT simulation at surface tension: γ = 0 and 20 mN m−1 (system III) | ||
| III | 0 | 0% |
| 10 | 0.49% | |
| 30 | 1.44% | |
| 60 | 2.84% | |
| 120 | 5.52% | |
| 240 | 10.46% | |
For simulation of the hydrocortisone containing LSM, five different models were set up. The rationale behind choosing this range of drug concentrations and the calculations used to translate an average drug dose of inhaled corticosteroids are outlined in the ESI section (S1.3).† The original configurations of the drug-containing system were collected from the final trajectories of the corresponding reference conformation from the fixed surface tension simulation (system I) and fixed APL simulation (system II). The hydrocortisone molecules were arbitrarily inserted in air phase of the system as shown in Fig. 1a. These five drugs containing systems were equilibrated 500 ns followed by a 2 μs production run in the NVT ensemble for fixed APL (system II) simulation. For fixed surface tension simulation, each of the five drug-LSM systems was equilibrated for 500 ns in an NVT ensemble, followed by further equilibration in an NPγT ensemble, and final 2 μs production run simulation for data analysis. The NPγT were carried out at 0 mN m−1 and 20 mN m−1 surface tension.
GROMACS version 5.1.4 (ref. 68) was used to simulate all the systems. Each system was energy minimised using the steep descent algorithm. The leapfrog algorithm69 with a 20 fs time step was used. The Martini standard cut-off for coulomb interaction potential and Lennard-Jones interaction were taken. Monolayer components (proteins, lipids and cholesterol), water and ions, and hydrocortisone were coupled independently at a temperature of 310 K with a V-rescale thermostat70 and a time constant of τ = 1.0 ps. The Berendsen barostat71 at temperature 310 K was used with τ = 4.0 ps. The compressibility along the xy-plane and z-axis were fixed 4.5 × 10−5 bar−1 and zero, respectively. The neighbour lists were updated for each 20 steps.
3 Results and discussion
The aims of this study were to examine the minimum and maximum compressibility for monolayer phase analysis and the concentration-dependent effect of hydrocortisone on the structural properties of the LSM. For this, the simulations were run in the absence and presence of five different hydrocortisone concentrations from 0% to ∼10.50% w/w (Table 1). The LSM was simulated at a range of fixed surface tension simulation (0 to 50 mN m−1) for the drug-free case to get the initial trajectories from fixed APL simulation. Each drug containing system was simulated in two different conditions inhalation (20 mN m−1) and exhalation (0 mN m−1) breathing. For fixed APL simulation, eight different APL values represent the range of compressibility of the LSM during the breathing cycle. The fixed surface tension simulations representing the expanded and compressed LSM were conducted at the extreme ends of breathing cycles; 20 mN m−1 for the end state of inhalation and 0 mN m−1 for the end state of exhalation. Hydrocortisone concentrations are chosen to those used in existing biophysical studies of corticosteroids.45,50,743.1 Monolayer compressibility and stability analysis
In the first part of the study, the monolayer's compressibility and stability have been analysed. This is done by calculating area per lipid (APL), phospholipids order parameter and density of the LSM components at different surface tensions. The APL value was calculated by measuring the average area of monolayer leaflet and dividing the number of lipid molecules.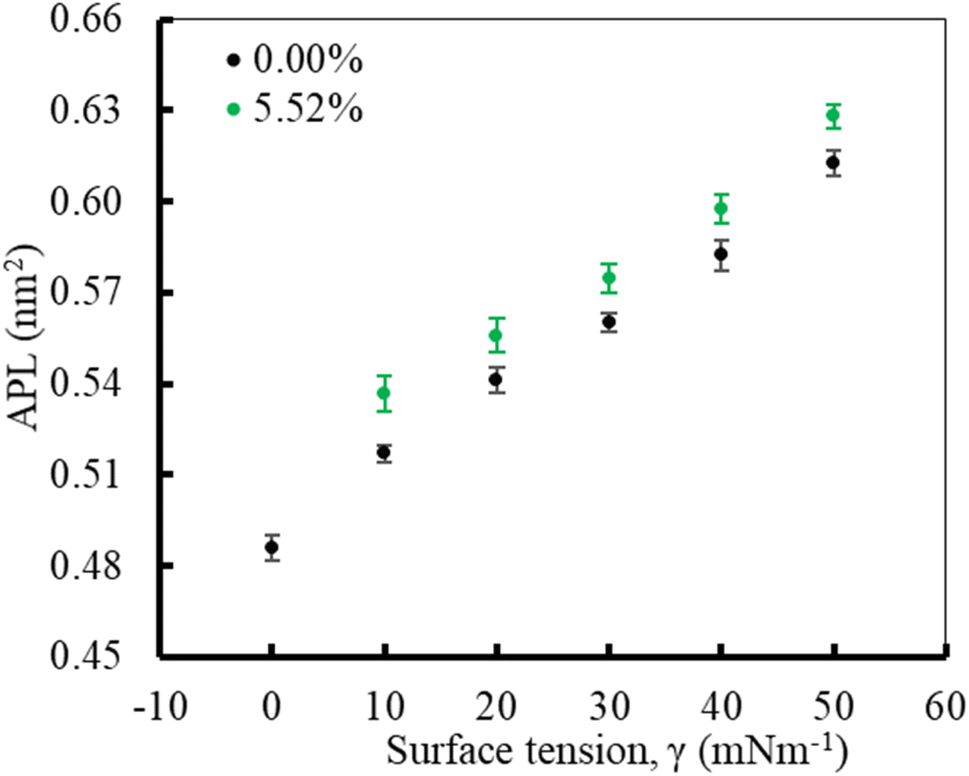 | ||
| Fig. 2 The APL values of the lung surfactant monolayer as a function of surface tension in the absence and presence of 5.52% w/w of glucocorticoid drug hydrocortisone for the surfactant protein containing monolayer. The error bars indicate standard deviation of the time evolution of APL data across the frames of the trajectories. | ||
These APL values for drug-free monolayer is in agreement with the value of APL found in previous studies of the comparable systems (surfactant protein-free monolayer) from atomistic and CG MD study investigated by Laing et al.,75 which are 0.527 ± 0.012 nm2 and 0.545 ± 0.007 nm2, respectively. In the presence of hydrocortisone, the APL values show similar concentration-dependent increase of APL from 0.537 ± 0.006 nm2 at 10 mN m−1 surface tension to 0.628 ± 0.004 nm2 at 50 mN m−1 surface tension with 99% confidence interval. It is notated that in the hydrocortisone-containing system at 0 mN m−1 surface tension, the APL is not presented since the drug causes the monolayer instability at this surface tension. The LSM starts engulfing the monolayer components by forming bilayer reservoir underneath the monolayer that induces the monolayer instability. In this state, the LSM shows small folding, eventually causing monolayer collapse. The previously reported study shows such monolayer-to-bilayer transformation in a study of an LSM composed of DPPC:POPC:POPG:CHOL:SP-B1–25:SP-C exposed to the corticosteroid drug mometasone.45
As the surface tension induces LSM expansion, this is also likely affecting the conformational mobility of the lipid tails. Fig. 3 illustrates the order parameter for the sn-1 and sn-2 chains of all phospholipids (DPPC, POPC and POPG) in the system calculated from simulations of surfactant protein containing monolayers in the absence of corticosteroid drug hydrocortisone at surface tensions 0 to 50 mN m−1. A consistent decrease of lipid order parameter with an increase of surface tension was observed for the saturated (DPPC, sn-1 and sn-2; POPC, sn-1 and POPG sn-1) and unsaturated (POPC, sn-2 and POPG sn-2) phospholipid chains. Thus, at 0 mN m−1 surface tension, the lipid tails are more ordered than at higher surface tension. This is consistent with the low APL values (Fig. 2) at low surface tension, reflecting highly packed lipids. This high order parameter shows that the monolayer is in liquid-condense (LC) phase.26 The comparison of order parameters between saturated and unsaturated tails of the phospholipids reveals the expected lower order parameter for the unsaturated tails (sn-2 chain) of POPC and POPG compared to saturated tails (DPPC, sn-1 and sn-2; POPC, sn-1 and POPG sn-1) due to the presence of double bonds at D2-C3 bead (Fig. 3) in the unsaturated chain. The change in local orientational tilt angle and the variation of local bond length at the unsaturated carbon chain contribute to reduce the order parameter at D2-C3 bead. The surface tension dependent decrease of order parameters is also observed in the presence of 5.52% w/w hydrocortisone (ESI Fig. S3†). Overall, the order parameter analysis suggests that at surface tension 0 mN m−1, the tails of the three phospholipids are highly ordered and become less ordered with increasing surface tension.
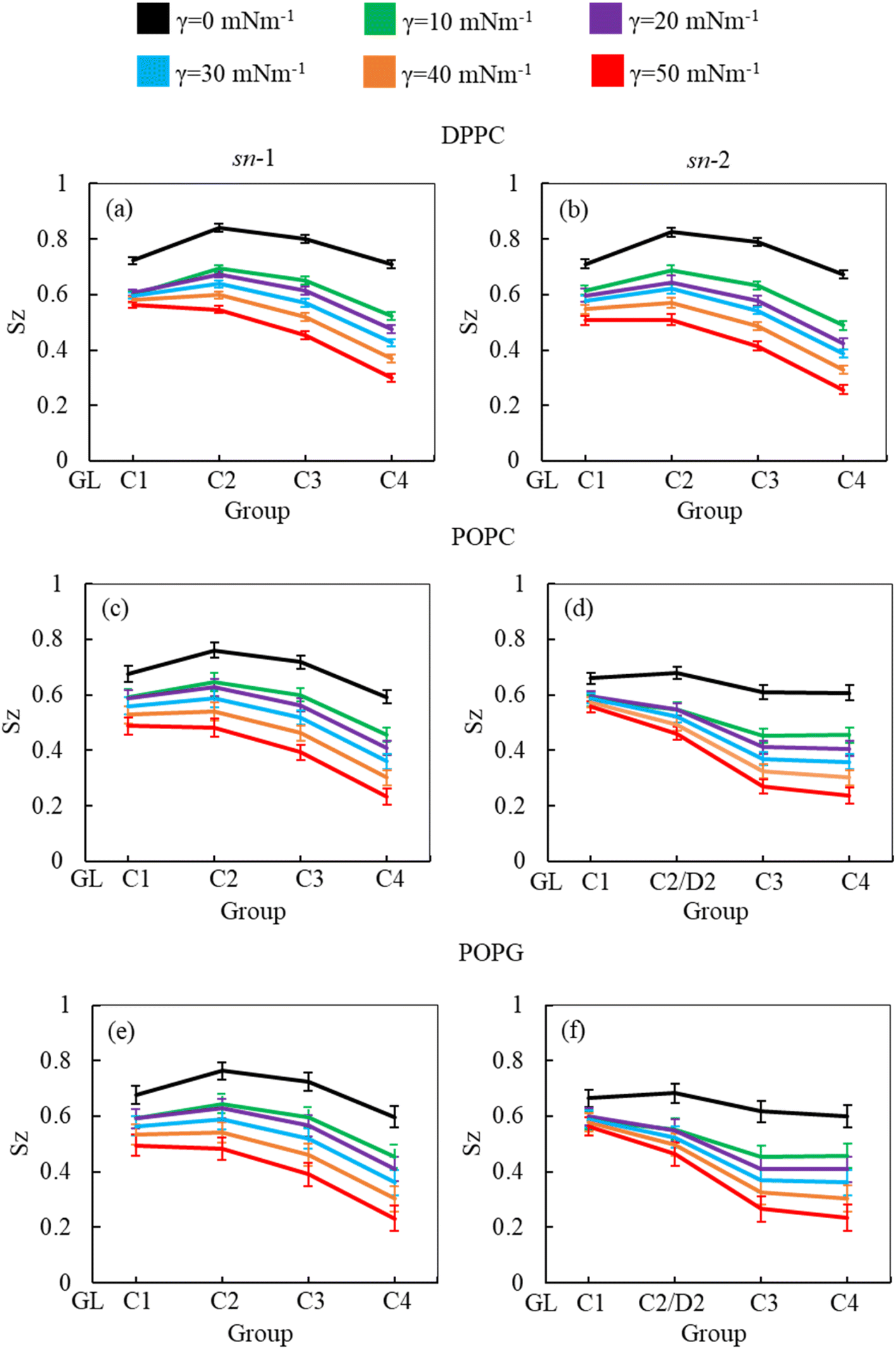 | ||
| Fig. 3 Order parameter of DPPC (a and b), POPC (c and d) and POPG (e and f) for sn-1 (a, c and e) and sn-2 (b, d and f) chains at different surface tension (0, 10, 20, 30, 40 and 50 mN m−1) of drug-free protein containing monolayers. Order parameters were estimated for last one microsecond of the two microsecond simulations. The error bars denote standard deviations across the frames of the trajectories. | ||
Mass density profiles of LSM components along the monolayer normal can be used to investigate whether the LSM is thinning or whether components are shrinking in height, leading to the apparent displacement of the monolayer components (lipids and proteins) toward/away from the lipid–water or lipid–air interface. Fig. 4 shows the mass density profiles for the phospholipids and surfactant proteins from simulations at different surface tensions in the absence and presence of 5.52% w/w hydrocortisone. The density profile of water is also shown in ESI Fig. S4.† For the drug-free monolayer, the comparison of density curves at the different surface tensions reveals that the peak heights of density curves at 0 mN m−1 surface tension are the furthest from the monolayer centre, while peak heights at 50 mN m−1 surface tension are the nearest. This means that the monolayer is shifting with the change of surface tension. These shifting of monolayer suggests that at high surface tension, the monolayer is compressed than at low surface tension, which is consistent with the APL and order parameter data showing a more expanded monolayer at higher surface tension. In both systems (absence and presence of drug), the density curves are lifted along the monolayer centre, confirming the monolayer condensing. However, no variations in monolayer thickness have been seen with changes in surface tension (ESI Table S4†).
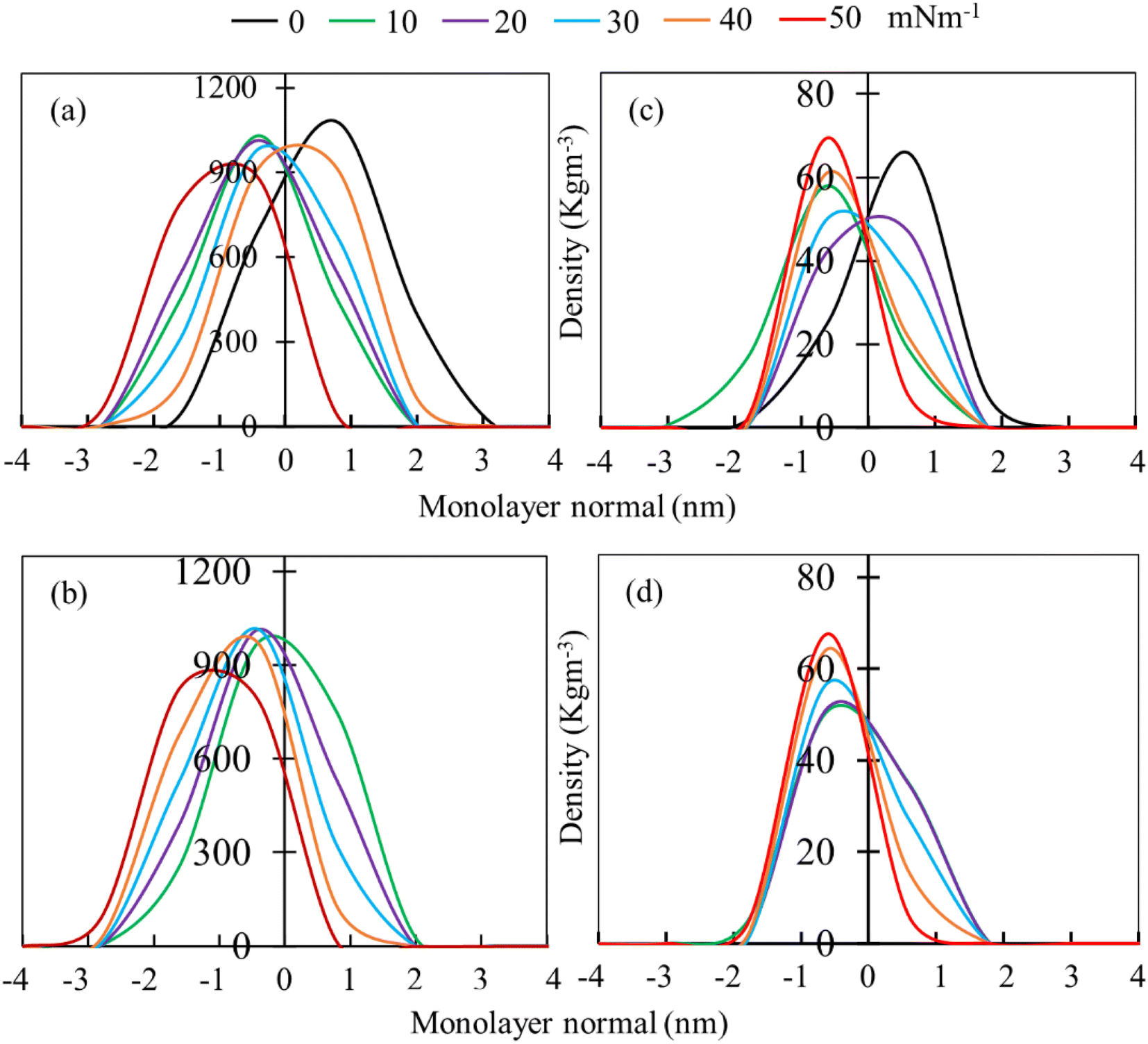 | ||
| Fig. 4 Averaged mass density profiles of phospholipid (left pannels: a and b) and surfactant protein (right pannels: c and d) of the LSM. Comparison of density curves of LSM model at different surface tension in the absence (a and c) and presence (b and d) of 5.52% w/w hydrocortisone concentration. | ||
The phospholipid density peak heights in the absence of drug (Fig. 4a) are 1.08, 1.03, 1.01, 0.98, 0.91 and 0.88 g cm−3 at surface tensions of 0, 10, 20, 30, 40 and 50 mN m−1, respectively, and in the presence of drug (Fig. 4b), the attained peak heights are 0.97, 1.01, 1.02 g, 0.97 and 0.83 g cm−3 at surface tensions of 10, 20, 30, 40 and 50 mN m−1, respectively. These variations of density curves are noticeable for drug-free monolayer, whereas drug-containing monolayer shows less effect. The effect of surface tension on the surfactant protein density profiles is also considerable in the drug containing monolayer, whereas there is no clear trend of protein density profiles for drug-free monolayer (Fig. 4c). These density fluctuations depend on surface tension, showing that surface tension has a noticeable impact on the lipid and protein density in the presence of drug (Fig. 4d). Overall, the density curves' findings imply that the protein and drug exacerbate the surface tension-induced monolayer shifting. This is in line with the study by Islam et al.,45 for the corticosteroid drug mometasone.
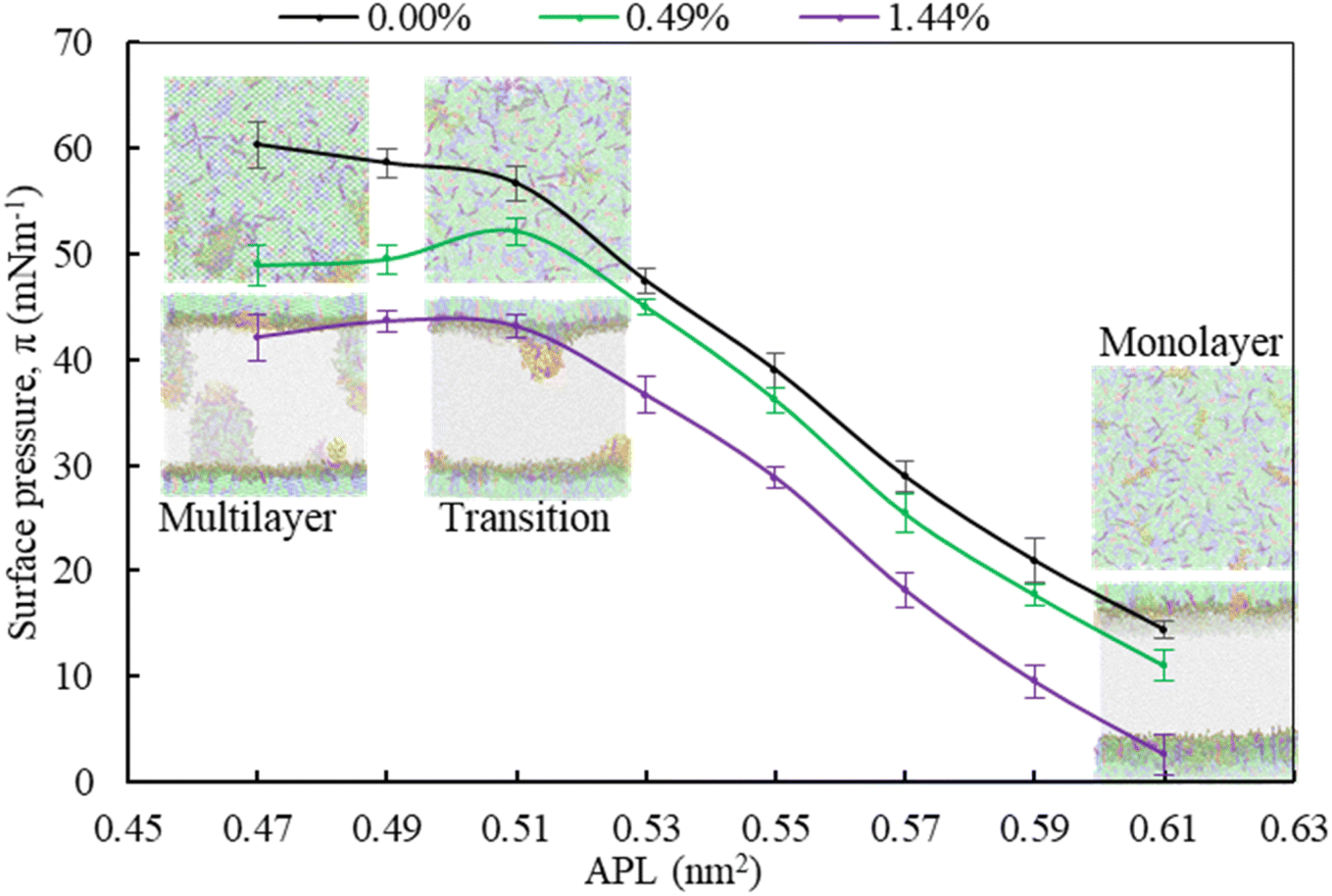 | ||
| Fig. 5 The effect of monolayer compression on the organisation of the lateral structure of LSM for increasing hydrocortisone concentrations by pressure-area (π-APL) isotherms calculation of the protein containing LSM of increasing hydrocortisone concentrations. | ||
Moving from the most expanded state (high APL) to the more condensed state (low APL), the monolayer passes through a monolayer-to-multilayer transition. As illustrated in the snapshots from the simulations, at lower APL, multilayered lipid folding starts occurring. If the monolayer is further compressed (<0.47 nm2), it is destabilised and transformed to lipid bilayer via the monolayer collapse. In the absence of drug, at an APL range of 0.47–0.51 nm2, the monolayer exists in the LC phase according to the Baoukina et al.,.58 For molecular area 0.51 < APL ≤ 0.61 nm2, the monolayer lies in the intermediate phase consisting of the combination of LC + LE phase. In this phase, the monolayer show different morphologies, structural and physicochemical features.26 As the isotherms in Fig. 5 are decreasing with the increase of drug concentration at a certain range, the molecular-level data from simulations can be used to gain insight into lipid film organisation. In the presence of hydrocortisone, an intercalating action might occur in which hydrocortisone intercalates between the phospholipid head groups, causing the monolayer components to pack more tightly. The decrease in APL shows that the phospholipid is becoming more compact, i.e., condensing the monolayer at increasing drug concentration.
In contrast to low APL, at APL > 0.61 nm2, the monolayer is so expanded that starts pore formation. These pores are more pronounced in the presence of drug. The number of pores is the same, but the size of the pores is increasing as it is observed from Fig. S5.† Wang et al.,35 reported similar concentration-dependent effects of corticosteroids on the exogenous lung surfactant (Infasurf).
3.2 Structural and dynamical properties of the monolayer at conditions mimicking exhalation and inhalation breathing
In the second part of the study, the systems were simulated at fixed surface tension of 0 and 20 mN m−1 at six different hydrocortisone contents from 0 to 10.46% w/w. These two surface tensions were considered to represent the extreme ends of exhalation (0 mN m−1) and inhalation (20 mN m−1) breathing conditions.9,76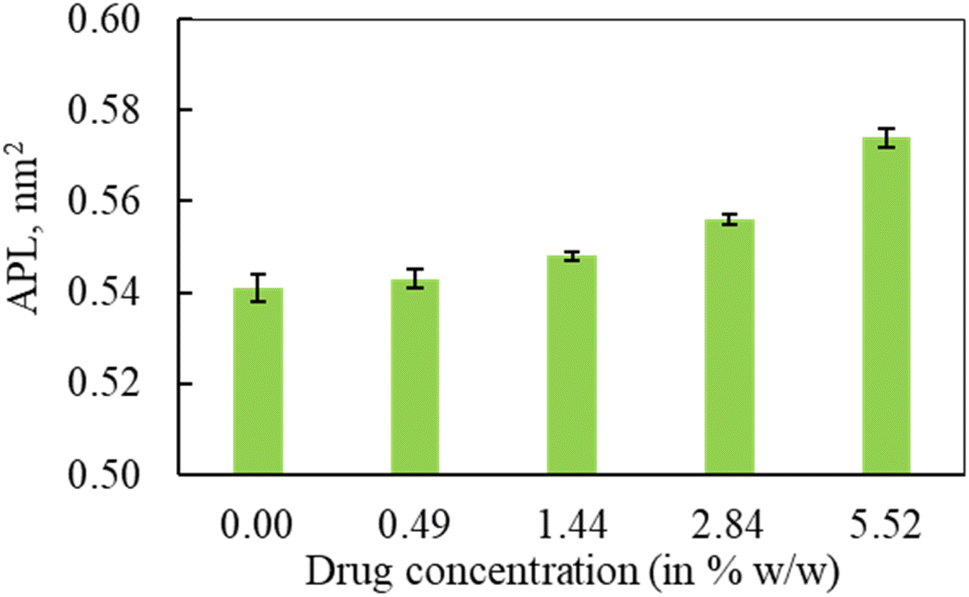 | ||
| Fig. 6 The effect of hydrocortisone concentration on the area per lipid (APL) from the simulation of protein containing system at 20 mN m−1. The data were estimated over the last 1 μs of the 2 μs simulation. The error bar refers to standard deviations of the APL data across the frames. | ||
The APL value for the drug-free monolayer agrees with the experimental value of APL (∼0.50–0.70 nm2) found in the literature as well as comparable system from atomistic77 and CG simulation studies75 at 20 mN m−1 surface tension producing the values ∼0.52 nm2 and 0.55 nm2, respectively. Hydrocortisone causes a considerable increase in the compressibility of the monolayer. Such increase in APL was previously reported for the corticosteroid drugs beclomethasone, budesonide and fluticasone.33 When the drug concentration exceeds >5.52% w/w, the additional drug molecules destabilise the monolayer and finally collapse (Fig. S6†). Such instability of the LSM is related to the increased fluidity in highly expanded monolayer, which was also reported by Zhang et al.,46 study.
In contrast to the compressibility (∼0.54 nm2) at 20 mN m−1 surface tension, the APL of LSM at 0 mN m−1 in the absence of drug and the presence of 0.49% w/w are estimated at 0.486 ± 0.004 nm2 and 0.480 ± 0.004 nm2, respectively. At 0 mN m−1 surface tension, the APL is not calculated for drug concentrations >0.49% because at this surface tension the drug causes LSM instability and monolayer collapse (Fig. 7).
 | ||
| Fig. 7 Snapshots of simulation system at surface tension 0 mN m−1 are shown in the absence and presence of increasing hydrocortisone concentrations of 0.49, 1.44, 2.84% w/w. Components are shown as DPPC (green), POPC (blue), POPG (cyan), CHOL (red), SP-B1–25 (orange), SP-C (yellow), cortisone (purple), water (silver), and phospholipids head groups (ochre). | ||
To examine the minimum surface tension where the drug molecules do not destabilise (i.e., monolayer collapse) the LSM at a given drug concentration, we repeated the simulation at surface tension from 1 to 5 mN m−1 at drug concentrations 0.49 to 5.52% w/w. The findings from these simulations imply that when surface tension ≥5 mN m−1, no LSM collapse has been observed at higher drug concentration, which is consistent with the other lung surfactant studies.78,79 To explore the collapsing mechanism at lower surface tension, the effect of surfactant protein was also investigated by simulation of the system in the absence and presence of surfactant protein. When APL values for drug-free and drug-containing monolayers are compared, it is evident that surfactant protein has a significant impact on monolayer compressibility. The effect of surfactant proteins can be found in the studies conducted by Islam et al.,45 and Duncan et al.,.23 The hydrophobic surfactant protein influences the lipid tails, potentially causing monolayer instability which is more pronounced in the presence of the drug hydrocortisone. The localisation and distribution of drug molecules in the lipid monolayer are affected by the hydrophobicity of the hydrocortisone molecule itself. The steric hindrance of the lipid head groups, the cationic interaction between the choline group of lipids, and the benzene ring of hydrocortisone are also responsible for monolayer instability. To gain insight into the interactions of the drug with its surrounding, we investigated its local environment using the pair correlation function g(r), between the hydrocortisone drug, and the lipid head and tail groups individually (see Fig. S7†). Drug adsorption to the hydrophilic region (lipid head at monolayer–water interface) is clearly preferred over the hydrophobic region (lipid tail group at monolayer–air interface) showing that the drug is less hydrophobic. By disrupting the drug distribution inside the monolayers, the preferential binding of the drug to the monolayer may have an impact on the long-range structure of the LSM. It appears that the drug aids in the expansion of monolayers as compared to drug-free monolayers for all the underlying surface tensions. Such monolayer expansion can also be found in our recent publication for another corticosteroid drug mometasone.45
In addition to APL calculation, the phospholipid order parameter (Sz) informs about the structural changes of the monolayer by reporting the orientation of phospholipid chains in different phases (LC and LE). The phospholipids in LC phase are highly ordered, producing higher-order parameters, whereas in LE phase the phospholipids show lower order parameter values (see ESI section S1.4† for detailed calculation). Fig. 8 shows the phospholipids order parameters for DPPC, POPC and POPG from the simulation systems of DPPC-POPC-POPG-CHOL-SP-B1–25-SP-C monolayer at surface tension 20 mN m−1 in the presence of increasing hydrocortisone concentrations. The order parameter for all three phospholipids (DPPC, POPC and POPG) decreases with increasing drug concentrations for increasing drug concentrations for both saturated and unsaturated phospholipid tails. Comparison between chain-1 (sn-1) and chain-2 (sn-2) reveals that all unsaturated tails (POPC sn-2 and POPG sn-2) are less ordered than saturated chains (DPPC sn-1, DPPC sn-2, POPC sn-1 and POPG sn-1). This disordering of the phospholipids is because of the expansion of the monolayer surface area as shown in Fig. 6. Low drug concentrations of 0.49% and 1.44% w/w show a negligible effect on the order parameters.
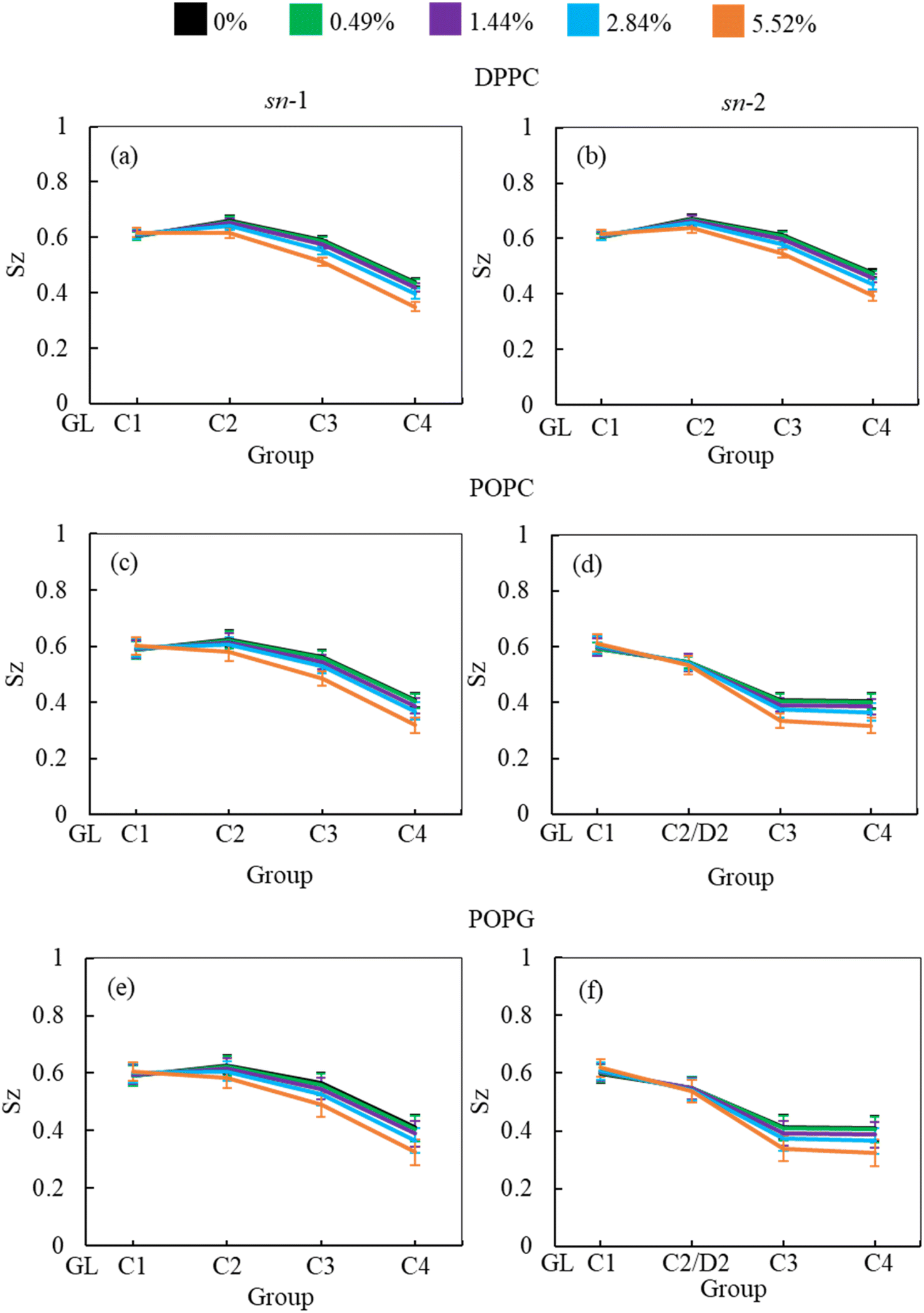 | ||
| Fig. 8 Concentration-dependent effect of hydrocortisone on the order parameter of (a and b) DPPC, (c and d) POPC, and POPG (e and f) for sn-1 (a, c and e) and sn-2 (b, d and f) chains at 20 mN m−1 surface tension. Order parameters were estimated for the last one microsecond of the two microsecond simulations. The error bars were calculated across the frames of the simulation. | ||
At 2.84% w/w of hydrocortisone, there is a decline in the order for DPPC, POPC and POPG at C2/D2-C3 and C3-C4 beads that gain drops for 5.52% w/w of hydrocortisone concentration. Simultaneously, the beads connecting GL-C1 and C1-C2/D2 show a slight rise. The results in Fig. 8 indicate that drug molecules alter the phospholipids systematic arrangement in the LSM, which substantially changes the physiological disordering at high drug concentrations (>5.52%) and causes the LSM to collapse. In contrast to inhalation breathing (20 mN m−1 surface tension), the incorporation of >0.49% hydrocortisone at exhalation breathing condition (0 mN m−1 surface tension) the drug molecules significantly cause the lipids disordering and destabilise the monolayer. Overall, the APL and order parameters data revealed that hydrocortisone causes a concentration-dependent monolayer expansion affecting the LSM disordering with the effect being strongest at low surface tension.
3.2.2.1 Diffusion of the monolayer components and drug molecules. As listed in Table 2, the lateral diffusion coefficients of phospholipids (DPPC, POPC and POPG), cholesterol and surfactant proteins (SP-B and SP-C) in the mixed DPPC-POPC-POPG-CHOL-SP-B1–25-SP-C monolayers for drug-free monolayers are (6.11 ± 0.61) × 10−7 cm2 s−1, (5.47 ± 0.01) × 10−7 cm2 s−1, (2.36 ± 0.71) × 10−7 cm2 s−1 at surface tension 20 mN m−1, respectively. The diffusion coefficients of phospholipids DPPC and POPC are (7.0 ± 2.0) × 10−7 cm2 s−1 and (8.0 ± 1.0) × 10−7 cm2 s−1, respectively, from existing in silico LSM modelling.50 To the best of our knowledge, there are no experimental data regarding lateral diffusion coefficients of phospholipids for monolayers.
| Drug (in % w/w) | Phospholipids | Cholesterol | Protein | Hydrocortisone |
|---|---|---|---|---|
| 0 | 6.11 ± 0.61 | 5.47 ± 0.01 | 2.36 ± 0.71 | — |
| 0.49 | 5.71 ± 0.12 | 5.30 ± 0.83 | 2.52 ± 0.52 | 6.59 ± 0.27 |
| 1.44 | 4.96 ± 0.34 | 4.96 ± 0.25 | 3.21 ± 0.45 | 5.03 ± 0.16 |
| 2.84 | 4.67 ± 0.48 | 4.46 ± 0.56 | 4.73 ± 0.26 | 4.79 ± 0.14 |
| 5.52 | 4.32 ± 0.30 | 3.51 ± 0.94 | 5.42 ± 0.41 | 4.41 ± 0.34 |
To assess the impact of corticosteroid drug hydrocortisone on the diffusion coefficient of phospholipids, cholesterol, protein, and hydrocortisone itself, the diffusion coefficient at four different concentrations of hydrocortisone (0.49, 1.44, 284 and 5.52% w/w) were calculated, respectively as shown in Table 2. As it is observed from Table 2, when the drug concentration is increasing then the diffusion coefficient of phospholipids and cholesterol decreases from (6.11 ± 0.61) × 10−7 cm2 s−1 at drug-free LSM to (4.32 ± 0.30) × 10−7 cm2 s−1 at 5.52% w/w drug concentration and (5.47 ± 0.01) × 10−7 cm2 s−1 at drug-free LSM to (3.51 ± 0.94) × 10−7 cm2 s−1 at 5.52% w/w drug concentration for phospholipids and cholesterol, respectively. The diffusion coefficient of cholesterol at 20 mN m−1 surface tension for drug-free LSM shows agreement with Laing et al.,75 study. In contrast to phospholipids and cholesterol diffusion coefficient, the diffusion coefficient of surfactant protein shows the opposite trend (increasing trend) with the increase of drug concentration. The estimated value of surfactant protein diffusion coefficient was (2.36 ± 0.71) × 10−7 cm2 s−1 at drug-free monolayer and (5.42 ± 0.41) × 10−7 cm2 s−1 at high drug concentration (5.52%). The incorporation of hydrocortisone molecules into the LSM hinders the spreading of the hydrocortisone itself that can be observed from the diffusion coefficient of hydrocortisone, which are amounted to (6.59 ± 0.27) × 10−7 cm2 s−1 at 0.49% and (4.41 ± 0.34) × 10−7 cm2 s−1 at 5.52% drug concentration. The diffusion coefficient of other corticosteroid prednisolone was found to be (6.0 ± 4.0) × 10−7 cm2 s−1 that are similar to the calculated diffusion coefficient of hydrocortisone.50 Overall, the dynamical properties of LSM components and corticosteroid drug by diffusion coefficient analysis demonstrates that the hydrophobic drug hinders the spreading rate of the phospholipids, cholesterol and the drug itself. On the other hand, the hydrocortisone influences the surfactant protein spreading.
3.2.2.2 Drug cluster formation. The adsorption of hydrocortisone drugs on the LSM can lead to cluster formation. The cluster criterion is set in such a way that two drug molecules will be in a cluster if the distance between the drug molecules ≤1.20 nm. The data was normalised to the number of drugs in the system due to show the effect of hydrcortisone concentration. To analyse the effect of hydrocortisone concentrations on the cluster formation of drug, the cluster size analysis of hydrocortisone was conducted for various hydrocortisone content at surface tension, γ = 20 mN m−1. The number of hydrocortisone molecules in the system are 10 (for 0.49%), 30 (for 1.44%), 60 (for 2.84%) and 120 (for 5.52%) per monolayer. To compare the clustering between the different systems, the number of clusters were normalised by using the number of drugs in the corresponding drug concentration with respect to the 0.49% w/w hydrocortisone concentration. Fig. 9 displays the normalised number of clusters and the number of hydrocortisone molecules in the largest cluster. For drug concentrations of 0.49, 1.44, 2.84, and 5.52% w/w, the time-averaged number of drug clusters were 9, 7, 6 and 3, respectively (Fig. 9a).
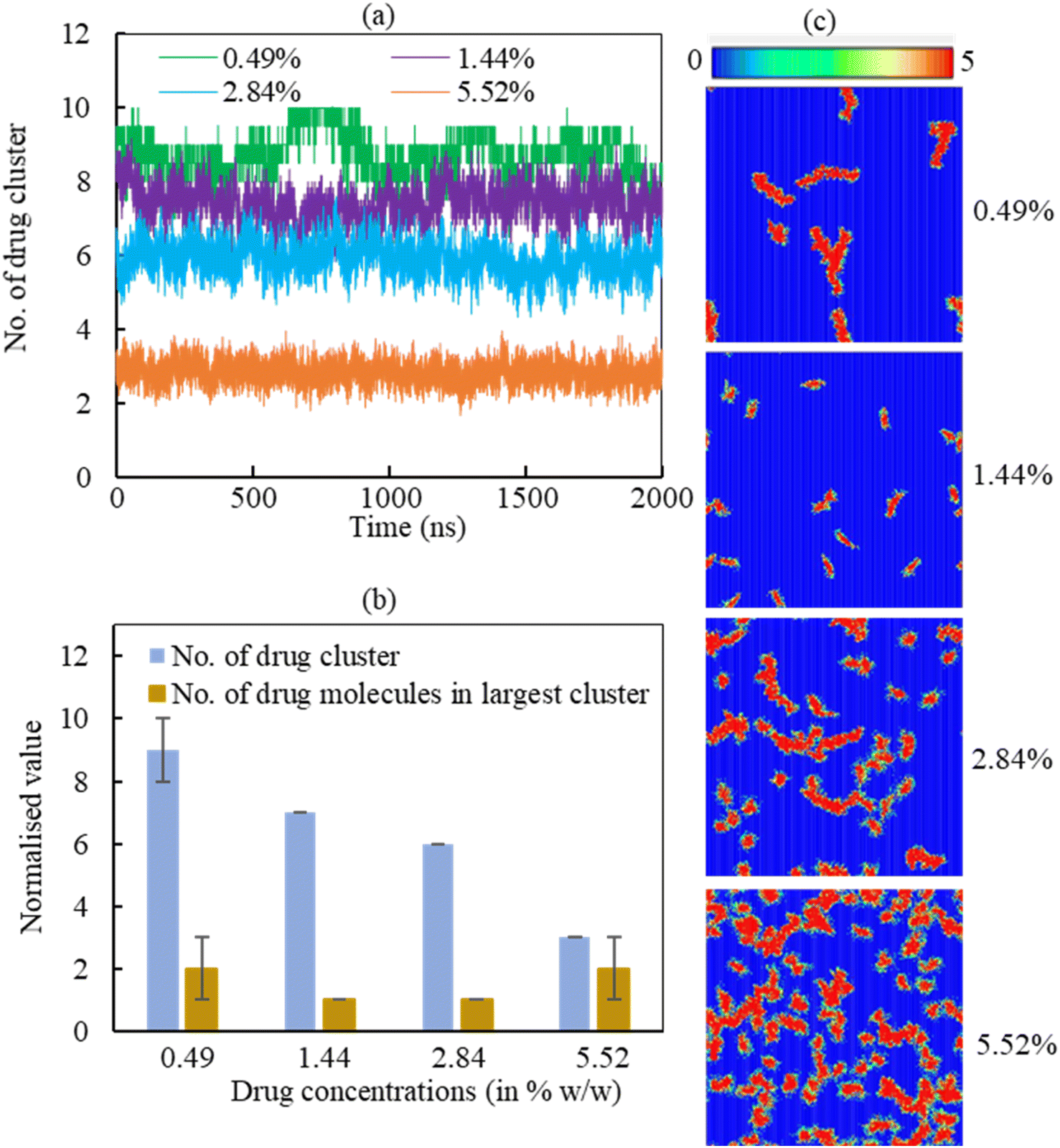 | ||
| Fig. 9 Normalised hydrocortisone cluster analysis of four drug concentrations (0.49, 1.44, 2.84, 5.52% w/w) in the LSM at surface tension γ = 20 mN m−1. (a) Number of normalised hydrocortisone cluster formation over the time. (b) Number of normalised hydrocortisone cluster and the number of hydrocortisone molecules in the largest cluster are shown for different hydrocortisone concentrations. (c) The two-dimensional (2D) density map of the hydrocortisone to show the cluster formation on the monolayer at different hydrocortisone concentrations. The data was normalised using the number of drugs in the corresponding drug concentration with respect to the 0.49% w/w hydrocortisone concentration. | ||
As seen in Fig. 9a, the number of clusters decreases with increasing drug concentration, indicating that drug–drug interactions are less pronounced with higher drug concentration. The normalised number of drug molecules in the largest cluster are calculated 2, 1, 1 and 2 for drug concentrations 0.49, 1.44, 2.84 and 5.52%, respectively (Fig. 9b). The clusters are often stable throughout the simulation time up to the hydrocortisone concentration 5.52% w/w.
As it is observed from the simulation at drug concentration >5.52% w/w, once the drug concentration exceeds 5.52%, the excess hydrocortisone molecules help to destabilise the LSM and finally collapse the monolayer as shown in Fig. S6 in the ESI.† Overall, the cluster analysis revealed that when drug concentration increases, the rate of change of the formation of drug clusters are reduced, and these clusters are of roughly the same size for all simulated drug concentrations (Fig. 9a). The number of drugs in the largest cluster remains constant during the simulation time, indicating that on the timescale of the simulations, the drugs cannot disperse once they have aggregated on the monolayer. Thus, the destabilising effect seen for concentrations >5.52% is not necessarily from an intrinsic change in clustering behaviour but the inability of breaking up such clusters in a comparatively short time scale. The lower number of cluster present at high drug concentrations results the instability of the monolayer by faster drug spreading compared to the rate of monolayer expansion. This behaviour of drug clustering has also been reported for the steroid drug prednisolone by Estrada-López et al.50 and mometasone by Islam et al.45
4 Implications
The interactions of corticosteroid drug hydrocortisone with LSM model appear to be very comparable to other corticosteroid drugs prednisone,28,50,81 cortisone82 or mometasone.45 As a result of the structural similarities in corticosteroid drugs, it may be possible to generalise the fates of corticosteroids in lung surfactants. It is also worth noting that the interaction of corticosteroids with the lipids in the LSM is strong enough to keep them in the hydrophobic tail region of the phospholipids for the whole duration of the simulations (2 μs). Hydrocortisone can form nanoaggregates causing the LSM to collapse at the end of the exhalation breathing condition. However, the proper spreading of the hydrocortisone molecules on the LSM can be observed in the inhalation breathing when the LSM is expanding. This spreading of hydrocortisone molecules is concentration-dependent and shows better spreading up to 5.52% w/w hydrocortisone concentration. According to this investigation, hydrocortisone given in a certain critical concentration (10.46% w/w) might cause the LSM damage. Lower concentrations of hydrocortisone, on the other hand, does not affect the LSM surface activity. The LSM has the characteristics of spreading hydrocortisone in a concentration-dependent manner and also dependent on the surface tension. The implications of this investigation will be used in the pharmaceutical industry to develop an effective corticosteroid dose formulation for respiratory distress syndrome and the people with asthma or other lung diseases.5 Limitations
In the simulation of various biomolecular processes, molecular modelling of biological systems using the molecular dynamics technique has shown significant progress.83 However, molecular simulations also have limitations, just like any experiment or analytical procedure.84 Considering the situation of lipid monolayers, there are issues with the computation of water surface tension and phospholipid monolayer surface pressure.85 The phase separation of phospholipids monolayer from the experiment cannot be seen from simulation.86 At low surface tension (<2 mN m−1), the phospholipid monolayers collapse, while at high surface tension (more than 50 mN m−1), a pore forms in the monolayer. The available experimental data is frequently limited or non-existent, difficult to obtain, and subject to significant fluctuations.87 Nevertheless, there are situations when the discrepancy is attributable to a deficiency in the coarse-grained approach itself.88 Our goal was to look at the partitioning and distribution of hydrocortisone in the LSM rather than the precise atomistic corticosteroid–lipid interactions. A CG model like MARTINI is appropriate for this application because it was designed with a specific focus on reproducing lipid partitioning behaviour.89 Furthermore, our research included over 136 with 3 μs simulations, which are impossible to do with atomistic models. Steroids are frequently overly hydrophobic, which is one of the weaknesses of the model. Hydrocortisone partition coefficient is underestimated, implying that the free energy of partitioning is excessively negative. The relative partition coefficients for cortisone, mometasone, prednisolone, and cholesterol that we utilised as a reference are, however, accurate (Table S3†).6 Conclusion
The stability of LSM interacting with corticosteroid drug hydrocortisone was effectively investigated using CG MD simulation. The structural details of the LSM were studied without and with hydrocortisone. In both the absence and presence of hydrocortisone, the drug appeared to assist in the enlargement of monolayers compared to drug-free monolayers for applied interfacial tension of the LSM. The findings from constant-APL simulations show a continuous compression of the monolayer, and a concentration-dependent decrease in pressure-area isotherm curves, which replicates the surface pressure during normal breathing. An intercalating effect may occur, in which the hydrocortisone intercalates between the phospholipid head groups, causing the monolayer constituents to pack more closely at highly compressed monolayer. On the other hand, the findings from constant surface tension simulation mimicking inhalation and exhalation breathing condition imply that hydrocortisone produces a compression of the monolayer in a concentration-dependent manner that is more significant at exhalation breathing causing the LSM to collapse at >0.49% w/w hydrocortisone concentration. For the inhalation case, the drug-induced collapse was observed at a higher hydrocortisone concentration (>5.52%). The accumulation of drug molecules limits drug capacity to spread over the LSM at maximum drug contents, resulting the monolayer collapse. The results from the dynamical properties analysis demonstrate that larger clusters signal a lower drug capacity to diffuse into the monolayer, which is significantly impeded when more drug molecules are present in the system. The findings from this investigation may help to better understand the interaction mechanism between inhaled corticosteroids and LSM, which will be helpful to pharmaceuticals to determine effective corticosteroid dosing for lung diseases.Author contributions
M. Z. I. performed the simulations and formal data analysis, participated in the study's conceptualization, and drafted the original manuscript. S. I. H. edited the manuscript. E. D. supervised M. Z. I. and critically revised the manuscript, Z. L. edited the manuscript and S. C. S. developed the concept, supervised M. Z. I. and edited the manuscript. All authors gave final approval for publication and agreed to be held accountable for the work performed therein.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted with the support of University of Technology Sydney (UTS) FEIT Research Scholarship and UTS International Research Scholarship (Mohammad Zohurul Islam). The UTS eResearch High-Performance Computer Cluster (HPCC) and National Computing Interface (NCI), Australia provided the computational facilities. Some of the simulations have also been conducted from High-Performance Computing (HPC) Lab at Jashore University of Science and Technology, Bangladesh. Zhen Luo was funded by the Australian Research Council (ARC) [ Funding ID ARC DP210101353] and the open access charge was supported by Zhen Luo.References
- P. T. Daley-Yates, Br. J. Clin. Pharmacol., 2015, 80, 372–380 CrossRef CAS PubMed
.
- T. Rhen and J. A. Cidlowski, N. Engl. J. Med., 2005, 353, 1711–1723 CrossRef CAS PubMed
- P. J. Barnes, Pharmaceuticals, 2010, 3, 514–540 CrossRef CAS PubMed
- K. J. Rademaker, L. S. de Vries, C. S. Uiterwaal, F. Groenendaal, D. E. Grobbee and F. van Bel, Arch. Dis. Child. Fetal Neonatal Ed., 2008, 93, F58–F63 CrossRef CAS PubMed
- I. P. Morris, N. Goel and M. Chakraborty, Eur. J. Pediatr., 2019, 178, 1171–1184 CrossRef CAS PubMed
- L. W. Doyle, R. A. Ehrenkranz and H. L. Halliday, Neonatology, 2010, 98, 111–117 CrossRef CAS PubMed
- M. W. Petersen, T. S. Meyhoff, M. Helleberg, M. B. N. Kjær, A. Granholm, C. J. S. Hjortsø, T. S. Jensen, M. H. Møller, P. B. Hjortrup and M. Wetterslev, Acta Anaesthesiol. Scand., 2020, 64, 1365–1375 CrossRef CAS PubMed
- C. S. Kow and S. S. Hasan, Cleveland Clin. Q., 2020, 87, 10–3949 Search PubMed
- J. Perez-Gil and T. E. Weaver, Physiology, 2010, 25, 132–141 CrossRef CAS PubMed
- J. Goerke, Biochim. Biophys. Acta, Mol. Basis Dis., 1998, 1408, 79–89 CrossRef CAS
- M. Chakraborty and S. Kotecha, Breathe, 2013, 9, 476–488 CrossRef
- C. Autilio and J. Pérez-Gil, Arch. Dis. Child. Fetal Neonatal Ed., 2019, 104, F443–F451 Search PubMed
- S. P. Caminiti and S. L. Young, Hosp. Pract., 1990, 26, 87–100 CrossRef PubMed
- E. J. Veldhuizen and H. P. Haagsman, Biochim. Biophys. Acta, Biomembr., 2000, 1467, 255–270 CrossRef CAS
- E. Parra and J. Pérez-Gil, Chem. Phys. Lipids, 2015, 185, 153–175 CrossRef CAS PubMed
- R. Veldhuizen, K. Nag, S. Orgeig and F. Possmayer, Biochim. Biophys. Acta, Mol. Basis Dis., 1998, 1408, 90–108 CrossRef CAS
- Y. Y. Zuo, R. A. Veldhuizen, A. W. Neumann, N. O. Petersen and F. Possmayer, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1947–1977 CrossRef CAS PubMed
- A. G. Serrano and J. Pérez-Gil, Chem. Phys. Lipids, 2006, 141, 105–118 CrossRef CAS PubMed
- E. Lopez-Rodriguez and J. Pérez-Gil, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 1568–1585 CrossRef CAS PubMed
- R. Guagliardo, J. Pérez-Gil, S. De Smedt and K. Raemdonck, J. Controlled Release, 2018, 291, 116–126 CrossRef CAS PubMed
- S. Orgeig, P. S. Hiemstra, E. J. A. Veldhuizen, C. Casals, H. W. Clark, A. Haczku, L. Knudsen and F. Possmayer, Respir. Physiol. Neurobiol., 2010, 173, S43–S54 CrossRef CAS PubMed
- J. Pérez-Gil, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1676–1695 CrossRef PubMed
- S. L. Duncan and R. G. Larson, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1632–1650 CrossRef CAS PubMed
- S. Schurch, H. Bachofen, J. Goerke and F. Possmayer, J. Appl. Physiol., 1989, 67, 2389–2396 CrossRef CAS PubMed
- S. Baoukina, E. Mendez-Villuendas and D. P. Tieleman, J. Am. Chem. Soc., 2012, 134, 17543–17553 CrossRef CAS PubMed
- S. L. Duncan, I. S. Dalal and R. G. Larson, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2450–2465 CrossRef CAS PubMed
- S. Baoukina, L. Monticelli, S. J. Marrink and D. P. Tieleman, Langmuir, 2007, 23, 12617–12623 CrossRef CAS PubMed
- M. Z. Islam, M. Krajewska, S. I. Hossain, K. Prochaska, A. Anwar, E. Deplazes and S. C. Saha, Langmuir, 2022, 38, 4188–4199 CrossRef CAS PubMed
- J. Hu, H. Liu, P. Xu, Y. Shang and H. Liu, Langmuir, 2019, 35, 13452–13460 CrossRef CAS PubMed
- K. M. W. Keough and H. W. Taeusch, Pediatr. Pathol. Mol. Med., 2001, 20, 519–536 CrossRef
- A. Cimato, G. Facorro and M. M. Sarrasague, Respir. Physiol. Neurobiol., 2022, 296, 103825 CrossRef CAS PubMed
- B. Baer, L. McCaig, C. Yamashita and R. Veldhuizen, Lung, 2020, 198, 909–916 CrossRef CAS PubMed
- A. Cimato, G. Facorro and M. M. Sarrasague, Respir. Physiol. Neurobiol., 2018, 247, 80–86 CrossRef CAS PubMed
- K. W. Lu, J. Goerke, J. A. Clements and H. W. Taeusch, Pediatr. Res., 2005, 57, 237–241 CrossRef CAS PubMed
- Y. E. Wang, H. Zhang, Q. Fan, C. R. Neal and Y. Y. Zuo, Soft Matter, 2012, 8, 504–511 RSC
- A. Cimato, G. Facorro and M. Martínez Sarrasague, Respir. Physiol. Neurobiol., 2022, 296, 103825 CrossRef CAS PubMed
- A. G. Dos Santos, J. C. Bayiha, G. Dufour, D. Cataldo, B. Evrard, L. C. Silva, M. Deleu and M.-P. Mingeot-Leclercq, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1930–1940 CrossRef CAS PubMed
- W. Daear, P. Lai, M. Anikovskiy and E. J. Prenner, J. Phys. Chem. B, 2015, 119, 5356–5366 CrossRef CAS PubMed
- M. Javanainen, A. Lamberg, L. Cwiklik, I. Vattulainen and O. S. Ollila, Langmuir, 2018, 34, 2565–2572 CrossRef CAS PubMed
- A. Stachowicz-Kuśnierz, T. Seidler, E. Rogalska, J. Korchowiec and B. Korchowiec, Chemosphere, 2020, 240, 124850 CrossRef PubMed
- J. Hu, X. Li, M. Li, Y. Shang, Y. He and H. Liu, Colloids Surf., B, 2020, 190, 110922 CrossRef CAS PubMed
- A. Bykov, G. Loglio, R. Miller, O. Milyaeva, A. Michailov and B. Noskov, Colloids Surf., A, 2019, 573, 14–21 CrossRef CAS
- K. Y. C. Lee, Annu. Rev. Phys. Chem., 2008, 59, 771–791 CrossRef CAS PubMed
- F. Ravera, R. Miller, Y. Y. Zuo, B. A. Noskov, A. G. Bykov, V. I. Kovalchuk, G. Loglio, A. Javadi and L. Liggieri, Curr. Opin. Colloid Interface Sci., 2021, 55, 101467 CrossRef CAS
- M. Z. Islam, S. I. Hossain, E. Deplazes and S. C. Saha, J. Mol. Graphics Modell., 2022, 111, 108084 CrossRef CAS PubMed
- H. Zhang, Y. E. Wang, C. R. Neal and Y. Y. Zuo, Pediatr. Res., 2012, 71, 316–323 CrossRef CAS PubMed
- S. Meng, W. Cui, S. Lin, G. Wang, Y. Hei, B. Deng, S. Ma, Z. Zhang, Y. Liu and Y. Xie, J. Chin. Pharm.
Sci., 2018, 27, 415–428 CrossRef CAS
- S. Ortiz-Collazos, E. D. Estrada-López, A. A. Pedreira, P. H. Picciani, O. N. Oliveira Jr and A. S. Pimentel, Colloids Surf., B, 2017, 158, 689–696 CrossRef CAS PubMed
- F. Souza, F. Fornasier, L. Souza, M. Peñafiel, J. Nascimento, A. Malfatti-Gasperini and A. Pimentel, Colloids Surf., A, 2020, 584, 124024 CrossRef CAS
- E. D. Estrada-López, E. Murce, M. P. Franca and A. S. Pimentel, RSC Adv., 2017, 7, 5272–5281 RSC
- S. Edsbäcker and C. J. Johansson, Basic Clin. Pharmacol. Toxicol., 2006, 98, 523–536 CrossRef PubMed
- T. A. Wassenaar, H. I. Ingólfsson, R. A. Böckmann, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2015, 11, 2144–2155 CrossRef CAS PubMed
- S. I. Hossain, M. Z. Islam, S. C. Saha and E. Deplazes, Methods Mol. Biol., 2022, 2402, 103–121 CrossRef PubMed
- S. J. Marrink, A. H. De Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed
- S. J. Marrink, A. H. de Vries, T. A. Harroun, J. Katsaras and S. R. Wassall, J. Am. Chem. Soc., 2008, 130, 10–11 CrossRef CAS PubMed
- S. O. Yesylevskyy, L. V. Schäfer, D. Sengupta and S. J. Marrink, PLoS Comput. Biol., 2010, 6, e1000810 CrossRef PubMed
- S. Baoukina, L. Monticelli, S. J. Marrink and D. P. Tieleman, Langmuir, 2007, 23, 12617–12623 CrossRef CAS PubMed
- L. Gordon, K. Lee, M. Lipp, J. Zasadzinski, F. Walther, M. Sherman and A. Waring, J. Pept. Res., 2000, 55, 330–347 CrossRef CAS PubMed
- J. Johansson, T. Szyperski, T. Curstedt and K. Wuethrich, Biochemistry, 1994, 33, 6015–6023 CrossRef CAS PubMed
- J. Johansson and T. Curstedt, J. Intern. Med., 2019, 285, 165–186 CrossRef CAS PubMed
- M. Sarker, A. J. Waring, F. J. Walther, K. M. Keough and V. Booth, Biochemistry, 2007, 46, 11047–11056 CrossRef CAS PubMed
- N. Biswas, S. Shanmukh, A. J. Waring, F. Walther, Z. Wang, Y. Chang, R. H. Notter and R. A. Dluhy, Biophys. Chem., 2005, 113, 223–232 CrossRef CAS PubMed
- V. Schram and S. B. Hall, Biophys. J., 2001, 81, 1536–1546 CrossRef CAS PubMed
- F. Baumgart, O. L. Ospina, I. Mingarro, I. Rodríguez-Crespo and J. Pérez-Gil, Biophys. J., 2010, 99, 3234–3243 CrossRef CAS PubMed
- J. Ding, D. Y. Takamoto, A. Von Nahmen, M. M. Lipp, K. Y. C. Lee, A. J. Waring and J. A. Zasadzinski, Biophys. J., 2001, 80, 2262–2272 CrossRef CAS PubMed
- M. Melo, H. Ingólfsson and S. Marrink, J. Chem. Phys., 2015, 143, 243252 CrossRef PubMed
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef
- M. A. Cuendet and W. F. van Gunsteren, J. Chem. Phys., 2007, 127, 184102 CrossRef PubMed
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed
- H. J. Berendsen, J. v. Postma, W. F. van Gunsteren, A. DiNola and J. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- S. J. Marrink, Martini Coarse Grain Forcefield for Biomolecules, 2020, http://cgmartini.nl/images/tools/do-order-gmx5.py, accessed November 2020 Search PubMed
- E. Boger and M. Fridén, J. Aerosol Med. Pulm. Drug Delivery, 2019, 32, 1–12 CrossRef CAS PubMed
- C. Laing, S. Baoukina and D. P. Tieleman, Phys. Chem. Chem. Phys., 2009, 11, 1916–1922 RSC
- S. Schürch, H. Bachofen and F. Possmayer, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2001, 129, 195–207 CrossRef
- S. L. Duncan and R. G. Larson, Biophys. J., 2008, 94, 2965–2986 CrossRef CAS PubMed
- R. E. Pattle, Nature, 1955, 175, 1125–1126 CrossRef CAS PubMed
- R. E. Pattle and G. R. Cameron, Proc. R. Soc. London, Ser. B, 1958, 148, 217–240 CAS
- A. Einstein, Ann. Phys., 1905, 17, 549–560 CrossRef CAS
- M. Z. Islam, S. I. Hossain, E. Deplazes, S. Bhowmick and S. C. Saha, AIP Conf. Proc., 2021, 2324, 060008 CrossRef CAS
- M. Z. Islam, S. I. Hossain, E. Deplazes and S. C. Saha, Mol. Simul., 2022, 48, 1627–1638 CrossRef CAS
- W. Wang and R. Gómez-Bombarelli, npj Comput. Mater., 2019, 5, 125 CrossRef
- F. R. Souza, L. M. P. Souza and A. S. Pimentel, J. Chem. Inf. Model., 2020, 60, 5881–5884 CrossRef CAS PubMed
- D. H. De Jong, S. Baoukina, H. I. Ingólfsson and S. J. Marrink, Comput. Phys. Commun., 2016, 199, 1–7 CrossRef CAS
- S. Baoukina and D. P. Tieleman, in Biomolecular simulations, Springer, 2013, vol. 924, pp. 431–444 Search PubMed
- W. F. van Gunsteren, J. Dolenc and A. E. Mark, Curr. Opin. Struct. Biol., 2008, 18, 149–153 CrossRef CAS PubMed
- R. Alessandri, P. C. Souza, S. Thallmair, M. N. Melo, A. H. De Vries and S. J. Marrink, J. Chem. Theory Comput., 2019, 15, 5448–5460 CrossRef CAS PubMed
- S. J. Marrink and D. P. Tieleman, Chem. Soc. Rev., 2013, 42, 6801–6822 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra05268g |
| ‡ Current address: Department of Mathematics, Faculty of Science, Jashore University of Science and Technology, Jashore-7408, Bangladesh. |
| This journal is © The Royal Society of Chemistry 2022 |