
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeciphering the role of faujasite-type zeolites as a cation delivery platform to sustain the functions of MC3T3-E1 pre-osteoblastic cells†
Gaëtan
Lutzweiler
 *a,
Yu
Zhang
b,
Fanny
Gens
a,
Aline
Echalard
a,
Guy
Ladam
a,
Jérémy
Hochart
a,
Théo
Janicot
a,
Nadine
Mofaddel
c and
Benoît
Louis
b
*a,
Yu
Zhang
b,
Fanny
Gens
a,
Aline
Echalard
a,
Guy
Ladam
a,
Jérémy
Hochart
a,
Théo
Janicot
a,
Nadine
Mofaddel
c and
Benoît
Louis
b
aNormandie Univ, UNIROUEN, INSA Rouen, CNRS, PBS (UMR 6270), 55 rue Saint-Germain, 27000 Évreux, France. E-mail: gaetanlutzweiler@gmail.com
bICPEES-UMR 7515, Université de Strasbourg, CNRS, 25 rue Becquerel, F-67087 Strasbourg, France
cNormandie Univ, UNIROUEN, INSA Rouen, CNRS, COBRA (UMR 6014), 55 rue Saint-Germain, 27000 Évreux, France
First published on 1st September 2022
Abstract
The control of cell fate assisted by synthetic drug-delivering materials is still a challenging task for spatiotemporal regulation of bioactive molecules in native tissues. In this study, the potential of faujasite type (FAU) zeolites for delivering bioactive ions (i.e. Ca2+ and Mg2+) to pre-osteoblastic MC3T3-E1 cells is unveiled. Zeolites are porous aluminosilicates with a large surface area suitable for the storage of biologically active compounds. However, only a few studies have reported the use of zeolites in tissue engineering; therefore, this study aims to correlate the release profile of zeolites with the resulting cell functions. The textural properties of zeolites were assessed via MEB, BET, XRF, and EDX analyses. This study shows that either Mg-loaded (Mg-Y) or Ca-loaded (Ca-Y) zeolites are able to gradually release ions over up to three weeks in classical culture media in contrast to the burst release profiles often described in the literature. The ion concentration can be adjusted simply through the mass of zeolite added in the system and was quantified by colorimetric methods and capillary electrophoresis. Three zeolite concentrations were tested (i.e. 1, 1.5, and 3 wt%) for both ion types in order to span biologically relevant ranges (from 5.6 to 19.6 mM). The cell responses against various ionic strengths were evaluated not only by the reduction of resazurin assay for viability/proliferation assessment, but also through immunostainings and collagen secretions (Picrosirius red staining). The cell proliferation was found to be proportional to the Ca2+ concentration whereas an upper limit seemed to have been reached for Mg 3% compared to Mg 1% and Mg 1.5%. Matrix secretions were delayed under conditions exposed to Ca2+ relative to Mg2+ after 7 days, but this trend reversed after 21 days. Overall, this study emphasizes the potential role of FAU as a simple, low cost, biocompatible, and versatile way to modulate the functions of MC3T3-E1 cells through ion delivery over up to three weeks. The ion concentration is correlated with the zeolite concentration, and the payload could be easily handled. Thus, this study will make it possible to broaden the applications of zeolites in tissue engineering which can also be implemented in multiple ways including surface coatings and nanocarriers.
1. Introduction
In recent years, the resurgence of innovative strategies to treat bone diseases or trauma has shown drastic progress especially with the design of a new generation of biomaterials. Most of the implants used nowadays for bone repair and orthopedics are still made of titanium or ceramics owing to their excellent resistance against corrosion and good biocompatibility. However, such devices lack osteoconductivity which hampers their regenerative potential,1 and drove scientists to seek bioactive materials instead, being able to either release chemotactic agents, or implement regeneration with topological or physical cues designed to elicit an appropriate cellular response.2Regarding bone regeneration, numerous studies have focused on osteogenesis driven by the exposure to growth factors such as bone morphogenic proteins 2 and 7 (BMP-2 and BMP-7), or recombinant human platelet-derived growth factors-BB (rhPDGF-bb),3 though side effects including ectopic bone formation or inflammation could be reported.4 Unlike conventional growth factors, smaller molecules including statins or flavonoids could also stimulate bone formation, displaying an intrinsic ability to diffuse through the cell membrane with higher shelf-life.5 Importantly, multiple ions including Ca2+, Mg2+, Cu2+ and Sr2+ appeared as potent and affordable inducers of the differentiation of pre-osteoblastic cells even at low doses, without the stability and conformational-dependence encountered with peptide-derived growth factors.6
Among the diverse ions involved in the bone mineralization process and homeostasis, two divalent elements, namely calcium (Ca2+) and magnesium (Mg2+), recur. Indeed, calcium is mostly found within the mineralized matrix of bones in the form of hydroxyapatite (HAp), and is also solubilized in the interstitial fluid, and plays a major role in the mineralization of the extracellular matrix (ECM).7 In addition, Ca2+ is also known to activate the extracellular signal-related kinase (ERK), subsequently involved in RUNX-2 expression, type I collagen secretion, and osteocalcin upregulation, further promoting the proliferation and differentiation of immature osteoblasts.8 Yeh et al.9 revealed recently that hematopoietic stem cells reside at locations with higher calcium concentrations (∼1.5 mM) compared to the blood serum, which later affects their clonal expansion. Similarly, magnesium, albeit less abundant, was also shown to contribute to bone mineralization via stabilizing effects of amorphous calcium phosphate precursors.10 The osteoblastic lineage of human bone marrow stromal cells (HBMSCs) could be targeted via the release of Mg2+ ions activating the Wnt-signaling pathway.11 These ions are usually delivered by biomaterials such as bioceramics, containing calcium phosphates, for instance β-wollastonite12 or BioGlass®, which show potential owing to their ability to promote the formation of an apatite layer at the interface with native bones under appropriate calcium concentration conditions.13 Equivalently, beta tri-calcium phosphate (β-TCP) is also widely studied as a bone substitute, due to its ability to trigger apatite nucleation upon cell-mediated resorption.14 For example, Mg2+ ions could also be delivered to the surroundings upon resorption of Mg-doped HA, serving as an osteoconductive coating applied onto the surface of titanium implants.15
However, the release kinetics and consequently the doses must be finely tuned to avoid either a rapid pH variation upon Mg dissolution,16 or the other associated dysfunctions of bone metabolism.17 Similarly, vascular calcification can result from high calcium levels in the blood serum.18 For this purpose, systems that could slow down or regulate the diffusion of manifold ions are highly attractive, for example core–shell microparticles synthesized by Lin et al.19 Indeed, they managed to gradually release Mg for 2 weeks, and a similar trend was observed for the Mg-containing metal organic framework (Mg-MOF-74).20 In another study, Castano et al.21 generated electrospun fibers made from a bioglass/polymer composite to control the calcium delivery for two weeks.
Porous materials such as clay minerals including the smectite family (e.g. montmorillonite and bentonite) are crystalline aluminosilicates and are attractive for drug release applications owing to their large surface area and tunable surface chemistry.22,23 Interestingly, zeolite is another class of crystalline aluminosilicates made from connected TO4 tetrahedra where T denotes either an Al or Si atom, which form a regular microporous structure during synthesis.24 Zeolites are wide-spread in the industry as catalysts, molecular sieves or sorbents25 since their pore dimensions, surface areas, and sorption capacity can be hierarchized according to the synthesis route.26,27 Surprisingly, their application in the biomedical field remains largely under-explored, except scarce reports. Zeolites can be used to produce composite scaffolds intended for tissue engineering, for instance the study by Ninan et al.28 reported a gelatin/hyaluronic acid composite loaded with faujasite-type (FAU) zeolites, and highlighted that the oxygen supply to cells was improved after desorption from the FAU cavities. Y-zeolites/Hap composites were also obtained from microwave-assisted precipitation, providing scaffolds which sustain the functions of osteoblastic cells. This method appeared as a versatile tool to modify the surface topology by changing the zeolite percentage.29 MFI-type zeolites are also quite stable in the biological environment (e.g. resistance against corrosion) and can be crystallized directly atop some surfaces by in situ crystallization which allows the crystalline structure of zeolites to be preserved. Likewise, MFI-coated titanium surfaces led to the reduction of the leaching out of toxic Al and V ions underneath, and enhanced cellular tethering.30 Hence, the surface features such as roughness can therefore be adjusted through zeolite coatings, and, in turn, promote the adhesion and differentiation of osteoblasts.31 More recently, nanosized zeolites were also synthesized for the local delivery of gadolinium and carbogen for tumor imaging and treatment in primates,32 but larger molecules such as cyclophosphamide could also be loaded in their channels.33 Regarding bone tissue engineering, the release of zinc ions on MC3T3-E1 pre-osteoblastic cells, from a zeolitic imidazole framework-8, i.e. a hybrid between zeolites and metal organic frameworks (MOFs), was described, and the authors concluded that the biological activity was dose-dependent.34
Considering the unique architectural features of zeolites (i.e., large surface area and hierarchized pore size), and the fact that these materials can be implemented in multiple ways including as nanocarriers,32 or as surface coatings,34–36 particular attention can be paid to their use in bone regeneration, which requires establishing their potential action towards bone cells.
Herein, we addressed the role of FAU zeolites as a reservoir to ensure a gradual release with a controlled dosage of two biologically relevant cations, namely calcium and magnesium. The ion release profile was defined, and considered in light of the biological response induced on MC3T3-E1 osteoblastic cells, which encompassed cell morphology, proliferation and collagen secretion.
2. Results and discussion
Both Mg- and Ca-activated zeolites (Mg-USY and Ca-USY) were characterized in order to correlate any biological effect due to their loading/release capacity. These materials exhibited a characteristic pyramidal morphology according to the SEM micrograph shown in Fig. 1(a). Individual crystals were about 0.7–1 μm in size and were aggregated into larger, 5–10 μm structures. For this reason, zeolites were placed in culture inserts rather than directly in contact with cells in order to prevent any potential damage resulting from the sedimentation of zeolite aggregates. Moreover, as the purpose of this study was to investigate the potential of FAU zeolites to induce an ion-based modulation of MC3T3-E1 pre-osteoblasts, any cellular response that could arise from direct contact with zeolites should be avoided. The adsorption of Mg ions was confirmed first by elemental EDX analysis (Fig. 1(b) and (c)), showing a homogenous distribution of Mg ions throughout the zeolite framework, with an atomic percentage estimated around 4% of the whole composition comprising oxygen, aluminum, and silicon (Fig. 1(b)–(e)). Likewise, calcium was also homogeneously distributed within the zeolite crystals (data not shown). One can observe from the N2 adsorption–desorption experiments a classical type IV isotherm for both Mg- and Ca-loaded FAU (denoted as Mg-Y and Ca-Y in Fig. 2(a), respectively) due to the presence of a hysteresis, which ascertains the coexistence of both micropores and mesopores. The presence of small mesopores (i.e. 2–3 nm) was a result of the dealumination process, and hierarchically organized micro-mesoporous zeolites were produced.37 Of note, this multiscale porosity can be used to load both bioactive ions and small molecules even though the latter aspect is beyond the scope of this study. | ||
| Fig. 1 Image taken by SEM depicting the morphology of FAU nanocrystals (a). The framed area was subjected to elemental analysis to determine the overall atomic composition (b), and the individual percentages of Mg (c), Si (d), O (e) and Al (f). | ||
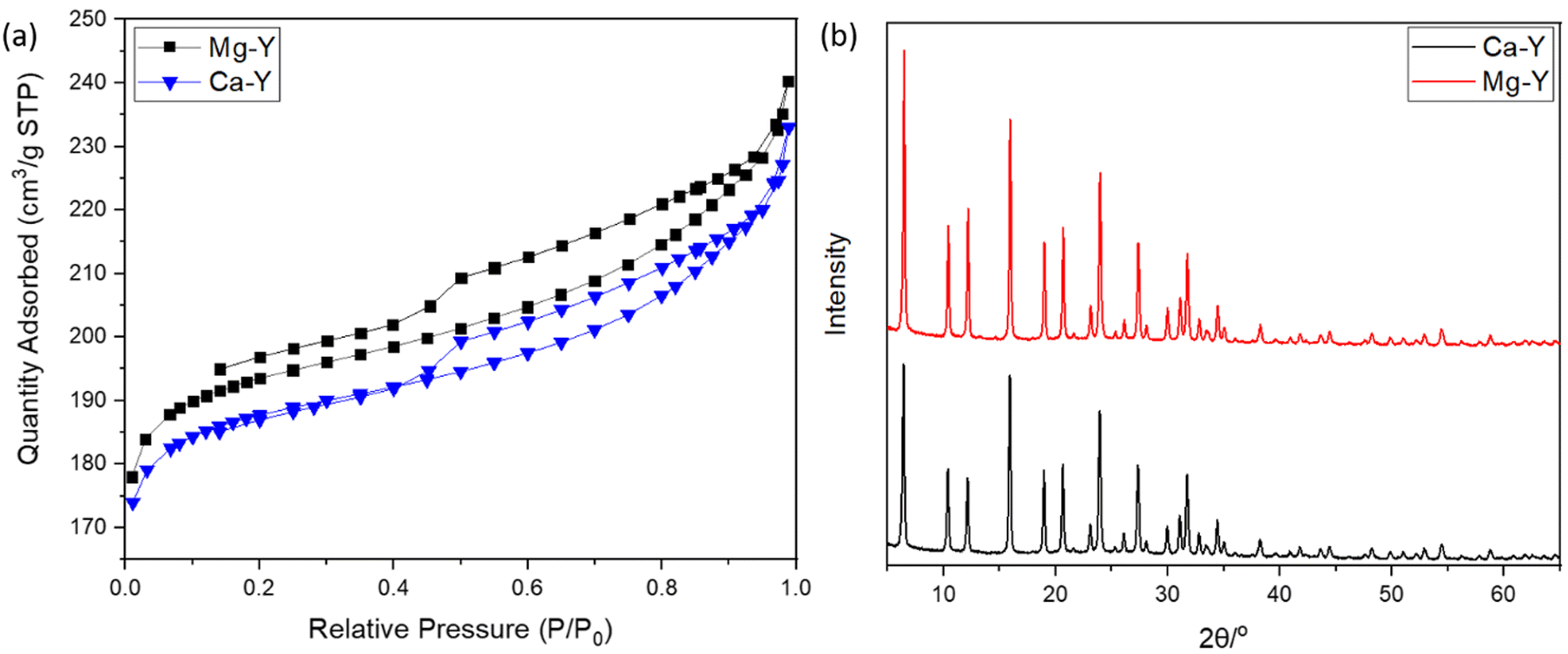 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms of Mg-exchanged FAU (Mg-Y) and Ca-exchanged FAU (Ca-Y). (b) Diffraction patterns of Ca- and Mg-exchanged FAU. | ||
The Mg-Y curve is shifted upward with respect to the Ca-Y curve, which indicates that the loading of Mg ions in the zeolite crystal was less important than for Ca-Y.
Van Mao et al.38 already reported that the ability of FAU zeolites (X- and Y-types) to exchange Mg2+ ions was less efficient than that to exchange Ca2+ ions, which was explained by the higher solvation degree of Mg, leading to larger complexes, and thus limiting the diffusion of these ions throughout zeolite channels. Moreover, Coker et al.39 also compared the diffusivity difference between Mg2+ and Ca2+ ions to the degree of the crystallinity of zeolites, but this argument could be discarded in our case since the XRD patterns of both Mg-Y and Ca-Y were nearly identical (Fig. 2(b)); hence, no structural modification was expected after our soft cation-exchange procedure. Though Mg2+ ions are significantly less concentrated in the culture medium (α-MEM) (i.e. 0.8 mM for Mg vs. 1.8 mM for Ca), the extent of Mg loading in the zeolite framework may still be large enough to induce a biological response.
The textural properties including the BET surface areas and microporous volumes are presented in Table 1. The amount of ions that can be stored in the zeolite structure is fairly correlated with the quantity of aluminum atoms present in a tetrahedral configuration, whose excess of negative charges is neutralized by compensating cations. The silica-to-aluminum ratio (Si/Al) was estimated to be 2.9 by energy-dispersive X-ray (EDX) (Fig. S1, ESI†) analysis, which is in agreement with the common values found for FAU.40 X-Ray fluorescence spectroscopy was also performed to obtain a global elemental composition of the solids. A Si/Al = 3.3 was confirmed, being in line with the EDX mapping analysis. More importantly, the Ca and Mg loadings were estimated to be 2.93 and 2.29 wt%, respectively. The cationic exchange in the FAU zeolites could therefore be calculated for Ca-Y and Mg-Y, being 48 and 39%, respectively. These results indicate a higher exchange of Ca2+ cations with respect to Mg2+-loaded Y zeolites, which is in line with the early studies from Breck.41
| Sample | BET surface area (m2 g−1) | Micropore surface area (m2 g−1) | Total pore volume (cm3 g−1) | Micropore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|---|---|
| Ca-Y | 599 | 524 | 0.35 | 0.26 | 2.3 |
| Mg-Y | 618 | 539 | 0.36 | 0.26 | 2.3 |
The ion release profile was established in order to determine the kinetics, and the concentrations of either Ca2+ and Mg2+ ions which can be delivered by the zeolites in the surroundings (Fig. 3(a) and (c)). Therefore, 100, 150 and 300 mg of Ca- and Mg-exchanged FAU were placed at the bottom of 15 mL tubes, being subsequently filled with 10 mL of complete α-MEM media to obtain solutions with 1, 1.5 and 3 wt% of zeolites, respectively. The samples were denoted as X Y%, where X stands for the metal ion (Ca or Mg), and Y is the weight percentage of zeolites. The ion release studies were performed for 21 days and at 37 °C to be consistent with cell culture experiments. The Mg2+ concentration was found to increase gradually over time for 14 days under all conditions, prior to level off, whilst the Ca2+ concentration reached a plateau already after 7 days. These results are analogous to the kinetic profile described by Lateef et al.,42 whose complete Mg2+ and Ca2+ delivery from nanosized zeolites was reached within the same delay. At each time-point, ion concentrations increased with the amount of zeolites; after 24 h, the concentrations of Ca2+ ions were 13.2, 16.6 and 20.6 mM for conditions with 1, 1.5 and 3 wt% of Ca-exchanged FAU, respectively (Fig. 3(d)). After 21 days, these values increased to 18.6, 19.9 and 28.2 mM, corresponding to increases of 36.6% (Ca 1%), 19.8% (Ca 1.5%) and 36.9% (Ca 3%), respectively.
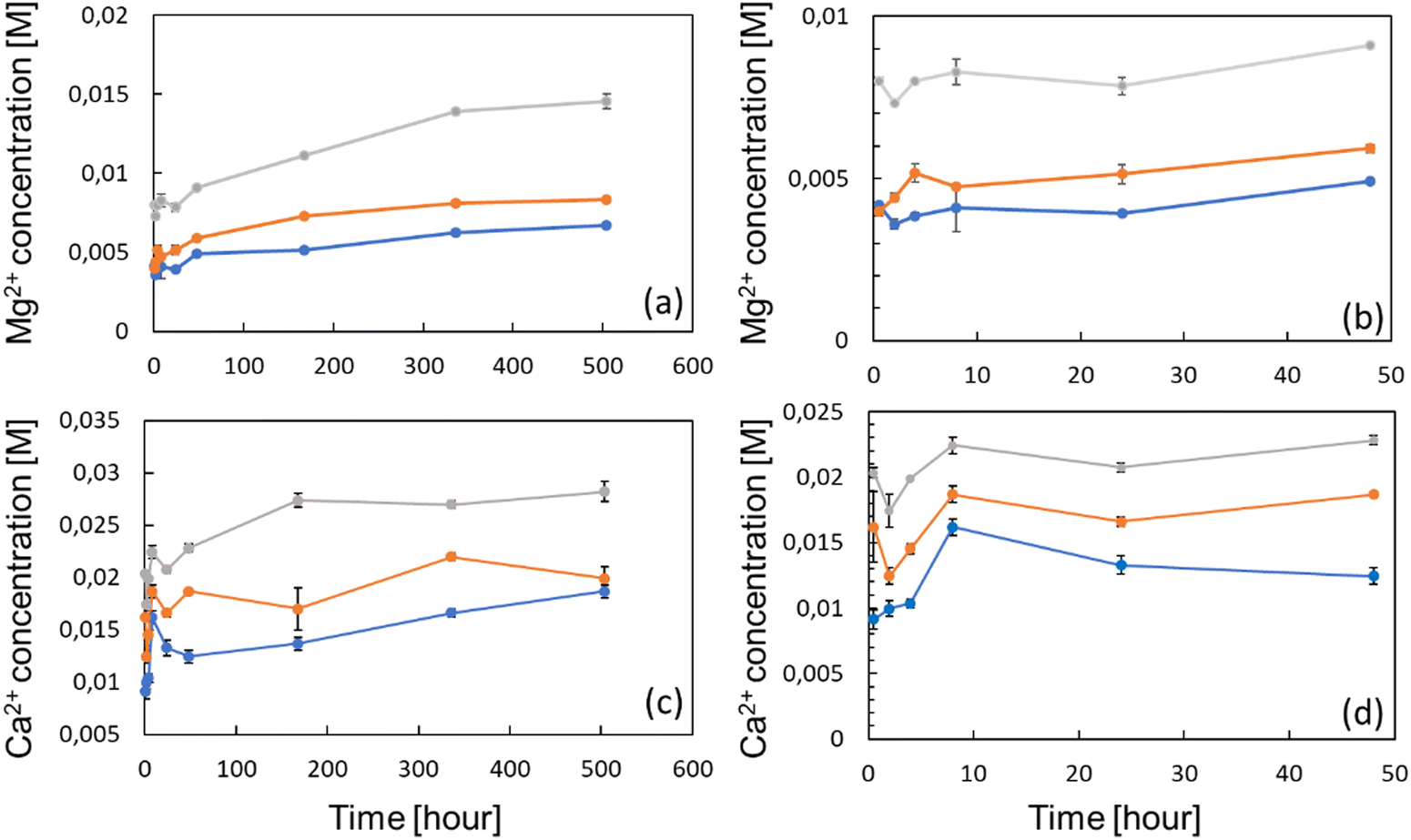 | ||
| Fig. 3 Global release profiles of Mg2+ ions from FAU zeolites from 0.5 to 504 h (a), and the corresponding profiles of Ca2+ ions (c). Same profiles focused on the first 48 h for (b) Mg2+ ions and (d) Ca2+ ions. Three zeolite concentrations were used, namely 1 wt% (blue), 1.5 wt% (orange), and 3 wt% (gray). The values are represented as mean SD, n = 3. | ||
Considering magnesium ions, the measured concentrations after 24 h were 3.9, 5.1 and 7.8 mM following the increasing order of zeolite concentrations. After 21 days, ion levels also increased to 6.7, 7.3 and 14.5 mM, consisting of 71.7% (Mg 1%), 69.8% (Mg 1.5%) and 85.9% (Mg 3%) increases (Fig. 3(a)). This slow cation release kinetics shows a quite different trend from the results presented in the work of Chen et al.,43 who found that the release profile of nanosized FAU exchanged with zinc, copper and iron ions, exhibited a plateau at around 1.5 h. Such a discrepancy can be explained by the fact that even if the diffusivity of both Ca2+ and Mg2+ ions should be enhanced by the nano-size of the crystals, the ion release experiments of Chen et al. were conducted in saline media only. Conversely, in this study, zeolites were immersed in protein-rich media (due to FBS supplementation) whose adsorption onto the zeolite mesopores or on the outer surface may hinder the mobility of species contained inside the pores and channels. This was highlighted by Bingre et al.44 who established a relationship between structural features of ZSM-5 crystals and the diffusivity of probe molecules. Furthermore, Titus et al.45 already found that protein monomers (i.e. amino acids) could strongly adsorb on ZSM-5 zeolites by hydrophobic interactions. Manifold proteins can form a “corona” on FAU nanocrystals including apolipoprotein, fibrinogen, or even albumin.46 Vinu and co-workers47 showed that mesoporous silicas (MCM-41 and SBA-15) displayed a strong affinity toward one of the heme proteins, and this adsorptive ability was further increased upon the introduction of aluminum atoms into the framework, which further reinforced the electrostatic interactions arising from the net negative charge harbored by aluminum atoms. According to this, it is worth mentioning here that as observed in Fig. 3(b) and (d), Mg2+ and Ca2+ concentrations remained fairly constant within the first 24 h. Between the first point taken after 0.5 h and after 24 h of the experiment, the concentrations of Mg2+ varied from 4.1 to 3.9 mM (Mg 1%), from 3.1 to 5.9 mM (Mg 1.5%) and from 7.9 to 7.8 mM (Mg 3%). The Mg basal concentration of the complete α-MEM solely was found to be around 1.0 mM, indicating that, for all experimental points, the Mg concentration was higher and above the detection threshold. Similarly, the Ca2+ concentration of the bare complete medium was 1.3 mM, below the smallest values found under all zeolite-exposed conditions. For Mg, the Ca variation within the first 24 h was anecdotal except for Ca 1% whose value increased from 9.1 to 13.2 mM; conversely, the two other cases displayed Ca2+ concentration variations from 16.1 to 16.5 mM for Ca 1.5%, and from 20.3 to 20.7 mM for Ca 3%.
The viability/proliferation of MC3T3-E1 pre-osteoblastic cells was assessed using the reduction of resazurin assay (i.e. Alamarblue™) and the results are shown in Fig. 4(a). The reduction percentages were normalized with respect to the results of day 3, in order to emphasize the proliferation in each condition. On the one hand, one can see that the proliferation of a positive control (Ctrl+) was not very pronounced, with only a slight increase of the average relative reduction percentage (0.84 to 1.04) between days 7 and 21, which was not found to be statistically significant. On the other hand, the proliferation of Mg- or Ca-exposed cells was more important especially between days 7 and 14, prior to stabilization. Globally, cells continuously proliferated from days 7 to 21 except for Ca 1% whose average normalized intensity values decreased from 1.60 to 1.15. Also, the proliferation of cells exposed to Mg 3% was slightly less at days 14 and 21 compared to Mg 1% and Mg 1.5% even though it was not found statistically different.
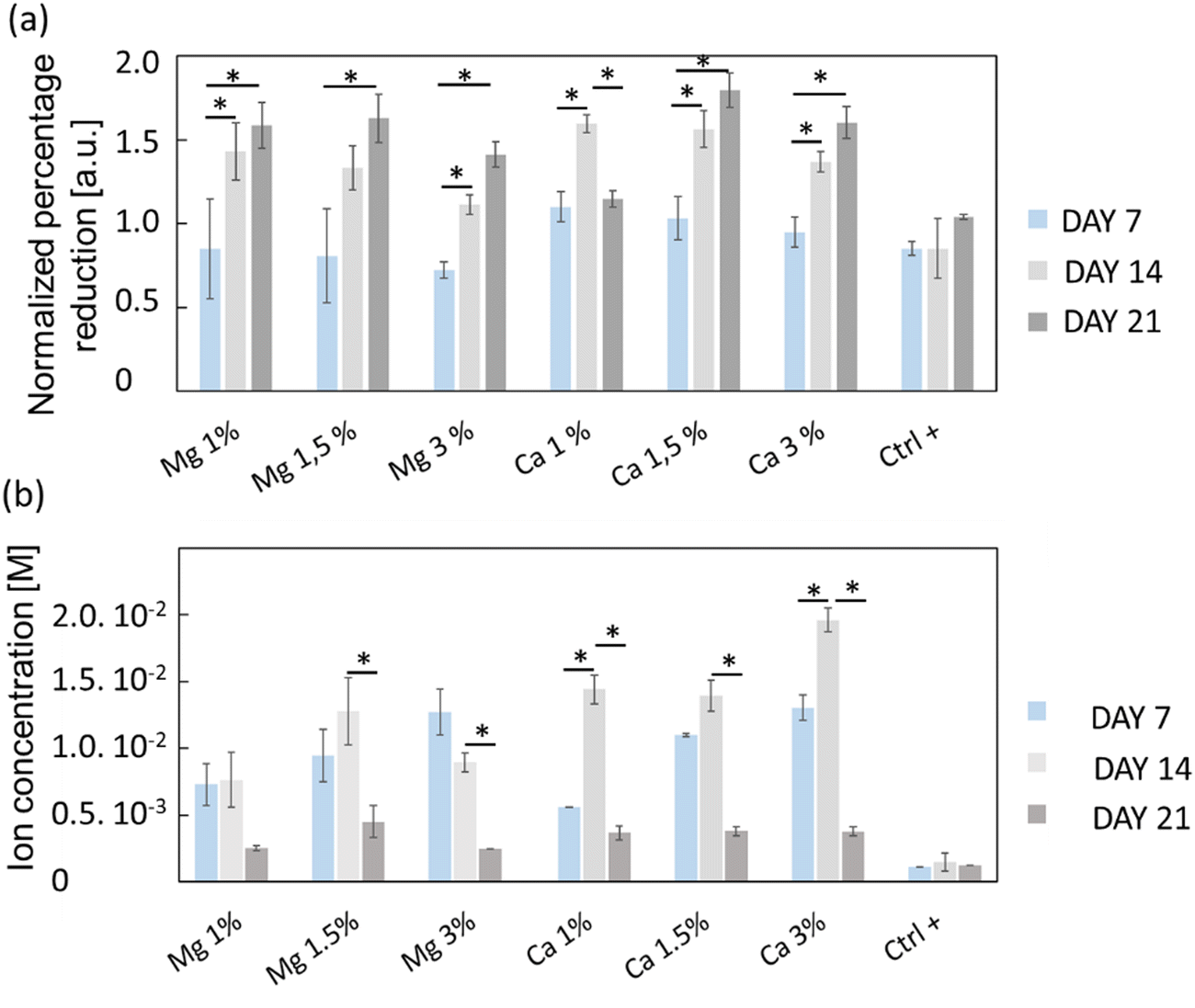 | ||
| Fig. 4 (a) Proliferative activity of MC3T3-E1 osteoblasts evaluated from the reduction percentage of resazurin (AlamarBlue®) over 21 days for both Mg-exchanged FAU (Mg 1%, Mg 1.5% and Mg 3%) and Ca-exchanged FAU (Ca 1%, Ca 1.5% and Ca 3%), and control conditions (Ctrl+) consisting of cell culture directly onto glass coverslips without zeolites. (b) Mg and Ca concentrations collected from culture media and measured by capillary electrophoresis. For each condition, n = 4 and the values are represented as mean ± SD. * for p < 0.05. | ||
The ion concentrations during culture experiments were measured using capillary electrophoresis (Fig. 4(b)); since the culture media were changed upon viability/proliferation assays, some ions were inevitably removed. The Mg2+ levels remained stable between days 7 and 14 under all conditions which indicates that the cation release kinetics over 7 days is conserved, but also that zeolites are suitable for their sustainable delivery over time. At day 21, the cation concentration decreased for all samples, likely because most of the cations adsorbed onto the zeolite surface were released. Conversely, the Ca2+ levels were increased from day 7 to day 14 in Ca 1% and Ca 3% samples. The measured concentrations were higher than that for Mg2+ especially at day 14, with values of 14.4 ± 1 mM (Ca 1%), 13.9 ± 1 mM (Ca 1.5%) and 19.6 ± 0.4 mM (Ca 3%), which were in line with the higher payload of Ca2+ cations in FAU displayed on N2 adsorption–desorption isotherms (Fig. 2(a) and Table 1).
For Mg 1%, the ion molarities increased from 7.3 ± 1.5 mM to 7.6 ± 2.0 mM between days 7 and 14, and decreased to 2.5 ± 0.2 mM at day 21. In the case of Mg 1.5%, the concentration increased from 9.5 ± 2 mM to 13 ± 3 mM between days 7 and 14, and varied from 13 ± 2 to 9.0 ± 0.4 mM for the last sample (Mg 3%) during the same time interval. According to Fig. 4(a), one can conclude that the proliferation of osteoblasts with Mg 3% was less pronounced than that with other conditions, albeit normalized intensities were close to that of Mg 1.5%. Wu and co-workers48 found that alkaline phosphatase (ALP) and tartrate-resistant acid phosphatase (TRAP) activities were decreased in osteoblasts incubated with Mg extracts above 14 mM, while the highest activity was measured between 3 and 10 mM at the same time-points. It is possible that an upper limit was reached and maintained for longer periods in Mg 3% samples, thus explaining this reduction. In contrast, Shen et al.49 claimed that, for MC3T3-E1 cells, the most appropriate concentration of Mg2+ ions was around 4.1 mM, and, above this value, adhesion, proliferation and migration regressed, whereas in our case the cell proliferation was the highest for Mg 1% and Mg 1.5% despite the corresponding maximal concentrations of 7.6 and 13 mM, respectively. In this study, the concentrations evolved due to the progressive release of cations from the zeolites, but also by the necessary changes of culture media. Thus, the concentrations measured at specific time points do not represent a constant value but must be rather smoothed, and according to the release profiles (Fig. 3), Mg concentrations reached 4 mM before 10 h for both Mg 1% and Mg 1.5% which indicates that MC3T3-E1 proliferation can be induced at larger ranges, and that the cell response may be different in time-evolutive Mg2+ concentrations compared to fixed concentrations, triggering some adaptative mechanisms. Of note, a 10 mM Mg content is thought to promote MC3T3-E1 binding to type I collagen through α2β1 integrins, further stimulating proliferation and differentiation via the FAK/ERK signaling pathway,50 which emphasizes the beneficial effect of a moderate ( 10 mM) Mg2+ concentration on cell responses. The slow release kinetics described herein may also avoid a Mg burst release, known to affect the pH value. Indeed, Wu et al.51 showed that progressive exposure to Mg2+ ions released from bioceramic substrates enhanced osteogenesis of MC3T3-E1 cells without pH-related damage to the cell integrity. After 7 days, the Ca2+ cation concentrations were fairly proportional to the mass of zeolites i.e. 5.6 ± 0.2 mM (Ca 1%), 11.0 ± 0.1 mM (Ca 1.5%), and 13.0 ± 1 mM (Ca 3%). Unlike Mg-loaded samples, the Ca2+ concentrations increased slightly after 14 days. It is therefore possible that different diffusion regimes may occur for Ca2+ ions, across either micropores or mesopores. Indeed, our team was able to decipher several effective diffusion contributions of various probes in zeolites.44,52
The highest values at day 14 were respectively 14.4 ± 1 mM (Ca 1%), 13.9 ± 1 (Ca 1.5%) and 19.6 ± 0.1 mM (Ca 3%). The effectiveness of Ca2+ ions to trigger the proliferation of osteoblastic cells via the activation of the calcium-sensing receptor (CaSR) was demonstrated by Hu et al.53 whereby the highest cell number was obtained for extracellular calcium concentrations of 5 and 10 mM. Cytotoxic effects were assigned to a Ca2+ concentration above 10 mM in mouse primary osteoblasts.54 In another study, Gabusi et al.55 found that the proliferation of human osteoblasts was promoted in media supplemented with 2.6 mM CaCl2, as confirmed by ERK and phospholipase C-β1 expression levels, but they did not probe higher concentrations. Nonetheless, Xiang et al.56 showed that the migration and adhesion of osteoblasts was enhanced at lower extracellular Ca2+ concentration (0.5 mM), whereas the osteoblastic activities assessed through YAP/TAZ localization in the nucleus were more pronounced at 1.2 mM. Interestingly, the ability of extracellular Ca2+ to regulate gene expression and protein secretions was also demonstrated for MSCs, whose secretory profiles of osteopontin and transforming growth factor beta 1 (TGF-β1) were elevated at Ca2+ concentrations within the range of 6–10 mM.57 Overall, the dynamic remodeling of bones induces significant ion fluctuations whereby the Ca2+ concentration can increase up to 40 mM.58 Furthermore, Ca2+ ions can also induce a conformational change of serum proteins such as albumin, which further modifies the binding receptors involved in cell adhesion and subsequently the related downstream signaling cascades.59 Such ion variations can arise from several factors such as changing media, but also Ca precipitation induced by phosphate-releasing materials60 including HAp. Atif et al.61 recently found that under static conditions, extracellular Ca can decrease to 0.8 mM which prevents MC3T3-E1 adhesion. In their study, they used microfluidic chips to perfuse fresh media continuously, and dampen Ca depletion. In a sense, the sustained release promoted by the presence of zeolites can also prevent the Ca decrease observed in static cultures.
The morphology of MC3T3-E1 cells was observed after 21 days of culture and in the absence of zeolites (Fig. 5, Ctrl+), whereby cells were elongated as evidenced by the longitudinal alignment of F-actin fibers, which is consistent with the actin organization on flat and moderately stiff glass coverslips.62 Stress fibers could be observed for Ca 1% and Mg 1% along with filopodia extensions, but without the characteristic alignment found in Ctrl+. The cell density was higher for Mg 1% compared to those for the other Mg samples. Shen et al.49 showed that the surface of osteoblastic cells increases with a Mg2+ concentration up to 8 mM (7.6 mM found at maximum in our study) prior to diminishing which is in accordance with another recent study that depicted the same dependence of the size on Mg2+ ions, based on the argument that a lot of proteins, proteases, or apolipoproteins are prone to adsorb onto material surfaces in the presence of less than 10 mM Mg2+.63 Likewise, protein adsorption may provide several sites to support cell adhesion and the establishment of focal contacts. Moreover, MC3T3-E1 cells in Mg 1% displayed the polygonal morphology whereas they appeared smaller and spindle-shaped in Mg 1.5%. The higher Mg loading may possibly favor the extension of long protein chains (microtubules) from the centrioles in all directions. At higher Mg loading, no difference could be detected in Mg 3% with respect to Ctrl+ based on the visual aspect. It is well-known that the cell adhesion depends on the magnesium concentration, until an upper limit (∼10 mM) which induces cellular damages.64 Abed et al.65 unveiled that the Mg influx in MG-63 osteoblastic cells relies on the melastatin-like transient receptor potential 7 (TRPM7), being itself regulated by the level of extracellular Mg (at concentrations higher than 0.1 mM), and subsequently, triggers cell migration and proliferation mechanisms. However, their study also suggested the prominent role of platelet-derived growth factors (PDGFs) in conjunction with TRPM7 which was not supplemented in the present work. Hence, the lack of a noticeable difference in the structural organization of actin filaments in Mg 1.5% and Mg 3% samples can be correlated to the absence of PDGF supplementation.
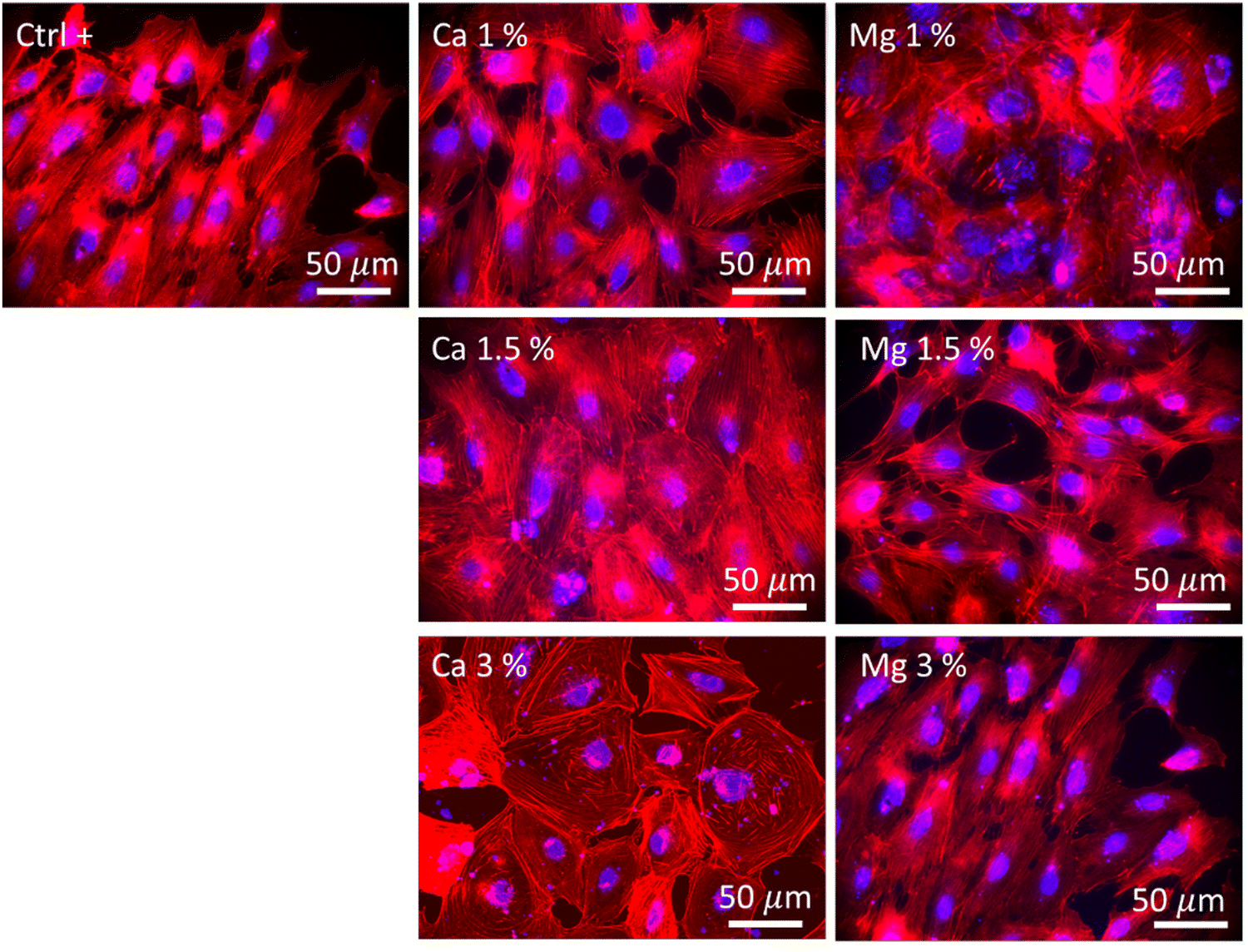 | ||
| Fig. 5 Images displaying the morphologies of MC3T3-E1 cells after 21 days under an epifluorescence microscope. F-actin filaments were stained using Alexa Fluor™ 568 Phalloidin (red), and nuclei were stained using DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) (blue). Ctrl+ corresponded to zeolite-free conditions. Both Mg- and Ca-loaded FAU were imaged at all zeolite concentrations. | ||
The highest cell population in samples with Ca2+ ions was found for Ca 1.5% with large flattened and cuboidal morphologies along with cell–cell contacts. This morphology was conserved in Ca 3% albeit more isolated, with lamellipodia outstretched, which either confirms the stable adhesion on the substrate,66 or can be associated with an early commitment in osteoblastic lineage.67 Moreover, the number of cell–cell and cell–matrix contacts is also regulated by the extracellular Ca, which was demonstrated by Nakamura et al.,68 whereby, connexin43, a marker of cell–cell contacts and integrin β1, a marker of cell–matrix contacts were both overexpressed in MC3T3 E1 cultured in 50 mM Ca compared to that in 6 mM. In this study, the largest concentrations were found to be between 14.4 and 19.6 mM (Fig. 4(b)), a range for which the Ca-dependence of the mode and the strength of adhesion is significant. As previously emphasized, it is likely that such a Ca-dependence of cell morphology and proliferation is related either to surface receptors (CaSR)51 or to G-coupled proteins such as calmodulin, which is sensitive to extracellular Ca, responsible for the Ca influx in the osteoblast cytoplasm, and in turn governs numerous cellular processes.66,69
The collagen secretions were visualized under a light microscope using Picrosirius red staining. In Fig. 6, one can observe poor collagen secretions after 7 days regardless of the conditions tested compared with the control. The smallest secretions were observed in Ca 1% and Ca 1.5% samples while they were a bit more marked in Ca 3%. In comparison, higher secretions could be observed in Mg-containing conditions, with a slight increase in Mg 1.5% samples. The influence of Mg to promote ECM deposition including collagen is clearly established;70 Liu et al.71 showed that ECM depositions by MC3T3-E1 were altered above 10 mM Mg. Here, these values were reached as early as at 7 days in Mg 3% (12.7 mM), which may explain the reduced collagen secretions and the associated lower proliferation (Fig. 4(a)). Of note, type I collagen is more secreted under a dual release of Cu and Mg than Mg alone.72 After 21 days, the trend is fully inverted, and the samples exposed to Ca2+ ions show a dramatic increase of protein secretions even larger than in control samples, suggesting a delayed production of collagen by MC3T3-E1 cells in the presence of Ca2+ ions. No distinction could be made between all the conditions of Ca2+ ions at day 21. Although the influence of Ca2+ ions on collagen secretions was well evidenced, the optimal molarity is still unclear since the Ca2+ concentration is rather transient in the bone microenvironment, and Ca2+ levels are often evolutive owing to their release from bioceramics for instance.73 Furthermore, the targeted concentration of Ca2+ may also vary between different cell types.74 Likewise, early works of Valerio et al.75 nuanced the correlation between the collagen deposition and Ca content in the range of 2.5–25 mM. The distinction between Mg-loaded samples was more pronounced as indicated by a moderate increase for Mg 1% between days 7 and 21, a marked deposition for Mg 1.5%, and a small decrease for Mg 3%.
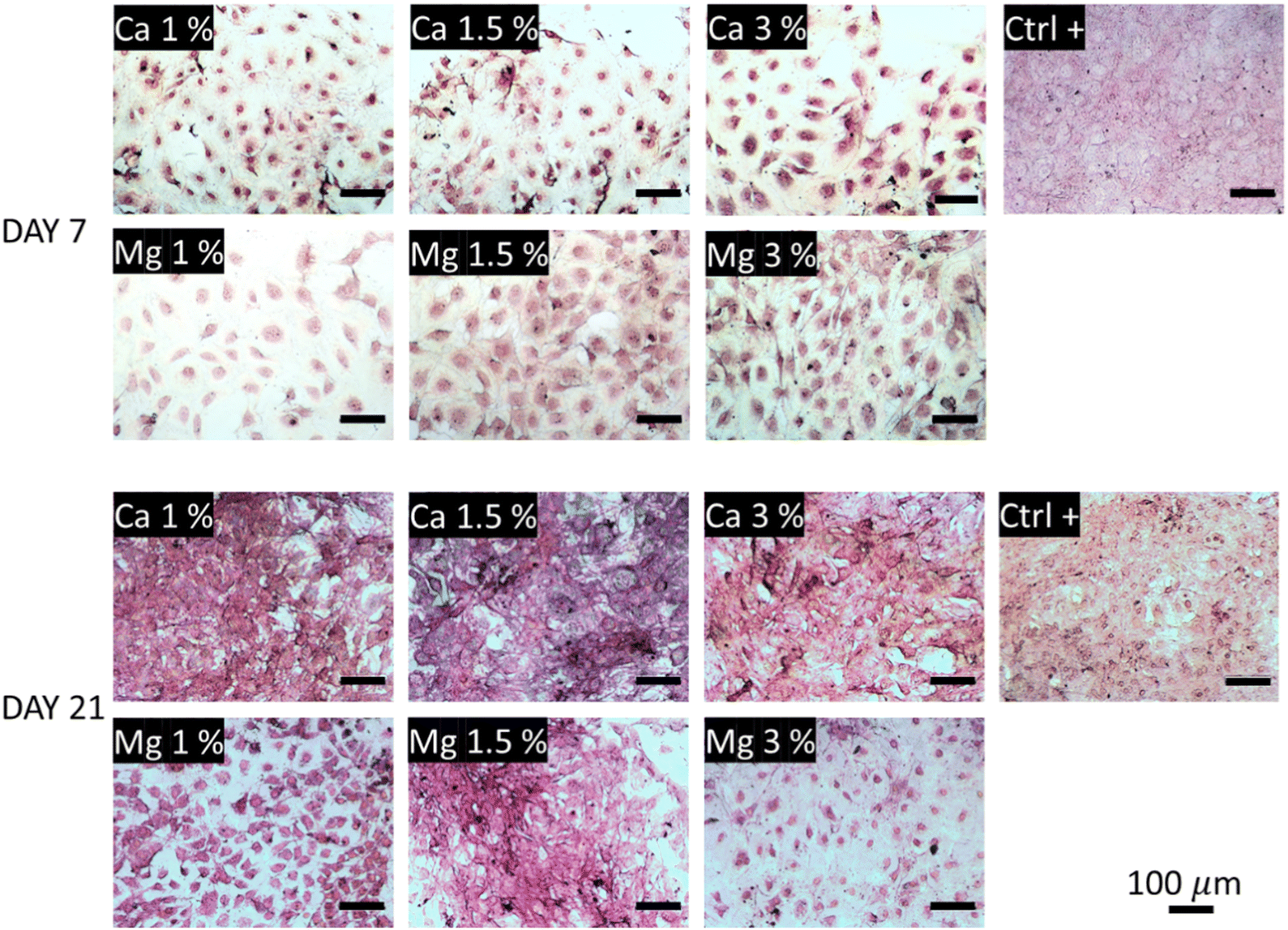 | ||
| Fig. 6 Collagen secretions visualized under a light microscope after Picrosirius red staining. The collagenous content appears in pink-red color. | ||
5. Conclusions
In summary, this study demonstrates that nanosized FAU zeolite crystals can be used as a reservoir for the sustained delivery of bioactive cations (i.e. Mg2+ and Ca2+), in a typical range of cell sensitivity, and without any cytotoxic effect. Owing to their large surface areas and adjustable pore sizes, zeolites could be investigated further as drug delivery systems considering their low cost and versatile functionalization process through ion-exchange. The ion delivery could be sustained over 21 days representative of classical cell culture experiments, and the concentrations can be adjusted by the amount of zeolites introduced in the culture medium. The prolonged release kinetics generated an intermediate, close to in vivo conditions, Ca and Mg concentrations, which allowed the cell behavior to be modulated. Proliferation was the most pronounced in Mg 1% and Mg 1.5%, and for conditions with the largest amount of Ca-exchanged FAU, namely Ca 1.5% and Ca 3%. Overall, the presence of high Ca content could stimulate cell spreading and collagen secretions. In future works, zeolites could be functionalized with other types of ions to gain antimicrobial (Ag+ or Cu2+), anti-inflammatory and/or angiogenic properties. Lastly, the mesopores of zeolites could be the leverage to introduce larger molecules in their channels. Lastly, one should note that the tight cell layers observed in Fig. 5 along with the increasing density of the collagenous matrix (Fig. 6) may act as a diffusion barrier for ions or molecules trapped into zeolites underneath, as already observed for the monolayers of epithelial cells.76 Therefore, the bioactivity of ions toward cells directly anchored to the material surface may be unaffected. Conversely, the diffusion profile of ions intended to diffuse further away from the surface (e.g. antibacterial effects) may require deeper examination, to ensure that the bioactivity of zeolite-based materials is sustained over time.4. Materials and methods
Preparation of Mg-Y and Ca-Y zeolites
Commercial ultra-stabilized Y-zeolites (USY) were kindly supplied by Zeolyst International (CBV500) bearing ammonium ions as the charge compensation of aluminum sites (NH4-Y). The ion exchange procedure was performed as follows: 1.5 g of NH4-USY were placed in a glass flask with 200 mL of magnesium chloride hexahydrate (LABOSI, 98% purity) at a concentration of 1 M. The flask was placed in a heating mantle, and then surmounted by a cooling tube. The mixture was allowed to stand at 80 °C for 2 h, and then separated by suction filtration. Afterwards, the supernatant was removed, and the ion exchange procedure was repeated twice. At the end, the resulting Mg-Y zeolites were placed in an oven at 80 °C for 48 h, and the dried powder was then used for experiments. The same procedure was used to obtain Ca-Y zeolites, except that calcium chloride dihydrate (Fluka, 99.5% purity) was mixed with 1.5 g of NH4-Y powder.Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) analyses
Scanning electron microscopy (SEM) images were acquired using a ZEISS GEMINI SEM 500 microscope at an electron high tension (EHT) voltage ranging from 2 to 6 kV. To determine the elemental distribution, EDX and mapping analyses were also performed using an EDAX SDD detector.X-ray diffraction (XRD) analysis of Mg-USY and Ca-USY
X-ray diffraction patterns were recorded using a Bruker D8 Advance diffractometer, with a Ni detector side filtered Cu Kα radiation (1.5406 Å) over a 2θ range of 5–65°.Measurements of specific surface areas (BET)
The nitrogen adsorption–desorption isotherms of all zeolites were recorded at 77 K using a Micromeritics ASAP 2420. The specific surface areas and pore volumes were calculated using the Brunauer–Emmett–Teller (BET) method. Prior to analysis, FAU zeolites were pre-treated at 250 °C under vacuum for 10 h.X-Ray flurorescence spectroscopy
Elemental analysis of the zeolites was performed by X-ray flurorescence spectroscopy using an Epsilon 3XL Panalytical apparatus. The fluorimeter holds a silver tube working at a maximum voltage of 50 kV. Samples were analyzed in the form of micrometer-sized pearls.Cell culture
Pre-osteoblastic cells MC3T3-E1 (ATCC, subclone 4) were grown in 75 T-flasks containg α-MEM (PAN Biotech) media supplemented with 10% v/v fetal bovine serum (FBS), 1% v/v L-glutamine, and 1% v/v penicillin/streptomycin, and placed in an incubator (37 °C, 5% CO2). Before seeding, culture media were changed every 2–3 days. When the cells reached about 80% confluency, they were washed with phosphate buffer saline (PBS), and detached with an EDTA-trypsin mixture for 5 min in an incubator (37 °C, 5% CO2), before deactivation with an equivalent volume of fresh media. Glass coverslips (∅ = 12 mm, Thermo scientific) were placed at the bottom of each well in order to be able to extract them at the end of the experiments; these coverslips could then be readily used for observation under an epifluorescence microscope. At passage number 15, cells were seeded at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per cm2 at the bottom of 24 well plates with 1 mL of the fresh medium. Mg-Y and Ca-Y were introduced at different weight percentages with respect to the volume of culture media (i.e. 1 mL per well), namely: 0 (control), 1, 1.5, and 3 wt%, in sterile inserts for cell culture (0.4 μm, 12 mm in diameter, PIHP01250, MilliCell®), and placed into each well to allow the diffusion of ions. The well-plates were placed in an incubator, and culture media were replaced at days 7, 14, 17 and 21 in order to quantify the concentration of both Ca2+ and Mg2+ ions released from zeolites.
000 cells per cm2 at the bottom of 24 well plates with 1 mL of the fresh medium. Mg-Y and Ca-Y were introduced at different weight percentages with respect to the volume of culture media (i.e. 1 mL per well), namely: 0 (control), 1, 1.5, and 3 wt%, in sterile inserts for cell culture (0.4 μm, 12 mm in diameter, PIHP01250, MilliCell®), and placed into each well to allow the diffusion of ions. The well-plates were placed in an incubator, and culture media were replaced at days 7, 14, 17 and 21 in order to quantify the concentration of both Ca2+ and Mg2+ ions released from zeolites.
The viability and proliferation of the cells were measured using the Alamar blue® (BUF012B, Bio-rad) kit, following the instructions provided by the manufacturer. Briefly, culture media were removed and replaced with a mixture of fresh culture media supplemented with the 10% v/v Alamar blue® reagent, and incubated for 4 hours in the dark. Afterwards, 100 μL were pipetted in each well and transferred into a 96 well-plate prior to measurements at 570 and 600 nm using a microplate reader (Synergy 2, Biotek Instruments, Inc.). The reduction percentage of Alamar blue® was calculated from the absorbance values at 570 and 600 nm using the following equation:
| Percentage reduction of Alamar blue:= ((O2·A1) − (O1·A2))/((R1·N2) − (R2·N1)) | (1) |
586 and O2 = 117216 are the molar extinction coefficients of oxidized Alamar blue at 570 and 600 nm, respectively (values provided by the manufacturer); R1 = 155617 and R2 = 14652 are the molar extinction coefficients of reduced Alamar blue also at 570 and 600 nm. A1 and A2 are the absorbance values of the samples at 570 and 600 nm, while N1 and N2 are the absorbance values of negative controls (i.e. media with Alamar blue without cells). The test was performed at days 7, 14, and 21, in order to quantify the proliferative potential of each conditions.
After 21 days, experiments were stopped, and cells were washed with PBS, and fixed with 500 μL of paraformaldehyde (4% v/v in PBS) for 30 min at room temperature, prior to storing at 4 °C. In order to assess the morphology of the cells, both nuclei and F-actin filaments were stained. First, the cell membrane was permeabilized using Triton® X-100 (0.1% v/v) in PBS for 10 min followed by two rinsing steps for 5 min in PBS. To increase the contrast, a saturation step was performed by incubation in a solution of 1% (v/v) bovin serum albumin (BSA) in PBS for 20 min. Nucleus staining was achieved by incubation for 1 hour at RT with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) (Thermofischer, D1306) prepared at 1/100 v/v in PBS. After two rinsing steps in PBS for 10 min, F-actin filaments were stained with Alexa Fluor™ 568 Phalloidin (Thermofischer, A12380) at a dilution of 1/40 v/v in PBS for 30 min at RT. Then, two more rinsing steps of 10 min each in PBS were performed, prior to storing the samples in the refrigerator at 4 °C.
The amount of collagen secreted by the cells was stained using the Picrosirius Red Stain Kit (Polysciences, Inc., 24901-250), following the supplier instructions. Briefly, samples were fixed with a solution of 4% v/v paraformaldehyde (prepared in PBS) for 30 min at RT prior rinsing with distilled water. Next, samples were incubated with 200 μL per well of solution B (Picosirius Red Stain) for 60 min, and subsequently incubated in 200 μL of solution C (HCl, 1 M) for 2 min. Finally, samples were dehydrated in 70% ethanol solution for 45 min keeping 200 μL per well, and placed in a refrigerator at 4 °C until observation.
Light microscopy
Collagenous secretions were visualized under a light microscope (LEICA, DM LM/P) mounted with an HD digital microscope camera (LEICA MC170 HD). Image acquisition was performed by using the Leica Application Suite (LAS.V4.12) software. The auto colour balance tool was applied before each acquisition, and the exposition time was set at 500 ms.Epifluorescence microscopy
The morphology of the MC3T3-E1 cells was analysed using an epifluorescence microscope (Zeiss Axio Scope A1) mounted with a digital microscope camera (LEICA MC170 HD). The glass coverslips recovered by cells after 21 days of culture were placed on microscope slides. Images were acquired using the LAS.V4.12 software, and taken each time twice with an excitation/emission wavelength of 578/600 nm for Phalloidin staining, and an excitation/emission wavelength of 358/461 nm for DAPI staining (nucleus).Capillary electrophoresis (CE)
The concentrations of both Ca2+ and Mg2+ ions released in the culture media were measured by capillary electrophoresis (Agilent CE, G1600A) at days 7, 14, 17 and 21 in accordance with viability/proliferation measurements. As the culture media were enriched with sodium ions, a 1/100 dilution of the harvested solutions was made in the background electrolytes (BEs) (i.e. 7 μL of culture media into 700 μL of BEs), prepared according to the methodology described by Shi et al.77 Typically, imidazole (Sigma, I-0250) was prepared at 20 mM in ultrapure water, and the pH was adjusted to 6 using a solution of 3 M sulfuric acid (Fischer Scientific, S/9220/PB15). Then, hydroxypropylmethylcellulose (Merck, 09963) was dissolved at 0.1 wt%. The indirect photometric detection was used to identify ions at a wavelength of 214 nm at the cathode. The concentration of each ion was calculated after the establishment of calibration curves for concentrations ranging from 10−3 to 10−6 M (data not shown), in order to assign the corresponding retention time of each ion, while concentrations were deduced from the integrated area under each peak. The separation/quantification procedure was conducted as follows: capillary tubes (fused silica, effective length 24.5 cm, total length 33 cm, ID: 50 μm) were rinsed with a solution of 1 M sodium hydroxide for 15 min and then rinsed with BEs for 3 min. Samples were injected at a pressure of 50 mbar for 30 s, and the separation was performed at an applied tension of 10 kV for 4 min, followed by further washing steps with sodium hydroxide 1 M.Colorimetric titrations of Ca2+ and Mg2+ ions
In conjunction with capillary electrophoresis, colorimetric titrations of Ca2+ and Mg2+ ions were also performed in order to establish the release profile of each ion over time in both ultrapure water and complete culture media. For the determination of Ca2+, calibration standards were prepared using calcium chloride dihydrate (Fluka, 99.5% pure) dissolved in both ultrapure water and culture media at concentrations of 10−5, 10−4, 10−3, 10−2, 5 × 10−2, 10−1, 0.15, and 1 M, and 1 mL of these standards were then added to 4 mL of sodium oxalate solution (Fischer Scientific, BP353-500) (4 wt%) in order to form a calcium oxalate precipitate. The solutions were allowed to stand at 4 °C overnight prior to centrifugation at 10000 rpm for 10 min. The supernatant was removed, and the calcium oxalate pellets were then dissolved in3 M sulfuric acid at 80 °C in a heating bath. The concentration of oxalic acid was then titrated under stirring with potassium permanganate (Sigma, 223468) at 0.1 M according to the following reaction:78| 2KMnO4 + 5H2C2O4 + 3H2SO4 → K2SO4 + 2MnSO4 + 8H2O + 10CO2 |
The titration end point was characterized by the appearance of a slightly pink coloration along with the brownish precipitate due to the presence of manganese compounds. Once the calibration curve was obtained, the unknown concentration of Ca2+ containing samples was determined from equivalent volumes, by using the fitting curve of the calibration standards.
Regarding Mg2+ ions, another colorimetric titration was performed using the classical Eriochrome black T (EBT) indicator.79 For Ca2+, Mg2+ standards were first prepared by dissolving magnesium chloride hexahydrate (LABOSI, 98% pure) at concentrations of 10−4, 10−3, 10−2, 10−1, 0.4, 0.6, 0.8 and 1 M, in both ultrapure water and complete culture media. 1 mL of the standard solution was then mixed with 5 mL of ammonia buffer (pH = 10) i.e. for 100 mL, 1.07 g of ammonium chloride (Prolabo, 21236) with 6.4 mL of ammonia solution 20 wt% (Prolabo, rectapur™, 21180), and then completed up to 100 mL with ultrapure water. The whole solution was placed on a magnetic stirrer and a spatula tip containing Eriochrome Black T was added until the color turned red. Of note, as the culture media used in this study was already red due to the pH indicator, the quantity of Eriochrome Black T needed was slightly higher compared to ultrapure water. The titration was conducted with a solution of ethylenediaminetetraacetic acid (EDTA, Sigma, ED-500G) at 10−2 M, whilst the end point was the characteristic color shift from red to blue. At pH = 10, the Mg fixation to EBT is not selective compared to Ca–EBT complexes, and the end point of the titration corresponds to both Mg and Ca concentrations.80 The amount of Mg ions in the culture media was determined by subtracting the basal Ca2+ concentration of pure media measured by capillary electrophoresis. The concentration of Mg2+ present in all samples was determined again by reporting the measured equivalent volumes on the calibration curve. All titrations were performed in triplicate.
Statistical analysis
All results relative to cell proliferation/viability and ion concentrations were performed in triplicate and expressed as mean ± SD. To assess significance between different conditions, the non-parametric Mann–Whitney–Wilcoxon test was used, and conditions with p-values < 0.05 were considered as statistically different.Author contributions
G. Lutzweiler – conceptualization, supervision, and writing; Y. Zhang – experiments and data curation; F. Gens – experiments; A. Echalard – experiments and investigation; G. Ladam – visualization; J. Hochart – investigation; T. Janicot – investigation; N. Mofaddel – supervision methodology; B. Louis – conceptualization, writing, and validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Thierry Romero for his technical assistance.Notes and references
- X. Liu, P. Chu and C. Ding, Mater. Sci. Eng., R, 2004, 47, 49–121 CrossRef.
- G. Lutzweiler, A. Ndreu Halili and N. Engin Vrana, Pharmaceutics, 2020, 12(7), 602 CrossRef CAS PubMed.
- A. Ho-Shui-Ling, J. Bolander, L. E. Rustom, L. A. W. Johnson, F. P. Luyten and C. Picart, Biomaterials, 2018, 180, 143–162 CrossRef CAS PubMed.
- J. N. Zara, R. K. Siu, X. Zhang, J. Shen, R. Ngo, M. Lee, W. Li, M. Chiang, J. Chung, J. Kwak, B. M. Wu, K. Ting and C. Soo, Tissue Eng., Part A, 2011, 17(9–10), 1389–1399 CrossRef CAS PubMed.
- N. Goonoo and A. Bhaw-Luximon, RSC Adv., 2019, 9(32), 18124–18146 RSC.
- E. O’Neill, G. Awale, L. Daneshmandi, O. Umerah and K. W. H. Lo, Drug Discovery Today, 2018, 23(4), 879–890 CrossRef PubMed.
- N. J. Lakhkar, I. H. Lee, H. W. Kim, V. Salih, I. B. Wall and J. C. Knowles, Adv. Drug Delivery Rev., 2013, 65(4), 405–420 CrossRef CAS PubMed.
- D. W. Liu, D. C. Genetos, Y. Shao, D. J. Geist, J. L. Li, H. Z. Ke, C. H. Turner and R. L. Duncan, Bone, 2008, 42, 644–652 CrossRef CAS PubMed.
- S. C. Yeh, J. Hou, J. W. Wu, S. Yu, Y. Zhang, K. D. Belfield, F. D. Camargo and C. P. Lin, Nat. Commun., 2022, 13(1), 1–13 Search PubMed.
- A. L. Boskey and A. S. Posner, Mater. Res. Bull., 1974, 9, 907–916 CrossRef CAS.
- C. C. Hung, A. Chaya, K. Liu, K. Verdelis and C. Sfeir, Acta Biomater., 2019, 98, 246–255 CrossRef CAS PubMed.
- T. Kokubo, M. Shigematsu, Y. Nagashima, M. Tashiro, T. Nakamura, T. Yamamuro and S. Higashi, Bulletin of the Institute for Chemical Research, Kyoto University, 1982, vol. 60, no. 3–4, pp. 260–268 Search PubMed.
- T. Kokubo and S. Yamaguchi, J. Biomed. Mater. Res., Part A, 2019, 107(5), 968–977 CrossRef CAS PubMed.
- M. Bohner, B. L. G. Santoni and N. Döbelin, Acta Biomater., 2020, 113, 23–41 CrossRef CAS PubMed.
- Z. S. Tao, W. S. Zhou, X. W. He, W. Liu, B. L. Bai, Q. Zhou, Z. L. Huang, K. Tu, H. Li, T. Sun, Y. Lv, W. Cui and L. Yang, Mater. Sci. Eng., C, 2016, 62, 226–232 CrossRef CAS PubMed.
- G. L. Koons, M. Diba and A. G. Mikos, Nat. Rev. Mater., 2020, 5(8), 584–603 CrossRef CAS.
- J. F. Navarro-González, C. Mora-Fernández and J. García-Pérez, Seminars in Dialysis, Blackwell Publishing Ltd, Oxford, UK, 2009, vol. 22, no. 1, pp. 37–44 Search PubMed.
- N. D. Toussaint, G. J. Elder and P. G. Kerr, Clin. J. Am. Soc. Nephrol., 2009, 4(1), 221–233 CrossRef CAS PubMed.
- Z. Lin, J. Wu, W. Qiao, Y. Zhao, K. H. Wong, P. K. Chu, L. Bian, S. Wu, Y. Zheng, K. M. C. Cheung, F. Leung and K. W. Yeung, Biomaterials, 2018, 174, 1–16 CrossRef CAS PubMed.
- X. Shen, Y. Zhang, P. Ma, L. Sutrisno, Z. Luo, Y. Hu, B. Tao, C. Li and K. Cai, Biomaterials, 2019, 212, 1–16 CrossRef CAS PubMed.
- O. Castano, N. Sachot, E. Xuriguera, E. Engel, J. A. Planell, J. H. Park, G. Z. Jin, T. H. Kim, J. H. Kim and H. W. Kim, ACS Appl. Mater. Interfaces, 2014, 6(10), 7512–7522 CrossRef CAS PubMed.
- M. Sandomierski, M. Zielińska, K. Adamska, A. Patalas and A. Voelkel, New J. Chem., 2022, 46(7), 3401–3408 RSC.
- M. A. Sakr, M. G. Mohamed, R. Wu, S. R. Shin, D. Kim, K. Kim and S. Siddiqua, Appl. Clay Sci., 2020, 199, 105860 CrossRef CAS.
- R. M. Barrer, Zeolites, 1981, 1(3), 130–140 CrossRef CAS.
- C. J. Rhodes, Sci. Prog., 2010, 93(3), 223–284 CrossRef CAS PubMed.
- E. S. Gomes, G. Lutzweiler, P. Losch, A. V. Silva, C. Bernardon, K. Parkhomenko, M. M. Peireira and B. Louis, Microporous Mesoporous Mater., 2017, 254, 28–36 CrossRef CAS.
- M. M. Pereira, E. S. Gomes, A. V. Silva, A. B. Pinar, M. G. Willinger, S. Shanmugam, C. Chizallet, G. Laugel, P. Losch and B. Louis, Chem. Sci., 2018, 9(31), 6532–6539 RSC.
- N. Ninan, M. Muthiah, I. K. Park, A. Elain, T. W. Wong, S. Thomas and Y. Grohens, ACS Appl. Mater. Interfaces, 2013, 5(21), 11194–11206 CrossRef CAS PubMed.
- N. Iqbal, M. R. A. Kadir, N. H. B. Mahmood, M. F. M. Yusoff, J. A. Siddique, N. Salim, G. R. A. Froemming, M. N. Sarian, H. R. B. Baghavendran and T. Kamarul, Ceram. Int., 2014, 40(10), 16091–16097 CrossRef CAS.
- R. S. Bedi, D. E. Beving, L. P. Zanello and Y. Yan, Acta Biomater., 2009, 5(8), 3265–3271 CrossRef CAS PubMed.
- R. S. Bedi, L. P. Zanello and Y. Yan, Adv. Funct. Mater., 2009, 19(24), 3856–3861 CrossRef CAS.
- C. Anfray, S. Komaty, A. Corroyer-Dulmont, M. Zaarour, C. Helaine, H. Ozcelik, C. Allioux, J. Toutain, K. Goldyn, E. Petit, K. Bordji, M. Bernaudin, V. Valtchev, O. Touzani, S. Mintova and S. Valable, Biomaterials, 2020, 257, 120249 CrossRef CAS PubMed.
- C. V. Uglea, I. Albu, A. Vatajanu, M. Croitoru, S. Antoniu, L. Panaitescu and R. M. Ottenbrite, J. Biomater. Sci., Polym. Ed., 1995, 6(7), 633–637 CrossRef PubMed.
- X. Zhang, J. Chen, X. Pei, J. Wang, Q. Wan, S. Jiang, C. Huang and X. Pei, ACS Appl. Mater. Interfaces, 2017, 9(30), 25171–25183 CrossRef CAS PubMed.
- S. Ivanova, B. Louis, M. J. Ledoux and C. Pham-Huu, J. Am. Chem. Soc., 2007, 129(11), 3383–3391 CrossRef CAS PubMed.
- B. Louis, F. Ocampo, H. S. Yun, J. P. Tessonnier and M. M. Pereira, Chem. Eng. J., 2010, 161(3), 397–402 CrossRef CAS.
- C. F. Imbachi-Gamba and A. L. Villa, Mater. Today Chem., 2021, 20, 100442 CrossRef CAS.
- R. Le Van Mao, N. T. Vu, S. Xiao and A. Ramsaran, J. Mater. Chem., 1994, 4(7), 1143–1147 RSC.
- E. N. Coker and L. V. Rees, Microporous Mesoporous Mater., 2005, 84(1-3), 171–178 CrossRef CAS.
- C. Bernardon, B. Louis, V. Bénéteau and P. Pale, ChemPlusChem, 2013, 78(9), 1134–1141 CrossRef CAS PubMed.
- D. W. Breck, Zeolite molecular sieves: structure, chemistry, and use, John Wiley & Sons, 1973 Search PubMed.
- A. Lateef, R. Nazir, N. Jamil, S. Alam, R. Shah, M. N. Khan and M. Saleem, Microporous Mesoporous Mater., 2016, 232, 174–183 CrossRef CAS.
- S. Chen, J. Popovich, N. Iannuzo, S. E. Haydel and D. K. Seo, ACS Appl. Mater. Interfaces, 2017, 9(45), 39271–39282 CrossRef CAS PubMed.
- R. Bingre, P. Losch, C. Megías-Sayago, B. Vincent, P. Pale, P. Nguyen and B. Louis, ChemPhysChem, 2019, 20(21), 2874–2880 CrossRef CAS PubMed.
- E. Titus, A. K. Kalkar and V. G. Gaikar, Colloids Surf., A, 2013, 223(1-3), 55–61 CrossRef.
- M. Rahimi, E. P. Ng, K. Bakhtiari, M. Vinciguerra, H. A. Ahmad, H. Awala, S. Mintova, M. Daghighi, F. B. Rostami, M. de Vries, M. M. Matazacker, M. P. Peppelenbosch, M. Mahmoudi and F. Rezaee, Sci. Rep., 2015, 5(1), 1–12 Search PubMed.
- A. Vinu, V. Murugesan, O. Tangermann and M. Hartmann, Chem. Mater., 2004, 16(16), 3056–3065 CrossRef CAS.
- L. Wu, F. Feyerabend, A. F. Schilling, R. Willumeit-Römer and B. J. Luthringer, Acta Biomater., 2015, 27, 294–304 CrossRef CAS PubMed.
- J. Shen, B. Chen, X. Zhai, W. Qiao, S. Wu, X. Liu, Y. Zhao, C. Ruan, H. Pan, P. K. Chu, K. M. C. Cheung and K. W. Yeung, Bioact. Mater., 2021, 6(2), 503–519 CrossRef CAS PubMed.
- X. Nie, X. Sun, C. Wang and J. Yang, Regener. Biomater., 2020, 7(1), 53–61 CAS.
- Q. Wu, S. Xu, F. Wang, B. He, X. Wang, Y. Sun, C. Ning and K. Dai, Regener. Biomater., 2021, 8(6), rbab016 CrossRef PubMed.
- R. Bingre, B. Vincent, Q. Wang, P. Nguyen and B. Louis, J. Phys. Chem. C, 2028, 123(1), 637–643 CrossRef.
- F. Hu, L. Pan, K. Zhang, F. Xing, X. Wang, I. Lee, X. Zhang and J. Xu, PLoS One, 2014, 9(9), e107217 CrossRef PubMed.
- S. Maeno, Y. Niki, H. Matsumoto, H. Morioka, T. Yatabe, A. Funayama, Y. Tomaya, T. Tagushi and J. Tanaka, Biomaterials, 2005, 26(23), 4847–4855 CrossRef CAS PubMed.
- E. Gabusi, C. Manferdini, F. Grassi, A. Piacentini, L. Cattini, G. Filardo, E. Lambertini, R. Piva, N. Zini, A. Facchini and G. Lisignoli, J. Cell. Physiol., 2012, 227(8), 3151–3161 CrossRef CAS PubMed.
- B. Xiang, Y. Liu, W. Zhao, H. Zhao and H. Yu, Cell Biol. Int., 2009, 43(10), 1125–1136 CrossRef PubMed.
- M. N. Lee, H. S. Hwang, S. H. Oh, A. Roshanzadeh, J. W. Kim, J. H. Song, E. S. Kim and J. T. Koh, Exp. Mol. Med., 2018, 50(11), 1–16 Search PubMed.
- I. A. Silver, R. J. Murrills and D. J. Etherington, Exp. Cell Res., 1988, 175(2), 266–276 CrossRef CAS PubMed.
- S. L. Haag, N. R. Schiele and M. T. Bernards, Biotechnol. Appl. Biochem., 2022, 69(2), 492–502 CrossRef CAS PubMed.
- U. Klammert, T. Reuther, C. Jahn, B. Kraski, A. C. Kübler and U. Gbureck, Acta Biomater., 2009, 5(2), 727–734 CrossRef CAS PubMed.
- A. R. Atif, M. Pujari-Palmer, M. Tenje and G. Mestres, Acta Biomater., 2021, 127, 327–337 CrossRef CAS PubMed.
- E. Huethorst, M. F. Cutiongco, F. A. Campbell, A. Saeed, R. Love, P. M. Reynolds, M. J. Dalby and N. Gadegaard, Biofabrication, 2020, 12(2), 025009 CrossRef CAS PubMed.
- A. Cerqueira, F. Romero-Gavilán, I. García-Arnáez, C. Martinez-Ramos, S. Ozturan, R. Izquierdo, M. Azkargorta, F. Elortza, M. Gurruchaga, J. Suay and I. Goñi, Mater. Sci. Eng., C, 2021, 125, 112114 CrossRef CAS PubMed.
- S. Mostofi, E. Bonyadi Rad, H. Wiltsche, U. Fasching, G. Szakacs, C. Ramskogler, S. Srinivasaiah, M. Ueçal, R. Willumeit, A.-M. Weinberg and U. Schaefer, PLoS One, 2016, 11(7), e0159879 CrossRef PubMed.
- E. Abed and R. Moreau, Am. J. Physiol.: Cell Physiol., 2009, 297(2), C360–C368 CrossRef CAS PubMed.
- Y. Zhang, K. Wang, K. Dong, N. Cui, T. Lu and Y. Han, Colloids Surf., B, 2020, 187, 110773 CrossRef CAS PubMed.
- N. Nabavi, Y. Urukova, M. Cardelli, J. E. Aubin and R. E. Harrison, J. Biol. Chem., 2008, 283(28), 19678–19690 CrossRef CAS PubMed.
- S. Nakamura, T. Matsumoto, J. I. Sasaki, H. Egusa, K. Y. Lee, T. Nakano, T. Sohmura and A. Nakahira, Tissue Eng., Part A, 2010, 16(8), 2467–2473 CrossRef CAS PubMed.
- M. Zayzafoon, J. Cell. Biochem., 2006, 97(1), 56–70 CrossRef CAS PubMed.
- B. Li, Y. Han and M. Li, J. Mater. Chem. B, 2016, 4(4), 683–693 RSC.
- W. Liu, S. Guo, Z. Tang, X. Wei, P. Gao, N. Wang, X. Li and Z. Guo, Biochem. Biophys. Res. Commun., 2020, 528(4), 664–670 CrossRef CAS PubMed.
- C. Liu, X. Fu, H. Pan, P. Wan, L. Wang, L. Tan, K. Wang, Y. Zhao, K. Yang and P. K. Chu, Sci. Rep., 2016, 6(1), 1–17 CrossRef PubMed.
- A. Nandakumar, A. Barradas, J. de Boer, L. Moroni, C. van Blitterswijk and P. Habibovic, Biomatter, 2013, 3(2), e23705 CrossRef PubMed.
- A. M. Barradas, H. A. Fernandes, N. Groen, Y. C. Chai, J. Schrooten, J. Van de Peppel, J. P. T. M. Van Leeuwen, C. A. Van Blitterswijk and J. De Boer, Biomaterials, 2012, 33(11), 3205–3215 CrossRef CAS PubMed.
- P. Valerio, M. M. Pereira, A. M. Goes and M. F. Leite, Biomaterials, 2004, 25(15), 2941–2948 CrossRef CAS PubMed.
- M. Mihajlovic, L. P. Van Den Heuvel, J. G. Hoenderop, J. Jansen, M. J. Wilmer, A. J. Westheim, W. A. Allebes, D. Stamatialis, L. B. Hilbrands and R. Masereeuw, Sci. Rep., 2017, 7(1), 1–14 CrossRef CAS PubMed.
- H. Shi, R. Zhang, G. Chandrasekher and Y. Ma, J. Chromatogr. A, 1994, 680(2), 653–658 CrossRef CAS.
- J. C. Witt, J. Phys. Chem., 2002, 26(5), 435–446 CrossRef.
- H. Diehl and F. Lindstrom, Anal. Chem., 1959, 31(3), 414–418 CrossRef CAS.
- G. P. Hildebrand and C. N. Reilley, Anal. Chem., 1957, 29(2), 258–264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00768a |
| This journal is © The Royal Society of Chemistry 2022 |