
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalently attached intercalators restore duplex stability and splice-switching activity to triazole-modified oligonucleotides†
Anna
Dysko
a,
Ysobel R.
Baker
 a,
Graham
McClorey
b,
Matthew J. A.
Wood
b,
Sabine
Fenner
c,
Glynn
Williams‡
c,
Afaf
El-Sagheer
ad and
Tom
Brown
*a
a,
Graham
McClorey
b,
Matthew J. A.
Wood
b,
Sabine
Fenner
c,
Glynn
Williams‡
c,
Afaf
El-Sagheer
ad and
Tom
Brown
*a
aDepartment of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: tom.brown@chem.ox.ac.uk
bDepartment of Physiology, Anatomy and Genetics, University of Oxford, Oxford, UK
cGSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire, SG1 2NY, UK
dChemistry Branch Department of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez 43721, Egypt
First published on 16th May 2022
Abstract
Oligonucleotides are rapidly emerging as powerful therapeutics for hard to treat diseases. Short single-stranded oligonucleotides can base pair with target RNA and alter gene expression, providing an attractive therapeutic approach at the genetic level. Whilst conceptually appealing, oligonucleotides require chemical modification for clinical use. One emerging approach is to substitute the phosphodiester backbone with other chemical linkages such as triazole. The triazole linkage is inherently resistant to enzymatic degradation, providing stability in vivo, and is uncharged, potentially improving cell-penetration and in vivo distribution. Triazole linkages, however, are known to reduce RNA target binding affinity. Here we show that by attaching pyrene or anthraquinone to the ribose sugar on the 5′-side of the triazole, it is possible to recover duplex stability and restore the splice switching ability of triazole-containing oligonucleotides.
Introduction
Antisense oligonucleotides (ASOs) are rapidly emerging as powerful agents for treating genetic diseases.1–3 These short single stranded analogues of DNA (typically 15–25 bases in length) can bind to their complementary RNA targets and alter gene expression by a variety of mechanisms.4 ASOs can inhibit translation of specific proteins by recruiting ribonuclease H1 (RNase H) to degrade the target messenger RNA (mRNA),5 or by binding to mRNA translational start sites, thereby blocking the binding of ribosomes.6 ASOs can also be designed to bind precursor mRNA (pre-mRNA) and modulate RNA splicing.7–9 This can introduce an out-of-frame deletion, triggering nonsense mediated decay of the transcript. Alternatively, it can correct aberrant splicing caused by splice point mutations, or truncate the mRNA by removing exons that contain premature stop codons or out-of-frame deletion mutations.Without chemical modification, oligonucleotides (ONs) are rapidly degraded by nucleases in biological environments rendering them ineffective as therapeutics. For this reason, chemical modifications have been developed to provide protection against nucleases and enhance pharmacokinetic properties.10 Commonly used chemical modifications for ASOs include replacing phosphodiester (PO) linkages in the backbone with phosphorothioate groups (PS)11,12 to improve duplex stability against enzymatic degradation and cellular uptake, and ribose modifications to improve target affinity and lipophilicity. These include 2′-F, 2′-OMe, 2′-O-(2-methoxyethyl) and locked nucleic acids (LNAs).11,13–15 These modifications have transformed the field, and the FDA has now approved several ASOs for clinical use.16 However, the widespread therapeutic applications of ASOs are still hampered by potential toxicity and their inability to efficiently cross cellular membranes. Hence, there remains a need for new ASO chemistries to reduce dosage, toxicity, cost and improve clinical efficacy.
Charge-neutral backbone modifications offer an intriguing alternative for ASO development.17–19 Here, the PO backbone is partially or fully replaced with linkages that have no charge (each phosphodiester linkage carries an anionic charge, Fig. 1A). These linkages can confer nuclease resistance (in vivo stability) and alter the overall anionic nature and lipophilicity of an ASO, potentially aiding cellular uptake and modulating protein interactions that influence in vivo distribution. The triazole linkage (Fig. 1B) has several characteristics that are desirable when developing new ASO candidates. It is biocompatible and completely stable to endogenous nucleases, it is simple to synthesise and is charge neutral.20,21 Despite these advantages, the triazole linkage reduces the affinity of an ON for target RNA, and a single triazole incorporation can significantly reduce the stability of ON![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :RNA duplexes.21–23 Several different approaches to overcome this have been reported including the use of an aminoethoxyphenoxazine nucleobase analogue (G-clamp) which increases duplex stability by a combination of increased base stacking and an extra hydrogen bond with guanine (Fig. 1C),23 and substitution of the flanking ribose sugars with a morpholino analogue (Fig. 1D),24 or an LNA derivative (Fig. 1E and F).21,25,26 Whilst several triazole linkage designs exist,22,27–29 this study focuses on the structure shown in Fig. 1B due to its ease of synthesis. In the current study, by incorporating various chemical moieties adjacent to the triazole linkage that are known to stabilise ON duplexes, we have significantly improved the RNA binding affinity of ONs.
:RNA duplexes.21–23 Several different approaches to overcome this have been reported including the use of an aminoethoxyphenoxazine nucleobase analogue (G-clamp) which increases duplex stability by a combination of increased base stacking and an extra hydrogen bond with guanine (Fig. 1C),23 and substitution of the flanking ribose sugars with a morpholino analogue (Fig. 1D),24 or an LNA derivative (Fig. 1E and F).21,25,26 Whilst several triazole linkage designs exist,22,27–29 this study focuses on the structure shown in Fig. 1B due to its ease of synthesis. In the current study, by incorporating various chemical moieties adjacent to the triazole linkage that are known to stabilise ON duplexes, we have significantly improved the RNA binding affinity of ONs.
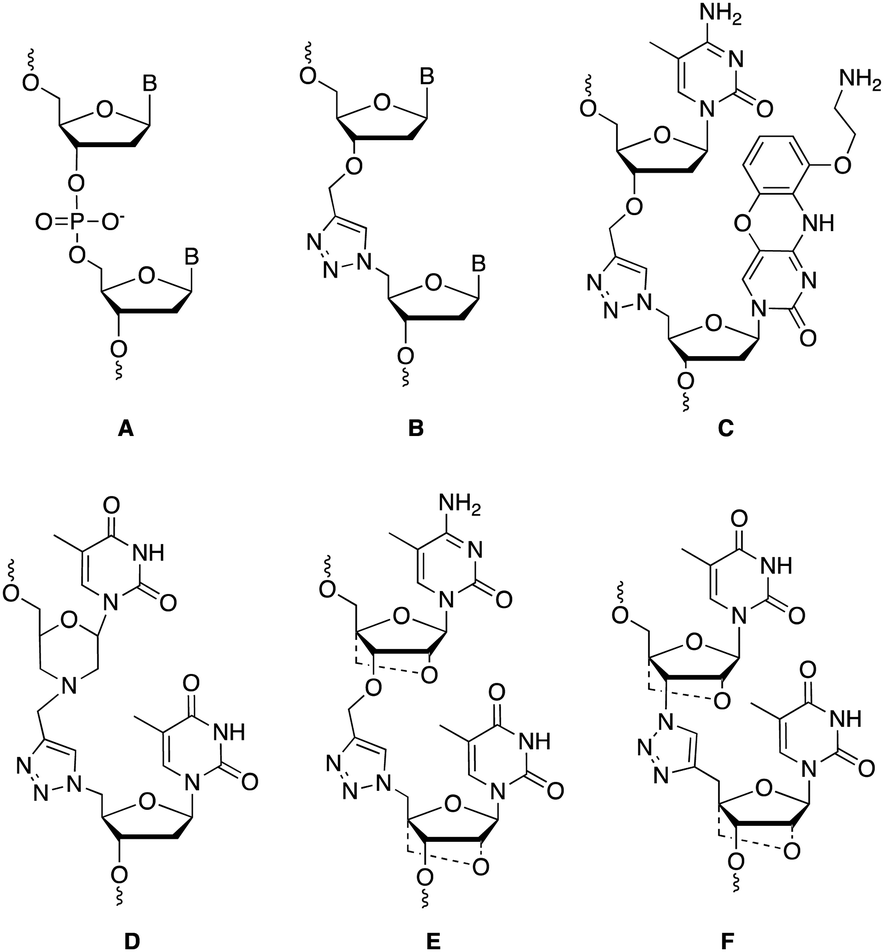 | ||
| Fig. 1 Comparison of the PO backbone (A) and the biocompatible triazole linkage (B). Current approaches to stabilise the linkage include using a G clamp (C) and replacing the ribose with a morpholino (D) or an LNA sugar (E and F), dashed bonds are used to indicate different combinations of LNA on the 5′ or 3′ were investigated. | ||
Results and discussion
Synthesis of triazole modified ONs
We envisaged the development of universal phosphoramidite building blocks that would enable multiple incorporations of the triazole and allow the post-synthetic attachment of various chemical functionalities to the 2′-position of the ribose on the 5′-side of the triazole. The phosphoramidites (Fig. 2) were designed to be compatible with standard solid-phase ON synthesis and contain an amino group that can be functionalised after ON synthesis. A key advantage to this approach is that one phosphoramidite monomer can be used to screen multiple modifications without requiring multistep synthesis of a new phosphoramidite for each modification of interest. In this study, three different moieties were investigated: pyrene (PY) and anthraquinone (AQ) which are known to increase duplex stability by intercalation and/or minor grove binding,30–37 and guanidine (Gu) which stabilises duplexes by masking the charge repulsion between opposite nucleic acid strands, and potentially improves ON cellular uptake.38–40 Although aromatic groups such as pyrene or anthraquinone stabilise unmodified duplexes due to intercalation or groove binding, in this study, we specifically wanted to determine if they could increase duplex stability when placed directly next to an artificial backbone. We also wanted to determine if cationic guanidine stabilises duplexes when placed adjacent to an uncharged artificial backbone.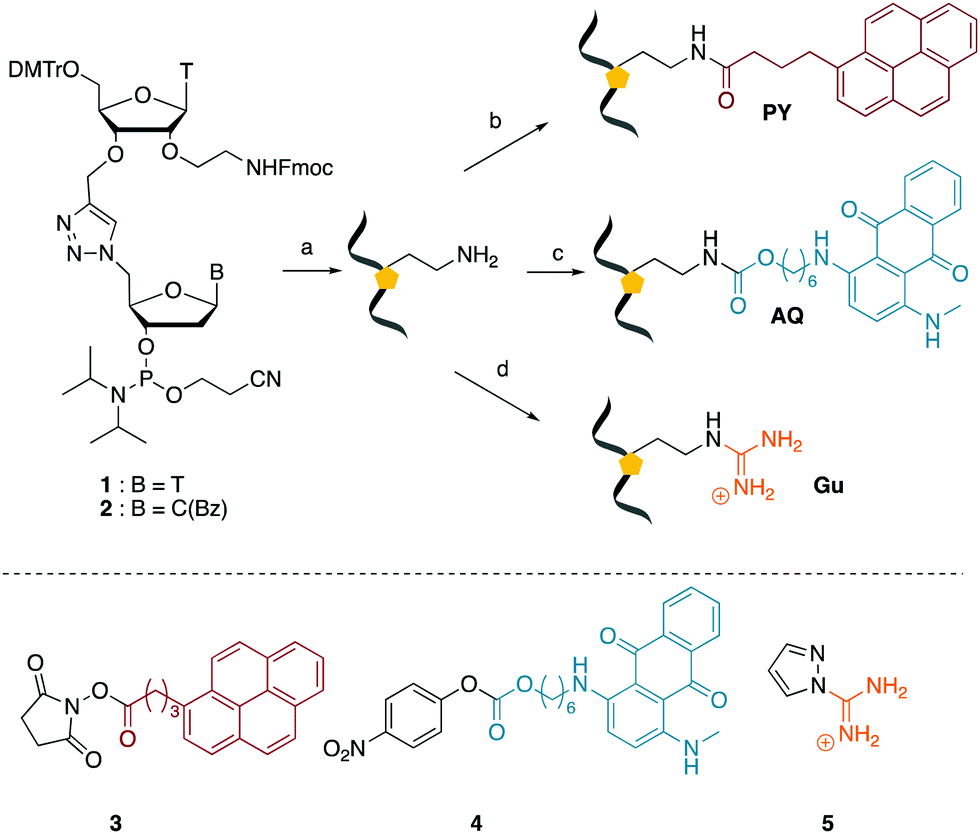 | ||
| Fig. 2 (Top) Overview of ON synthesis strategy via amino-modified ONs. (Bottom) reagents used to functionalise the ONs. Reagents and conditions: (a) solid phase ON synthesis, (b) N-hydroxysuccinimide (NHS) labelling with 3, (c) nitrophenyl active ester (oNp) labelling with 4, (d) guanidine addition with 5. DMT = 4,4′-dimethoxytrityl. | ||
The synthesis of phosphoramidites 1 and 2 started with propargylation of the 2′-hydroxy group in ribonucleoside 641 (Scheme 1). Staudinger reduction of the azide in 7 followed by protection of the amine in 8 using Fmoc-succinimide (Fmoc-OSu) gave nucleoside 9. This was then joined to 5′-azidothymidine 1042 or benzoyl protected 5′-azidodeoxycytidine 1143 using the CuI-catalysed alkyne-azide cycloaddition (CuAAC) reaction44 to give the T–T dinucleoside 12 and the T–C dinucleoside 13 respectively. These were then phosphitylated to give the phosphoramidite building blocks 1 and 2.
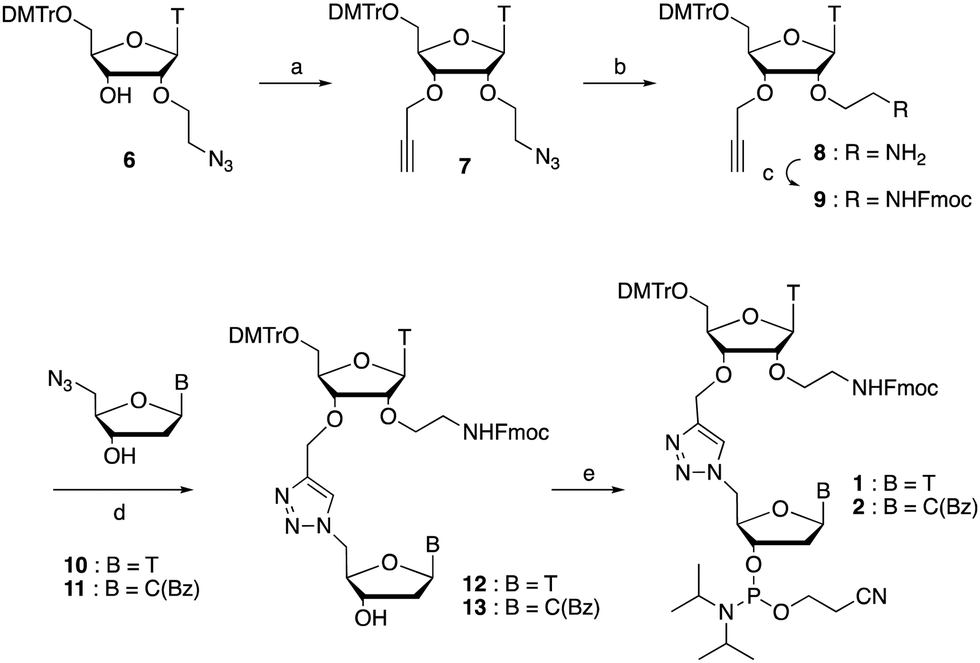 | ||
| Scheme 1 Synthesis of phosphoramidites 1 and 2. (a) NaH, propargyl bromide, THF, reflux, 14 h, 72% (b) Ph3P, H2O, THF, 45 °C, 2 h (c) Fmoc-OSu, CH2Cl2, pyridine, r.t., 2 h, 65% over 2 steps (d) sodium ascorbate, CuSO4, (tris-(benzyltriazolylmethyl)amine ligand (TBTA), DMF, H2O, r.t., 4 h, T = 86%, C(Bz) not fully purified. (e) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, DIPEA, CH2Cl2, r.t., 2.5 h, T = 34%, C(Bz) = 55% over 2 steps. | ||
We then evaluated the labelling strategy shown in Fig. 2. ON1 (Table 1) was synthesised using phosphoramidite 1 and further functionalised. Commercially available 1-pyrenebutyric acid N-hydroxysuccinimide (NHS) ester 3 was used to attach pyrene with a butyryl linker to yield ON2. ON1 was converted to ON3 using the active carbonate derivative of AQ 4 which was prepared from the previously reported alcohol45,46 using 4-nitrophenyl chloroformate. ON1 was guanadinylated using 1H-pyrazole-1-carboxamidine hydrochloride to give ON4. Details for the labelling conditions and synthesis of 4 are given in the supporting information. A second set of ONs with a phosphodiester instead of a triazole linkage (ON5-8, Table 1) was prepared for use as controls in the biophysical studies. ON5 was synthesised using a 2′-aminoethoxy thymidine (AE–T) phosphoramidite41 and labelled with NHS ester 3, active carbonate 4 and carboxamidine 5, as described above, to give ONs 6–8 respectively.
| ON | Sequence 5′ to 3′ | DNA target Tm (ΔTm) °C | RNA target Tm (ΔTm) °C |
|---|---|---|---|
ΔTm = change with respect to the unmodified duplex; X = 2′-aminoethoxy thymidine (AE–T);  = pyrene labelled AE–T; = pyrene labelled AE–T;  anthraquinone labelled AE–T; anthraquinone labelled AE–T;  = guanidinylated AE–T; = guanidinylated AE–T;  = position of a triazole in place of the phosphodiester; = position of a triazole in place of the phosphodiester;  = triazole dimer 1; Melting temperatures were determined at 4 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of DNA target = GCTGCAAGCGTCG; Sequence of RNA target = GCUGCAAGCGUCG. = triazole dimer 1; Melting temperatures were determined at 4 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of DNA target = GCTGCAAGCGTCG; Sequence of RNA target = GCUGCAAGCGUCG. |
|||
| 1 |

|
53.0 (−8.9) | 54.6 (−7.4) |
| 2 |

|
66.0 (+4.1) | 61.3 (−0.7) |
| 3 |

|
66.1 (+4.2) | 64.6 (+2.6) |
| 4 |

|
53.4 (−8.5) | 54.2 (−7.8) |
| 5 |
|
59.0 (−1.9) | 60.5 (−1.5) |
| 6 |

|
68.4 (+6.5) | 64.5 (+2.6) |
| 7 |

|
69.5 (+7.6) | 69.0 (+7.0) |
| 8 |

|
61.5 (−0.4) | 62.8 (+0.8) |
| 9 |

|
61.9 | 62 |
Biophysical evaluation
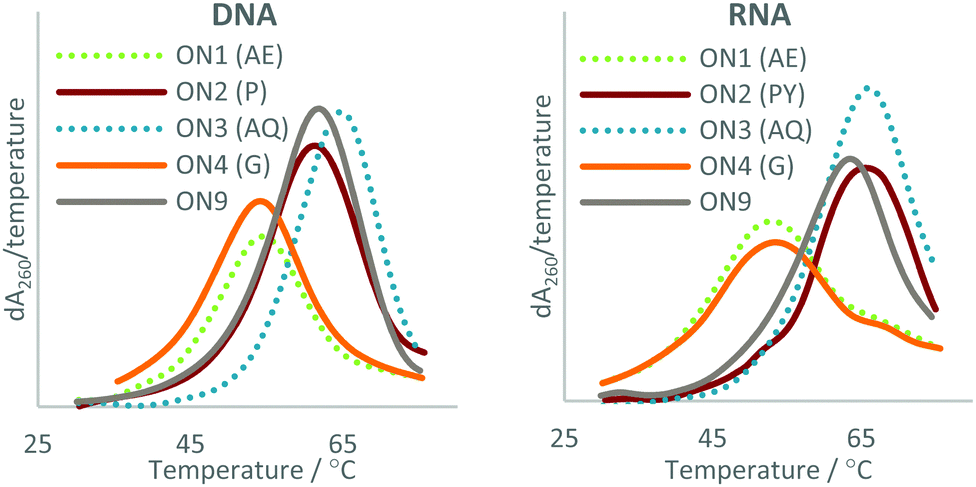 | ||
| Fig. 3 UV melting studies of the triazole-modified ONs against DNA (Left) and RNA (Right). The first derivatives of the melting curves are shown. AE = 2′-aminoethoxy, P = pyrene, AQ = anthraquinone, G = guanidine. For sequences see Table 1. | ||
The data show that this approach can be used to overcome the loss in duplex stability associated with backbone modifications47 such as the charge-neutral triazole. In agreement with previous reports, the triazole is destabilising compared with the phosphodiester linkage,21–23 a ΔTm of −8.9 °C was observed in duplexes with DNA and a ΔTm of −7.4 °C in duplexes with RNA (compare ON1 with ON9). This indicates that the 2′-aminoethoxy (AE) modification alone does not significantly improve the duplex stability of triazole modified ONs. Attachment of PY and AQ resulted in DNA duplexes that were 4.1 and 4.2 °C more stable than the unmodified DNA:DNA duplex (compare ON2:DNA and ON3:DNA with ON9:DNA). PY and AQ also regained the loss in thermal stability of duplexes with a complementary RNA target, albeit to a lesser degree. The addition of PY to the triazole resulted in a duplex with RNA that was 0.7 °C less stable than the unmodified duplex (compare ON9:RNA with ON2:RNA) but 6.7 °C more stable than the triazole modified ON1 (compare ON2:RNA with ON1:RNA). AQ improved the stability of the triazole modified DNA:RNA duplex to a greater degree than PY, with an increase in Tm of 2.6 °C compared to unmodified DNA (compare ON3:RNA with ON9:RNA) and an increase of 10 °C compared to the triazole modified DNA (compare ON3:RNA with ON1:RNA). It is worth noting that the Gu modification had little effect on the stability of duplexes with and without a triazole linkage, and was therefore not considered further. Guanidine is cationic at neutral pH, and stabilises unmodified duplexes by masking charge repulsion between opposite nucleic acid strands. However, in the modified duplexes guanidine is close to an uncharged triazole linkage. It is likely that stabilisation does not occur because there is no ‘anion–anion’ charge repulsion to neutralise at triazole site.
These findings suggest that it is not the addition of the PY and AQ modifications alone, but synergy between the triazole and the PY/AQ modifications that give rise to the observed stabilisation. For example, looking at destabilisation resulting from triazole (ON1:DNA ΔTm = −8.9 °C) and the stabilising effect of the PY without the triazole (ON6:DNA ΔTm = +6.5 °C) it might be expected that a combination of the two modifications would produce a duplex with slightly lower stability than the unmodified control duplex; however, a significant increase in stability is observed (+4.1 °C). The triazole only destabilises a pyrene modified ON by −2.4 °C (compare ON6:DNA with ON2:DNA) which is much lower than the −8.9 °C difference between triazole-modified ON1:DNA and the control ON9:DNA. Similar results were observed for the AQ modification against DNA. This effect was also observed for the modified ONs with RNA but to a lesser degree.
The duplex stabilisation conferred by the triazole-pyrene or triazole-anthraquinone oligonucleotides against DNA (Table 1) is slightly greater than against RNA. However, in these examples the majority of the oligonucleotide backbone is deoxyribose-phosphodiester (i.e., natural DNA), which is relevant to a number of diagnostic applications including real-time PCR. However, the application described below involves cell studies, where the target is RNA, and for which stability to nucleases is essential. For this reason, we used a 2′-OMe RNA-phosphorothioate backbone which is known to favour binding to RNA targets over DNA targets, so in vivo selectivity of the triazole-modified oligonucleotides for RNA over DNA should not be an issue.
Circular dichroism (CD) studies show that triazole combined with PY or AQ has greater effect on the structure of DNA:DNA duplexes than the corresponding DNA:RNA duplexes (Fig. 4) suggesting that the mode of interaction in the duplexes with a DNA target could be different to that for duplexes with an RNA target. This could explain the additional stability observed when duplexed with DNA compared to RNA. When hybridised with complementary RNA, ON1–ON3 show similar CD spectra, and they overlap with the unmodified ON9:RNA spectra almost perfectly (Fig. 4A). These constructs display characteristic A-type signals with strong maxima at 270 nm and minima at 240 nm. In contrast, whilst the CD spectra for ON1-3 retain B-type features (maxima at 280 nm and minima at 250 nm) variations in the shift and peak intensity were observed compared to the unmodified duplex. These observations suggest that the triazole perturbs the B form structure and that the AQ and PY modifications restore the B form (as observed by the slight blue shift in the peak at 280 nm). Further structural studies are required to determine whether this is a result of intercalation or groove binding.
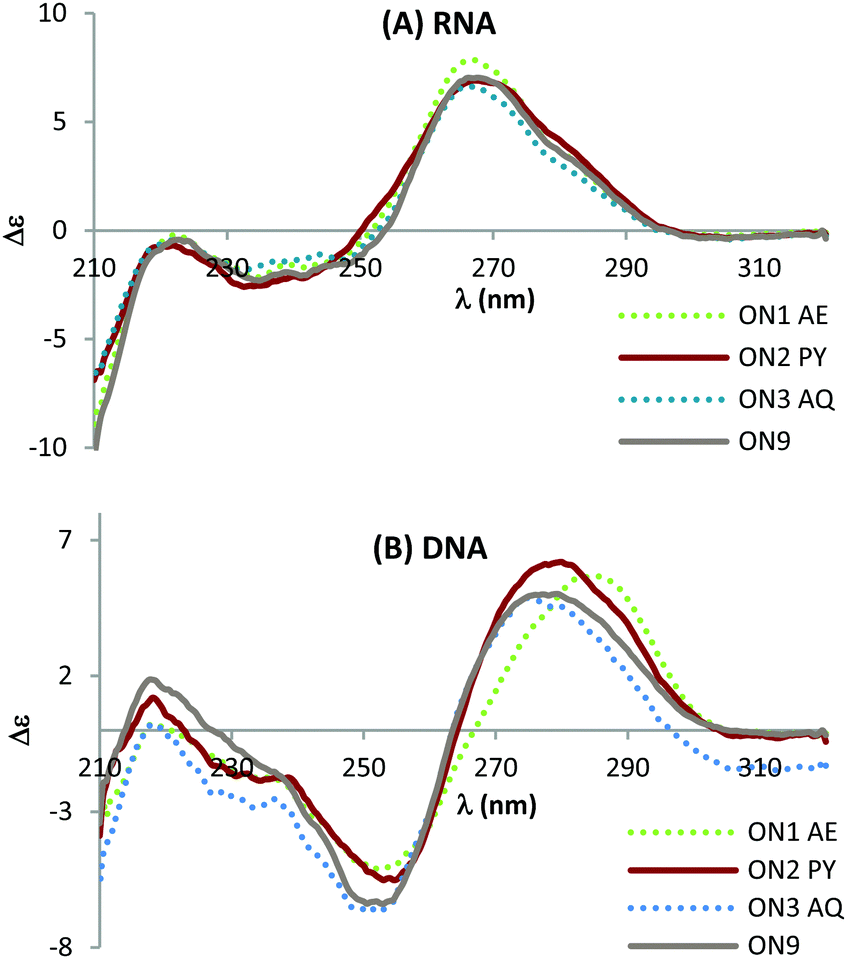 | ||
| Fig. 4 CD spectra of the modified DNA:RNA duplexes (top) and modified DNA:DNA duplexes (bottom). For sequences see Table 1. Melting temperatures were determined at 4 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of DNA target = GCTGCAAGCGTCG; sequence of RNA target = GCUGCAAGCGUCG. | ||
| ON | ON sequence (5′ to 3′) | T m °C |
|---|---|---|
X = AE–T;  = pyrene labelled AE–T; = pyrene labelled AE–T;  anthraquinone labelled AE–T; anthraquinone labelled AE–T;  = position of a triazole in place of the phosphorothioate linkage; = position of a triazole in place of the phosphorothioate linkage;  = triazole dimer 1, = triazole dimer 1, 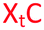 = triazole dimer 2. Melting temperatures were determined at 2 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of RNA target = UGUAACUGAGGUAAGAGG. = triazole dimer 2. Melting temperatures were determined at 2 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of RNA target = UGUAACUGAGGUAAGAGG. |
||
| 10 |
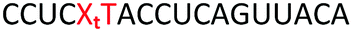
|
65.9 |
| 11 |

|
55.5 |
| 12 |
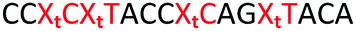
|
49.4 |
| 13 |
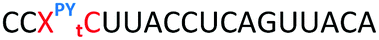
|
71.1 |
| 14 |

|
60.5 |
| 15 |

|
56.7 |
| 16 |

|
74.1 |
| 17 |

|
51.1 |
| 18 |

|
45.1 |
| 19 |

|
70.2 |
The Tms of the ONs with a complementary RNA strand were determined by UV-melting (Table 2). Incorporation of a single triazole linkage (ON10) decreased the thermal stability of the duplex by 4.3 °C, triple addition (ON11) reduced the Tm by 14.7 °C and quadruple addition (ON12) dropped the Tm by 20.8 °C. These results might suggest that the triazole is better tolerated in an A-form PS/2′-OMe backbone than a B-form DNA backbone (compare ON1:RNA ΔTm = −7.4 °C with ON10:RNA ΔTm = −4.3); however, as these data were recorded in different sequences caution must be applied when drawing comparisons. As before, addition of PY and AQ to the triazole modified ONs restored some of the lost stability. ON13 with a single triazole linkage and PY showed a Tm increase of 5.2 °C compared to the triazole alone in ON10 and exceeded the Tm of the PS/2′-OMe control ON19 without a triazole by 0.9 °C. Similarly, addition of AQ to ON10 increased the stability by 8.2 °C (Compare ON10 with ON16), which is 3.9 °C higher than the unmodified control ON19.
The stabilising effect of AQ and PY with triple and quadruple additions of the triazole linkage was not as great as anticipated. Adding PY to ON11 with three triazole linkages only increased the Tm by 5.0 °C (ON14), and the resulting duplex was 9.7 °C less stable than the control (ON19). ON15 with four PY functionalised triazole linkages was only 7.3 °C more stable than ON12 without PY, and 13.5 °C less stable than the control. More surprisingly, addition of three and four anthraquinone moieties next to triazole linkages (ON17 Tm = 51.1 °C and ON18 Tm = 45.1 °C) caused a decrease in duplex stability compared to the triazole containing ON11 (Tm = 55.5 °C) and ON12 (Tm = 49.4 °C).
Biological evaluation of the pyrene triazole combination
Having confirmed that pyrene is more capable than anthraquinone of compensating for the loss of duplex stability incurred with multiple triazoles in PS/2′-OMe ASOs, we evaluated efficacy of the modified ASOs in a commonly used splice-switching reporter cell system.48 This model system is based on a mutation that causes the blood disorder β-thalassemia. The HeLa pLuc705 cells used had been stably transfected with a plasmid that encodes for luciferase interrupted by a mutated human β-globin intron 2 (IVS2-705). This single point mutation causes aberrant splicing, preventing translation of active luciferase. Masking the aberrant splice site with an ASO redirects the splicing machinery and restores luciferase production. ONs used in the Tm studies (Table 2) were designed to be complementary to the splice site, and ON19 is the standard control sequence used with this assay.The splice switching assay confirmed that replacement of the PS backbone with more than one triazole is detrimental to activity and showed that the addition of pyrene to the triazole modified ASO re-establishes splice switching. The addition of PY also improved the response of the ASO with a single triazole relative to the parent ASO (Fig. 5). Here, the cells were treated with ON10-15 complexed with Lipofectamine 2000 in the concentration range 7.8 nM to 180 nM. At the higher concentration a maximal effect is observed for ON10, ON13, ON15 and ON19 (around 40-fold increase in activity). Higher doses of oligonucleotide-lipofectamine complexes caused significant cell toxicity. A clear dose response was observed for all oligonucleotides except for ON12 with four triazoles and no PY, which was inactive. These dose responses indicate that the most to least active oligonucleotides are ON13 (Tm = 71.1 °C) > ON10 (Tm = 65.9 °C) > ON19 (Tm = 70.2 °C) > ON14 (Tm = 60.5 °C) > ON15 (Tm = 56.7 °C) > ON11 (Tm = 55.5 °C) > ON12 (Tm = 49.4 °C) suggesting that duplex stability correlates with splice switching efficacy. It is clear that the addition of pyrene to triazole modified ONs improves their splice switching activity; a higher response is observed when comparing ON13 with ON10 at the lower concentrations, and ON14 and ON15 with PY modifications are significantly more active than their triazole-only equivalents ON11 and ON12.
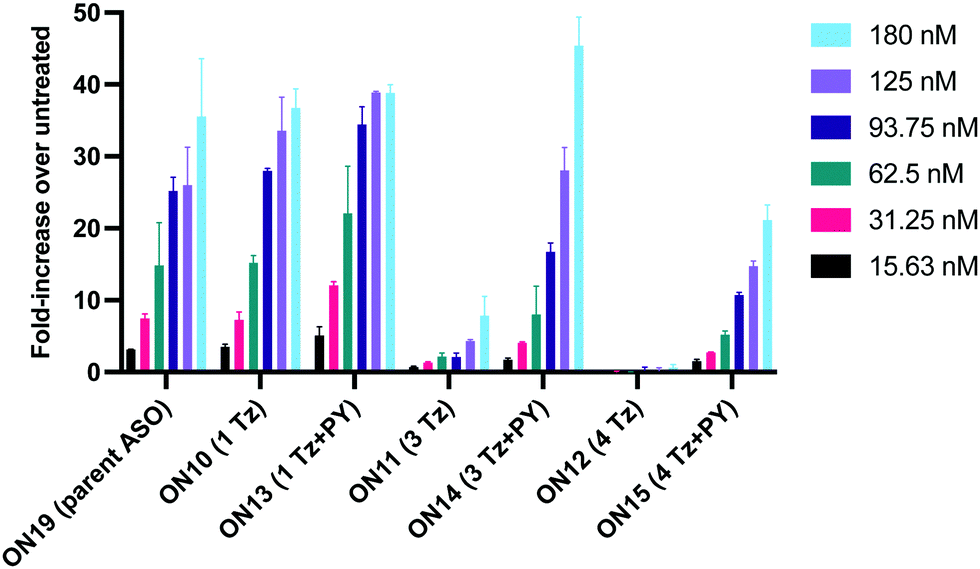 | ||
| Fig. 5 Splice-switching activity of transfected antisense oligonucleotides in the stably transfected HeLa Luc/705 cell line (assay 1) using Lipofectamine 2000. Cells were treated for 24 h. Experiments were replicated with data showing standard deviation of the mean (SD). | ||
In order to substantiate these findings, a second luciferase exon skipping assay was performed, targeting the C to T nucleotide mutation at position 654 in human β-globin intron 2, a mutation that also causes β-thalassemia. In this assay, MCF7 cells are transduced with a pHTBV1 plasmid encoding luciferase, interrupted with IVS2-695, using BacMam one day prior to treatment with the ONs. The sequences of the ONs used and their duplex melting temperatures are shown in Table 3. As before, PY restored the duplex stability lost from the triazole incorporation, although some sequence dependence was observed. Lipofection with ON20-24 in a concentration range of 91 pM to 22.2 nM resulted in a dose dependent response for all ONs tested (Fig. 6, top graph) and toxicity was observed at higher concentrations. Again, the data show that the triazole alone reduces the efficacy of the ASO and that adding a PY restores the activity of the ASO. As before, a correlation between the Tm and activity was observed, with the exception of ON24 (Tm = 70.7 °C) appearing slightly more active that ON20 (Tm = 72.8 °C).
| ON | ON sequence (5′ to 3′) | T m °C |
|---|---|---|
X = AE–T;  = pyrene labelled AE–T; = pyrene labelled AE–T;  = position of a triazole in place of the phosphorothioate; = position of a triazole in place of the phosphorothioate;  = triazole dimer 1. Melting temperatures were determined at 2 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of RNA target = CUGGGUUAAGGUAAUAGC. = triazole dimer 1. Melting temperatures were determined at 2 μM duplex concentration in 10 mM NaH2PO4/Na2HPO4 buffer with 200 mM NaCl at pH 7.2. Sequence of RNA target = CUGGGUUAAGGUAAUAGC. |
||
| 20 |

|
72.8 |
| 21 |

|
60.5 |
| 22 |

|
68.1 |
| 23 |

|
66.1 |
| 24 |

|
70.7 |
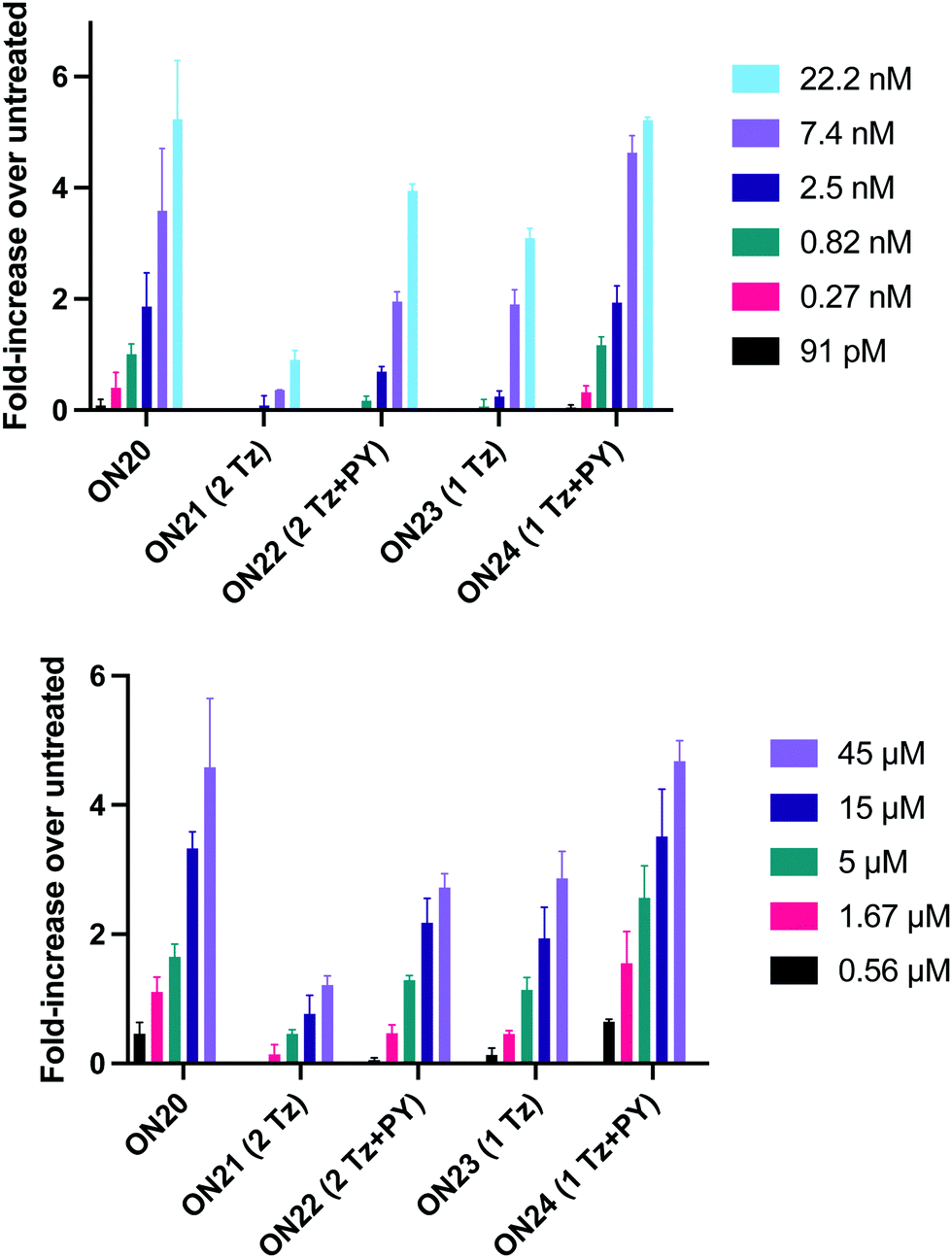 | ||
| Fig. 6 Splice-switching activity of triazole-modified ASOs in MCF7 cells transduced with a pHTBV1 plasmid encoding luciferase interrupted with IVS2-695 (assay 2). (Top) Cells were treated for 24 h with lipofectamine:ASO complexes. (Bottom) Gymnotic delivery experiments. Cells were treated with the ASO for 24 without a transfection agent. Experiments were performed in triplicate with data showing standard deviation (SD). | ||
The effect of modifications on gymnotic (naked) delivery was also determined using this assay (Fig. 6 bottom graph). As with the lipofection experiments PY improved the activity of the triazole modified ONs. Double addition of the triazole without PY (ON21) also had a less detrimental effect on activity when compared to the parent ASO (ON20) than was observed with lipofection. This could be a result of increased lipophilicity aiding endosomal escape or cell uptake. Further investigations are required to clarify this.
Conclusions
We have prepared two novel dinucleotide phosphoramidite monomers containing a triazole internucleoside linkage and a 2′-aminoethoxy modification on the 5′-side of the triazole. The building blocks were readily incorporated into oligonucleotides during solid phase oligonucleotide synthesis and provided a convenient point for post-synthetic functionalisation with electrophilic reagents. The effects on duplex stability of cationic and intercalating agents positioned adjacent to the triazole linkage were then compared. Having identified that addition of the AQ or PY modifications restores the stability lost by inclusion of the triazole, we evaluated these linkages in exon-skipping ASOs using two splice switching assays. As anticipated, the triazole alone resulted in lower activity; however, addition of PY to the ONs fully or partially restored the lost activity and, in some cases, the combination resulted in more potent ASO than the parent ASO with a standard 2′-OMe-phosphorothioate backbone. Taken together, these results indicate that the triazole linkage combined with PY or AQ could be a useful addition to the ASO chemical toolkit and is worth further exploration. Placing the modifications in the gap or the wings of antisense oligonucleotides, is also worth investigating to determine if they interfere with the activity of RNase-H. Use of the modifications in siRNA might also feasible. In general, future research, including optimisation of the linkage between the nucleoside and the PY or AQ, might produce oligonucleotides with increased potency.The approach used here can also potentially be applied to other backbone modifications which may improve an ASO's pharmacokinetic properties but have previously taken a backseat due to duplex stability challenges. It also provides a route for the rapid screening of chemical attachments, beyond the three functionalities presented here, and could be used to evaluate a plethora of moieties for endosomal escape and/or enhanced duplex stability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by UK BBSRC grants BB/J001694/2 (Extending the boundaries of nucleic acid chemistry), BB/R008655/1 (New and versatile chemical approaches for the synthesis of mRNA and tRNA) and BB/S018794/1 (New oligonucleotide analogues for therapeutic applications). The authors would like to thank Dr Samir El-Andaloussi for the HeLa pLuc/705 cell line and Dr Susan Merrihew for valuable discussions. This research utilised equipment funded by the John Fell Oxford University Press Research Fund and an EPSRC Strategic Equipment Grant (EP/T019190/1).References
- C. I.-E. Smith and R. Zain, Therapeutic Oligonucleotides: State of the Art, Annu. Rev. Pharmacol. Toxicol., 2019, 59, 605–630 CrossRef CAS PubMed.
- A. Khvorova and J. K. Watts, The chemical evolution of oligonucleotide therapies of clinical utility, Nat. Biotechnol., 2017, 35, 238–248 CrossRef CAS PubMed.
- S. T. Crooke, X.-H. Liang, B. F. Baker and R. M. Crooke, Antisense technology: A review, J. Biol. Chem., 2021, 296, 100416 CrossRef CAS PubMed.
- S. T. Crooke, Molecular Mechanisms of Antisense Oligonucleotides, Nucleic Acid Ther., 2017, 27, 70–77 CrossRef CAS PubMed.
- X.-H. Liang, H. Sun, J. G. Nichols and S. T. Crooke, RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus, Mol. Ther., 2017, 25, 2075–2092 CrossRef CAS PubMed.
- C. Rinaldi and M. J. A. Wood, Antisense oligonucleotides: the next frontier for treatment of neurological disorders, Nat. Rev. Neurol., 2018, 14, 9–21 CrossRef CAS PubMed.
- Z. Dominski and R. Kole, Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8673–8677 CrossRef CAS PubMed.
- H. Sierakowska, M. J. Sambade, S. Agrawal and R. Kole, Repair of thalassemic human beta-globin mRNA in mammalian cells by antisense oligonucleotides, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12840–12844 CrossRef CAS PubMed.
- A. Aartsma-Rus, et al., Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients, Hum. Mol. Genet., 2003, 12, 907–914 CrossRef CAS PubMed.
- L. K. McKenzie, R. El-Khoury, J. D. Thorpe, M. J. Damha and M. Hollenstein, Recent progress in non-native nucleic acid modifications, Chem. Soc. Rev., 2021, 50, 5126–5164 RSC.
- B. P. Monia, J. F. Johnston, H. Sasmor and L. L. Cummins, Nuclease resistance and antisense activity of modified oligonucleotides targeted to Ha-ras, J. Biol. Chem., 1996, 271, 14533–14540 CrossRef CAS PubMed.
- F. Eckstein, Phosphorothioates, essential components of therapeutic oligonucleotides, Nucleic Acid Ther., 2014, 24, 374–387 CrossRef CAS PubMed.
- L. L. Cummins, et al., Characterization of fully 2'-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity, Nucleic Acids Res., 1995, 23, 2019–2024 CrossRef CAS PubMed.
- P. Martin, A New Access to 2′-O-Alkylated Ribonucleosides and Properties of 2′-O-Alkylated Oligoribonucleotides, Helv. Chim. Acta, 1995, 78, 486–504 CrossRef CAS.
- A. A. Koshkin, et al., LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition, Tetrahedron, 1998, 54, 3607–3630 CrossRef CAS.
- F. Wang, T. Zuroske and J. K. Watts, RNA therapeutics on the rise, Nat. Rev. Drug. Discov., 2020, 19, 441–442 CrossRef CAS PubMed.
- D. Sheehan, et al., Biochemical properties of phosphonoacetate and thiophosphonoacetate oligodeoxyribonucleotides, Nucleic Acids Res., 2003, 31, 4109–4118 CrossRef CAS PubMed.
- B. R. Meade, et al., Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications, Nat. Biotechnol., 2014, 32, 1256–1261 CrossRef CAS PubMed.
- Y. Shoji, S. Akhtar, A. Periasamy, B. Herman and R. L. Juliano, Mechanism of cellular uptake of modified oligodeoxynucleotides containing methylphosphonate linkages, Nucleic Acids Res., 1991, 19, 5543–5550 CrossRef CAS PubMed.
- A. H. El-Sagheer, A. P. Sanzone, R. Gao, A. Tavassoli and T. Brown, Biocompatible artificial DNA linker that is read through by DNA polymerases and is functional in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11338–11343 CrossRef CAS PubMed.
- P. Kumar, A. H. El-Sagheer, L. Truong and T. Brown, Locked nucleic acid (LNA) enhances binding affinity of triazole-linked DNA towards RNA, Chem. Commun., 2017, 53, 8910–8913 RSC.
- Y. R. Baker, et al., Searching for the ideal triazole: Investigating the 1,5-triazole as a charge neutral DNA backbone mimic, Tetrahedron, 2020, 76, 130914 CrossRef CAS.
- A. H. El-Sagheer and T. Brown, Combined nucleobase and backbone modifications enhance DNA duplex stability and preserve biocompatibility, Chem. Sci., 2014, 5, 253–259 RSC.
- M. J. Palframan, R. D. Alharthy, P. K. Powalowska and C. J. Hayes, Synthesis of triazole-linked morpholino oligonucleotides via Cu(I) catalysed cycloaddition, Org. Biomol. Chem., 2016, 14, 3112–3119 RSC.
- P. Kumar, L. Truong, Y. R. Baker, A. H. El-Sagheer and T. Brown, Synthesis, Affinity for Complementary RNA and DNA, and Enzymatic Stability of Triazole-Linked Locked Nucleic Acids (t-LNAs), ACS Omega, 2018, 3, 6976–6987 CrossRef CAS PubMed.
- V. K. Sharma, et al., Synthesis and biological properties of triazole-linked locked nucleic acid, Chem. Commun., 2017, 53, 8906–8909 RSC.
- A. M. Varizhuk, et al., Synthesis of triazole-linked oligonucleotides with high affinity to DNA complements and an analysis of their compatibility with biosystems, J. Org. Chem., 2013, 78, 5964–5969 CrossRef CAS PubMed.
- H. Isobe, T. Fujino, N. Yamazaki, M. Guillot-Nieckowski and E. Nakamura, Triazole-linked analogue of deoxyribonucleic acid ((TL)DNA): design, synthesis, and double-strand formation with natural DNA, Org. Lett., 2008, 10, 3729–3732 CrossRef CAS PubMed.
- R. Lucas, et al., Microwave-assisted synthesis of a triazole-linked 3 '-5 ' dithymidine using click chemistry, Tetrahedron Lett., 2008, 49, 1004–1007 CrossRef CAS.
- K. Yamana, et al., 2′-Pyrene modified oligonucleotide provides a highly sensitive fluorescent probe of RNA, Nucleic Acids Res., 1999, 27, 2387–2392 CrossRef CAS PubMed.
- P. Grünefeld and C. Richert, Synthesis of a 1‘-Aminomethylthymidine and Oligodeoxyribonucleotides with 1‘-Acylamidomethylthymidine Residues, J. Org. Chem., 2004, 69, 7543–7551 CrossRef PubMed.
- M. E. Ostergaard and P. J. Hrdlicka, Pyrene-functionalized oligonucleotides and locked nucleic acids (LNAs): Tools for fundamental research, diagnostics, and nanotechnology, Chem. Soc. Rev., 2011, 40, 5771–5788 RSC.
- M. Nakamura, et al., Pyrene is highly emissive when attached to the RNA duplex but not to the DNA duplex: the structural basis of this difference, Nucleic Acids Res., 2005, 33, 5887–5895 CrossRef CAS PubMed.
- N. Ben Gaied, Z. Zhao, S. R. Gerrard, K. R. Fox and T. Brown, Potent Triple Helix Stabilization by 5 ',3 '-Modified Triplex-Forming Oligonucleotides, ChemBioChem, 2009, 10, 1839–1851 CrossRef CAS PubMed.
- Z. Zhao, G. Peng, J. Michels, K. R. Fox and T. Brown, Synthesis of anthraquinone oligonucleotides for triplex stabilization, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 921–925 CrossRef CAS PubMed.
- M. B. Gholivand, S. Kashanian and H. Peyman, DNA-binding, DNA cleavage and cytotoxicity studies of two anthraquinone derivatives, Spectrochim. Acta, Part A, 2012, 87, 232–240 CrossRef CAS PubMed.
- K. Mori, C. Subasinghe and J. S. Cohen, Oligodeoxynucleotide analogs with 5′-linked anthraquinone, FEBS Lett., 1989, 249, 213–218 CrossRef CAS PubMed.
- G. Deglane, et al., Impact of the guanidinium group on hybridization and cellular uptake of cationic oligonucleotides, ChemBioChem, 2006, 7, 684–692 CrossRef CAS PubMed.
- T. P. Prakash, et al., 2′-O-2-(Guanidinium)ethyl-modified oligonucleotides: Stabilizing effect on duplex and triplex structures, Org. Lett., 2004, 6, 1971–1974 CrossRef CAS PubMed.
- K. Ohara, et al., Amine-guanidine switch: A promising approach to improve DNA binding and antiproliferative activities, J. Med. Chem., 2007, 50, 6465–6475 CrossRef CAS PubMed.
- J. A. Richardson, M. Gerowska, M. Shelbourne, D. French and T. Brown, Six-Colour HyBeacon Probes for Multiplex Genetic Analysis, ChemBioChem, 2010, 11, 2530–2533 CrossRef CAS PubMed.
- W. Bannwarth, Solid-Phase Synthesis of Oligodeoxynucleotides containing phosphoramidate internucleotide linkages and their specific chemical cleavage, Helv. Chim. Acta, 1988, 71, 1517–1527 CrossRef CAS.
- I. Yamamoto, M. Sekine and T. Hata, One-step synthesis os 5'-azido-nucleosides, J. Chem. Soc., Perkin Trans. 1, 1980, 306–310 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
- K. S. Chamberlin, Acid-aided selectivity in bromination of 1-aminoathraquinones, Synth. Commun., 1995, 25, 27–31 CrossRef CAS.
- R. Ohrlein and G. Baisch, in US pat. 2009/0203931A11-26, Ciba Corporation, USA, 2009.
- S. M. Freier and K.-H. Altmann, The ups and downs of nucleic acid duplex stability: Structure-stability studies on chemically-modified DNA:RNA duplexes, Nucleic Acids Res., 1997, 25, 4429–4443 CrossRef CAS PubMed.
- S.-H. Kang, M.-J. Cho and R. Kole, Up-Regulation of Luciferase Gene Expression with Antisense Oligonucleotides: Implications and Applications in Functional Assay Development, Biochemistry, 1998, 37, 6235–6239 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods and supplementary data. See DOI: https://doi.org/10.1039/d2cb00100d |
| ‡ Current address: Evotec, Abingdon, UK. |
| This journal is © The Royal Society of Chemistry 2022 |