
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorine-induced polarity increases inhibitory activity of BPTI towards chymotrypsin†
Jakob
Leppkes
 a,
Nicole
Dimos
b,
Bernhard
Loll
b,
Thomas
Hohmann
a,
Michael
Dyrks
a,
Ariane
Wieseke
a,
Bettina G.
Keller
c and
Beate
Koksch
*a
a,
Nicole
Dimos
b,
Bernhard
Loll
b,
Thomas
Hohmann
a,
Michael
Dyrks
a,
Ariane
Wieseke
a,
Bettina G.
Keller
c and
Beate
Koksch
*a
aDepartment of Biology, Chemistry and Pharmacy, Institute of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 20, 14195, Berlin, Germany. E-mail: beate.koksch@fu-berlin.de
bDepartment of Biology, Chemistry and Pharmacy, Institute of Chemistry and Biochemistry, Structural Biochemistry, Freie Universität Berlin, Takustr. 6, 14195 Berlin, Germany
cDepartment of Biology, Chemistry and Pharmacy, Institute of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 19th May 2022
Abstract
Substituting the P1 position in bovine pancreatic trypsin inhibitor (BPTI) is known to heavily influence its inhibitory activity towards serine proteases. Side-chain fluorinated aliphatic amino acids have been shown to alter numerous properties of peptides and proteins and thus are of interest in the context of BPTI. In our study, we systematically investigated the site-specific incorporation of non-canonical amino acids into BPTI by microwave-assisted solid-phase peptide synthesis (SPPS). Inhibitor activity of the variants was tested towards the serine protease α-chymotrypsin. We observed enhanced inhibition of two fluorinated BPTIs compared to wild type and hydrocarbon variants. To further investigate the complexes, we performed X-ray structure analysis. Our findings underline the power fluorine offers as a tool in protein engineering to beneficially alter the effects on phenomena as protein–protein interactions.
Introduction
In nature fluorine is a rare find in organic molecules.1 Due to the unique properties of fluorine in an organic context, chemists increasingly harness its power in fields like medicinal or agrochemistry. In the past 20 years, a trend was visible culminating in the observation that 23% of all new drugs contain at least one fluorine atom.2 This number more than doubles for agrochemicals: 53% of newly registered compounds contain fluorine.3 In the past decades, the incorporation of fluorine into peptides and proteins has been of great interest.4 Previous work of our group studied the impact of fluorination on peptides and proteins;5 for this purpose different model systems were utilized ranging from small amyloid core sequences6 to helical HIV inhibitors.7Introducing C–F bonds into these biopolymers is accompanied by changes in their intra- and intermolecular interactions. Fluorine is quite unique compared to other halogen atoms, as the C–F bond stability is superior to those of other carbon–halogen bonds and due to its size fluorine is considered bioisosteric to hydrogen. Specific predictions regarding the impact of fluorination on the molecule's properties are difficult, due to the complexity of the underlying physicochemical factors. Fluorination can influence steric properties, the local electrostatics of the peptides and proteins and their hydrophobicity, as well as facilitate interactions with other functional groups through hydrogen or halogen bonding.8,9 These changes can result in varying overall properties such as the secondary structure propensity,10 folding behavior11 and proteolytic stability of peptides12–14 and protein–protein interactions as for instance association kinetics.15
Studies have shown that the degree of fluorination is crucial for all these properties of peptides and proteins. A single fluorine atom is able to drastically alter the properties of the entire molecule.10,13,16,17 For these systematic studies, the fluorinated derivatives of aminobutanoic acid (Abu), monofluoroethylglycine (MfeGly), difluoroethylglycine (DfeGly) and trifluoroethylglycine (TfeGly) have been established. They have proven to be a powerful tool for studying the influence of varying degrees of fluorination on the physico-chemical properties of peptides and proteins, as for example their secondary structure propensities, proteolytic stability and protein–protein interactions.5,15
Synthetic access to side-chain fluorinated amino acids is key for introducing fluorine into peptides and proteins. Methodologies for obtaining fluorinated α-amino acids were recently reviewed by our group.18
In the past, access to MfeGly has been especially limited.19 In consequence there are no structures described in the protein data bank (PDB) containing this non-canonical amino acid and only a limited number of studies including this amino acid have been published.10 We recently reported the gram-scale synthesis of Fmoc-protected MfeGly for the application in solid phase peptide synthesis (SPPS).20 Abu and its fluorinated variants have mainly been employed in model peptides and few works with globular proteins are reported.15,21,22 A reason for this might be the difficulty of site-specific incorporation of these class of amino acids by in vitro methods. The majority of the studies dealt with either a global substitution of aliphatic amino acids with their fluorinated variant,23,24 or the site-specific incorporation of side-chain fluorinated aromatic amino acids.25,26
As a model for globular proteins, BPTI has been and still is of pronounced interest. When it comes to fundamental investigations, it is used as a model for understanding protein folding,27 protein–protein binding,28 and chemical protein synthesis.29 Due to its relatively small size of 58 amino acids, and its chemical and thermal stability, it is an ideal system for protein chemistry and structural biology. This small protein-based inhibitor was one of the first globular proteins to be crystallized and its structure has been solved in detail by X-ray diffraction analysis30 and NMR spectroscopy.31
Position 15 in wild type BPTI is a Lys. Like in many other trypsin-like (Kunitz type) inhibitors, this is conserved or replaced by an arginine residue; another conservation are six cysteine residues, which form three defined disulfide bridges (5–55, 14–38, 30–51), resulting in an overall stabilization of the protein. In a previous work, we showed that replacing Lys15 in the binding loop of BPTI with Abu, DfeGly or TfeGly had a significant impact on the inhibitor activity of the protein towards β-trypsin. The BPTI–trypsin is too strong to be characterized by inhibition assay. Therefore, we seek to reproduce the effect from Ye et al.15 with the BPTI–chymotrypsin complex, which has an inhibitory constant of 110 nM for the wild type system.32
The underlying phenomenon of the restored inhibition has not been elucidated so far. The crystal structure analysis of the enzyme–inhibitor complexes indicated that an interplay between the fluorinated groups and structural water molecules in the binding pocket of trypsin could be involved in restoring inhibitor activity.
In this study, we present the chemical protein synthesis of an expanded scope of P1-substituted BPTI variants, bearing various non-canonical, side-chain fluorinated amino acids. Their inhibitor properties towards α-chymotrypsin were tested, association constants determined and structural investigations of the enzyme–inhibitor complexes were performed by X-ray crystallography.
Results and discussion
Full-length synthesis and refolding of BPTI variants
Our initial synthesis strategy for BPTI included the synthesis and condensation of two fragments, Arg1–Gly37 and Cys38–Ala58, by native chemical ligation. This strategy was initially described by Lu et al.33 and further described by our group in 2015.15In contrast to previous studies, the total chemical synthesis of eight BPTI mutants was achieved by full-length solid-phase peptide synthesis (Fig. 1).15,33 The use of microwave-assisted deprotection and coupling cycles led to a remarkable decrease in reaction time. The previous 4.8 days synthesis was achieved in 11.5 h, with only using 19% of DMF compared to the synthesis strategy before. Full-length synthesis, furthermore, reduced the required reaction and purification steps, since no two fragments had to be synthesized, purified and chemically ligated, thus our new approach led to significantly increase in yields of BPTI variants.15,33 Additionally, microwaved-assisted SPPS allowed the coupling of fluorinated non-canonical amino acids using 1.2 equiv. excess instead of 5 equiv. for canonical amino acids. With respect to the amount of the amino acids employed, this result was found to be highly advantageous, since the fluorinated aliphatic amino acids are not commercially available and require extensive efforts in organic synthesis.20,34,35
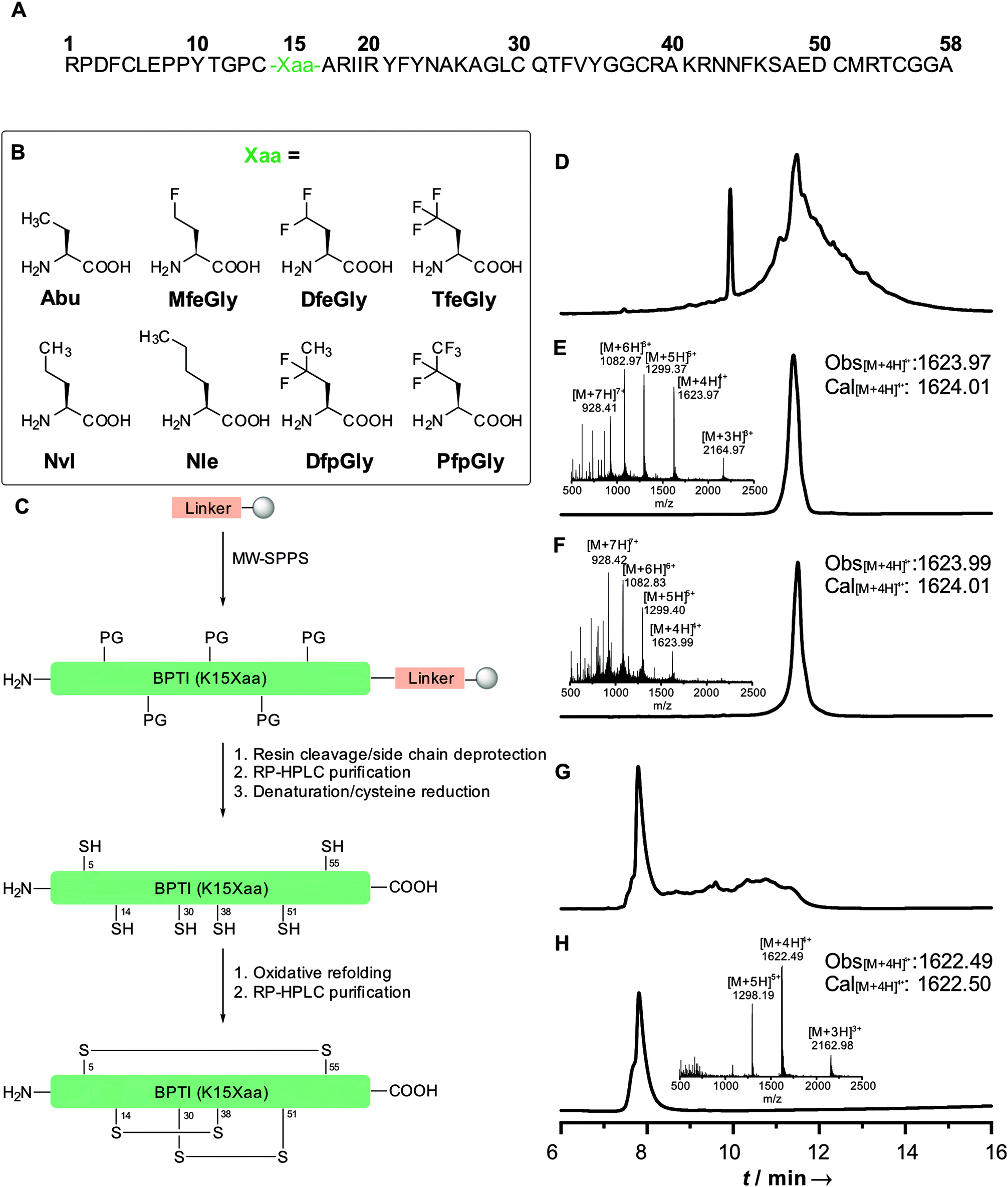 | ||
| Fig. 1 Primary sequence of BPTI (A); non-canonical amino acids incorporated at position 15 (B); full-length synthesis and refolding strategy of BPTI(K15Xaa) (C); HPLC chromatograms and ESI-ToF mass of BPTI K15MfeGly: crude full-length (D); purified full-length (E); chemically denatured and reduced (F); crude refolded (G); purified refolded (H). | ||
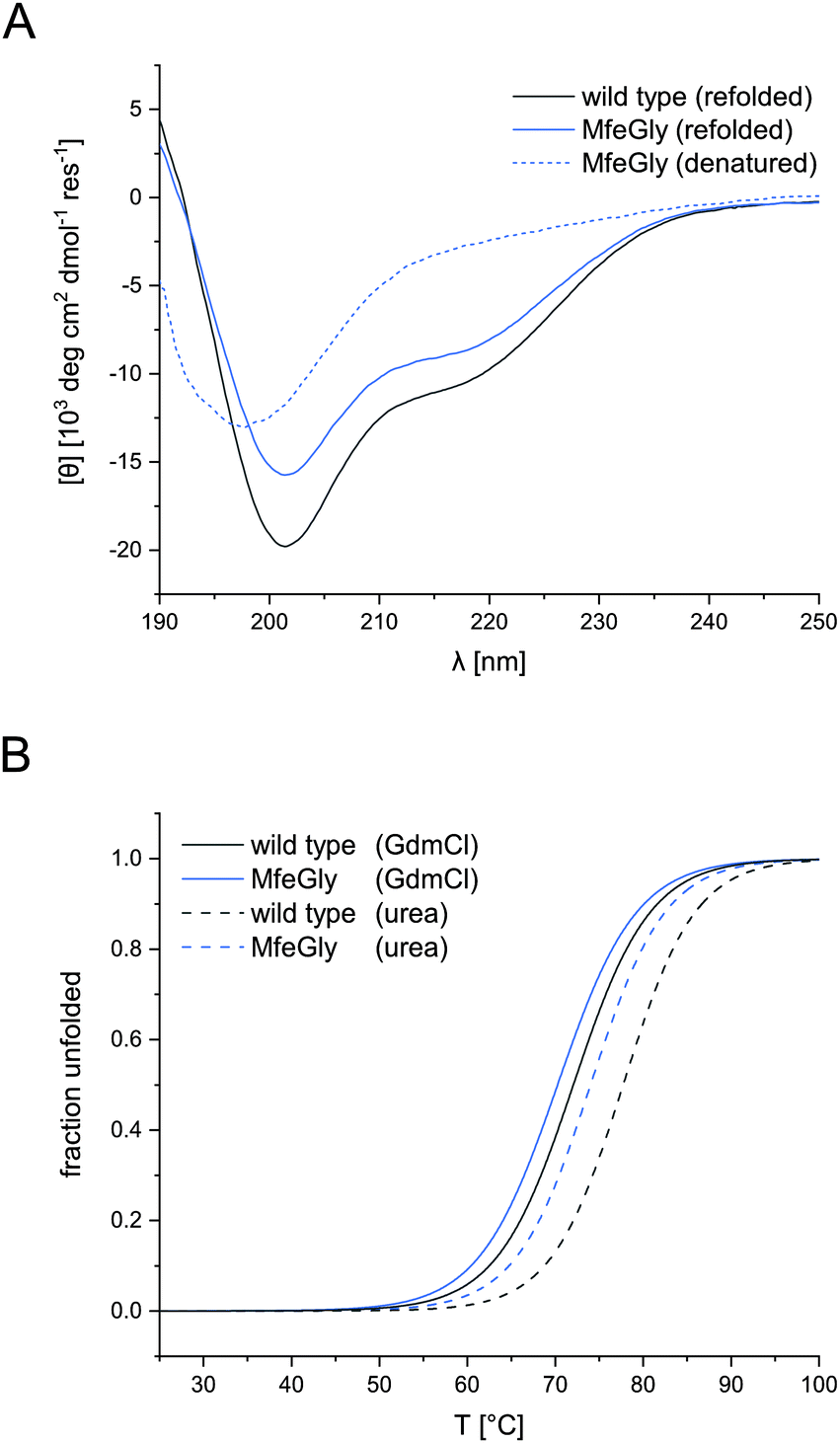 | ||
| Fig. 2 CD spectra of BPTI K15MfeGly and wild type (A) and thermal denaturation of BPTI K15MfeGly and wild type (B). CD spectra of denatured (dashed, A) and refolded (solid, A) BPTI K15MfeGly and wild type. Measurements were performed in 10 mM glycine–HCl buffer, pH 2.0 (denatured) or 10 mM phosphate buffer, pH 7.4 (refolded) with an overall protein concentration of 20 μM. All spectra are normalized and represent the mean of three independent measurements. Thermal denaturation curves of BPTI variants with different denaturants, 8 M urea (dashed, B) and 6 M GdmCl (solid, B). Measurements were performed in 10 mM glycine–HCl, pH 2.0 with an overall protein concentration of 20 μM. All spectra depicted in terms of fraction unfolded and represent the mean of three independent measurements. | ||
Different resins were tested for the full-length synthesis of BPTI(K15Abu). Syntheses on Wang and TGA resin did not afford the 58 amino acid long chain. The full-length sequence was successfully isolated after syntheses on Cl–TCP(Cl), Cl–MPA and rink amide resin. Further approaches with the rink amide resin were not pursued, due to the undesired amide functionality after full-cleavage at the C-terminal end of the chain. In case of the Cl–TCP(Cl) resin, the isolated yield was unsatisfactorily low at 1.5%. Due to the acid lability of this resin, there might be partial cleavage of the amino acid sequence from the resin during the synthesis. This yield could be significantly increased to 12.6% by employing the more acid-stable Cl-MPA resin, which was then established as standard procedure for full-length synthesis of BPTI variants (Table 1).
| Mutant | Yielda [%] | Literature yieldb [%] |
|---|---|---|
| a Yields represent obtained purified lyophilized protein. b Yields refer to synthesis of two peptide fragments, followed by native chemical ligation. c Not described in literature. | ||
| K15Abu | 12.6 | 1.56 |
| K15MfeGly | 10.2 | —c |
| K15DfeGly | 11.5 | 1.0 |
| K15TfeGly | 11.4 | 1.4 |
| K15Nvl | 10.8 | —c |
| K15Nle | 11.2 | —c |
| K15DfpGly | 12.1 | —c |
| K15PfpGly | 11.7 | —c |
Refolding of BPTI mutants was achieved in two steps. First, denaturation of the secondary structure and simultaneous reduction of disulfide bridges with 20 mM TCEP and 6 M guanidinium chloride (GdmCl) led to the unfolding of the protein chain. Secondly, treatment with GSH/GSSG redox buffer including non-denaturing 1 M GdmCl allowed the oxidative refolding to the desired BPTI. Separation from misfolded products was achieved by RP-HPLC.
Secondary structure analysis
CD spectroscopy confirmed the native conformation of the refolded variants (spectra in ESI†). Unfolded BPTI K15MfeGly (Fig. 2A) showed no defined secondary structure. In contrast, refolded BPTI K15MfeGly displayed spectra analogous to the wild type with regard to secondary structure content. In addition to that CD spectroscopy of all BPTI variants showed that overall global conformation is not perturbed (Fig. S2, ESI†).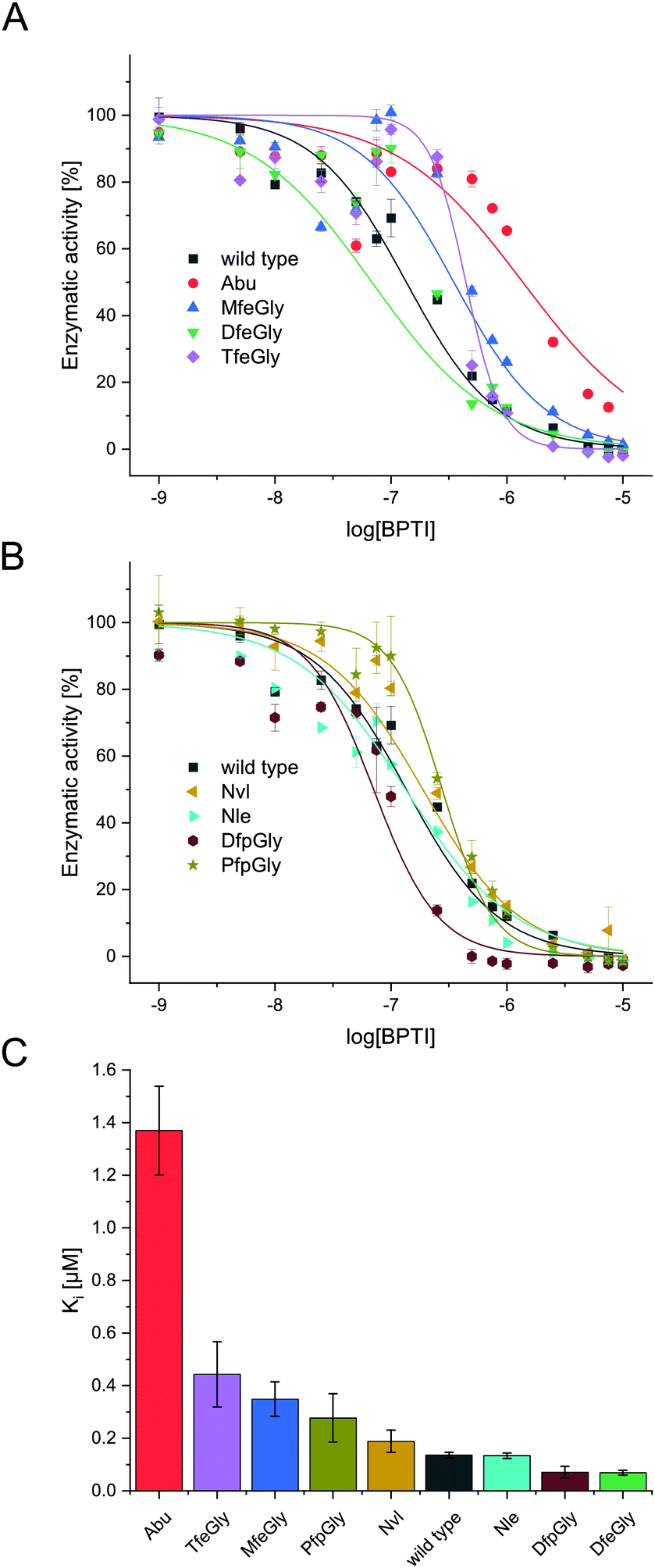 | ||
| Fig. 3 Inhibition of α-chymotrypsin with BPTI variants. Enzymatic activity plotted against log[BPTI] showing sigmoidal curve, wild type, Abu, MfeGly, DfeGly, TfeGly (A), wild type, Nvl, Nle, DfpGly, PfpGly (B). Comparision of Ki [μM] of BPTI variants; Abu, TfeGly, MfeGly, PfpGly, Nvl, wild type, Nle, DfpGly, DfeGly (C). | ||
The high thermal stability of BPTI, as well as its resistance towards denaturants, is well documented.15,36–38 Denaturation of the protein at neutral pH does not occur at temperatures below 100 °C.39 Only at low pH values and in the presence of high concentrations of denaturation agents like GdmCl or urea allows complete unfolding of the protein.
Therefore, denaturation experiments were performed at pH 2.0 with either 6 M GdmCl or 8 M urea as denaturants. Depending on the denaturant we obtained different melting temperatures for BPTI derivatives (Fig. 2B). In the case of K15MfeGly, we measured 74.0 ± 0.1 °C with urea as a denaturant, whereas the TM was 70.2 ± 0.1 °C under GdmCl conditions. Wild type BPTI showed melting temperatures of 78.0 ± 0.2 °C and 71.8 ± 0.2 °C with urea and GdmCl, respectively, showing a difference in 6.2 °C compared to 3.8 °C for K15MfeGly. This is due to the dependence of protein stability on electrostatic interactions. GdmCl's ionic nature has an electrostatic masking effect, making it a better reporter for hydrophobic contributions to stability, while this phenomenon is absent when uncharged urea is used.40
Inhibition of enzymatic activity
In the past decades, numerous studies investigated the inhibitory properties of native BPTI towards a broad range of serine proteases.41 In particular the Otlewski and Smalås groups structurally characterized as well as tested the inhibition activity of an extensive library of P1-substituted BPTI variants in complex with trypsin-like proteases.42–46 They showed, that BPTI has a relatively broad specificity, inhibiting trypsin- as well as chymotrypsin- and elastase like serine proteases. In complex with β-trypsin, the replacement of Lys15(P1) residue with apolar amino acids, like Nvl or Nle, pushes the hydrophobic side chains in the S1 specificity pocket, which hosts Asp189, leading to a six to nine orders of magnitude drop in inhibitory activity. Whereas in case of α-chymotrypsin, the affinity of wild type BPTI is quite similar to that of semisynthetic BPTI bearing Nvl or Nle instead of Lys. Our studies focus on proteases from the trypsin family, like α-chymotrypsin and β-trypsin. Ye et al. showed fluorination of the P1 position in BPTI can lead to increased inhibition as compared to the hydrocarbon species Abu. We extended this model to a wider range of side-chain fluorinated P1-modified inhibitors and also tested their activity towards α-chymotrypsin (Fig. 3).In natural substrates α-chymotrypsin shows specificity in cleaving at bulky, hydrophobic amino acids like Trp, Tyr, Phe, and Leu in the P1 position.47 This is accounted by the hydrophobic pocket formed by Ser189, Gly216 and Gly226, which is spacious enough for bulky residues. Whereas in trypsin a negatively charged active site is comprised of Asp189, Gly216 and Gly226, explaining its specificity towards cleavage at arginine and lysine at the P1 position. It is important to note though, that these small structural changes, a difference of a Ser and an Asp at position 189, do not alone determine the specificity of serine proteases, as studies have shown that transposition of these residues does not shift S1 specificity in serine proteases.47
In terms of inhibitor activity towards α-chymotrypsin, we observed significant differences among the non-natural variants. All showed sub micromolar activity except K15Abu. In our attempt of explaining the differences in inhibition, we focused on two important properties of the side-chains of fluorinated amino acids: their varying hydrophobicity and the different sterical demand expressed in their vdW volume of the amino acid side-chains (Fig. 4). For the hydrocarbon derivatives, we can see the trend of increasing vdW volume and hydrophobicity (Fig. 6) from Abu to Nvl to Nle is also reflected in their Ki decreasing from 1.37 ± 0.17 μM over 0.18 ± 0.04 μM to 0.13 ± 0.01 μM respectively (Fig. 3C). When looking at Ki for the fluorinated variants of BPTI, we cannot transfer the trends in hydrophobicity and vdW volume as easily to inhibition like in their non-fluorinated analogues.
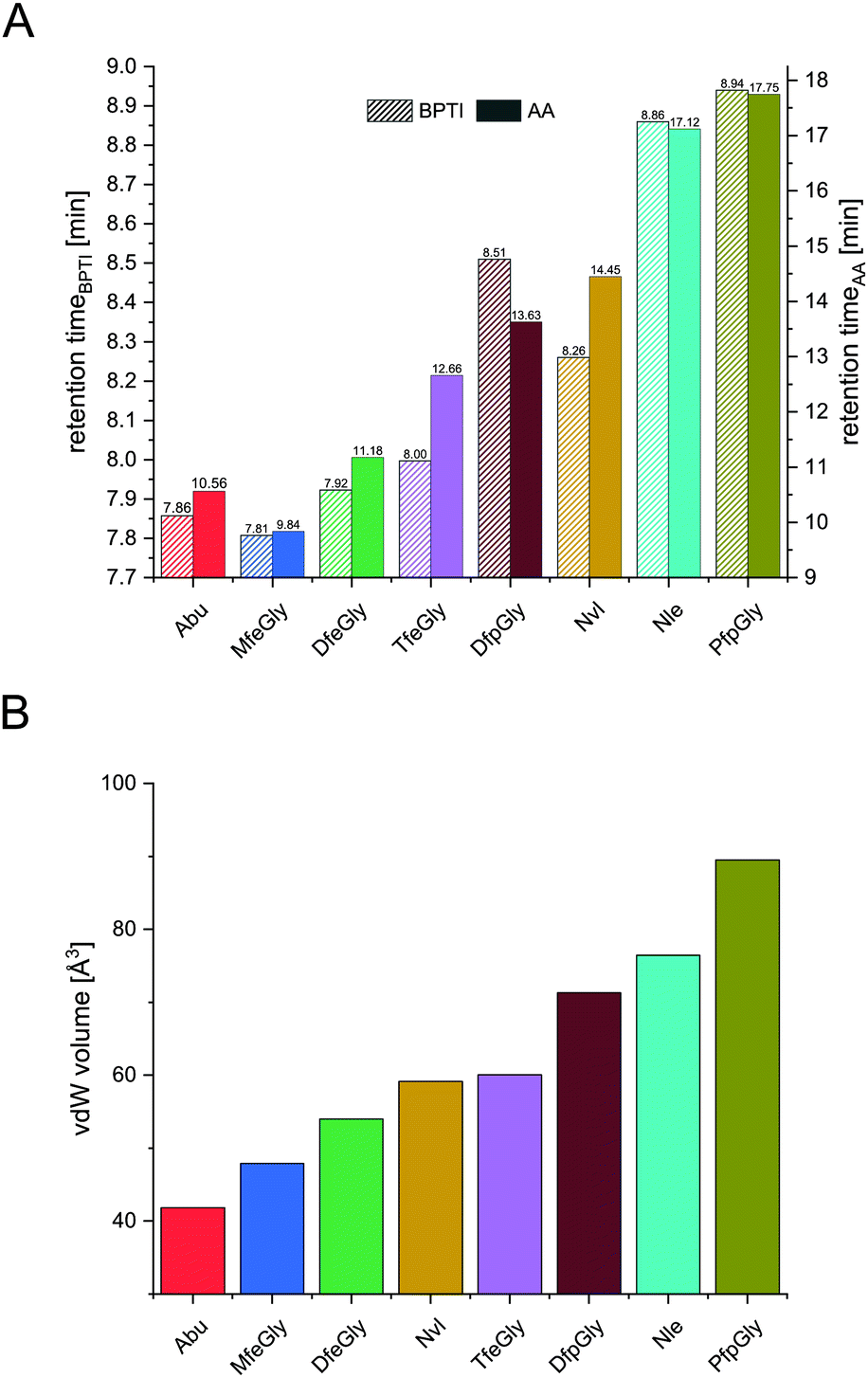 | ||
| Fig. 4 Retention times of BPTI variants (A, left y-axis) and retention times of Fmoc amino acids (A, right y-axis), and their vdW volume (B). | ||
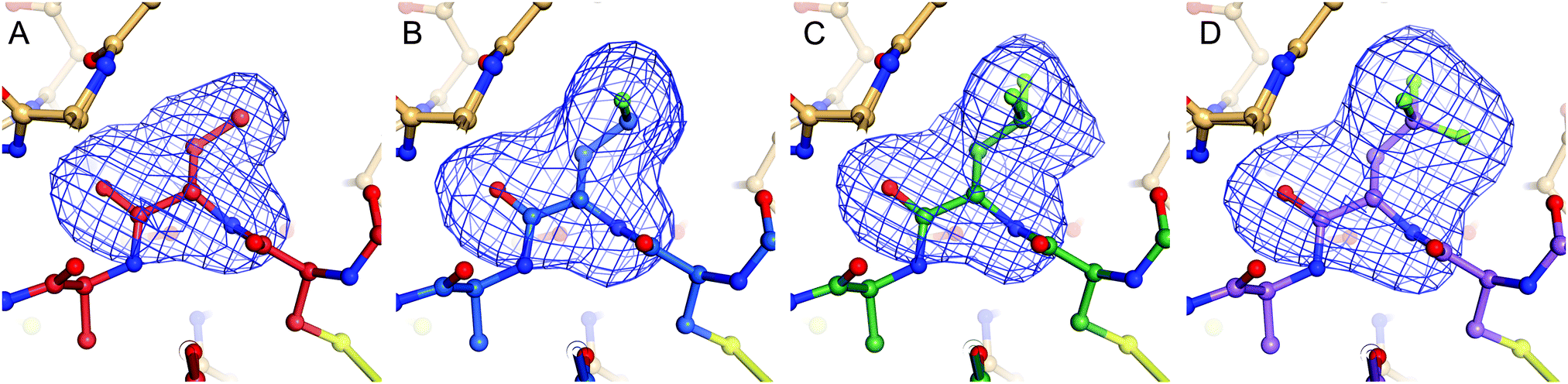 | ||
| Fig. 5 Sideview of the P1 residue of BPTI that has been omitted for calculation of a simulated annealing omit map contoured at 3.0 σ shown as blue mesh; K15Abu (A), K15MfeGly (B), K15DfeGly (C), K15TfeGly (D). | ||
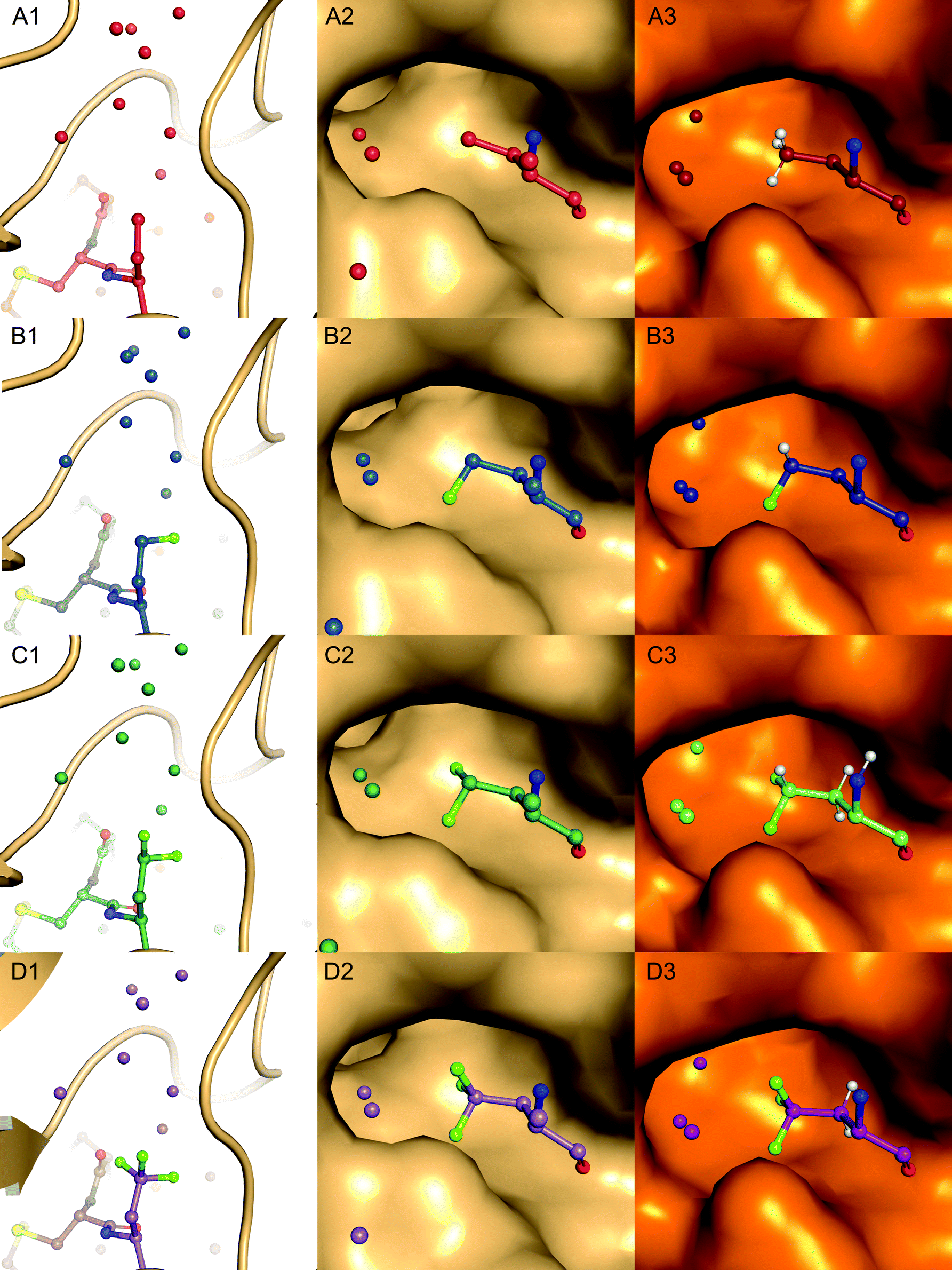 | ||
| Fig. 6 Pocket filling residues Abu (red, A1–3), MfeGly (blue, B1–3), DfeGly (green, C1–3) and TfeGly (purple, D1–3) at P1 position of BPTI, represented in balls and sticks, in the S1 specificity pocket of α-chymotrypsin (lightorange, A2–D2) and β-trypsin (orange, A3–D3), shown as surface. Water molecules are depicted as spheres coloured according to the BPTI residue at position 15. Only water molecules with a distance cut-off of 5.0 Å are shown. | ||
Firstly, when comparing fluorinated derivatives K15MfeGly (–CH2–CH2F), K15DfeGly (–CH2–CHF2) and K15TfeGly (–CH2–CF3) to K15Abu (–CH2–CH3), we see in all cases lower Ki than in the non-fluorinated variant K15Abu, thus fluorination of BPTI is showing enhanced inhibitor activity towards the protease. However, increasing degree of fluorination does not automatically result in improved inhibition. K15TfeGly shows a slightly higher Ki of 0.44 ± 0.12 μM than K15MfeGly with 0.34 ± 0.07 μM. CHF2-bearing K15DfeGly shows with Ki = 68 ± 9 nM the highest inhibitor activity of all variants we investigated. K15DfpGly shows a similar Ki value of 70 ± 22 nM compared to K15DfeGly, whereas bulky K15PfpGly has a Ki of 277 ± 42 nM. Under these circumstances explaining the inhibition simply with steric and hydrophobic properties of the side chains at P1 position is not possible, because no correlation could be observed. Therefore, we argue that varying inhibitor properties might be due to fluorine-induced differences in the polarity of the side chain. Vila Verde and coworkers approached this issue in a theoretical study highlighting the differences in physico-chemical properties of the side chain.48 They calculated the electrostatic potential of various fluorinated amino acids, containing MfeGly, DfeGly, DfpGly and TfeGly and could show that MfeGly, DfeGly as well as DfpGly posses a much higher polarity in the side chain than TfeGly. When arguing with polarity, we are able to explain the differences in the Ki of DfeGly and TfeGly as well as the trend, in which the higher fluorinated less polar PfpGly (–CH2–CF2–CF3) shows a higher Ki than DfpGly (–CH2–CF2–CH3). Overall, the results show that the presence of a CXF2-functionality (X = H, CH3) in the γ-position of the amino acid side chain at the P1 residue of the inhibitor seems to favorably influence its inhibitor activity towards α-chymotrypsin.
If we compare our data to association constants for K15Phe, we can see, that DfpGly and DfeGly show even better inhibitory properties towards chymotrypsin, then the bulky aromatic side-chain of phenylalanine (PDB: 1P2Q).46
We can see that a fluorine-induced polarity in the side-chains of the amino acids at the P1 position of BPTI increases the inhibitory activity towards chymotrypsin.
High resolution crystal structures of chymotrypsin–BPTI complexes
Complexes between BPTI K15 variants and the serine protease α-chymotrypsin were crystallized and the structures solved. We were able to obtain structures for all four complexes at high resolution, ranging from 1.83 to 1.99 Å (Table S13, ESI†). The asymmetric unit of all complexes is composed by two chymotrypsin–BPTI variant complexes. The electron densities are of excellent quality, which allowed unambiguous modelling of the polypeptide chains.The electron densities obtained, allowed to explicitly determine the orientation of the fluorinated amino acid side chains in the P1 position of the inhibitor (Fig. 5).
BPTI K15MfeGly in complex with trypsin and chymotrypsin are the first crystal structures reported in the PDB with MfeGly incorporated in their protein sequence.
As displayed in Fig. 6, we observe that all residues at position 15 are pointing into the specificity pocket of chymotrypsin, blocking the active site of the protease. This explains the inhibitory properties of the BPTI variants towards chymotrypsin. We were not able to determine significant structural differences between the BPTI variants, which can explain the differences in Ki values.
Experimental section
General methods
HRMS was measured on an Agilent 6230 ESI-TOF LC/MS spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA). Data analysis was carried out using MassHunter Workstation Software VersionB.04.00 (Agilent Technologies Inc., Santa Clara, CA, USA) and MestreNova Version 14.1.0 (Mestrelab Research S. L. Santiago de Compostela, Spain). Fmoc-L-amino acids were purchased at ORPEGEN Peptide Chemicals GmbH (Heidelberg, Germany) and Carbolution Chemicals GmbH (St. Ingbert, Germany). Fluorinated amino acids were synthesized according to literature.20,49Peptide synthesis
All proteins were synthesized by Fmoc based SPPS on a Liberty Blue automated microwave-assisted peptide synthesizer (CEM Corporation, Matthews, NC, USA) in a 0.1 mmol scale on a Cl–MPA ProTide LL resin (0.22 mmol g−1 resin substitution). Resin loading was carried out according to standard protocol with 1.0 M DIEA and 0.125 M KI in DMF. Fmoc-deprotection was performed with 10% (w/v) piperazine in NMP/ethanol (9/1, v/v) containing 0.1 M HOBt. Standard couplings conditions were 5-fold excess of 0.2 M Fmoc-AA with 1.0 M DIC and 1.0 M Oxyma containing 0.1 M DIEA in DMF. First 19 couplings were performed as single couplings. Starting with AA 20 and for all arginine residues, double couplings were employed. A special coupling cycle for fluorinated AA was used, reducing AA equivalents to 1.25 and elongating reaction time to 10 min. Resin cleavage and side-chain deprotection was accomplished by treatment with TFA/water/phenol/thioanisole/EDT (82.5/5/5/5/2.5, v/v) for 4 h at room temperature. Resin was washed thrice with TFA and CH2Cl2. Excess of solvent was removed under reduced pressure. Crude protein was precipitated with ice-cold diethyl ether and centrifuged. Precipitate was triturated twice more with ice cold diethyl ether and centrifuged. Supernatant was discarded, protein was dissolved in water and lyophilized overnight. Purification was achieved by using a reversed-phase HPLC LaPrep∑ low-pressure HPLC system (VWR, Darmstadt, Germany) equipped with a Kinetex RP-C18 end-capped LC column (5 μm, 100 Å, 250 × 21.2 mm, Phenomenex, Torrance, CA, USA) and a SecurityGuard PREP Cartridge Holder (21.2 mm, Phenomenex, Torrance, CA, USA) as precolumn. Both eluents, water (solvent A) and acetonitrile (solvent B), contained 0.1% (v/v) TFA. HPLC runs were performed over 20 min. After a 5 min hold at 10% B, a linear gradient ramped to 80% B in 15 min. The flow rate was kept constant for 5 min at 10 mL min−1 and linearly increased to 20 mL min−1 over 15 min. Absorbance was monitored at 220 nm. Purity of BPTI was analyzed by analytical RP-HPLC and ESI-MS (SI).Protein refolding
The purified protein sample was dissolved in deionized water containing 6 M GdmCl, 0.1 M Na2HPO4, 20 mM TCEP at pH 7.0, to obtain a concentration of 1.5 mM. The mixture was shaken overnight at 25 °C. Salts were removed with a PD-10 desalting column (Sephadex G-25 M, GE Healthcare). The obtained product was lyophilized and subsequently dissolved in 10 mM HCl and slowly added to degassed buffer containing 0.1 M Tris, 0.2 M KCl, 1 mM EDTA, 1 M GdmCl, 150 μM GSSG and 300 μM GSH at pH 8.7 with a final protein concentration of 30 μM. The refolding reaction was carried out at 37 °C for 24 h. The sample was concentrated with Amicon® Ultra-15 3 kDa Centrifugal Filter Unit (Merck, Darmstadt, Germany), followed by lyophilization. Refolded protein was further purified by RP-HPLC with a linear gradient starting with 10% to 80% B over 18 min at a flowrate of 15 mL min−1. Purity of fractions was analyzed by analytical RP-HPLC and ESI-MS (SI).Circular dichroism (CD) spectroscopy
CD spectrum measurements were performed on a Jasco J-810 spectropolarimeter (JASCO Deutschland GmbH, Pfungstadt, Germany) with a HAAKE WKL recirculation chiller (Karlsruhe, Germany). The temperature was set to a constant temperature of 25 °C with a Jasco PTC-423S Peltier-type thermocouple (JASCO Deutschland GmbH, Pfungstadt, Germany). Measurements were performed in the far-UV range (190–240 nm) using a 1 mm Quartz Suprasil cuvette (Hellma, Müllheim, Germany). Wavelength scans were performed from 250 to 190 nm. The protein concentration was adjusted to 20 μM in 10 mM phosphate buffer at pH 7.4 for refolded BPTI variants and 10 mM glycine–HCl buffer at pH 2.0. For denaturation experiments, measurements at 222 nm in a temperature range from 25 to 100 °C were performed with a protein concentration of 20 μM in 10 mM glycine–HCl buffer with either 6 M GdmCl or 8 M urea. The heating rate was set to 1 °C min−1. All measurements were performed in triplicate and averaged.Inhibitory activity assay
For inhibitory activity assays increasing concentrations of BPTI variants (0, 1, 5, 10, 25, 50, 75, 100, 250, 500, 750 nM, 1, 2.5, 5, 7.5, 10 μM) were incubated with 20 μL α-chymotrypsin (100 nM, Sigma-Aldrich) in 80 mM Tris–HCl, 20 mM CaCl2, pH 7.8 in a 96-well plate for 1 h. 20 μL Nα-benzoyl-L-tyrosine ethylester (BTEE, Sigma-Aldrich) (1 mM) were added to 180 μL preincubated enzyme/inhibitor mixture (all concentrations refer to measuring conditions). Hydrolysis of BTEE was monitored by measuring absorbance at 256 nm in an Infinite M200 microplate reader (Tecan Group AG, Männedorf, Switzerland) for 30 min at 25 °C. Initial velocities were determined by plotting absorbance against reaction time. IC50 values were determined by plotting residual enzyme activity against logarithmic inhibitor concentration using OriginPro 2020b (OriginLab Corporation, Northampton, MA, USA). Ki was calculated according to Cheng and Prusoff.50,51Protein crystallography
For complex formation of trypsin and BPTI variants, 60 μL of 2.0 mM BPTI in 25 mM Tris–HCl, 10 mM CaCl2, pH 7.4 were added to 50 μL of bovine trypsin in 25 mM Tris–HCl, 10 mM CaCl2, pH 7.4. Solution was incubated for 12 h at 4 °C. Chymotrypsin–BPTI complexes were purified by dialysis. Initial crystals of Chymotrypsin–BPTI variants were obtained by the sitting-drop vapor-diffusion method at 18 °C with a reservoir solution containing 1.8 M ammonium sulfate and 0.1 M MES/NaOH (pH 6.5). Inter-grown crystals were used to prepare a seed stock. With a cat whisker, seeds were transferred to a freshly prepared crystallization drop for each Chymotrypsin–BPTI variant. For cryo-protection, crystals were soaked in 30% glycerol in addition to the reservoir solution and flash frozen in liquid nitrogen. Diffraction data were collected at Berlin BESSY II at beamline 14-2. Diffraction data were processed with the XDS (Table S13, ESI†).52 The structure of the Chymotrypsin–BPTI–TfeGly was solved by molecular replacement with the coordinates of Chymotrypsin–BPTI (PDB: 1T8N),45 downloaded from the PDB–REDO53 server as search models using PHASER.54 All remaining structures were determined by isomorphous replacement. The structures were refined by maximum-likelihood restrained refinement and TLS refinement55 using PHENIX followed by iterative, manual model building cycles with COOT.56 Model quality was evaluated with MolProbity.57 Figures were prepared using PyMOL (Schroedinger Inc). The atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under the accession code 7QIQ (Chymotrypsin–BPTI–Abu), 7QIR (Chymotrypsin–BPTI–MfeGly), 7QIS (Chymotrypsin–BPTI–DfeGly), and 7QIT (Chymotrypsin–BPTI–TfeGly). Diffraction images have been deposited at proteindiffraction.org: XXXX (Chymotrypsin–BPTI–Abu), XXXX (Chymotrypsin–BPTI–MfeGly), XXXX (Chymotrypsin–BPTI–DfeGly), and XXXX (Chymotrypsin–BPTI–TfeGly). [THESE CODES WILL BE AVAILABLE, ONCE PDB RELEASES THE STRUCTURES.]Conclusions
We were able to establish a full-length synthesis of P1-substituted BPTI variants in high yields, allowing an in-depth analysis of their structural characteristics and inhibition properties towards α-chymotrypsin. The systematic analysis of BPTI derivatives activity towards α-chymotrypsin revealed useful insights on fluorine's impact on protein–protein interactions. We observed enhanced inhibition towards chymotrypsin by the two fluorinated BPTI variants bearing a CXF2-functionality (X = H, CH3) compared to wild-type BPTI.Those fluorinated amino acid side chains can be harboured by the natural protease chymotrypsin in its S1 binding pocket better than the natural canonical amino acid Phe, for which this binding pocket has a pronounced specificity.
We argue that fluorine induces polarity in otherwise hydrophobic aliphatic side chains of amino acids. Obviously, fluorine not being used by nature as building block in amino acid biosynthesis, offers the potential of creating amino acid functionalities that are compatible with natural amino acid properties and, thus, fluorinated amino acids can be accepted by natural protein binding pockets. Using these functions skillfully enables the establishment of fluorinated amino acids as bioorthogonal tools in protein engineering and drug development.
Author contributions
J. L. designed and performed syntheses of BPTI variants, synthesized fluorinated amino acids, performed inhibition and CD measurements analyzed the data, prepared Figures and wrote the manuscript and the ESI.† N. D. performed crystallization experiments and performed structural refinement. B. L. performed X-ray diffraction measurements and performed structural refinement. T. H. provided fluorinated amino acids, DfpGly and PfpGly, and measured hydrophobicity of Fmoc-protected amino acids. M. D. and A.W. assisted in the protein synthesis and the refolding process. B. G. K. helped writing the manuscript and evaluating inhibition data. B. K. designed and supervised the entire project and the writing of the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors gratefully thank Dr Allison A. Berger for proofreading the manuscript. This project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 387284271 – SFB 1349. We would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. We accessed beamlines of the BESSY II (Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung II) storage ring (Berlin, Germany) via the Joint Berlin MX-Laboratory sponsored by the Helmholtz Zentrum Berlin für Materialien und Energie, the Freie Universität Berlin, the Humboldt-Universität zu Berlin, the Max-Delbrück-Centrum, the Leibniz-Institut für Molekulare Pharmakologie and Charité – Universitätsmedizin Berlin. We are grateful to Elvenstar's Pukipon for the kind donation of cat whiskers.References
- D. O’Hagan and D. B. Harper, J. Fluorine Chem., 1999, 100, 127–133 CrossRef.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- M. Salwiczek, E. K. Nyakatura, U. I.-M. Gerling, S. Ye and B. Koksch, Chem. Soc. Rev., 2012, 41, 2135–2171 RSC.
- A. A. Berger, J.-S. Völler, N. Budisa and B. Koksch, Acc. Chem. Res., 2017, 50, 2093–2103 CrossRef CAS PubMed.
- S. Chowdhary, J. Moschner, D. J. Mikolajczak, M. Becker, A. F. Thünemann, C. Kästner, D. Klemczak, A.-K. Stegemann, C. Böttcher, P. Metrangolo, R. R. Netz and B. Koksch, ChemBioChem, 2020, 21, 3544–3554 CrossRef CAS PubMed.
- S. Huhmann, E. K. Nyakatura, A. Rohrhofer, J. Moschner, B. Schmidt, J. Eichler, C. Roth and B. Koksch, ChemBioChem, 2021, 3443–3451 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- N. K. Shinada, A. G. de Brevern and P. Schmidtke, J. Med. Chem., 2019, 62, 9341–9356 CrossRef CAS PubMed.
- U. I.-M. Gerling, M. Salwiczek, C. D. Cadicamo, H. Erdbrink, C. Czekelius, S. L. Grage, P. Wadhwani, A. S. Ulrich, M. Behrends, G. Haufe and B. Koksch, Chem. Sci., 2014, 5, 819–830 RSC.
- C. Jäckel, M. Salwiczek and B. Koksch, Angew. Chem., Int. Ed., 2006, 45, 4198–4203 CrossRef PubMed.
- K. M. Sicinski, V. Montanari, V. S. Raman, J. R. Doyle, B. N. Harwood, Y. C. Song, M. P. Fagan, M. Rios, D. R. Haines, A. S. Kopin, M. Beinborn and K. Kumar, ACS Cent. Sci., 2021, 7, 454–466 CrossRef CAS PubMed.
- S. Huhmann and B. Koksch, Eur. J. Org. Chem., 2018, 3667–3679 CrossRef CAS.
- R. Smits and B. Koksch, Curr. Top. Med. Chem., 2006, 6, 1483–1498 CrossRef CAS PubMed.
- S. Ye, B. Loll, A. A. Berger, U. Mulow, C. Alings, M. C. Wahl and B. Koksch, Chem. Sci., 2015, 6, 5246–5254 RSC.
- S. Huhmann, A.-K. Stegemann, K. Folmert, D. Klemczak, J. Moschner, M. Kube and B. Koksch, Beilstein J. Org. Chem., 2017, 13, 2869–2882 CrossRef CAS PubMed.
- S. Huhmann, E. K. Nyakatura, H. Erdbrink, U. I.-M. Gerling, C. Czekelius and B. Koksch, J. Fluorine Chem., 2015, 175, 32–35 CrossRef CAS.
- J. Moschner, V. Stulberg, R. Fernandes, S. Huhmann, J. Leppkes and B. Koksch, Chem. Rev., 2019, 119, 10718–10801 CrossRef CAS PubMed.
- S. Kröger and G. Haufe, Amino Acids, 1997, 12, 363–372 CrossRef.
- J. Leppkes, T. Hohmann and B. Koksch, J. Fluorine Chem., 2020, 232, 109453 CrossRef CAS.
- J.-S. Völler, M. Dulic, U. I.-M. Gerling-Driessen, H. Biava, T. Baumann, N. Budisa, I. Gruic-Sovulj and B. Koksch, ACS Cent. Sci., 2017, 3, 73–80 CrossRef PubMed.
- H.-P. Chiu, B. Kokona, R. Fairman and R. P. Cheng, J. Am. Chem. Soc., 2009, 131, 13192–13193 CrossRef CAS PubMed.
- D. Alexeev, P. N. Barlow, S. M. Bury, J.-D. Charrier, A. Cooper, D. Hadfield, C. Jamieson, S. M. Kelly, R. Layfield, R. J. Mayer, H. McSparron, N. C. Price, R. Ramage, L. Sawyer, B. A. Starkmann, D. Uhrin, J. Wilken and D. W. Young, ChemBioChem, 2003, 4, 894–896 CrossRef CAS PubMed.
- T. H. Yoo, A. J. Link and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13887–13890 CrossRef CAS PubMed.
- Y.-J. Lee, M. J. Schmidt, J. M. Tharp, A. Weber, A. L. Koenig, H. Zheng, J. Gao, M. L. Waters, D. Summerer and W. R. Liu, Chem. Commun., 2016, 52, 12606–12609 RSC.
- B. J. Wilkins, S. Marionni, D. D. Young, J. Liu, Y. Wang, M. L. Di Salvo, A. Deiters and T. A. Cropp, Biochemistry, 2010, 49, 1557–1559 CrossRef CAS PubMed.
- R. Mousa, S. Lansky, G. Shoham and N. Metanis, Chem. Sci., 2018, 9, 4814–4820 RSC.
- U. Kahler, A. S. Kamenik, F. Waibl, J. Kraml and K. R. Liedl, Biophys. J., 2020, 119, 652–666 CrossRef CAS PubMed.
- A. J. Donovan, J. Dowle, Y. Yang, M. A. Weiss and S. B.-H. Kent, Pept. Sci., 2020, 112, e24166 CAS.
- J. Deisenhofer and W. Steigemann, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 238–250 CrossRef.
- G. Wagner and K. Wüthrich, J. Mol. Biol., 1982, 155, 347–366 CrossRef CAS PubMed.
- M. J.-M. Castro and S. Anderson, Biochemistry, 1996, 35, 11435–11446 CrossRef CAS PubMed.
- W. Lu, M. A. Starovasnik and S. B.-H. Kent, FEBS Lett., 1998, 429, 31–35 CrossRef CAS PubMed.
- T. Tsushima, K. Kawada, S. Ishihara, N. Uchida, O. Shiratori, J. Higaki and M. Hirata, Tetrahedron, 1988, 44, 5375–5387 CrossRef CAS.
- D. Winkler and K. Burger, Synthesis, 1996, 1419–1421 CrossRef CAS.
- G. I. Makhatadze, K.-S. Kim, C. Woodward and P. L. Privalov, Protein Sci., 1993, 2, 2028–2036 CrossRef CAS PubMed.
- D. Krowarsch and J. Otlewski, Protein Sci., 2001, 10, 715–724 CrossRef CAS PubMed.
- J.-Y. Chang and A. Ballatore, FEBS Lett., 2000, 473, 183–187 CrossRef CAS PubMed.
- E. Moses and H.-J. Hinz, J. Mol. Biol., 1983, 170, 765–776 CrossRef CAS PubMed.
- O. D. Monera, C. M. Kay and R. S. Hodges, Protein Sci., 1994, 3, 1984–1991 CrossRef CAS PubMed.
- P. Ascenzi, A. Bocedi, M. Bolognesi, A. Spallarossa, M. Coletta, R. De Cristofaro and E. Menegatti, Curr. Protein Pept. Sci., 2003, 4, 231–251 CrossRef CAS PubMed.
- R. Helland, J. Otlewski, O. Sundheim, M. Dadlez and A. O. Smalås, J. Mol. Biol., 1999, 287, 923–942 CrossRef CAS PubMed.
- D. Krowarsch, M. Dadlez, O. Buczek, I. Krokoszynska, A. O. Smalas and J. Otlewski, J. Mol. Biol., 1999, 289, 175–186 CrossRef CAS PubMed.
- D. Krowarsch, M. Zakrzewska, A. O. Smalas and J. Otlewski, Protein Pept. Lett., 2005, 12, 403–407 CrossRef CAS PubMed.
- H. Czapinska, R. Helland, A. O. Smalås and J. Otlewski, J. Mol. Biol., 2004, 344, 1005–1020 CrossRef CAS PubMed.
- R. Helland, H. Czapinska, I. Leiros, M. Olufsen, J. Otlewski and A. O. Smalås, J. Mol. Biol., 2003, 333, 845–861 CrossRef CAS PubMed.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4524 CrossRef CAS PubMed.
- J. R. Robalo and A. Vila Verde, Phys. Chem. Chem. Phys., 2019, 21, 2029–2038 RSC.
- T. Hohmann, M. Dyrks, S. Chowdhary, M. Weber, D. Nguyen, J. Moschner and B. Koksch, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-wt95d.
- C. Yung-Chi and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef.
- J. Smirnovienė, L. Baranauskienė, A. Zubrienė and D. Matulis, Eur. Biophys. J., 2021, 50, 345–352 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- R. P. Joosten, F. Long, G. N. Murshudov and A. Perrakis, IUCrJ, 2014, 1, 213–220 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- M. D. Winn, M. N. Isupov and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 122–133 CrossRef CAS PubMed.
- A. Casanal, B. Lohkamp and P. Emsley, Protein Sci., 2020, 29, 1069–1078 CrossRef CAS PubMed.
- C. J. Williams, J. J. Headd, N. W. Moriarty, M. G. Prisant, L. L. Videau, L. N. Deis, V. Verma, D. A. Keedy, B. J. Hintze, V. B. Chen, S. Jain, S. M. Lewis, W. B. Arendall, 3rd, J. Snoeyink, P. D. Adams, S. C. Lovell, J. S. Richardson and D. C. Richardson, Protein Sci., 2018, 27, 293–315 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cb00018k |
| This journal is © The Royal Society of Chemistry 2022 |