
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrating DNA damage response and autophagy signalling axis in ultraviolet-B induced skin photo-damage: a positive association in protecting cells against genotoxic stress
Sheikh Ahmad Umar
 ab and
Sheikh Abdullah Tasduq
*ab
ab and
Sheikh Abdullah Tasduq
*ab
aAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, 201002, India
bPharmacokinetics-Pharmacodynamics and Toxicology Division, CSIR-Indian Institute of Integrative Medicine, Jammu Tawi, Jammu and Kashmir, India. E-mail: stabdullah@iiim.ac.in; tasduq11@gmail.com; Tel: +91-1912569000-10 ext.332
First published on 1st October 2020
Abstract
The skin acts as both physical as well as an immunological barrier against hazardous agents from the outside environment and protects the internal organs against damage. Skin ageing is a dynamic process caused by the influence of various external factors, including damage from ultraviolet (UV-B) radiation, which is known as photo-ageing, and due to internal chronological mechanisms. A normal ageing process requires several orchestrated defense mechanisms to diverse types of stress responses, the concomitant renewal of cellular characteristics, and the homeostasis of different cell types that directly or indirectly protect the integrity of skin. Cumulative oxidative and endoplasmic reticulum (ER) stress responses and their adverse impact on biological systems in the skin are a common mechanism of the ageing process, negatively impacting DNA by causing mutations that lead to many physiological, functional, and aesthetic changes in the skin, culminating in the development of many diseases, including photo-damage and photo-carcinogenesis. Exposure of the skin to ultraviolet-(B) elicits the activation of signal transduction pathways, including DNA damage response, autophagy, and checkpoint signal adaptations associated with clearing radiation-induced DNA damage. Recent experimental reports suggest that autophagy is involved in maintaining skin homeostasis upon encountering different stresses, notably genotoxic stress. It has also been revealed that autophagy positively regulates the recognition of DNA damage by nucleotide excision repair and that skin ageing is associated with defects in the autophagy process. Moreover, autophagy is constitutively active in the skin epithelium, imparting protection to skin cells against a diverse range of outside insults, thus increasing resistance to environmental stressors. It has also been found that the stress-induced suppression of the autophagy response in experimental settings leads to enhanced apoptosis during photo-ageing upon UV-B exposure and that the maintenance of homeostasis depends on cellular autophagy levels. More recent reports in this domain claim that relieving the oxidative-stress-mediated induction of the ER stress response upon UV-B irradiation protects skin cells from photo-damage effects. The integration of autophagy and the DNA damage response under genotoxic stress is being considered as a meaningful partnership for finding novel molecular targets and devising suitable therapeutic strategies against photo-ageing disorders. Here, we summarize and review the current understanding of the mechanisms governing the intricate interplay between autophagy and the DNA damage response and its regulation by UV-B, the roles of autophagy in regulating the cellular response to UV-B-induced photodamage, and the implications of the modulation of autophagy as a meaningful partnership in the treatment and prevention of photoaging disorders.
 Sheikh Ahmad Umar | Sheikh Umar Ahmad is currently a PhD student of Biological Sciences and is working as a Senior Research Fellow (DST INSPIRE) at the CSIR-Indian Institute of Integrative Medicine, Jammu, Jammu & Kashmir, India, in the laboratory of Dr Sheikh Tasduq Abdullah. He obtained his BS in Biotechnology, Chemistry, and Zoology from the University of Kashmir in 2013. In 2015, he obtained his MS in Biotechnology with a Gold Medal of Excellence from Baba Ghulam Shah Badshah University, Rajouri, Jammu and Kashmir, India, after completing his post-graduation dissertation training at the CSIR-Institute of Himalayan Bioresource Technology, Palampur, Himachal Pradesh, India. In 2019, he received a visiting fellowship offer from the Department of Dermatology, University of Alabama at Birmingham (USA) in the laboratory of Dr Nabiha Yusuf. His research focuses on understanding the interplay of autophagy and DNA damage response in UV-B-induced skin photo-damage. |
 Sheikh Abdullah Tasduq | Sheikh Tasduq Abdullah is a Principal Scientist/Associate Professor at the CSIR-Indian Institute of Integrative Medicine, Jammu, Jammu & Kashmir, India. He received his PhD in Biochemical Toxicology in 2004 from Dr H. S. Gour University, Sagar, Madhya Pradesh, India. He then did his post-doctoral training at the Department of Gastroenterology and Hepatology, Mayo Clinic, Rochester, Minnesota, United States of America, in the laboratory of Dr (Professor) Micheal R. Charlton. In 2006, he joined the CSIR-Indian Institute of Integrative Medicine, Jammu, Jammu & Kashmir, India, as a scientist. His primary research interests lie in understanding skin photo-damage (photo-carcinogenesis and photo-aging), melanogenesis, and melanoma in experimental animal models. |
1. Introduction
Skin is the largest organ in body and is made up of three layers, the epidermis, dermis and the hypodermis, which vary significantly in their anatomy and function. The epidermis comprises keratinocytes, melanocytes, Langerhans cells and Merkel's cells. Sweat glands, hair, hair follicles, muscles, sensory neurons, and blood vessels are housed by the dermis. The hypodermis is the deepest layer of skin and contains adipose lobules along with some skin appendages such as the hair follicles, sensory neurons and blood vessels.1 Skin is the first site of immunological defense by the action of the Langerhans cells in the epidermis and is part of the adaptive immune system.2 Skin maintains cellular homeostasis by regulating temperature and water loss, while also serving both endocrine and exocrine functions, including the production of vitamin D in the epidermis by the keratinocytes, which are responsible for converting 7-dehydrocholesterol into vitamin D with the assistance of UV light from the sun.3 Skin also imparts mechanical strength to the whole body, which is largely provided by a supporting framework of extracellular matrix that is mainly secreted by fibroblasts, and has basic requirements that must be satisfied for almost every tissue.4 Skin requires a network of blood vessels lined with endothelial cells to bring nutrients and oxygen and remove waste products and carbon dioxide and also to provide access routes for cells of the immune system to provide defenses against infection and thus mediate more sophisticated adaptive immune system responses.5 Nerve fibers help in conveying sensory information to and from the central nervous system by delivering signals and assisting in glandular secretion and smooth muscle contraction.6 Being external to the body, the epidermis suffers more direct, frequent and damaging encounters than any other tissue in the body; therefore its repair and renewal is central to its organization.5 The intricate structure and network of skin cells serve as the body's initial barrier against pathogens, UV light, chemicals and mechanical injury.7 Exposure to ultraviolet (UV) radiation is a significant risk factor for skin photo-ageing and photo-carcinogenesis.8 Changes to the skin due to either chronological or photo-ageing mechanisms are among the most recognizable signs of ageing and can lead to various types of skin perturbations if left untreated. Skin ageing is a growing area of interest for the cosmetic, aesthetic and skincare industries due to its clinical, social and strategic importance.9 Ageing is associated with various health problems including the loss of natural skin tone, fragility, aesthetic appearance and the rise in the occurrence of highly lethal malignant skin cancers in humans.10 Management of the skin ageing process depends on a thorough understanding of the natural aging mechanisms, which are defined as the chronological decline in tissue function caused by various internal and external factors that cumulatively contribute to the degradation of natural skin tone, and the signal transduction pathways that control the morphological and pathological changes to skin,11 and as well as an understanding of the functional interplay among the specific cell types that make up the skin.12 It has long been recognized that skin ageing is determined by diverse intrinsic and extrinsic factors such as UV and other environmental toxicants, but recent advances in gerontologic research have helped to demonstrate the domains pertaining to the molecular, cellular and physiological aspects of skin that underlie the ageing process.13–15 The ageing process is triggered mainly by the presence of damaged molecules within the cells and due to exposure to external threats like ultraviolet radiation, and the turnover of these damaged molecules is controlled partly by autophagy.16 Nutrient sensing, genomic instability, and oxidative and endoplasmic reticulum stress responses directly or indirectly activate the autophagy machinery, suggesting that it acts as a primary key regulatory process in maintaining skin homeostasis and healthy ageing.17 Solar ultraviolet (UV) rays are an external factor that can induce biological and clinical damage to the skin, including photoaging, immunosuppression, angiogenesis, mutations and subsequent photo-carcinogenesis, via acute as well as chronic exposure.18 UV-B accounts for around 5% of the total UV energy that reaches the Earth's surface, whereas long wavelength UV-A is the main component of terrestrial UV radiation (around 75% of total energy received on Earth).19 UV-A is believed to exert its pathologic effect through the generation of ROS with singlet oxygen as the major ROS species, which in turn causes oxidative damage to macromolecules including DNA and other cellular structures.20 Studies in mouse models have shown UV-A to be a relatively weak initiator of carcinogenesis, but a relatively potent tumor promoter, although both UV-A and UV-B can act as complete carcinogens. UV-B rays have shorter wavelengths and higher energy levels and damage the outermost layers of the skin, thereby directly damaging DNA. UV-B rays cause most skin cancers and also contribute to premature skin aging.21 The degree and type of DNA damage heavily depends on the wavelength of UV light that reaches the Earth's surface.22 UV-A-induced ROS-dependent lesions are repaired more quickly and also have lower mutagenicity than UV-B-inflicted lesions, which are repaired more slowly and lead to the majority of genomic mutations.23 The differential mutagenic properties of UV-A and UV-B are also reflected by their potential in the induction of carcinomas in experimental animals and human studies.24 It is therefore the UV-B part of the spectrum that has a more pronounced effect on the skin than UV-A, although both contribute separately to photo-aging and the critical mechanisms through which UV-A and UV-B irradiation affect the function of human skin cells are a growing topic of discussion.In the epidermis, UV-B radiation is efficiently absorbed by DNA and damages it directly by forming photoproducts, most commonly cyclopyrimidine dimers (CPDs) and to a lesser extent, pyrimidine-(6-4)-pyrimidone photoproducts (6-4PPs).25 6-4PPs are bulkier, but more efficiently repaired than CPDs and are responsible for most (∼80%) UV-B-induced mutations.26,27 Exposure of skin cells to UV-B activates autophagy directly by rapidly activating AMPK, UVRAG and p53.28 Upon its activation, p53 mediates the transcriptional activity of autophagy activators AMPK, TSC2, sestrin 1 and sestrin 2 and interacts with AMPK, TSC1 and TSC2 to inhibit mTOR signaling in response to genotoxic stress.29,30 Conversely, genotoxic stress is a trigger for autophagy whereas repair of UV-induced DNA damage has been found to be regulated by autophagy.31 Recent experimental findings suggest that the knockdown of key autophagy genes AMPK, Atg5, Atg7, Atg12 and Atg14 impairs the repair of UVB-induced DNA damage and support the hypothesis that autophagy is critical in maintaining cellular homeostasis, but works in tandem with DNA damage response pathways.32,33 More recent work has identified multiple pathways through which autophagy regulates UV-B-induced DNA damage repair.34 Multiple pathways induce autophagy in the epidermis as a defense response upon the exposure of skin to UV-irradiation. In contrast to the roles of the autophagy process in skin cells, different autophagy-related proteins have been found to play different and diverse roles in response to UV-B exposure. A recent study reported that autophagy gene Atg7 promotes UV-induced inflammation and skin tumorigenesis, regulates cytokine expression and secretion, and promotes Ptgs2/Cox-2 expression through a CREB1/CREB-dependent cell-autonomous mechanism and an IL1B/IL1β-dependent non-cell-autonomous mechanism. A similar study reported that ATG5 knockdown inhibits the UVB-induced expression of PTGS2 and cytokines. Furthermore, the loss of ATG7 increases the activation of the AMPK pathway and leads to a reduction in the ER stress response. These findings demonstrate that Atg7 deletion leads to suppression of the carcinogen-induced pro-tumorigenic inflammatory microenvironment of the epithelium in UV-B exposed skin cells.35 In another such study, it was found that the mRNAs of some key ATG genes such as ULK1, ATG5 and ATG7 exhibit significantly lower expression levels in certain skin tissues, particularly those of the face and chest, upon UV exposure compared to perineal skin. Interestingly, UV-B irradiation inhibits the autophagic flux via an mTOR-independent pathway in human keratinocytes; the autophagic flux cannot be restored by treatment with an mTOR-dependent autophagy inducer such as rapamycin. This study demonstrates that UV-B radiation impairs the autophagy response by down-regulating several autophagy-related genes in keratinocytes and demonstrates a linkage between autophagy and skin disorders associated with ultraviolet exposure.36 Other promising studies have found that ATG7 deletion results in increased baseline oxidative stress, reduced ability to degrade cross-linked proteins after oxidative stress and a strong increase in γH2AX positive nuclei (DNA damage response initiator protein) within the basal layer of Atg7-deficient epidermis, suggesting that autophagy deficiency significantly impairs the resistance of the keratinocytes to intrinsic and environmental oxidative stress and results in DNA damage, cell cycle arrest and a disturbed lipid phenotype, which are all typical of a premature cell aging response.37 Further research in this domain has revealed that anti-aging strategies involving the use of natural plant-based anti-oxidants alleviate the cellular damage to UV-B exposed skin cells by either adjusting the adaptation of cells to stresses or reducing the stress response through improving autophagy levels and thereby restoring cellular homeostasis upon exposure of skin to UV-B.38,39 Based on these preliminary clues from recent experimental works, we propose the integration of the DNA damage and autophagy response mechanisms as a meaningful partnership and describe the potential of exploring these twin signalling axis to devise suitable therapeutic targets against UV-B-induced photo-damage disorders and to be further explored in the development of novel skincare products of both therapeutic and cosmeceutical importance.
2. Autophagy: an overview
Macro-autophagy (hereafter referred to as autophagy) is a cellular conservative process acting as catabolic machinery entrusted to deliver damaged organelles and residual materials of the cell for lysosomal degradation.40 It involves the double-membrane autophagosome, which degrades the unwanted cellular materials contained within it after fusing with a lysosome. It is very energy conserving and ubiquitous in nature among a diverse range of eukaryotes. Autophagy is activated by a variety of stimuli, some of which are internal to cells, as well as external agents such as nutrient starvation and energy deprivation, oxidative and ER stress responses, blockage of growth factors, and pathogen infection, among others.41–47 The most commonly described type of autophagy is referred to as starvation-induced macro-autophagy, which occurs under nutrient or energy deprivation conditions and helps eukaryotic cells to recover nutrients.48 It ensures cellular survival by recycling internal residual reserves through their compartmentalization in double-membrane vesicles in the lysosome known as autophagosomes at phagophore assembly sites (PAS) in the cytosol for final energy release to maintain cellular homeostasis42 (Fig. 1). Autophagy is considered to be a double-edged sword. It is context-dependent, and can exert either pro- or anti-apoptotic mechanisms in response to external damage depending on the cellular needs.49 As is already known, numerous factors can lead to DNA damage and elicit damage response signalling in skin cells, including environmental mutagens, chemical mutagens and reactive metabolic byproducts, among others. Previous reports have mainly focused on autophagy triggered by DNA damage resulting from chemical substances, ionizing radiation (IR), and reactive oxygen species that induce alterations in the DNA structure.50 Previously, it has been revealed that UV induces on-rate autophagic flux in epithelial cells, and autophagy inhibition via knockdown of beclin-1 and Atg5 has been found to reduce cell viability and enhance apoptosis.51 Autophagy is also involved in regulating the DNA damage response under genotoxic stress conditions. ATR is a protein in the DNA damage response pathway and responds to a diverse spectrum of DNA damage, notably damage that interferes with genomic replication. ATR/Chk1 signaling is usually activated by single-stranded DNA or bulky DNA lesions and therefore has a crucial role in stabilizing the genome during the replication process and is essential for cell survival. However, the underlying mechanism by which the DNA damage response activates autophagy, especially the mechanism by which ATR/Chk1 signaling triggers autophagy, remains elusive.52–54 Preliminary findings have revealed that ATR phosphorylation during the early phases following UV-B irradiation is accompanied by microtubule-associated protein 1 light chain 3B II (LC3B-II) expression, which has been demonstrated to be a well-established inducer of ATR. ATR knockdown attenuates LC3B-II expression at early stages in response to UV treatment, implying a positive correlation with the induction of autophagy upon UV-B irradiation of skin cells, and contributes greatly to reducing the damaged cells.55 It has been determined that reduced ATR expression does not affect late-phase autophagy induction (24 and 48 h after UV treatment) despite the potential role of ATR in the autophagic response,56 but the initiation of autophagic response at this stage is instead activated via a distinct pathway that is yet to be deciphered. These studies demonstrated that autophagy acts as a cytoprotective mechanism against UV-induced apoptosis and that at late stages autophagy induction accompanied with apoptosis is independent of ATR activation. More recent reports attempted to investigate UV-radiation-induced DNA damage accompanied by autophagy and the biological role of autophagy in regulating genotoxic stress. ATM, a member of the PIKK (PI3K-like protein kinase) family with functional relationships to two other PIKKs, ATR (ATM and RAD3-related) and DNA-PKcs (DNA-dependent protein kinase catalytic subunit), is involved in the cellular response to genotoxic stress, is vigorously activated following double strand breaks, and fine-tunes a complex signalling network by phosphorylating a multitude of substrates in numerous branches of DNA damage response signalling.57 It has been found that ATM knockdown attenuates the autophagy response, demonstrating the involvement of ATM in genotoxic stress-induced autophagy, and that the integration of ATM with DNA damage signaling pathways is associated with cytoprotective events.58 ATR may also play a role in regulating the UV-induced autophagic response, because UV-B irradiation can induce ATR activation and ATM shares a high level of similarity and overlapping substrates with ATR.56 Moreover, natural-product-based anti-oxidants that have the potential to enhance cellular autophagy levels have been found to protect skin cells from ultraviolet-B-induced oxidative-stress-mediated modulation of the DNA damage response.39 Thus, improving cellular autophagy levels is thought to contribute to imparting skin with protection against various types of environmental insults. Environmental toxicants including ultraviolet radiation exposure have been found to induce autophagy through the oxidative DNA damage response as a conservative measure to protect cellular integrity under genotoxic stress.56 The exposure concentration and duration of a toxicant can variably induce autophagy in cells via a coordinated signalling response that determines the cell death or survival effect. Metal pollutants, most of which are persistent in the environment, have also been shown to modulate autophagy via similar or different response mechanisms.59–63 Autophagy has been extensively investigated elsewhere and is the major process controlling cellular homeostasis. In the present review, we will mainly focus on macro-autophagy (autophagy) and the various proteins that are implicated in the execution of the autophagy process in lower eukaryotes as well as their homologs in higher eukaryotes.64 The interpretation of the diverse roles of the various autophagy factors specifically involved in regulating homeostasis under genotoxic stress conditions is an open debate, and further work is required to clarify the positive associations involved.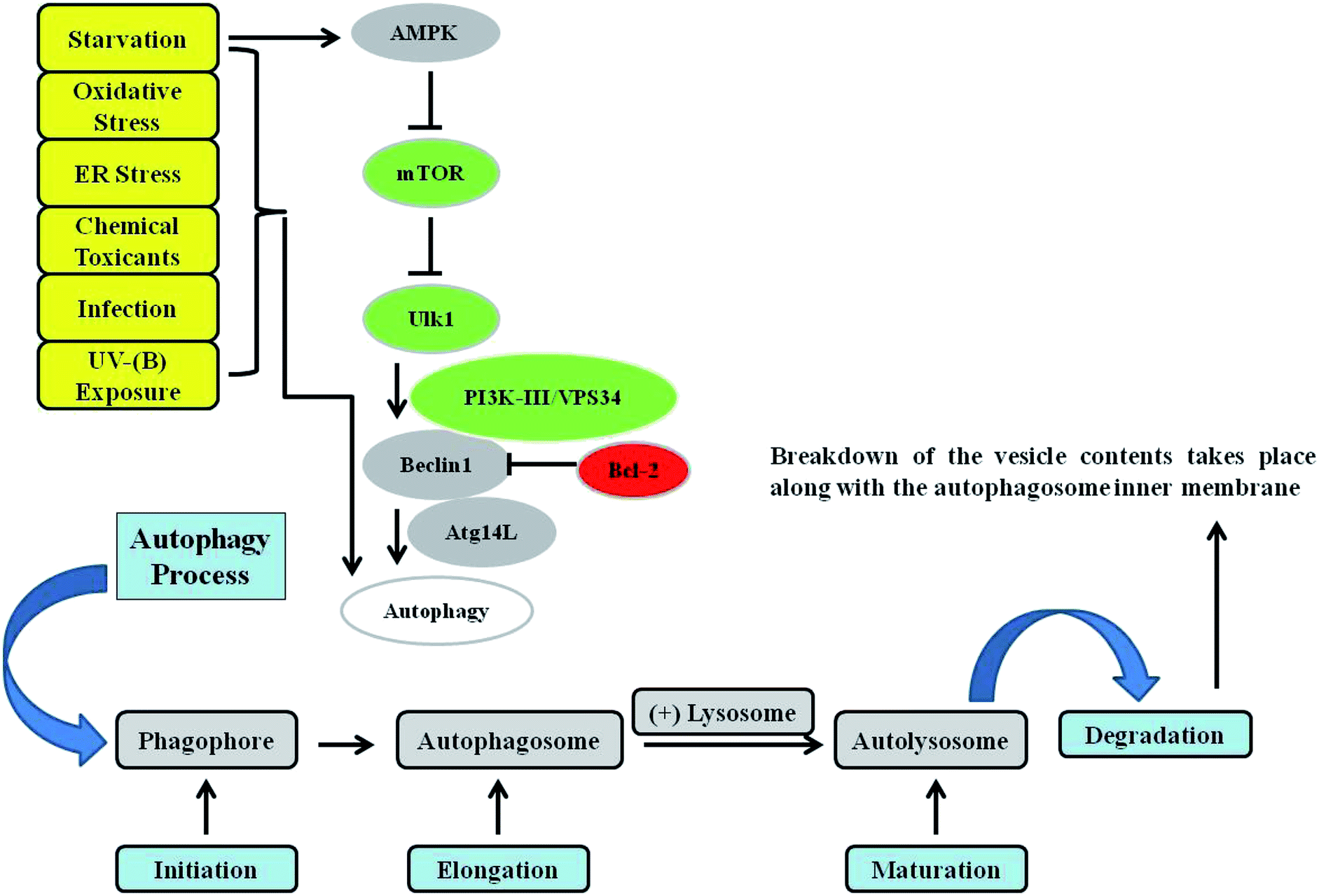 | ||
| Fig. 1 Autophagy signalling. Autophagy is mainly controlled by the upstream protein kinase signalling pathways of AMPK, PI3K-I, and MAPK/ERK. Phagophore initiation is regulated by the serine/threonine protein kinase ULK1/2 complex directly and by the activity of the class III phosphatidylinositol-3-phosphate kinase (PtdIns3K) VPS34 (lipid kinase activity), which forms a complex with Beclin 1 and ATG14L. Cytoplasmic constituents are enclosed in an isolation membrane upon the initiation of the process, and elongation into a double-membraned autophagosome occurs through the action of two ubiquitin-like conjugation systems. Maturation of the autophagosome occurs upon its fusion with a lysosome to form an autolysosome, in which breakdown of the vesicle contents take place. Various kinds of stressors, such as starvation, toxins, oxidative stress, endoplasmic reticulum (ER) stress, infections, and UV-B exposure, have been found to induce the autophagy response in cells. | ||
3. DNA damage response: an overview
The DNA damage response (DDR) is a complex set of comprehensive cellular pathways and specific molecular events activated in response to different types of damage caused by various intrinsic/extrinsic factors, including ultraviolet radiation exposure; the DDR facilitates repair and thereby prevents tumour development. The process has been thoroughly evaluated and well-reviewed elsewhere owing to the growing demand to find suitable target-based treatment regimens against various cancers.65–68 We will comment here on its functional aspects concerning UV-B-induced skin photo-damage and the relevant components necessary to understand the interplay between the DNA damage response and autophagy pathways. DNA damage can be concomitantly caused by several exogenous factors (e.g. UV-B irradiation) or endogenous agents, e.g. reactive oxygen species (ROS)/reactive nitrogen species (RNS).69 Various types of DNA damage can be induced depending upon the nature of the geno-toxicant as well as that of the mutation it causes; however, the most common types of lesions are single and double strand breaks (SSBs and DSBs) and inter-strand cross-links (ICLs). The network of crucial pathways that sense, stall and resolve damaged DNA in response to external insults can either prevent the cell death effect by recruiting molecular players that are indispensable in protecting the genomic integrity of cells or cause concomitant cell death to maintain the normal homeostatic state.70 In general, UV irradiation results in the formation of DNA photoproducts, mainly cis-syn-cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6-4) pyrimidone photoproducts (6-4PPs). The protein ATR of the repair response pathway is the major and crucial sensor of these UV-induced DNA lesions.71 Additionally, because ATM is involved in DNA-damage-induced autophagy, it may also be crucially involved in UV-induced autophagy. Additional evidence has nevertheless shown that ATM can be phosphorylated by ATR after UV irradiation through an independent mechanism and that these proteins may share a great deal of responsibility for initiating the autophagy response.72 However, it has been found that ATR can be recruited to the site of DSBs indirectly mediated by ATM and that ATR may transmit a signal to ATM and its downstream targets resulting in autophagy induction to initiate a cascade of other reactions that repair the damage.73,74 This interactome of ATM and ATR provides clues regarding the possible involvement of the ATM/ATR axis in DNA-damage-induced autophagy. These findings provide a comprehensive understanding of the linkage between ATM and ATR, their significant roles in DNA-damage-induced metabolic regulation and the exclusive role of ATR in UV-induced autophagy, but more studies in this regard will reveal the biological significance of ATM/ATR roles in stress response behaviour. Cells are capable of responding promptly to any mutagenic change upon radiation exposure, but if left unrepaired, these changes may result in cell death due to the genomic instability caused by the accumulation of replicative yet unrepaired photo-products produced in response to damage,31 particularly when typical cell death responses to protect cellular integrity are suppressed, such as cell cycle checkpoint kinases including p53, UVRAG, p73, ATM, E2F1, Parkin, HDAC, and PARP (Table 2). The DNA damage response employs a cascade of protein molecules involved in sensing, signalling, and repair. The initial activation events following damage are well elucidated, but the inter-regulatory signal that triggers damage response remains to be clarified.75 However, it has been proposed that when a lesion occurs, it induces conformational changes in chromatin at the site of DNA damage through post-translational histone modifications including ribosylation, phosphorylation and acetylation, among other molecular events that invariably attempt to resolve the damage, and that this sensing results in some initial responses in the form of cell cycle arrest, checkpoint activation, senescence and DNA damage repair aimed at either resolving the damage or likely causing autophagy-mediated apoptosis.76–78 Chromatin relaxation at the site of the lesion created by the intoxicant provides access to DDR sensor proteins that bind lesions specifically. DSBs initially promote the binding of Mre11 complexes (MRN) including Mre11/Rad50-Nbs1 at the damaged sites and recruit the kinase ATM.79 Other post-translational events subsequently induce ATM activation, which triggers activation of additional Chk2 kinases controlling the cell cycle, the tumour suppressor p53, which regulates cell survival, and histone modulators (HDACs) involved in chromatin modeling and remodelling to orchestrate and amplify the DSB signalling response mechanism. Phosphorylation of the typical histone variant γ-H2AX flanks the DBS sites induced by the ATM-DNA PK pathway, favouring the activation of the ATM and ATR Rad3-related kinases recruited by the replication protein A (RPA) complex. This complex array of molecular changes and recruitment of DNA damage marker proteins through the involvement of several other factors leads to signal amplification that helps in damage sensing and clearing.80 Five main DNA repair systems operate in cells, including mismatch repair (MMR), nucleotide excision repair (NER), base excision repair (BER), homologous recombination (HR) and non-homologous end joining (NHEJ). NER, MMR and BER are involved in the repair of various types of base lesions requiring single strand incision, whereas NHEJ and HR are employed in DSB repair.77,78 The DNA repair machinery is tightly regulated by a number of enzymes, including nucleases, polymerases, recombinases, topoisomerases, ligases, glycosylases, and demethylases, that chemically amplify the DNA damage response signalling and work in tandem to repair the damage.81 Ubiquitination and ubiquitin-like modifiers (UBLs) are essentially important in the regulation of enzymes through diverse post-translational events that ensure genome integrity upon damage.82 Defective DNA repair pathways resulting from mutations or chronological dysfunctions have been found to be implicated in certain neurological and immunological defects, ageing, and even cancer.83 DNA repair pathways also play a critical role in maintaining nervous system homeostasis given the fact that neurons can experience enhanced oxidative stress and subsequent DNA damage as they have limited capacity for replacement and because of high oxygen consumption by mitochondrial respiration.84 Deficiencies or impairments in multiple repair pathways have been linked to premature ageing, and defects in these pathways due to exogenous or endogenous causes have been associated with the potentiation of the UV-B-mediated DNA damage response.85 The indispensable role of repair pathways in the maintenance of genomic integrity is therefore critical in preventing skin tumorigenesis in response to mutagenic insults, as has been elucidated in several DDR syndromes found to result in a strong predisposition towards cancer.86 Moreover, further studies showed the involvement of the stability of mRNAs and proteins in UV-exposed cells, as well as the preferential sensitivity of UV-exposed cells to alteration of gene expression and transcriptional regulation of genes at the G1-S phase of the cell cycle and to the components of the DNA damage checkpoint ATR pathway. These studies established that E2f-1 is regulated at the G1-S transition by the phosphorylation of the retinoblastoma (RB) gene under the control of the Trp53 pathway and plays a dual role, promoting cell cycle progression and stimulating programmed cell death upon UV-B exposure in cells. In contrast, forced E2F-1 expression induces quiescent cells to enter the S phase, demonstrating the ability of E2F-1 to activate the transcription of molecules such as cdc2, cyclin E, cyclin A, and E2f-1 itself, whereas the ectopic expression of E2f-1 can induce apoptosis in several cell types through the transcriptional activation of proapoptotic molecules, APAF-1 (apoptotic protease activating factor 1, a human homolog of the C. elegans CED-4 gene), caspases, and Fhit. These findings clearly indicate that DNA-damage-related checkpoint proteins are tightly intertwined with regard to their function and establish cellular homeostasis under genotoxic stress conditions.87,88 Checkpoint adaptation is also an important evolutionarily conserved function of key damage response proteins that maintain the cellular integrity of cells. p53 plays an important role in the adaptive response of the skin to UVB damage through cell cycle arrest, repair and apoptosis, predominantly but not exclusively through transactivating genes with an active role in arrest, global genomic repair and apoptotic events.89 Chk2 and p53 are well characterized for their functional aspects in transducing the ATM-initiated DNA damage signal and in controlling the cell cycle and apoptosis response upon UV-B activation. The coordination of the responses of a number of ATM substrates such as p53, Chk2, and Mdm2 is critical for sustaining the cell cycle arrest in order to allow efficient repair. Several other ATM substrates, including breast cancer 1 (Brca1, early onset), Rad55, and Nbs1 are also known to be important for promoting DNA damage repair processes.90 The presence of p53 response elements in key regulatory genes of melanogenesis indicates that p53 may also mediate these adaptive responses against UV-B damage and that its activity is essential in clearing damaged foci upon UV-B irradiation in cells. As it is a sensor of UV-B damage, reduced p53 accumulation may reflect reduced stress in the first place as a counterfeit response mechanism. However, pharmacological suppression of p53 is also unwanted as it can lead to increased tumorigenesis.91 While at the moment, there is no clear evidence as to whether p53-targeted agents can increase protective capacity against photodamage, this attractive speculation could be further investigated in the future.4. Autophagy as the effector and player in the DNA damage response of cells to genotoxicants
Autophagy is regarded as an important mediator and effector in maintaining cellular homeostasis under genotoxic stress conditions. Recent work has unraveled diverse pathways implicated in the autophagy-mediated regulation of the UV-B-induced DNA damage response and their effect on efficient repair.92 However, further clarifications are needed to define the interrelationship between apoptosis and autophagy, notably in the case of UV-B induced genotoxic stress. Based on what has been identified and reported so far in this context, oxidative stress conditions simultaneously induce antioxidant response and autophagy mechanisms to concomitantly decrease upstream causes such as ROS and downstream effects to reduce the oxidative damage to DNA. This finely designed repair system in which autophagy promotes DNA repair and vice versa through a vicious on/off cycle in an attempt to find a new homeostatic state depending upon the extent of damage can perfectly fit the needs of the cell. ROS-induced oxidative changes to DNA activate DDR signalling, in which different classes of proteins that sense and activate repair pathways are switched on as a response signalling mechanism in order to maintain genomic integrity. The damage sensors preferentially identify the DNA lesions, the mediators and effectors help to co-transduce the nuclear response signal to the cytosol, and several other processes such as protein phosphorylation and dephosphorylation contextually remediate the potential tumour-evoking conditions68 (Table 1). Checkpoint proteins arrest the cell cycle instantly in the current phase of division and therefore halt further proliferation until repair is completed. However, severe damage or unrepaired lesions promote cellular quiescence culminating in cell death to reduce the chances of tumorigenesis.93 Autophagy is thought to act bi-contextually as either a pro-survival or cell death mechanism with many lines of evidence arguing that autophagy can delay or suppress apoptosis by fulfilling the energy requirements of the cell by recycling residual materials to support the DNA repair process. Conversely, damage-induced modulation of the autophagy response has been demonstrated and contributes to death in cells where DNA is left unrepaired; this switching process depends solely on the interplay of the autophagy pathway and the core DDR machinery.70 Among many other responses, autophagy is thought to promote a vast array of chromatin modifications in response to diverse genotoxic insults in order to expose the damaged part for repair to safeguard the genomic integrity of cells. Transcription and replication processes are frozen around the site of the DNA lesions, mostly double-strand breaks (DSBs), through chromatin responses including remodelling to facilitate subsequent repair by providing access to DDR sensors. ATM kinase and replication protein A (RPA) complexes, which predominantly respond to single-strand lesions, are also recruited and in turn recruit ATR kinase to spread the signal response.94 This response is induced immediately upon genotoxic insult and is of pivotal importance in reducing the chances of replication errors. These accumulating shreds of evidence with regard to the intertwined and diverse roles of ATM/ATR/RPA proteins suggest that autophagy can be activated by DNA damage and that damage-specific binding of these protein kinases triggers the recruitment of downstream proteins through cycles of phosphorylation to transduce a signalling response that positively orchestrates replication and repair and controls the cell cycle, transcription, and survival versus death mechanisms through various, albeit not exclusive, routes.95 Genotoxic stress induces ATM, which is a major sensor protein of DSBs. Recent reports have shown the direct link of ATM to the DDR in the regulation of the AMPK-mediated autophagy response, which in turn phosphorylates TSC2 and thereby diminishes the suppressive effect of TORC1 on autophagy.96 The aforementioned signalling-axis-mediated suppression of TORC1 by ATM-AMPK-TSC2 is thought to operate in response to oxidative and nitrosative stress,97 both of which induce the DNA damage response concurrently involved in the mobilization/translocation of ATM to the cytoplasm. ULK1, a serine/threonine-protein kinase involved in autophagy induction in response to starvation conditions, promotes autophagosome formation upon activation by AMPK.98 Recent evidence has shown that autophagy positively regulates the recognition of damaged sites by NER and enhances the repair of lesions induced by UV-B by upregulating the expression levels of the damage recognition proteins XPC, UVRAG, and DDB1/DDB2, which are involved in the NER pathway, at the site of DNA damage.99 UVRAG, which is upregulated by radiation, localizes at photo-lesions, leading to XPC and NER recruitment at the damage sites by associating with DDB1, which promotes the assembly of the DDB2–DDB1–Cul4A–Roc1 ubiquitin ligase complex. UVRAG has been reported to be involved in activating the autophagy response upon radiation insult when bound to the Beclin-1/Vps34 complex, which increases the catalytic activity of Vps34 and is of particular significance in the regulating the damage response through a multifunctional approach.100,101 Conversely, deficiencies in autophagy have been associated with the transcriptional suppression of XPC and reduction in UV-B-induced XPC ubiquitination, which is critical for the DNA damage recognition and repair mechanism, and impairing the recruitment of DDB1/2 to DNA damage sites.99 In vivo studies that underscore the importance of autophagy induction at DNA damage sites have been conducted in mice; in these studies, the inhibition of autophagy by spautin-1 promoted tumorigenesis whereas the mTOR inhibitor and autophagy-inducer rapamycin reduced tumour development.99,102 In accordance with this, various pharmacological agents have been used to activate the autophagy response in UV-exposed skin cells as a therapeutic strategy to combat the effects of irradiation on the skin. Commonly used agents include rapamycin, (mTORC1 inhibitor), everolimus (mTORC1 inhibitor), Torin1 (selective ATP-competitive inhibitor of mTORC1/2), and metformin (AMPK activator); these are currently being explored as possible therapeutic agents for the prevention of UV-B-induced photo-damage to skin in various experimental settings.103 Given the role of energy intermediates in the repair process, the fact that DSB repair requires ATP-dependent chromatin remodelling, and the ATP-dependence of the helicase unwinding process in NER, autophagy likely plays a generic role in DNA repair by sustaining the supply of the key intermediates ATP, NADC, and dNTPs, demonstrating the critical impact of autophagic pathways on the turnover of the DNA response mechanism.104 The function of PARP1 in base-excision repair is also dependent on the levels of NADC.105 Moreover, autophagy-mediated ribonucleotide reductase subunit degradation also requires a pool of dNTPs as important mediators and effectors for DNA replication and repair.106 Likewise, micronuclei, which are nuclear-membrane-enclosed chromosome fragments containing damaged DNA, are also removed by damage-induced autophagy; this process contributes to genomic integrity.107 Overall, the above findings suggest that autophagy plays a prominent role in coordinating DNA damage response signalling upon external mutagenic insults, but further studies are required to precisely interpret how the distinct classes of autophagy-related mechanisms control the various repair systems and conversely, how the different autophagic pathways interact and work in consonance to finely orchestrate the balance in DNA response systems via a vicious cycle of reinforcement effects.| Proteins involved in autophagy | Yeast | Mammals | Proteins involved in DNA damage check-point signalling | Mammals | Yeast |
|---|---|---|---|---|---|
| Sensors | |||||
| Atg1/ULK complex | Atg1 | ULK1/2 | 9-1-1 clamp and clamp loader | Rad17 | Rad24 |
| Atg13 | ATG13 | Rad9 | Ddc1 | ||
| Atg17 | FIP200 | Rad1 | Rad17 | ||
| (Functional homolog) | Hus1 | Mec3 | |||
| Atg29 | — | ||||
| Atg31 | |||||
| Atg11 | |||||
| Atg2 | ATG2 | ||||
| Atg9 and its cycling system | Atg9 | ATG9A/B | BRCT-containing | BRCA1 | Rad9 |
| Atg18 | WIPI1/2 | TopBP1 | Dpb11 | ||
| PtdIns3K complexes | Vps34 | PIK3C3/VPS34 | MRX complexes | Mre11 | Mre 11 |
| Vps15 | PIK3R4/VPS15 | Rad50 | Rad50 | ||
| Vps30/Atg6 | BECN1 | Nbs1 | Xrs2 | ||
| Atg14 | ATG14 | ||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Transducers | |||||
| Atg8 Ubl conjugation system | Atg8 | LC3A/B/C | PI3-kinases (PIKK) | ATR | Mec1 |
| GABARAP | ATM | Tel1 | |||
| GABARAPL1/2 | |||||
| Atg7 | ATG7 | ||||
| Atg3 | ATG3 | ||||
| Atg4 | ATG4A/B/C/D | ||||
| Atg12 Ubl conjugation system | Atg12 | ATG12 | PIKK binding partner | ATRIP | Ddc2/Lcd1 |
| Atg7 | ATG7 | ||||
| Atg10 | ATG10 | ||||
| Atg16 | ATG16L1 | ||||
| Atg5 | ATG5 | ||||
| Effector kinases | Chk1 | Chk1 | |||
| Chk2 | Rad53 | ||||
| Protein | Role in DNA damage response (DDR) | Role in autophagy and/or interplay with DDR |
|---|---|---|
| P53 | Regulates DNA repair, cell cycle arrest, and apoptosis in response to DNA damage | Induces autophagy in response to DNA damage through transcription of ULK1, ULK2, DRAM, and sestrins 1/2. Also inhibits autophagy through AMPK inhibition in cytoplasm |
| P73 | Promotes apoptosis in response to chemotherapeutic-induced DNA damage. Involved in transcription of glycosylases in response to bile acid-induced DNA damage | Induces autophagy in a DRAM-independent manner. Binds to genomic sites near the autophagy-related genes atg5, atg7, and Ambra1 |
| UVRAG | Partially complements the sensitivity of XPC transformed cells to UV-(C). Binds to and activates DNA-PK complexes, thereby promoting the repair of DNA double strand breaks through non-homologous end joining (NHEJ) | Participates in the multiprotein complex Bcl-2-Beclin1-PI(3)KC3-UVRAG, which in turn regulates autophagosome formation |
| E2F1 | Promotes DNA repair and survival or apoptosis in response to DNA damage. Recruits nucleotide excision repair factors to sites of UV-induced DNA damage to augment repair activity | Activates autophagy in response to etoposide and up-regulates transcription of atg1, LC3, atg5, and DRAM |
| Parkin | Associates with PCNA in the nucleus and enhances the NER-mediated resolution of UV-induced lesions and BER-mediated resolution of H2O2-induced lesions | Involved in mitophagy and recruits damaged mitochondria (by PINK1) to promote their degradation |
| ATM | Responds to and senses DNA double strand breaks, and regulates cell cycle arrest, DNA repair, and apoptosis | Inhibits mTORC1 in response to ROS and induces autophagy through the activation of TSC2. Involved in autophagy activation in response to DNA damage induced by the N-mustard derivative BO-1051 |
| HDAC | Downregulates the expression of apoptotic genes and may influence the repair of damaged DNA by regulating the accessibility of DNA repair enzymes to sites of lesions | Inhibition of HDAC by valproic acid has been found to promote the autophagic degradation of acetylated Sae2 and further reduction of DNA double strand break repair in yeast. Also found to impair autophagy activation |
| PARP | Involved in poly ADP-ribosylation and recruits BER proteins to the sites of DNA single strand breaks | Involved in AMPK activation, which in turn activates autophagy as PARP activation consumes NAD+, which results in ATP depletion. |
5. Autophagy, anti-oxidant response and melanogenesis in the modulation of DNA repair pathways upon UV-B irradiation to skin
The modulation of the autophagy response is regarded as a primary event in maintaining homeostasis upon external disturbances that compromise the genomic integrity, including oxidative and ER stress. To this end, autophagy controls the repair machinery via a vast array of molecular mechanisms through a specialized repository of effector and executioner proteins. Extensive evidence suggests that autophagy is a critical pathway in the DSB repair process, with most recent reports showing that key autophagy proteins are required for the efficient removal of damage-induced DNA lesions in response to various external insults including chemical carcinogens and radiation exposure.108 Consequently, autophagy-related mechanisms play an important role in distinct DNA repair pathways in cell defense. When genotoxic stress is induced by various environmental factors, including ultraviolet-radiation-induced stress, autophagy-related mechanisms mitigate the oxidative damage to biological macromolecules.109 Defective autophagy is associated with increased oxidative- and ER-stress-mediated DNA damage, gene amplification, and aneuploidy that concurrently induce DNA damage and cell death responses mediated in part by autophagy, very likely due to the depletion of metabolic activists in the absence of autophagy affecting the cellular energy levels and thereby impeding homeostasis.110 The transcription level of various autophagy and lysosomal genes is regulated at least in part through the stabilization of p53, which guards the cell against various genotoxic perturbations.110,111 Similarly, basal ROS act as important signalling molecules in healthy cells, but elevated levels can cause oxidative damage to biomolecules, as is seen in response to UV-B in cells. Oxidative DNA damage induces autophagy as a preventive mechanism to sustain the cellular energy demand for efficient repair68,112 by clearing the damaged proteins, lipids and DNA, and thus restoring the metabolic homeostasis.113 Nuclear factor Nrf2 triggers antioxidant-response-linked autophagy through the feedback loop mechanism of adaptor protein P62 bound to KEAP1, preventing the degradation of Nrf2. The adaptor protein leads to autophagy-mediated stabilization of Nrf2 and its degradation by KEAP1, in turn activating the P62 transcription levels by binding to the antioxidant response element within the p62 promoter.114,115 Nrf2 has also been found to positively regulate the expression of many autophagy genes, such as Atg3, Atg5, Atg7, SQSTM1 and GABARAPL1 during cellular stress,116 suggesting its positive role in autophagy induction. Other findings have proved that Nrf2 also inhibits AMPK and thereby directly controls the mTOR activity, demonstrating the direct involvement of Nrf2-mediated autophagy induction in regulating stress and restoring cellular homeostasis.117 Moreover, oxidative-stress-mediated modulation of autophagy elicits other cellular responses that concurrently help to defend cells against the effects of radiation damage. A network of keratinocyte- and fibroblast-derived factors interact to modulate the function of melanocytes and to reduce oxidative damage upon radiation exposure. Particularly important among them is the keratinocyte-derived paracrine factor for melanocytes, endothelin-1, whose synthesis is increased upon exposure to UVR. Similar to α-MSH, endothelin-1 promotes repair of UV-induced DNA damage, including oxidative damage, by inhibiting the generation of hydrogen peroxide in UV-irradiated melanocytes and is thought to trigger after the immediate induction of autophagy in cells upon radiation exposure.118,119 This fine modulation of the state of autophagy upon genotoxic stress clearly demonstrates that autophagy plays a broader role in maintaining cellular intactness and homeostasis once activated. Additionally, there is convincing evidence that dermal fibroblasts synthesize a panel of factors including fibroblast-derived basic fibroblast growth factor, hepatocyte growth factor, and stem cell factor, which are upregulated upon UV exposure and might be driven by an autophagy response that regulates, among other factors, the melanogenic response of melanocytes and positively impacts cellular viability.1206. The interplay between autophagy and DNA repair: a therapeutic approach
Recent trends in autophagy research have paved direct pathways for understanding the intricate crosstalk between autophagy and the DNA repair mechanism. Defects in autophagy signalling have been proven to render cells susceptible to nutrient stress, increasing the chances of tumorigenesis; these findings were the first line of evidence linking autophagy to the DNA damage response and provided a path-breaking approach in the quest for a suitable treatment option against a wide range of diseases caused due to the impairment of repair systems. Mathew and colleagues reported in 2007 that autophagy unequivocally acts to protect the genomic integrity of cells. They found that knockdown of ATG5 and Beclin1, which are key players in the autophagy machinery, results in gene amplification and chromosomal instability, facilitating tumour progression in response to external stimuli.110 Increases in the levels of the damage sensor γ-H2AX and other associated proteins in autophagy-deficient cells suggest that stress-driven autophagy is constitutively important in damage response and in maintaining the integrity of the genome by either halting cell progression or removing the damaged cells through apoptosis-mediated mechanisms to prevent tumour development.100,121–123 Interestingly, knockout of FAK-family interacting protein (FIP200), which is an essential element of autophagy activation as an important component of the ULK1 complex, results in a significant decrease in the repair mechanism in response to radiation and chemotherapeutic agents in murine embryonic fibroblasts (MEFs).124 The sensitization to apoptosis-inducing agents and possible impairment of the repair mechanism of autophagy-deficient cells confirms the potential tumour suppressive function of FIP200. Conversely, FIP200-null cells were experimentally shown to have increased sensitivity to camptothecin- and etoposide-induced cell death, which correlates to the increased DNA damage and impaired autophagy promoting tumorigenesis in FIP200-null cells. These studies also justified the critical role of P62 as a mediator of FIP200 in regulating the intricate balance between DNA damage repair and cell survival upon genotoxic stress.124 Other studies have demonstrated that autophagy participates in DNA repair pathways via diverse related mechanisms, in particular mitophagy, which selectively removes ROS-producing damaged mitochondria as they could directly or indirectly modify DNA to generate various lesions and affect cellular viability; thus, mitophagy prevents genomic instability.125 Furthermore, if DNA damage persists, mitophagy is negatively affected and the NAD+-SIRT1-AMPK pathway to activate DDR signalling is up-regulated, leading to an increase in P53 acetylation and AKT activity.126 Chemo-resistance mechanisms in cancer cells are induced in part by delayed apoptotic cell death upon DNA damage due to autophagy, which is necessary for energy conservation and for an efficient repair process, whereas the non-availability of metabolic precursors has been found to impair autophagic flux and affect the autophagy-mediated DNA damage response homeostasis.127 DNA-damaging agents used in chemotherapeutic settings, such as CPT, etoposide, temozolomide and ionizing radiation (IR), are known to induce cell cycle arrest, and have also been reported to initiate autophagy, making it clear that DNA damage agents can activate autophagy at multiple levels.128–130 Regulation of the intricate balance of different effector proteins (NAD+-SIRT1-AMPK pathway activating DDR signalling and leading to an increase in P53 acetylation and AKT activity) occupies a central position in the efficient repair of damage induced in response to different toxicants. However, it is thought that ATM kinase is a key regulator in the DDR response, as it increases the transcription level of the factor FOXO3a, which is a member of the FOXO subfamily of forkhead transcription factors that mediate a variety of cellular processes including apoptosis, proliferation, cell cycle progression, DNA damage and tumorigenesis under UV-B-irradiation and oxidative stress, thus leading to its activation and thereby promoting repair.97 Consistently, overexpression of FOXO3a has been found to promote ATM-mediated signalling, enhance the repair of damaged DNA, and increase the expression profile of S and G2-M phase cell-cycle checkpoints, while its depletion leads to defects in DDR functions and adversely impacts the repair capacity of damaged cells.131 In general, both ATM and FOXO3a have been linked to autophagy. Whereas FOXO3a essentially antagonizes FOXM1-dependent transcription, ATM leads to autophagy progression via induction of the energy sensor AMP-activated protein kinase (AMPK).132 AMPK directly phosphorylates ULK1, initiating the autophagy response, and interacts with the mTORC1 complex via a pathway involving tuberous sclerosis complex 1 and 2; this pathway also leads to autophagy induction, albeit through negative regulation mechanisms.133 Other reports have shown that AMPK activation through CaMKKβ sustains autophagy under non-starvation conditions, giving an indication that AMPK induces autophagy under both energy-starved and non-starved conditions to meet the needs of cells in response to various insults compromising the integrity of the genome.32 Via the activation of Che-1ATM, ATM also mediates the RNA polymerase II-binding protein Redd1 (regulated in development and DNA damage responses), which is a recently described component of the stress response gene induced by hypoxia and DNA damage, and Deptor, a DEP domain-containing protein that interacts with mTOR and is involved in the mTOR signaling pathway as an endogenous regulator.134 FOXO3a also controls the transcription of LC3 and Bnip3, which are essential proteins in the autophagy pathway, demonstrating the diverse roles of these proteins in regulating the autophagy machinery upon damage.135–137 Recent experimental evidence has demonstrated that hyper-activated poly[ADP ribose] polymerase 1 (PARP1) depletes ATP levels when a DNA lesion is created upon genotoxic insult, which leads to AMPK activation and subsequently to a damage-mediated autophagy response that further synthesizes poly[ADP ribose] chains and recruits damage sensor proteins for an efficient repair process.138Tumour suppressor protein p53, which is regarded as the guardian of the cell, is another crucial regulator of DNA repair pathways and is suggested to have a dual role in autophagy.29,139 On one hand, p53 transcriptionally regulates autophagy-related proteins concurrently with other members of its family (p63 and p73). It also directly affects AMPK signalling to promote the autophagy response under severe stress. Moreover, HDACs also influence the damage response through acetyl modification of crucial damage-related and checkpoint proteins, and thus represent a significant link between the autophagy and repair pathways. Roberts and colleagues found that HDACs regulate chromosomal stability by coordinating the DSB processing and ATR checkpoint signalling with autophagy; in particular, the inhibition of HDACs triggers degradation of the recombination protein Sae1 by promoting autophagy. It was also reported in the same study that it affects the HDAC hda1 and rpd3 mutants sensitivity towards DNA damage by modulating the autophagy machinery.76
Sirtuin deacetylases, another family of NAD+-dependent proteins that are involved in autophagy by promoting autophagosome formation, have been suggested to play an essential role in the autophagy-mediated DNA repair process through the deacetylation of ATG5, ATG7, and ATG8. Sirtuins regulate the transcriptional activity of p53 under DNA damage conditions and thereby affect the cell cycle progression, demonstrating that p53 has diverse roles associated with the autophagy process in protecting cells against genotoxic stress.140 ATM dysfunctions in patients with ataxia-telangiectasia, which is characterized by defective DNA damage repair in the nucleus, lead to impairments in mitophagy through deregulated Sirtuin1 activity. In turn, this deregulation affects the mitochondrial uncoupling protein 2 (UCP2), which is responsible for the modulation of a critical molecule in mitophagy induction, PINK1, by affecting its import, cleavage, and removal.141 Intriguingly, autophagy has been found to play direct roles in the HR (homologous recombination), NER (nucleotide excision repair), and MMR (mismatch repair) error-proofing mechanisms. NER, an adaptive pathway for correcting helix-distortions in DNA induced by environmental carcinogens, is reported to be directly regulated by autophagy via the downregulation of the transcription levels of XPC, which leads to defective DDB2 recruitment to UV-B-induced lesion sites through EP300 inhibition by TWIST1.99 DDB2 has been found to execute the initial step of damage recognition in UV-B-induced photolesions in NER, and has been shown to be rapidly degraded after cellular UV insult.142 Mispairs induced by the nucleoside analogues 6-thioguanine and 5-fluorouracil have been shown to induce a p53-mTOR-dependent autophagy response via up-regulation of BNIP3, whereas defects in MMR caused by chemotherapeutic drugs impair autophagy signalling.143 These combined studies suggest that targeted inhibition of autophagic pathways may enhance the cytotoxicity of chemotherapeutic agents by the MMR system, suggesting the broader interplay of autophagy in regulating DNA damage response systems in cells. Moreover, the stress-induced autophagy regulator DRAM (damage-regulated autophagy modulator) is a p53 target gene that encodes a lysosomal protein that induces macroautophagy and is crucial to the induction of autophagy-mediated apoptotic cell death by p53, indicating that p53 is involved in the regulation of autophagy; however, the exact nature of this signal remains elusive.144 UVRAG (UV radiation resistance-associated gene protein), which complements the ultraviolet sensitivity in xeroderma pigmentosum, also plays a dual role in both autophagosome formation and chromosomal stability,145 and facilitates the recruitment of DDB1/2 to sites of UV-induced DNA damage when bound to DDB1, but in an autophagy-independent manner. In contrast, the interaction of Beclin 1 with UVRAG is essential for the regulation and activation of class III PI 3-kinase through autophagy-mediated mechanisms. Although preliminary evidence suggested that UVRAG directly promotes DSB repair, UVRAG-mediated autophagy has been found to positively regulate DNA repair upon its activation by UV-B. In the NHEJ (non-homologous end joining) pathway, UVRAG has been reported to help in the assembly of the upstream protein kinase DNA-PK by directly interacting with it, demonstrating its broader role in DNA damage response signalling.146 Notably, autophagy and DNA repair pathways have been thought to be tightly controlled through different classes of initiator and executioner protein molecules in response to diverse genotoxic insults. Conversely, it has been shown in autophagy-defective mice models characterized by genomic instability that autophagy-deficient cells rely on diverse repair systems, underlining the importance of the repair machinery in protecting cellular integrity and thereby indicating that cooperative DNA repair mechanisms are involved in the concurrent activation of autophagy-related pathways. Recent findings have shown that treatment with the TOR inhibitor and autophagy inducer rapamycin delays skin aging in mice. Metformin, which is also an autophagy inducer, has been shown to retard aging in Caenorhabditis elegans by modulating the microbial folate and methionine metabolism, and is also reported to increase lifespan in C. elegans when co-cultured with Escherichia coli. Metformin is known to slightly reduce food consumption and in turn lead to starvation-induced autophagy mechanisms, complementing other studies that postulate autophagy as a promising therapeutic strategy against various aging-related disorders.
Taken together, the above findings demonstrate that the autophagy pathway has the potential to be explored as a suitable therapeutic target in devising appropriate treatment strategies to reduce the DNA damage effects induced by UV-B and chemotherapeutic agents. Various therapeutic strategies are currently being explored or are already in use against UV-induced photoaging/carcinogenesis, and autophagy is a recent entry to this list (Table 3). Some experimental evidence supporting the role of autophagy as a potential therapeutic strategy is summarized here. Both natural- and chemical-based autophagy modulators have been found to be of significant clinical interest for the treatment and prevention of skin cancer and many other photo-damage-related disorders. However, the context-dependent and wide-ranging functionality of autophagy makes it difficult to predict the clinical response to autophagy modulators and their potential application in therapeutics. Preliminary experimental findings have revealed that AMPK is activated by UV-B to induce autophagy, which promotes repair and inhibits apoptosis under stress conditions. AMPK activation has been found to be reduced in human skin cancer samples. Therefore, targeting AMPK, which is directly involved in autophagy activation, in skin cancers could provide an opportunity to block tumor growth and suppress tumorigenesis. This data supports the use of AMPK activators such as metformin and TORC1 activators like rapamycin and everolimus that activate autophagy to block the growth of UV-B-induced skin tumors in vivo.33 Furthermore, the autophagy activator rapamycin has been found to reduce UV-induced skin tumor formation and progression, and consequently reduces the number of UV-induced signature mutations in p53 in skin tumors.147 Collectively, this work suggests that autophagy has a crucial tumor-suppressive function in the UV-B response and could be explored as a potential therapeutic strategy against photo-damage. In our own lab, we have recently reported that the natural plant-based anti-oxidant glycyrrhizic acid modulates the autophagy process in response to UV-B exposure in primary dermal fibroblasts and is involved in the alleviation of UV-B-induced oxidative DNA damage to skin cells;39 these results also support the conclusion that autophagy has a promising role in UV-B therapeutics and in the development of suitable drug targets against UV-B-induced photo-damage disorders.
| Therapeutic strategy | Mechanism of action | References |
|---|---|---|
| Autophagy | Autophagy promotes the degradation of metabolic adducts following the exposure of skin to UV and inhibits aging- and aging-related diseases by removing potentially toxic metabolites from cells and preventing photo-carcinogenesis | 161 |
| Natural plant-based antioxidants | Natural plant-based antioxidants protect skin by absorbing UV radiation, inhibiting free-radical reactions induced by UV radiation in cells, and by modulating endogenous antioxidant and inflammatory systems | 162 and 163 |
| Oral photo-protectants | Oral photo-protectant substances usually contain one or more active principles that activate different mechanisms of photoprotection and act by increasing the antioxidant efficacy of the body following the loss of endogenous anti-oxidants after UV exposure | 164 |
7. Damage-induced autophagy pathway: a conservative approach of cells
Autophagy is known to be regulated by diverse DNA damage sensors such as FOXO3a, ATM, ATR, and p53 in response to diverse genotoxic insults, including UV and chemotherapeutic agents.148 DNA damage induces autophagy to initiate a cascade of response mechanisms to repair the lesions; however, if the damage is severe and the cell cannot repair itself, cell-specific responses such as cell death and senescence are triggered to remove the damaged material to prevent tumorigenesis, possibly via autophagy-mediated apoptosis.15 Conversely, damage-causing agents like etoposide or staurosporine have been reported to induce autophagy in mouse embryonic Bax−/− Bak−/− fibroblasts, which exhibit autophagy-dependent non-apoptotic cell death.149 At its core, subsequent work elsewhere has established that higher eukaryotes display autophagy responses under genotoxic stress that are directly dependent on the ATM-p53-mTOR signaling axis; these responses are referred to as geno-toxin stress-induced autophagy (GTA). p53 binds to the promoters of numerous autophagy-related genes (ULK1/ULK2, ATG7, ATG4A, and UVRAG) under genotoxic stress conditions and mediates their transcriptional up-regulation in an ATM/ATR-dependent manner, thereby directly combating stress by causing cell cycle arrest, senescence or autophagy-mediated apoptotic degradation of damaged materials. It has been found that p53-deficient cells are unable to induce the autophagy response after DNA damage and essentially require transcriptional up-regulation of autophagy-related genes for its execution.111 Another unique protein, DRAM1, connects DNA damage to the autophagy pathway via a specific DNA-damage-responsive branch and appears to be mediated in part by p53.28 While further work is required to clarify whether autophagy regulation by DRAM1 is mediated in part by other cues, such as nutrient deprivation, other reports have proven its involvement in other autophagic processes in which pathogens are selectively destroyed by the autophagic machinery, which is referred to as xenophagy.150 Interestingly, other roles of p53 in specific types of autophagy other than bulk autophagy have been reported, with the DRAM1 locus encoding multiple splice variants inducible by p53 that are not localized to lysosomes but instead appear at peroxisomes and ER via the activation of the protein death-associated protein kinase-1, suggesting that p53 may control specific types of autophagy151,152. p73, a closely related ally of the p53 family, induces autophagy independent of p53 by regulating the transcriptional levels of autophagy-related genes, which in turn initiate a cascade of relevant pathway response mechanisms in protecting cellular integrity.153 It is now clear that autophagy activation in response to cellular DNA damage is mediated in part via ATM and nuclear p53 levels, which activate the transcription of multiple pro-autophagic genes including PTEN, AMPK, and SESTRINS. Although PTEN suppresses AKT, regulating the tumour-suppressive conditions in the cellular environment, AMPK-mediated phosphorylation activates TSC2, promoting TSC inactivation.96 ATM, in addition to promoting p53 activation, also leads to autophagy induction through inhibition of mTOR signaling by phosphorylation of the Lkb1 kinase, which in turn activates the AMP kinase and promotes Tsc2-mediated inhibition of mTORC1. The damage-induced activation of the repair enzyme PARP-1 consumes NAD+ molecules and senses AMPK levels in cells, thereby increasing its activity toward mTORC1.132,154 Moreover, suppression of mTOR in response to other damage-causing agents such as etoposide indicates that TOR inhibition is specific and context dependent. Although it has diverse effects, the exact role of mTOR signalling in GTA remains enigmatic and will require further studies to clearly elucidate its exact role in regulating damage response. With regard to the role of ATM, its effect appears to be largely ROS specific and requires a cytoplasmic pool of ATM. More recent reports in mouse fibroblasts suggest that various genotoxins can induce chaperone mediated autophagy (CMA). However, it is still unclear whether a selective or bulk pathway process is induced after encountering damage due to these genotoxins.155 Stress-induced CMA is known to prevent proteo-toxicity from dysfunctional proteins via their selective degradation, whereas suppressed CMA activity contributes to decreased proteome quality in various diseases and in response to various genotoxic insults.156 Here, it is worth noting that impaired autophagy may result in DNA damage, leading to the assumption that the interplay between the two is complex and suggesting that many molecular players exist to link many associated processes. Therefore, any dysfunction in this regard might be involved in evoking various pathological states, while the prime role of any alterations in metabolic demand has yet to be reported. Recently, research in budding yeast suggested that DSBs induced by endo-nucleases and genotoxins preferentially lead to a selective autophagy pathway under starvation conditions, as judged by the Atg11 requirement.157 The DDR proteins Mec1, Tel1, Ddc2/Lcd1, Chk1, and Rad53 are thought to be strongly implicated and specific for GTA, in contrast to the roles of such proteins in higher eukaryotes. Autophagy induction upon treatment with rapamycin occurs irrespective of these kinases, suggesting that GTA in yeast is distinct from nutrient-deprivation-induced autophagy and is instead mediated by autophagy-related gene Rph1 (KDM4 in mammals) and regulated by CHK2, suggesting that GTA is a unique pathway involved in the degradation of specific substrates and is distinct in lower and higher eukaryotes.158ATM/ATR signalling is context dependent and largely depends on the extent of DNA damage, demonstrating that it may either promote or inhibit autophagy, indicating its broader role. There is an optimum requirement for the induction of autophagic flux to promote DNA repair upon encountering damage, and depending upon the severity of the damage, a context-dependent autophagy response may be induced in the cell. Autophagy activation is a time-specific event following genotoxic stress. Upon genomic damage, autophagy is instantly induced (denoted by the red line in Fig. 2A) in cells, which in turn induces checkpoint signalling activation (denoted by the blue line), which promotes cell cycle arrest and subsequent repair of the damaged DNA. After the damage is fixed, the autophagy again declines to basal levels. Cellular senescence, on the other hand, is a delayed response and a function of time event that directly depends on the fate of the autophagic flux. Senescence is a state of durable cell cycle arrest with metabolic activities distinct from those of the proliferative state and was originally reported to be induced by various genotoxic stressors, such as chronic UV-B response, and other factors that are intrinsic to cells, such as telomere erosion and oncogenic signaling. It is proposed to play a pivotal role in aging-related changes and as an antitumorigenic barrier, and is thus essentially important in checkpoint activation (Fig. 2A).159 Similarly, the unfolded protein response (UPR) is a cellular stress response related to the endoplasmic reticulum (ER) and activated in response to an accumulation of unfolded or misfolded proteins in the lumen of the endoplasmic reticulum.160 Autophagy regulates the cell survival vs. death effect in response to various stress conditions including UV-B irradiation by intricately balancing the UPR and apoptotic mechanisms. UV-B irradiation of skin cells induces the oxidative-stress-mediated manifestation of endoplasmic reticulum stress (UPR), which induces autophagy. The autophagy may switch the expression of DNA damage response proteins on or off depending upon the extent of DNA damage and regulates the expression of pro/anti-apoptotic proteins in order to maintain cellular homeostasis and in turn prevent tumorigenesis (Fig. 2B). Accordingly, genetic defects in autophagy genes increase ROS production in cells and the accumulation of damaged organelles, which in turn promotes metabolic reprogramming and induces tumorigenesis. Several possible mechanisms have been proposed to define the interplay between oxidative stress, the damage response and autophagy, but a precise mechanistic link for the tuning of autophagy is still to come. Understanding the fine regulation of the oxidative-damage-induced autophagy signalling response would be valuable information that could be of scientific importance in future for improving treatment strategies and developing new target-based selective therapies as new findings unfold. Preliminary findings have proven that autophagy likely prevents oxidative DNA changes in UV-induced skin photo-damage, but there is currently no significant indication that interventions in autophagy pathways would be effective in finding target-specific treatment regimens. Future research in this context will certainly unravel the functional aspects of autophagy in the UV response in skin to prevent photo-damage, and will certainly provide a molecular basis for targeting autophagy in regulating skin and photo-damage-induced disorders.
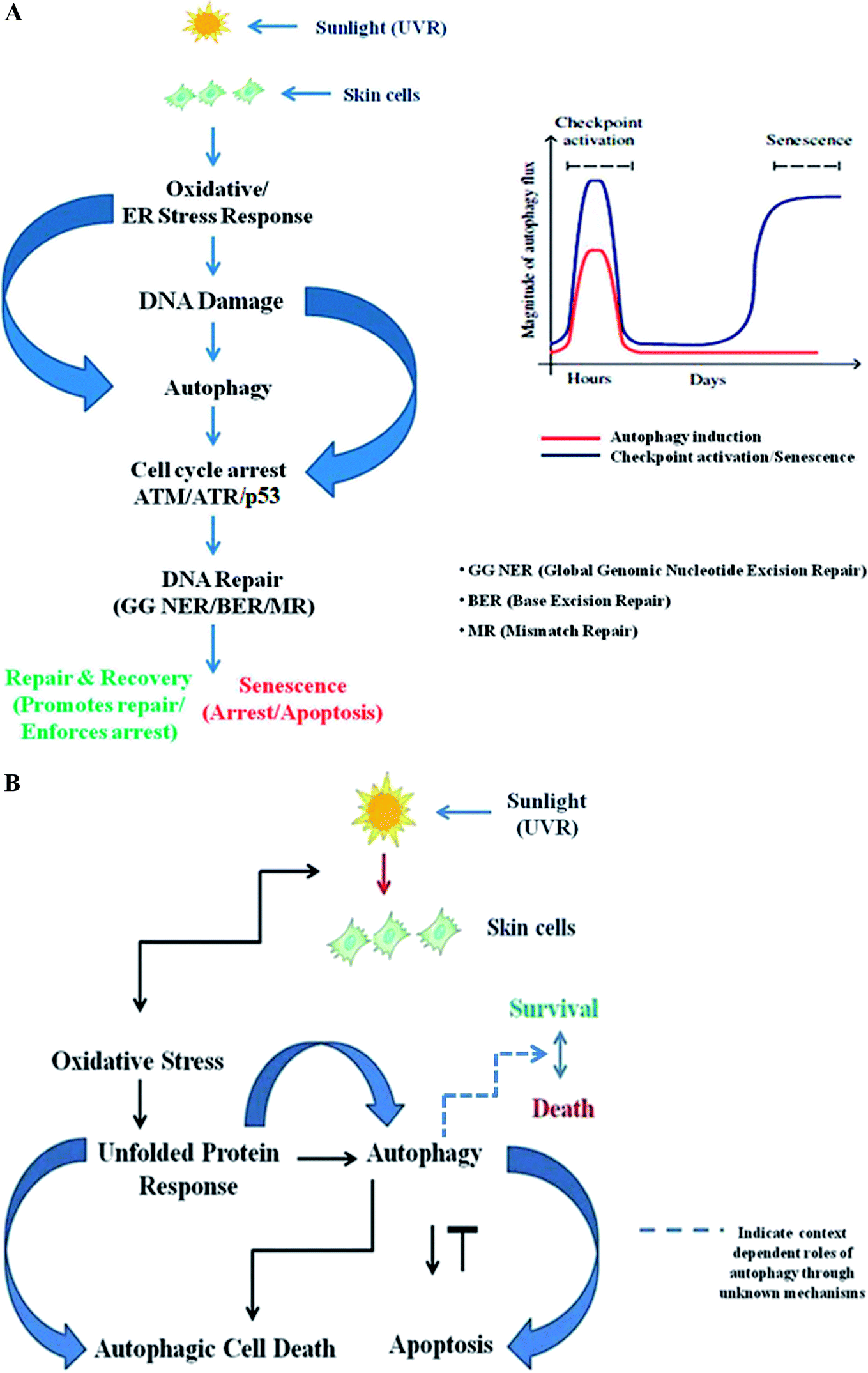 | ||
| Fig. 2 (A) The interplay of autophagy and DNA damage response signalling. ATM/ATR signaling may either promote or inhibit autophagy, depending on the amount and extent of DNA damage. There is an optimum requirement for the induction of autophagic flux to promote DNA repair upon encountering damage, and depending upon the severity of the damage, a context-dependent autophagy response may be induced in the cell. Autophagy activation is a time-specific event following genotoxic stress. Upon genomic damage, autophagy is instantly induced (red line) in cells, which in turn induces checkpoint signalling activation (blue line), which promotes cell cycle arrest and subsequent repair of the damaged DNA. After the damage is fixed, the autophagy again declines to basal levels. Cellular senescence, on the other hand, is a delayed response and a function of time event that directly depends on the fate of the autophagic flux. Senescence is a state of durable cell cycle arrest with metabolic activities distinct from those of the proliferative state and it was originally reported to be induced by various genotoxic stressors, such as chronic UV-B response, and other factors that are intrinsic to cells, such as telomere erosion and oncogenic signaling. It is proposed to play a pivotal role in aging-related changes and as an antitumorigenic barrier, and it is thus essentially important in checkpoint activation. (B) The interplay of the unfolded protein response (UPR)-autophagy-apoptosis signalling axis. Autophagy regulates the cell survival vs. death effect in response to various stress conditions, including UV-B irradiation, by intricately balancing the unfolded protein response (UPR) and apoptotic mechanisms. UV-B irradiation of skin cells induces the oxidative-stress-mediated manifestation of endoplasmic reticulum (ER) stress, culminating in UPR, which induces autophagy. The autophagy may switch the expression of DNA damage response proteins on or off depending upon the extent of DNA damage and it regulates the expression of pro/anti-apoptotic proteins in order to maintain cellular homeostasis and in turn prevent tumorigenesis. | ||
8. Conclusions
The interplay between autophagy and the DNA damage response has evolved as a tightly regulated quality control mechanism to respond to many stressful conditions, including radiation-induced damage. Autophagy functions as the main pathway in mammalian homeostasis and is involved in the physiology and pathophysiology of many diseases. Human health is regulated by autophagy via various mechanistic pathways, mostly concerning cancer progression, immune response, and the ageing process, which can be either chronological or due to UV-B induced photo-damage. The transcriptional activity of some DNA repair enzymes is defined by the rate of basal autophagy levels, thereby directly regulating genomic integrity and playing a central role in preventing tumorigenesis. Aberrant DNA damage responses and autophagy signalling are particularly relevant in cancer therapy, as cells cannot recuperate from chemically induced damage when there is no exact communication in the interplay of these pathways. Dysfunctional repair systems and defects in autophagy-related genes have been linked to clinical phenotypes causing a vast array of pathological responses, including premature ageing, developmental defects, and neuro-degeneration, and to the general failure of DNA repair, especially HR and NER. The accumulation of misfolded proteins as potential toxic intermediates due to an impaired autophagy response contributes to DNA damage and increases the chances of genomic instability in cells. Chemotherapy and genotoxic protocols could emerge as effective strategies for enhancing anticancer treatment when combined with autophagy, and better understanding of this crosstalk may help to provide an understanding of how chronological ageing occurs and will certainly prove helpful in finding the origin of phenotypes such as premature aging in patients with defective autophagy and DNA damage response signaling. Other reports concerning the roles of autophagy in DNA repair in cancer cells have suggested that chemotherapy resistance may be a result of the positive effect of the autophagy process in the repair of DNA lesions, but more studies in this regard will definitely establish a concerted base of results to prove the autophagy-mediated regulation of cellular homeostasis under genotoxic stress conditions. Likewise, stimulating autophagy via the DNA response signalling axis holds great promise for devising novel therapeutic strategies against various damage-induced responses. While the current approach of preventing photo-aging either by the avoidance of sunlight exposure or through antioxidant mechanisms offers a viable option in combating radiation-induced skin pathologies, more recent studies have suggested that repair-related enzymes could be used as therapeutic ingredients or as adjuncts to sunscreens to potentiate their potential protective nature against sunlight-induced skin aging. Despite the great progress in elucidating the roles of autophagy in photo-aging resistance, much more must be unraveled in order to overcome this problem more effectively. More refined approaches for understanding the mechanisms of the autophagy-mediated regulation of damage underlying the switching response will provide valuable insights into UV-induced skin disorders and could be developed into promising therapeutic strategies offering a molecular platform for interventional autophagy to slow skin aging and related chronic diseases of the skin. This could include UV-induced oxidative/ER-stress-mediated skin cancers such as melanoma.Conflicts of interest
The authors declare that no conflict of interest exists.Acknowledgements
Senior Research Fellowship (SRF) awarded to author SAU by the Department of Science and Technology (DST), Government of India Vide No. IF-160982 is acknowledged. The authors are also thankful to Director CSIR-Indian Institute of Integrative Medicine, Jammu for funding this work vide Project No. MLP-1003 and to the Department of Biotechnology (DBT), Ministry of Science and Technology, New Delhi, Government of India, vide project No. GAP-2166.References
- J. Fenner and R. Clark, Anatomy, physiology, histology, and immunohistochemistry of human skin, Skin tissue engineering and regenerative medicine, 2016, vol. 1 Search PubMed.
- N. Romani, B. E. Clausen and P. Stoitzner, Langerhans cells and more: langerin-expressing dendritic cell subsets in the skin, Immunol. Rev., 2010, 234(1), 120–141 CrossRef CAS.
- B. Lehmann, K. Querings and J. Reichrath, Vitamin D and skin: new aspects for dermatology, Exp. Dermatol., 2004, 13, 11–15 CrossRef CAS.
- L. D. Muiznieks and F. W. Keeley, Molecular assembly and mechanical properties of the extracellular matrix: a fibrous protein perspective, Biochim. Biophys. Acta (BBA) - Mol. Basis Dis., 2013, 1832(7), 866–875 CrossRef CAS.
- B. Alberts, et al., Epidermis and its renewal by stem cells, in Molecular Biology of the Cell, Garland Science, 4th edn, 2002 Search PubMed.
- L. Sherwood, Human physiology: from cells to systems, Cengage Learning, 2015 Search PubMed.
- G. L. Flynn, Cutaneous and Transdermal Delivery—Processes and Systems of Delivery, in Modern pharmaceutics, CRC Press, 2002. pp. 314–385 Search PubMed.
- A. Sample and Y. Y. He, Autophagy in UV damage response, Photochem. Photobiol., 2017, 93(4), 943–955 CrossRef CAS.
- L. Baumann, Skin ageing and its treatment, J. Pathol., 2007, 211(2), 241–251 CrossRef CAS.
- P. C. Durai, et al., Aging in elderly: chronological versus photoaging, Indian J. Dermatol., 2012, 57(5), 343 CrossRef.
- A. M. Leidal, B. Levine and J. Debnath, Autophagy and the cell biology of age-related disease, Nat. Cell Biol., 2018, 20(12), 1338–1348 CrossRef CAS.
- S. R. Pandi-Perumal, et al., Physiological effects of melatonin: role of melatonin receptors and signal transduction pathways, Prog. Neurobiol., 2008, 85(3), 335–353 CrossRef CAS.
- B. Gilchrest and J. Krutmann, Photoaging of skin, Skin Aging, 2006, pp. 33–44 Search PubMed.
- A. L. S. Chang, Expanding our understanding of human skin aging, J. Invest. Dermatol., 2016, 136(5), 897–899 CrossRef CAS.
- H. Rodriguez-Rocha, et al., DNA damage and autophagy, Mutat. Res., 2011, 711(1–2), 158–166 CrossRef CAS.
- K. Ghosh and B. C. Capell, The senescence-associated secretory phenotype: critical effector in skin cancer and aging, J. Invest. Dermatol., 2016, 136(11), 2133–2139 CrossRef CAS.
- L. Eckhart, E. Tschachler and F. Gruber, Autophagic Control of Skin Aging, Frontiers in cell and developmental biology, 2019, vol. 7, p. 143 Search PubMed.
- F. Afaq and H. Mukhtar, Botanical antioxidants in the prevention of photocarcinogenesis and photoaging, Exp. Dermatol., 2006, 15(9), 678–684 CrossRef CAS.
- C. Marionnet, et al., Diversity of biological effects induced by longwave UVA rays (UVA1) in reconstructed skin, PloS One, 2014, 9(8), e105263 CrossRef.
- G. T. Bowden, Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling, Nat. Rev. Cancer, 2004, 4(1), 23–35 CrossRef CAS.
- V. Chaturvedi, et al., Abnormal NF-κB signaling pathway with enhanced susceptibility to apoptosis in immortalized keratinocytes, J. Dermatol. Sci., 2001, 26(1), 67–78 CrossRef CAS.
- N. Chouinard, et al., UVB-mediated activation of p38 mitogen-activated protein kinase enhances resistance of normal human keratinocytes to apoptosis by stabilizing cytoplasmic p53, Biochem. J., 2002, 365(1), 133–145 CrossRef CAS.
- M. D'Errico, et al., Cell type and DNA damage specific response of human skin cells to environmental agents, Mutat. Res., 2007, 614(1–2), 37–47 CrossRef.
- S. Lisby, R. Gniadecki and H. C. Wulf, UV-induced DNA damage in human keratinocytes: quantitation and correlation with long-term survival, Exp. Dermatol., 2005, 14(5), 349–355 CrossRef CAS.
- T. Matsunaga, K. Hieda and O. Nikaido, Wavelength dependent formation of thymine dimers and (6-4) photoproducts in DNA by monochromatic ultraviolet light ranging from 150 to 365 nm, Photochem. Photobiol., 1991, 54(3), 403–410 CrossRef CAS.
- S. Nakajima, et al., UV light-induced DNA damage and tolerance for the survival of nucleotide excision repair-deficient human cells, J. Biol. Chem., 2004, 279(45), 46674–46677 CrossRef CAS.
- T. Budden and N. A. Bowden, The role of altered nucleotide excision repair and UVB-induced DNA damage in melanomagenesis, Int. J. Mol. Sci., 2013, 14(1), 1132–1151 CrossRef CAS.
- D. Crighton, et al., DRAM, a p53-induced modulator of autophagy, is critical for apoptosis, Cell, 2006, 126(1), 121–134 CrossRef CAS.
- Z. Feng, et al., The coordinate regulation of the p53 and mTOR pathways in cells, Proc. Natl. Acad. Sci. U.S.A., 2005, 102(23), 8204–8209 CrossRef CAS.
- A. V. Budanov and M. Karin, p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling, Cell, 2008, 134(3), 451–460 CrossRef CAS.
- W. P. Roos, A. D. Thomas and B. Kaina, DNA damage and the balance between survival and death in cancer biology, Nat. Rev. Cancer, 2016, 16(1), 20 CrossRef CAS.
- M. Zhao and D. J. Klionsky, AMPK-dependent phosphorylation of ULK1 induces autophagy, Cell Metabol., 2011, 13(2), 119–120 CrossRef CAS.
- C. Wu, et al., Role of AMPK in UVB-induced DNA damage repair and growth control, Oncogene, 2013, 32(21), 2682–2689 CrossRef CAS.
- S. Meloche and P. P. Roux, F-box proteins elongate translation during stress recovery, Sci. Signal., 2012, 5(227), pe25 Search PubMed.
- L. Qiang, et al., Autophagy gene ATG7 regulates ultraviolet radiation-induced inflammation and skin tumorigenesis, Autophagy, 2017, 13(12), 2086–2103 CrossRef CAS.
- X. Chen, et al., Ultraviolet B radiation down-regulates ULK1 and ATG7 expression and impairs the autophagy response in human keratinocytes, J. Photochem. Photobiol. B Biol., 2018, 178, 152–164 CrossRef CAS.
- X. Song, et al., Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress in vitro and in vivo, Redox Biol., 2017, 11, 219–230 CrossRef CAS.
- S. Lin, et al., Raffinose increases autophagy and reduces cell death in UVB-Irradiated keratinocytes, J. Photochem. Photobiol. B Biol., 2019, 201, 111653 CrossRef CAS.
- S. A. Umar, et al., Glycyrrhizic Acid Prevents Oxidative Stress Mediated DNA Damage Response through Modulation of Autophagy in Ultraviolet-B-Irradiated Human Primary Dermal Fibroblasts, Cell. Physiol. Biochem., 2019, 53, 242–257 CrossRef.
- R. L. Deter and C. De Duve, Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes, J. Cell Biol., 1967, 33(2), 437–449 CrossRef CAS.
- C. Kang and L. Avery, To be or not to be, the level of autophagy is the question: dual roles of autophagy in the survival response to starvation, Autophagy, 2008, 4(1), 82–84 CrossRef.
- B. Ravikumar, et al., Regulation of mammalian autophagy in physiology and pathophysiology, Physiol. Rev., 2010, 90(4), 1383–1435 CrossRef CAS.
- R. Mathew and E. White, Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night, Curr. Opin. Genet. Dev., 2011, 21(1), 113–119 CrossRef CAS.
- J. Lee, S. Giordano and J. Zhang, Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling, Biochem. J., 2011, 441(2), 523–540 CrossRef.
- Z. Tallóczy, et al., Regulation of starvation-and virus-induced autophagy by the eIF2α kinase signaling pathway, Proc. Natl. Acad. Sci. U.S.A., 2002, 99(1), 190–195 CrossRef.
- V. Deretic, T. Saitoh and S. Akira, Autophagy in infection, inflammation and immunity, Nat. Rev. Immunol., 2013, 13(10), 722–737 CrossRef CAS.
- M. Chen, et al., Essential role for autophagy in the maintenance of immunological memory against influenza infection, Nat. Med., 2014, 20(5), 503 CrossRef CAS.
- R. C. Scott, O. Schuldiner and T. P. Neufeld, Role and regulation of starvation-induced autophagy in the Drosophila fat body, Dev. Cell, 2004, 7(2), 167–178 CrossRef CAS.
- W. Martinet, et al., Autophagy in disease: a double-edged sword with therapeutic potential, Clin. Sci., 2009, 116(9), 697–712 CrossRef CAS.
- M. D. Sutton, et al., The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance, Annu. Rev. Genet., 2000, 34, 479–497 CrossRef CAS.
- B. Levine and G. Kroemer, Autophagy in the pathogenesis of disease, Cell, 2008, 132(1), 27–42 CrossRef CAS.
- B. Shiotani and L. Zou, ATR signaling at a glance, J. Cell Sci., 2009, 122(3), 301–304 CrossRef CAS.
- K. A. Cimprich and D. Cortez, ATR: an essential regulator of genome integrity, Nat. Rev. Mol. Cell Biol., 2008, 9(8), 616–627 CrossRef CAS.
- J. Smith, et al., The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer, in Advances in cancer research, Elsevier, 2010, p. 73–112 Search PubMed.
- L.-H. Chen, et al., Targeting protective autophagy exacerbates UV-triggered apoptotic cell death, Int. J. Mol. Sci., 2012, 13(1), 1209–1224 CrossRef CAS.
- L. F. Batista, et al., How DNA lesions are turned into powerful killing structures: insights from UV-induced apoptosis, Mutat. Res., 2009, 681(2–3), 197–208 CAS.
- Y. Shiloh and Y. Ziv, The ATM protein kinase: regulating the cellular response to genotoxic stress, and more, Nat. Rev. Mol. Cell Biol., 2013, 14(4), 197–210 CrossRef CAS.
- A. V. Knizhnik, et al., Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage, PloS One, 2013, 8(1), e55665 CrossRef CAS.
- S. Chatterjee, S. Sarkar and S. Bhattacharya, Toxic metals and autophagy, Chem. Res. Toxicol., 2014, 27(11), 1887–1900 Search PubMed.
- M. Di Gioacchino, et al., Autophagy as an ultrastructural marker of heavy metal toxicity in human cord blood hematopoietic stem cells, Sci. Total Environ., 2008, 392(1), 50–58 CrossRef CAS.
- A. M. Bolt, R. M. Douglas and W. T. Klimecki, Arsenite exposure in human lymphoblastoid cell lines induces autophagy and coordinated induction of lysosomal genes, Toxicol. Lett., 2010, 199(2), 153–159 CrossRef CAS.
- A. Chargui, et al., Cadmium-induced autophagy in rat kidney: an early biomarker of subtoxic exposure, Toxicol. Sci., 2011, 121(1), 31–42 CrossRef CAS.
- D. M. Smith, et al., Arsenic trioxide induces a beclin-1-independent autophagic pathway via modulation of SnoN/SkiL expression in ovarian carcinoma cells, Cell Death Differ., 2010, 17(12), 1867 CrossRef CAS.
- V. Rogov, et al., Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy, Mol. Cell, 2014, 53(2), 167–178 CrossRef CAS.
- A. Ciccia and S. J. Elledge, The DNA damage response: making it safe to play with knives, Mol. Cell, 2010, 40(2), 179–204 CrossRef CAS.
- A. Maréchal and L. Zou, DNA damage sensing by the ATM and ATR kinases, Cold Spring Harbor Perspect. Biol., 2013, 5(9), a012716 CrossRef.
- F. Ribezzo, Y. Shiloh and B. Schumacher, Systemic DNA damage responses in aging and diseases, Semin. Canc. Biol., 2016 Search PubMed.
- G. Filomeni, D. De Zio and F. Cecconi, Oxidative stress and autophagy: the clash between damage and metabolic needs, Cell Death Differ., 2015, 22(3), 377 CrossRef CAS.
- B. Poljšak and R. Dahmane, Free radicals and extrinsic skin aging, Dermatology Research and Practice, 2012, 2012, 1–4 CrossRef.
- Z. Jin and W. S. El-Deiry, Overview of cell death signaling pathways, Canc. Biol. Ther., 2005, 4(2), 147–171 CrossRef.
- R. P. Rastogi, et al., Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair, J. Nucleic Acids, 2010, 2010, 1–32 CrossRef.
- D. Speidel, The role of DNA damage responses in p53 biology, Arch. Toxicol., 2015, 89(4), 501–517 CrossRef CAS.
- A. Jazayeri, et al., ATM-and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks, Nat. Cell Biol., 2006, 8(1), 37–45 CrossRef CAS.
- K. Adams, et al., Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of the MRN protein complex, Oncogene, 2006, 25(28), 3894–3904 CrossRef CAS.
- J. Lukas, C. Lukas and J. Bartek, More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance, Nat. Cell Biol., 2011, 13(10), 1161 CrossRef CAS.
- T. Robert, et al., HDACs link the DNA damage response, processing of double-strand breaks and autophagy, Nature, 2011, 471(7336), 74 CrossRef CAS.
- S. P. Jackson and J. Bartek, The DNA-damage response in human biology and disease, Nature, 2009, 461(7267), 1071 CrossRef CAS.
- T. D. Halazonetis, V. G. Gorgoulis and J. Bartek, An oncogene-induced DNA damage model for cancer development, Science, 2008, 319(5868), 1352–1355 CrossRef CAS.
- Y. Shiloh, The ATM-mediated DNA-damage response: taking shape, Trends Biochem. Sci., 2006, 31(7), 402–410 CrossRef CAS.
- D. Cortez, et al., ATR and ATRIP: partners in checkpoint signaling, Science, 2001, 294(5547), 1713–1716 CrossRef CAS.
- P. J. O'Brien, Catalytic promiscuity and the divergent evolution of DNA repair enzymes, Chem. Rev., 2006, 106(2), 720–752 CrossRef.
- P. Schwertman, S. Bekker-Jensen and N. Mailand, Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers, Nat. Rev. Mol. Cell Biol., 2016, 17(6), 379 CrossRef CAS.
- D. M. Wilson III and V. A. Bohr, The mechanics of base excision repair, and its relationship to aging and disease, DNA Repair, 2007, 6(4), 544–559 CrossRef.
- M. K. Shigenaga, T. M. Hagen and B. N. Ames, Oxidative damage and mitochondrial decay in aging, Proc. Natl. Acad. Sci. U.S.A., 1994, 91(23), 10771–10778 CrossRef CAS.
- A. Sancar, et al., Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints, Annu. Rev. Biochem., 2004, 73(1), 39–85 CrossRef CAS.
- S. Lagerwerf, et al., DNA damage response and transcription, DNA Repair, 2011, 10(7), 743–750 CrossRef CAS.
- H. Soehnge, A. Ouhtit and O. Ananthaswamy, Mechanisms of induction of skin cancer by UV radiation, Front. Biosci., 1997, 2, D538–D551 CrossRef CAS.
- H. Ishii, et al., Components of DNA Damage Checkpoint Pathway Regulate UV Exposure-Dependent Alterations of Gene Expression of FHIT and WWOX at Chromosome Fragile Sites, Mol. Canc. Res., 2005, 3(3), 130–138 CrossRef CAS.
- L. Verschooten, L. Declercq and M. Garmyn, Adaptive response of the skin to UVB damage: role of the p53 protein, Int. J. Cosmet. Sci., 2006, 28(1), 1–7 CrossRef CAS.
- H.-J. Cha and H. Yim, The accumulation of DNA repair defects is the molecular origin of carcinogenesis, Tumor Biol., 2013, 34(6), 3293–3302 CrossRef CAS.
- J. J. Bernard, R. L. Gallo and J. Krutmann, Photoimmunology: how ultraviolet radiation affects the immune system, Nat. Rev. Immunol., 2019, 19(11), 688–701 CrossRef CAS.
- M. Uhl, et al., Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells, Cell Death Differ., 2009, 16(7), 991 CrossRef CAS.
- W. K. Kaufmann and R. S. Paules, DNA damage and cell cycle checkpoints, Faseb. J., 1996, 10(2), 238–247 CrossRef CAS.
- N. I. Orlotti, et al., Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage, Autophagy, 2012, 8(8), 1185–1196 CrossRef CAS.
- C. J. Norbury and B. Zhivotovsky, DNA damage-induced apoptosis, Oncogene, 2004, 23(16), 2797 CrossRef CAS.
- A. Alexander, et al., ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS, Proc. Natl. Acad. Sci. U.S.A., 2010, 107(9), 4153–4158 CrossRef CAS.
- A. Alexander, J. Kim and C. L. Walker, ATM engages the TSC2/mTORC1 signaling node to regulate autophagy, Autophagy, 2010, 6(5), 672–673 CrossRef.
- J. Kim, et al., AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1, Nat. Cell Biol., 2011, 13(2), 132 CrossRef CAS.
- L. Qiang, et al., Autophagy positively regulates DNA damage recognition by nucleotide excision repair, Autophagy, 2016, 12(2), 357–368 CrossRef CAS.
- C. Liang, et al., Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG, Nat. Cell Biol., 2006, 8(7), 688 CrossRef CAS.
- X. Yin, et al., UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation, EMBO Rep., 2011, 12(7), 727–734 CrossRef CAS.
- Y. Yang, et al., Autophagic UVRAG promotes UV-induced photolesion repair by activation of the CRL4DDB2 E3 ligase, Mol. Cell, 2016, 62(4), 507–519 CrossRef CAS.
- H. Vakifahmetoglu-Norberg, H.-g. Xia and J. Yuan, Pharmacologic agents targeting autophagy, J. Clin. Invest., 2015, 125(1), 5–13 CrossRef.
- Y. Bao and X. Shen, Chromatin remodeling in DNA double-strand break repair, Curr. Opin. Genet. Dev., 2007, 17(2), 126–131 CrossRef CAS.
- J. M. De Murcia, et al., Requirement of poly (ADP-ribose) polymerase in recovery from DNA damage in mice and in cells, Proc. Natl. Acad. Sci. U.S.A., 1997, 94(14), 7303–7307 CrossRef CAS.
- D. Kumar, et al., Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools, Nucleic Acids Res., 2010, 39(4), 1360–1371 CrossRef.
- S. Rello-Varona, et al., Autophagic removal of micronuclei, Cell Cycle, 2012, 11(1), 170–176 CrossRef CAS.
- B. Van Houten, S. E. Hunter and J. N. Meyer, Mitochondrial DNA damage induced autophagy, cell death, and disease, Front. Biosci., 2016, 21, 42 CrossRef CAS.
- A. Vessoni, et al., Autophagy and genomic integrity, Cell Death Differ., 2013, 20(11), 1444 CrossRef CAS.
- R. Mathew, et al., Autophagy suppresses tumor progression by limiting chromosomal instability, Genes Dev., 2007, 21(11), 1367–1381 CrossRef CAS.
- D. K. Broz, et al., Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses, Genes Dev., 2013, 27(9), 1016–1031 CrossRef CAS.
- E. McAdam, R. Brem and P. Karran, Oxidative stress-induced protein damage inhibits DNA repair and determines mutation risk and therapeutic efficacy, Mol. Cancer Res., 2016, 14(7), 612–622 CrossRef CAS.
- X. Wen, et al., Deconvoluting the role of reactive oxygen species and autophagy in human diseases, Free Radic. Biol. Med., 2013, 65, 402–410 CrossRef CAS.
- A. Jain, et al., p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription, J. Biol. Chem., 2010, 285(29), 22576–22591 CrossRef CAS.
- M. Komatsu, et al., The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1, Nat. Cell Biol., 2010, 12(3), 213 CrossRef CAS.
- G. Kroemer, G. Mariño and B. Levine, Autophagy and the integrated stress response, Mol. Cell, 2010, 40(2), 280–293 CrossRef CAS.
- O. Kapuy, et al., Systems-level feedbacks of NRF2 controlling autophagy upon oxidative stress response, Antioxidants, 2018, 7(3), 39 CrossRef.
- A. F. Nahhas, et al., The potential role of antioxidants in mitigating skin hyperpigmentation resulting from ultraviolet and visible light-induced oxidative stress, Photodermatol. Photoimmunol. Photomed., 2019, 35(6), 420–428 CrossRef CAS.
- M. Brenner and V. J. Hearing, The protective role of melanin against UV damage in human skin, Photochemistry and photobiology, 2008, 84(3), 539–549 CrossRef CAS.
- D. Kulms and T. Schwarz, Molecular mechanisms of UV-induced apoptosis, Photodermatol. Photoimmunol. Photomed., 2000, 16(5), 195–201 CrossRef CAS.
- Z. Yue, et al., Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor, Proc. Natl. Acad. Sci. U.S.A., 2003, 100(25), 15077–15082 CrossRef CAS.
- V. Karantza-Wadsworth, et al., Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis, Genes Dev., 2007, 21(13), 1621–1635 CrossRef CAS.
- A. Takamura, et al., Autophagy-deficient mice develop multiple liver tumors, Genes Dev., 2011, 25(8), 795–800 CrossRef CAS.
- H. Bae and J.-L. Guan, Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents, Mol. Cancer Res., 2011, 9(9), 1232–1241 CrossRef CAS.
- M. Scheibye-Knudsen, et al., Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy, J. Exp. Med., 2012, 209(4), 855–869 CrossRef CAS.
- N. B. Fakouri, et al., Toward understanding genomic instability, mitochondrial dysfunction and aging, FEBS J., 2019, 286(6), 1058–1073 CrossRef CAS.
- M. Abedin, et al., Autophagy delays apoptotic death in breast cancer cells following DNA damage, Cell Death Differ., 2007, 14(3), 500 CrossRef CAS.
- D. L. Bordin, et al., DNA alkylation damage and autophagy induction, Mutat. Res., 2013, 753(2), 91–99 CAS.
- M. Rieber and M. Strasberg Rieber, Sensitization to radiation-induced DNA damage accelerates loss of bcl-2 and increases apoptosis and autophagy, Canc. Biol. Ther., 2008, 7(10), 1561–1566 CrossRef CAS.
- M. Katayama, et al., DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells, Cell Death Differ., 2007, 14(3), 548 CrossRef CAS.
- G. Nestal de Moraes, et al., Insights into a critical role of the FOXO3a-FOXM1 axis in DNA damage response and genotoxic drug resistance, Curr. Drug Targets, 2016, 17(2), 164–177 CrossRef CAS.
- A. Alexander, et al., Erratum: ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS (Proceedings of the National Academy of Sciences (2010) 107 (4153-4158)), Proc. Natl. Acad. Sci. U. S. A., 2012, 109(21), 4153–4158 Search PubMed.
- K. Inoki, T. Zhu and K.-L. Guan, TSC2 mediates cellular energy response to control cell growth and survival, Cell, 2003, 115(5), 577–590 CrossRef CAS.
- A. Desantis, et al., Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy, EMBO J., 2015, 34(9), 1214–1230 CrossRef CAS.
- C. Mammucari, et al., FoxO3 controls autophagy in skeletal muscle in vivo, Cell Metabol., 2007, 6(6), 458–471 CrossRef CAS.
- A. Salminen and K. Kaarniranta, Regulation of the aging process by autophagy, Trends Mol. Med., 2009, 15(5), 217–224 CrossRef CAS.
- F. Chiacchiera and C. Simone, The AMPK-FoxO3A axis as a target for cancer treatment, Cell Cycle, 2010, 9(6), 1091–1096 CrossRef CAS.
- J. A. Muñoz-Gámez, et al., PARP-1 is involved in autophagy induced by DNA damage, Autophagy, 2009, 5(1), 61–74 CrossRef.
- A. Valbuena, S. Castro-Obregon and P. A. Lazo, Downregulation of VRK1 by p53 in response to DNA damage is mediated by the autophagic pathway, PloS One, 2011, 6(2), e17320 CrossRef CAS.
- J. Yi and J. Luo, SIRT1 and p53, effect on cancer, senescence and beyond, Biochim. Biophys. Acta Protein Proteonomics, 2010, 1804(8), 1684–1689 CrossRef CAS.
- E. F. Fang, et al., Defective mitophagy in XPA via PARP-1 hyperactivation and NAD+/SIRT1 reduction, Cell, 2014, 157(4), 882–896 CrossRef CAS.
- M. A. El-Mahdy, et al., Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC, J. Biol. Chem., 2006, 281(19), 13404–13411 CrossRef CAS.
- X. Zeng and T. J. Kinsella, BNIP3 is essential for mediating 6-thioguanine-and 5-fluorouracil-induced autophagy following DNA mismatch repair processing, Cell Res., 2010, 20(6), 665 CrossRef CAS.
- A. Garufi, et al., p53-dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions, J. Exp. Clin. Canc. Res., 2017, 36(1), 126 CrossRef.
- Z. Zhao, et al., A dual role for UVRAG in maintaining chromosomal stability independent of autophagy, Dev. Cell, 2012, 22(5), 1001–1016 CrossRef CAS.
- J. M. Park, et al., Beclin 1 and UVRAG confer protection from radiation-induced DNA damage and maintain centrosome stability in colorectal cancer cells, PloS One, 2014, 9(6), e100819 CrossRef.
- P. Voskamp, et al., Rapamycin impairs UV induction of mutant-p53 overexpressing cell clusters without affecting tumor onset, Int. J. Canc., 2012, 131(6), 1267–1276 CrossRef CAS.
- S. Galati, et al., Autophagy: a player in response to oxidative stress and DNA damage, Oxid. Med. Cell. Longev., 2019, 2019, 1–12 CrossRef.
- S. Shimizu, et al., Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes, Nat. Cell Biol., 2004, 6(12), 1221 CrossRef CAS.
- M. van der Vaart, et al., The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense, Cell Host Microbe, 2014, 15(6), 753–767 CrossRef CAS.
- L. Y. Mah, et al., DRAM-1 encodes multiple isoforms that regulate autophagy, Autophagy, 2012, 8(1), 18–28 CrossRef CAS.
- A. Martoriati, et al., dapk1, encoding an activator of a p19 ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53, Oncogene, 2005, 24(8), 1461 CrossRef CAS.
- Z. He, et al., p73 regulates autophagy and hepatocellular lipid metabolism through a transcriptional activation of the ATG5 gene, Cell Death Differ., 2013, 20(10), 1415 CrossRef CAS.
- J. M. Rodríguez-Vargas, et al., ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy, Cell Res., 2012, 22(7), 1181 CrossRef.
- C. Park, Y. Suh and A. M. Cuervo, Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage, Nat. Commun., 2015, 6, 6823 CrossRef CAS.
- F. Dotiwala, et al., DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase, Proc. Natl. Acad. Sci. U.S.A., 2013, 110(1), E41–E49 CrossRef CAS.
- V. V. Eapen, et al., A pathway of targeted autophagy is induced by DNA damage in budding yeast, Proc. Natl. Acad. Sci. U.S.A., 2017, 114(7), E1158–E1167 CrossRef CAS.
- K. d. A. Mesquita, Role of mitochondria and DNA damage responses in cancer stem cells resistance to chemotherapy, 2016 Search PubMed.
- Y. Johmura and M. Nakanishi, Multiple facets of p53 in senescence induction and maintenance, Canc. Sci., 2016, 107(11), 1550–1555 CrossRef CAS.
- C. Hetz and F. R. Papa, The unfolded protein response and cell fate control, Mol. Cell, 2018, 69(2), 169–181 CrossRef CAS.
- O. Yamaguchi and K. Otsu, Role of autophagy in aging, J. Cardiovasc. Pharmacol., 2012, 60(3), 242–247 CrossRef CAS.
- N. Saewan and A. Jimtaisong, Natural products as photoprotection, J. Cosmet. Dermatol., 2015, 14(1), 47–63 CrossRef.
- V. Kostyuk, et al., Natural substances for prevention of skin photoaging: screening systems in the development of sunscreen and rejuvenation cosmetics, Rejuvenation Res., 2018, 21(2), 91–101 CrossRef CAS.
- C. Parrado, et al., Oral photoprotection: effective agents and potential candidates, Front. Med., 2018, 5, 188 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |