
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a multi-branched dandelion-like SAPO-11 by an in situ inoculating seed-induced-steam-assisted conversion method (SISAC) as a highly effective hydroisomerization support†
Zhou Chen,
Wenjing Song,
Shaohong Zhu,
Weikun Lai,
Xiaodong Yi * and
Weiping Fang
* and
Weiping Fang
National Engineering Laboratory for Green Chemical Productions of Alcohols, Ethers and Esters, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: xdyi@xmu.edu.cn
First published on 16th January 2017
Abstract
A dandelion-like SAPO-11 with multiple branches was fabricated by seed-induced steam-assisted conversion (SISAC), which combined an in situ inoculating seed method (in this case, the initial gel was pre-crystallized at 433 K for 24 h) and steam-assisted conversion (SAC) process. The dandelion-like SAPO-11 was fully crystalline and exhibited more acidity, external surface and mesopore volume compared with conventional hydrothermally crystallized SAPO-11, confirmed by Ar adsorption–desorption, pyridine-adsorbed infrared (Py-IR), 2,6-dimethylpyridine-adsorbed infrared (2,6-DMPy-IR) and NH3 temperature-programmed desorption (NH3-TPD). After loading Pt nanoparticles by incipient wetness impregnation, the morphological modification (benefiting from smaller nanobranches and introducing more external surface active sites) leads to enhanced heptane hydroisomerization activity of the dandelion-like Pt/SAPO-11 as great as 15.4% higher in comparison with a conventional Pt/SAPO-11 catalyst.
Introduction
The catalytic hydroisomerization of n-alkanes to their corresponding branched isomers has generated a great amount of interest in the modern petroleum industry. This process is considered to be an effective strategy to increase the octane number of gasoline and to improve the cold-flow properties of middle-distillate fuels and lube base oils.1 In particular, searching for catalysts to isomerize C5–C8 n-alkanes of low octane rating into branched isomers of high octane rating has been the subject of many investigations around the world. Such catalysts could satisfy the ever-increasing demand for high-octane gasoline due to the rapid development of the automobile industry and increasingly rigorous environmental regulation.In the hydroisomerization process, catalytic systems have been successfully constructed for hydroisomerization evolution. These systems typically use bifunctional catalysts that contain noble metal particles as a hydro-/dehydrogenation component in conjunction with an acid component. Recently, among the numerous catalytic materials explored for n-alkane isomerization, it has been reported that zeolite with large pores, such as Pt/HBEA(BEA),2 mesoporous aluminosilicate3,4 and strongly acidic zeolite, such as ZSM-5(MFI)5 and ZSM-22(TON),6 can produce a large yield of cracked products because these catalysts have either spatial constraints that might induce shape selectivity or excessive acid sites that would induce cracking. On the other hand, SAPO-11 (AEL) with one-dimensional 10-membered ring channels has attracted considerable interest in the field of hydroisomerization due to its shape selectivity and moderate acidity.7,8
However, under the same metal loadings, considering the mass transport viewpoint, the challenge is its diffusion efficiency for which the interior active sites of the support are less accessible to reactants, especially for bulkier molecules.9,10 This is due to the predominance micropores. To date, extensive efforts using hydrothermal synthesis conditions11–14 have been devoted to synthesize SAPO-11 with more exposed external active sites to solve this problem. These strategies have the disadvantage of relying on the addition of a secondary template, such as carbon,15 CTAB,13,16 organosilane17 or glucose.18 However, the use of a secondary porogen results in a complicated synthetic process and extra energy consumption during the calcination procedures. Therefore, a simple and cost-effective manufacturing procedure to construct SAPO-11 with a large number of exposed external active sites is yet to be developed.
An alternative to the conventional hydrothermal synthesis (CHT) method is steam-assisted conversion (SAC), one of the typical dry gel conversion (DGC) methods for zeolite synthesis. This method has recently attracted increasing interest for the synthesis of hierarchical zeolites (HSZs)19–21 and superfine zeolites.22–24 Under these conditions, the zeolite precursor mixtures are completely dried before they are converted by steam, and such a quasi-solid-state has proven helpful against phase separation and provides an ideal condition for massive nucleation. Because the massive nucleation of zeolite facilitates the generation of nano-zeolite, HSZs are generated by controlling the assembly of the precursor nanoparticles (NPs), which are often observed in the early stages of the zeolite crystallization process.25 However, most literature that reports the construction of HSZs using the SAC-based method usually describes the use of some surfactant such as CTAB,26 F127 (ref. 27) or an organosilane,28 and a hard template29 to manipulate the regular assembly of the NPs and their crystallization process. Similar mechanisms and methods have been reported for a variety of morphologies and structures of SAPO-11. Lei et al.23 used CTAB to synthesize a superfine SAPO-11 by the SAC method as an FCC catalyst promoter. In addition, in the presence of fluoride ions in the synthesized dry gel, SAPO-11 has been reported to be an excellent promoter for hydroisomerization due to the existence of superfine particles.22 For all of the examples mentioned above, a large amount of additive is still needed for the formation of a hierarchical network or superfine particles. So the challenge considered for the synthesis of smaller SAPO-11 particles with more external active sites by the SAC method is to reduce the doping during the synthetic process.
On the other hand, engineering a hierarchical structure on conventional zeolites by a seed-assisted process is another encouraging way to address the aforementioned problems by promoting nucleation. Seeds that act as a nucleation promoting agent have been widely studied to improve the yield or shorten the crystallization time of the product.30 Accordingly, it is expected that the combination of SAC and the seeding technology would constitute a high yield, low-cost, and flexible method for constructing HSZs without a secondary template. Very recently, Cheng et al.31 obtained a rich intercrystalline mesoporosity by a seeding-steam-assisted conversion method. Foreign seeds were exposed to the pre-treated dry gel and the crystallization of beta zeolite commenced derived from the interfaces between the seeds (as nucleation centers) and the amorphous compound. The beta zeolite thus obtained possessed a high surface area and pore volume. The high LDPE cracking activity was attributed to the synergistic effects of the TEAOH and extraneous zeolite seeds. There is no doubt that the seed play a vital role in the morphology and physical properties of the obtained zeolites.
In the present study, our aim is to develop an effective method to synthesize SAPO-11 with more external acid sites. To combine the advantages of SAC and a seed-induced method and intensify the interaction between the seed and dry gel, we have designed a novel synthetic route by inoculating the seed in situ and then growing the crystals during a subsequent SAC process, considering the advantage of both low temperature treatment and the dry gel conversion system which facilitated the crystal nucleation. In this novel in situ inoculating seed-induced-SAC (SISAC) method, as illustrated in Scheme S1, ES1.† The aluminosilicophosphate gel was pre-crystallized at 433 K for 24 h, and the intermediate with some embryonic AEL structure (acting as the seed structure) was obtained during this low-temperature hydrothermal process. The intermediates were then transformed into SAPO-11 crystals via a non-classical crystallization mechanism32–34 in the course of a further SAC crystal growth process. As a result, the dandelion-like SAPO-11 was formed.
To the best of our knowledge, no prior work regarding the application of an in situ inoculating seed method to obtain dry gel for constructing SAPO-11 has been reported to date, and the dandelion-like SAPO-11 is first reported here. More importantly, such a larger external surface, mesopore volume and greater number of acid sites endowed with the dandelion-like Pt/SAPO-11 catalyst exhibited an excellent catalytic performance in heptane hydroisomerization.
Experimental section
Synthesis samples
Tetraethyl orthosilicate (TEOS, C12H28O4Si), aluminum isopropoxide (AIP, 24% Al2O3), ortho-phosphoric acid (85% H3PO4), heptane (99.9%), chloroplatinic acid hexahydrate (H2PtCl6·6H2O) were obtained from Sinopharm Chemical Reagent (China). Di-n-propylamine (DPA, C6H15N) was supplied from Aladdin Chemical Reagent Company. All of the above chemicals were used as purchased without any further purification. The SAPO-11 samples were hydrothermally synthesized using TEOS, AIP, ortho-phosphoric acid and DPA as the Si source, Al source, P source, and template, respectively.The typical gel ratio for each SAPO-11 was 1.0Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0P2O5:2.0DPA:0.3TEOS:50H2O. For the initial gel, the synthesis was carried out as follows: 6.81 g of AIP was hydrolyzed in 15 ml of deionized water with stirring for 12 h. Then, 3.84 g of H3PO4 was added to the mixture and homogenized for 4 h. Afterwards, 4.6 ml of DPA was added to the mixture, which was followed by the addition of 1.3 ml of TEOS. For the C-HTC-SAPO-11 (C-HTC stands for the conventional hydrothermal conversion method), the initial gel was transferred into a Teflon-lined stainless steel autoclave and hydrothermally crystallized at 433 K for 24 h.
:1.0P2O5:2.0DPA:0.3TEOS:50H2O. For the initial gel, the synthesis was carried out as follows: 6.81 g of AIP was hydrolyzed in 15 ml of deionized water with stirring for 12 h. Then, 3.84 g of H3PO4 was added to the mixture and homogenized for 4 h. Afterwards, 4.6 ml of DPA was added to the mixture, which was followed by the addition of 1.3 ml of TEOS. For the C-HTC-SAPO-11 (C-HTC stands for the conventional hydrothermal conversion method), the initial gel was transferred into a Teflon-lined stainless steel autoclave and hydrothermally crystallized at 433 K for 24 h.
For the C-SAC-SAPO-11 (C-SAC stands for the conventional steam-assisted method), the initial gel was further stirred in a water bath at 333 K to evaporate the solvent, followed by adding 25 ml ethanol with vigorous stirring. The solution was stirred until it could no longer rotate before being transferred to a Petri dish and placed in an oven at 333 K for 48 h where the residual solvent was allowed to evaporate. Then, the obtained dry gel (denoted as DG-C) was ground to a powder using a mortar and pestle before being charged into a 25 ml crucible and placed inside a Teflon-lined stainless steel autoclave (100 ml). A total of 20 ml water was added outside the crucible to create steam for the steam-assisted conversion synthesis conditions. The autoclave was placed in an oven set at 473 K and maintained at that temperature for up to 24 h.
For comparison, seed-SAC-SAPO-11 was also synthesized. An amount of 1 wt% C-HTC-SAPO-11 powder was added to the initial gel under stirring conditions, followed by the same procedure as the C-SAC-SAPO-11.
For the SISAC-SAPO-11, the initial gel was pre-crystallized at 433 K for 24 h. In such cases, some embryonic AEL structure was implanted into the amorphous gel, which was deemed to be the in situ inoculating seed method (denoted as SI) and provides an intimate and large contact interface between the seed and amorphous gel. This can shorten the induction period and promote the nucleation rate of the SAPO-11 crystal. Under this condition, the obtained gel was denoted as DG-160, which was then subjected to the same procedure as the C-SAC-SAPO-11.
The obtained product was recovered by filtration, washed thoroughly with deionized water, dried at 373 K overnight and then calcined at 873 K for 6 h using a ramp rate of 2 K min−1 in air.
Preparation of Pt/SAPO-11 catalysts
All the calcined samples were extruded, crushed, and sieved to obtain 20–40 mesh particles. Then, the 0.5 wt% Pt was supported on the shaped samples via the incipient wetness impregnation method using an aqueous solution of H2PtCl6. After impregnation, the samples were dried overnight at 373 K and calcined in a muffle furnace at 723 K for 3 h.Characterization
Powder X-ray diffraction (XRD) characterization was performed using an X'Pert Pro automatic powder diffractometer operated at 35 kV and 15 mA using CuKα (K = 0.15406 nm) monochromatized radiation in all cases. Ar physisorption was measured with a Micromeritics ASAP 2020 M instrument. Prior to the adsorption, the sample was evacuated at 573 K for 3 h. Scanning electron microscopy (SEM) images were obtained on a field emission SEM (FESEM) instrument (Zeiss SIGMA, Germany). Transmission electron microscopy (TEM) experiments were performed using an FEI Tecnai 30 high-resolution transmission electron microscope (Philips Analytical) operated at an accelerating voltage of 300 kV. The 27Al and 31P solid-state NMR spectra were measured on an Avance III Bruker device at a frequency of 500 MHz. The powdered samples were packed in the rotor and spun at a rate of 12 kHz. The experiments were conducted at a contact time of 2 ms and a total of 20 scans were recorded with a 5 s recycle delay for each sample.The X-ray fluorescence (XRF) and X-ray photoelectron spectroscopy (XPS) characterizations were carried out to obtain information about the bulk and surface compositions of the SAPO-11, respectively. XRF measurements were conducted on a S8 TIGER (BRUKER) operated at 60 kV and 50 mA. XPS measurements were made with a Qtac100 spectrometer using the incident radiation Al Ka of 1486.6 eV at 250 W, 20 mA, and 50 eV of pass energy. The C1s peak at 284.6 eV was used as the internal standard to compensate for sample charging. The atomic ratios were quantitatively analyzed by determining the elemental areas of the core-level peaks for O1s, Si2p, Al2p, and P2p.
Raman spectra were recorded on Renishaw Invia Raman systems equipped with CCD detectors. The line at 532 nm from a He–Cd laser was used as an excitation source with an output of 5.6 mW. The power of the laser at the samples was approximately 0.56 mW.
The FT-IR spectra of pyridine and 2,6-dimethylpyridine adsorption were recorded on a Thermo Nicolet Nexus spectrometer with a resolution of 4 cm−1. The samples were pressed into self-supporting discs, which were treated in H2 at 673 K in an IR quartz cell with a CaF2 window for 1 h, followed by evacuation at the same temperature for 5 min to remove the gas-phase H2. After cooling to 373 K, the samples were exposed to pyridine vapor for 10 min. Then, the spectra were recorded after evacuation at 423 K. For calculating the number of total Brønsted acid sites, Lewis acid sites and external Brønsted acid sites, the molar extinction coefficient of Py-IR was used according to the report of Zeng et al.,35 and 2,6-DMPy-IR was used according to Onfroy et al.36 reported.
NH3 temperature-programmed desorption (NH3-TPD) measurements were performed on a Micromeritics AutoChem II 2920 instrument. Typically, the sample was pretreated in a quartz reactor with an O2–He gas (20 vol% O2) flow at 673 K for 1 h, followed by purging with high-purity He. The adsorption of NH3 was performed at 373 K in an NH3–He mixture (10 vol% NH3) for 1 h, and TPD was performed in a He flow by raising the temperature to 1173 K at a rate of 10 K min−1.
CO chemisorption was performed at 308 K with a micromeritics ASAP 2020 apparatus. Prior to the measurement, the catalysts (0.1 g) were reduced in situ for 1 h at 573 K in H2 (heating rate of 10 K min−1), and evacuated at 573 K to remove chemisorbed hydrogen, and cooling to 308 K under vacuum. Analysis was performed by collecting a CO adsorption isotherm at 308 K, evacuating the sample at 308 K for 1 h to remove the physisorption of CO, and collecting a second isotherm at 308 K.
H2-temperature programmed reduction (H2-TPR) analyses were carried out with the GC-TPR apparatus. Prior to reduction, the catalyst was first treated under O2 flow of 40 ml min−1 at 673 K for 30 min. After cooled to room temperature, a flow of 5% H2/Ar (50 ml min−1) was switched and the sample was heated to 973 K with a rate of 10 K min−1. The H2 consumption of the catalyst reduction was determined by a thermal conductivity detector (TCD).
Catalytic tests
The activity evaluation was carried out in a fixed-bed continuous-flow stainless steel reactor with an internal diameter of 4.0 mm, using n-heptane (n-C7, Sinopharm chemical reagent) hydroisomerization as a model reaction. In each run, 0.5 g of the catalyst was loaded into the reactor and the reactant n-heptane was fed into the reactor by a syringe pump. Prior to the reaction, the catalyst was reduced in situ at 673 K for 4 h under flowing purity H2. The temperature was then lowered to the reaction temperature. The measurement was carried out at a total pressure of 1.5 MPa with a H2/n-heptane volumetric ratio of 400. The hydrocarbon products were collected online after a steady state had been reached and analyzed by a gas chromatograph (GC-9560) equipped with a flame ionization detector (FID) and a HP-PONA capillary column (50 m × 0.2 mm).Result and discussion
Physicochemical properties of the dry gel
To study the difference in the dry gel obtained by the above method and to confirm that some embryonic seed structure was embedded into the dry gel compound in situ during the pre-crystallization process, the power XRD patterns, Raman spectra, N2 adsorption–desorption isotherms, and the 31P and 27Al solid-state MAS NMR spectra of the dry gel were studied.As shown in Fig. 1A, the XRD pattern of the conventional dry gel contains an extremely broad halo near 2θ = 23°, indicating the predominant existence of an amorphous phase. In the low-angle region, the two peaks with 2θ values of 6.2° and 8.1° suggest that a layered intermediate with a certain degree of long-range ordering exists.37 In the present study, the layered phase is probably partially held by weak forces because the inorganic SAPO sheets are held together by the protonated amines via van der Waals forces or very weak hydrogen bonding.37,38 A similar phenomenon was reported for other aluminophosphate dry gels in some previous studies.39–43 The layered intermediate structure was also confirmed by 31P solid-state MAS NMR. As shown in Fig. 1C, the 31P spectra of DG-C has a sharp peak at −19 ppm, this was attributed to the crystalline layered phase.42 Interestingly, upon treating the aluminosilicophosphate gel under hydrothermal synthesis at 433 K for 24 h, the diffraction peak with 2θ = 6.2° which is attributed to the (100) layered structure reflection37 became spiculate, implying that the layered intermediate material has become more orderly. It is worth noting that some weak diffraction peaks at 8.1, 21.2, 22.1–23.2° and 26° corresponding to (110), (002), (240) and (400) are emphasized (labelled with ◆). These are vested in the reflections of the AEL structure and suggest that the periodic framework of the crystalline SAPO-11 embryo begins to form in the initial DG-160. The diffraction peaks at 12.3° and 18.4° (labelled with □) are assigned to the transitional intermediate structure of SAPO-type zeolite, which is also observed in other reports.37,39 However, this situation was not observed and those reflections due to the embryonic AEL phase are almost nonexistent in DG-C, which was ascribed to the pre-crystallization process.
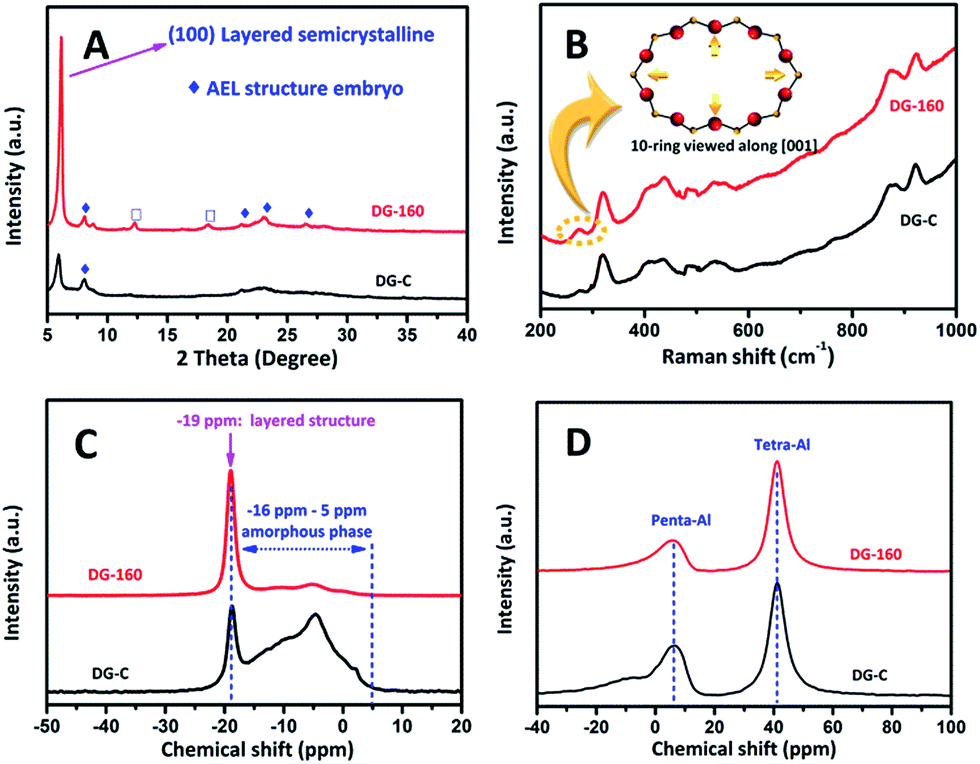 | ||
| Fig. 1 XRD patterns (A), Raman spectra (B), 31P (C) and 27Al MAS NMR spectra (D) of synthesized DG-C and DG-160. | ||
To analyze the ring systems of the dry gel, we recorded the Raman spectra because of the T–O–T (T = tetrahedral site) bending region being very sensitive to the ring systems in the zeolite frameworks.44–46 As shown in Fig. 1B, the observation of a relatively strong peak in the Raman shift at 262 cm−1 suggested that a large number of 10-ring structure existed in DG-160, and the Raman shift band in the region at 400–500 cm−1 is assigned to the bending vibrations of the 4- and/or 6-membered rings in the zeolite framework. Compared to DG-C, the enhanced intensity of the peak area in this region is relatively large, suggesting the existence of a large number of 4- and/or 6-membered rings in DG-160, which was attributed to the implantation of a large amount of AEL embryonic structure during the first step. The Raman results indicated that the DG-160 contains some similarity to the AEL structure. The rings existing in DG-160 will facilitate the transformation of amorphous materials into a 3D AEL structure. Intriguingly, the BET surface area determined by N2 adsorption–desorption isotherms (Fig. S1, ES1†) show that DG-160 has a large value (57.3 m2 g−1) compared with the DG-C (28.9 m2 g−1), which was most likely due to a different form of the as-synthesized dry gel.
To characterize the local environments of the P and Al atoms in the layered intermediate compound, 31P and 27Al solid-state MAS NMR spectra were recorded. In the 31P MAS NMR spectra (Fig. 1C), both dry gel exhibits a sharp peak at −19 ppm (denoted as P−19), which implies the presence of a layered intermediate,42 while the broad superimposed peak between −16 ppm and 5 ppm (denoted as Pa) is due to the existence of an amorphous phase.42 There is no distinct peak at around −30 ppm, indicating that the P in the layered intermediate is not fully condensed to a P(OAl)4 environment. However, the signal intensity ratio of P−19 to Pa follows the order of DG-160 > DG-C, implying that the precursor P species was transformed into a layered semi-crystalline phase with long-range ordering during the pre-crystallization process. Similarly, the 27Al MAS NMR spectra in Fig. 1D show that the above mentioned two dry gels both have peaks at approximately 40 ppm (denoted as Al40) and 6 ppm (denoted as Al6) manifesting the presence of tetra-coordinated Al and penta-coordinated Al, respectively. The signal intensity ratio of Al40 to Al6 follows the order of DG-160 > DG-C, consistent with the order of the 31P MAS NMR spectra and the result of XRD and Raman spectroscopy. The slight difference emphasizes that the pre-crystallization treatment accelerated a deeper transformation of AlV into AlIV in the dry gel. This trend is in line with the coordination changes during crystallization.47 This phenomenon is attributed to the generation of more nuclei and thus the formation of smaller SAPO-11 during the subsequent SAC process (see below).
Collectively, these data suggest that under pre-crystallization treatment, the induction stage of the SAPO-11 crystal nuclei during the dry gel preparation process is enhanced by this step, as synthesis at low temperature tends to promote the nucleation process of zeolite. Some embryonic AEL phase was subsequently incorporated and initially formed (the featured peaks were emphasized in the XRD and Raman spectra) in the dry gel. The embryonic AEL structure was well-proportioned and implanted in a layered semi-crystalline form. It will act in a seeding role, which was denoted an in situ inoculating seed method. The strong interaction between the AEL seed and the amorphous compound may influence the morphology and physical properties of the SAPO-11 and will accelerate the crystallization growth rate of SAPO-11. Thanks to the nanoscale intimacy of the seed structure and amorphous compound, the merits of the alleged in situ inoculating seed method are: (1) promoting the nucleation of SAPO-11 from pre-crystalline initial aluminosilicophosphate gel, thus creating a mass of growth centers by lowering the activation energy; (2) distinguishing the conventional seed-assisted method, the ultra-strong interaction (nanoscale intimacy) between the seed structure and amorphous compound will assume a pivotal role in forming a distinct morphology.
Crystallization and morphology evolution under different SAC methods
The above characterizations clearly demonstrate that the pre-crystallization has a significant impact on the structure of dry gel. To gain a clearer understanding of the structural evolution of the two SAC methods, the crystallization process was thoroughly investigated by XRD and SEM characterization. XRD patterns of as-synthesized samples (unwashed samples) at different crystallization times prepared by different methods are shown in Fig. 2, which indicate the crystalline evolution of the C-SAC-SAPO-11 and SISAC-SAPO-11. For the initial dry gel mixtures (DG-160 and DG-C), both the XRD patterns contain a strong reflection with 2θ = 6.2°, which was attributed to the layered intermediate structure.37,39 Similar to the above analysis in the section on the physicochemical properties of the dry gel, a similar phenomenon was reported for other aluminophosphate dry gel structures in some previous studies.39–43 Because of the pre-crystallization process, the initial XRD patterns of DG-160 show some weak diffraction peaks at 8.1, 21.2, 22.1–23.2° and 26° which are assigned to the reflections of the AEL structure. In fact, for the C-SAC method, the AEL phase emerged after 3 h under crystallization conditions (Fig. 2A). For the SISAC method, the layered semi-crystalline intermediate was maintained until the period of the crystallization stage exceeded 3 h. Compared with a crystallization time of 2 h, the typical diffraction peaks of SAPO-11 molecular sieve, at 2θ = 8.1, 9.4, 13.1, 15.6, 20.3, 21.2 and 22.1–23.2° were detected, suggesting the solid sample was mainly composed of the AEL structure mixed with some semi-crystalline intermediate structure. The intensity of the (100) reflection became weaker and broader because of the consumption of the semi-crystalline intermediate phase and gradually evolved into the AEL crystalline phase. The obvious peak at 6.2° disappeared after the crystallization period reached 4 h, while the fully developed three-dimensional covalent AEL crystalline structure kept on growing in the subsequent SAC process. The structural evolution tendency for the C-SAC method is similar to that of the SISAC method. However, a complete transformation of the amorphous phase to the AEL structure occurs in 6 h. This fact indicates that the degree of embedding of the embryonic AEL structure affects the further crystallization rate of SAPO-11. | ||
| Fig. 2 XRD patterns of SAPO-11 at different crystallization time prepared by conventional SAC method (A) and SISAC method (B). | ||
The more clearly to compare the crystallization rate of both methods, the crystallization curve is plotted in Fig. S2 ES2.† It is immediately obvious from the curve that the SISAC method has a faster crystallization rate, as shown in the red curve in Fig. S2 ES2.† This is to be expected, since the seed structure was introduced by the SI method and was able to facilitate nucleation and growth under the same SAC conditions.
To elucidate the formation process of the SAPO-11 under the two methods, several unwashed samples were collected during each SAC procedure and measured by SEM (Fig. 3). For the C-SAC method at t = 0–3 h, the amorphous morphology was mainly observed, in which the XRD patterns mostly showed the amorphous phase. That is, no marked crystallization occurs at these times. After 6 h (Fig. 3B), these amorphous gel areas disperse and re-associate to form some well-defined regions, and a few cubes appear on the profile of the agglomerate. After being treated for 8 h (Fig. 3C), the cubical region becomes even clearer, and it forms interconnected micro-spheres with diameters of approximately 1–2 μm. Meanwhile, some smaller particles are observed around the surfaces of the micro-spheres, and these particles seem to attach to the surfaces of the larger micro-spheres and become more crystalline under the subsequent SAC process. As the heating time is further prolonged to 12 h, the micro-spheres becomes even larger and the particles around it become fewer, which indicates the evolution of crystallization from particles merging into the crystalline AEL-type structure under this C-SAC method. After crystallization for 24 h, the final SAPO-11 crystals are present in Fig. 3E, and the amorphous gel or fine particles have been completely consumed. These changes are consistent with the trend of the XRD pattern evolution shown in Fig. 2A.
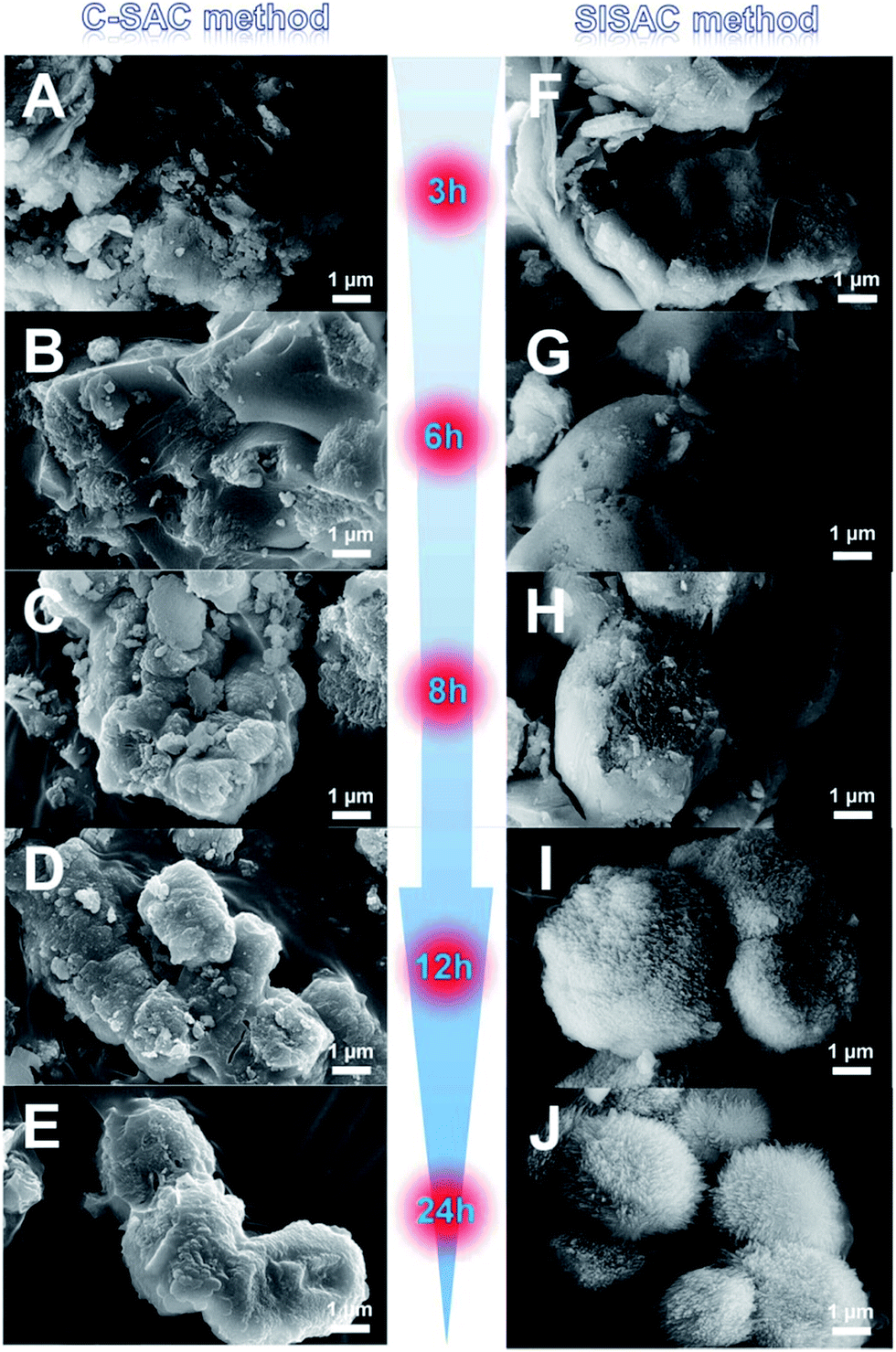 | ||
| Fig. 3 Evolution of SEM images as a function of time in the formation of SAPO-11 obtained by the conventional SAC method (left) and the SISAC method (right). | ||
For the SISAC method, at the initial 2 h, the aggregation of the layered semi-crystalline intermediate with relatively smooth surfaces was observed, almost the same as the morphology of DG-160 (Fig. S3B ES3†). The semi-crystalline intermediate with a layered structure does not have a regular shape and size, although the embryonic AEL phase existed in these compounds. This must be due to the embryonic AEL being infrequent and not growing and stacking into primary crystals. With an increase in the crystallization time to 3 h, the bulk gel appears to aggregate and fuse together (Fig. 3F). Meanwhile, the margins of the crystals progressively emerged. A visible cavity with branches emerged (Fig. 3F and G), and the SAPO-11 crystallinity increases. This is explained by the enhanced intensity of the peaks assigned to the typical AEL structure with regard to the layered structure at 6.2° becoming weaker, but still not being fully converted to the regular morphology. It was clarified that the SISAC condition afforded a maximized local concentration for massive nucleation because of the higher supersaturation. In addition, the embryonic AEL structure which was generated during the pre-crystallization process can act in a seed role to accelerate the nucleation process of SAPO-11. Interestingly, the SEM images at 8 h (Fig. 3H) of crystallization time reveal that the bulk materials exhibit a multi-branched surface with branch lengths of approximately 200 nm and identical crystal orientations. It seems that these branches of dandelion-like SAPO-11 derived from the shell. It has been previously stated that the seed structure existed in the DG-160 and served as the growth center. It is unambiguous that the formation of the branches was attributed to the seed-induced amorphous compound transforming into AEL crystals. After prolonging the crystallization period to 12 h (Fig. 3I), there is a slight increase in the branched width and well-defined branches on the outer surface were observed. These phenomena demonstrated that using the SASAC method, crystallization preferentially takes place on the outer surface, and extends from the shell to the core accompanied by the inner amorphous gel gradually being consumed. This is in good agreement with the enhanced intensity of the AEL diffraction peaks. After 24 h of SAC treatment, the branches on the outer surface finally result in the formation of a fully oriented and assembled dandelion-like SAPO-11.
It is well-known that zeolite crystal growth mechanism does not occur via the classic Lamer mechanism,48 but instead is via the non-classic orientated growth (OA) mechanism suggested by Banfield et al.49 The OA mechanism50 of crystal growth has been observed in zeolites of different structures such as SSZ-13,34 Beta,51 SAPO-11,52 and SAPO-5.53 A similar OA crystal-growth phenomenon was also observed in our system using a different SAC method. However, the existence of the seed structure in DG-160 results in a different morphology of SISAC-SAPO-11, and we will further study the physicochemical properties of three SAPO-11 samples.
Physicochemical properties of the SAPO-11 samples
The SEM and TEM images of SAPO-11 prepared by different methods are shown in Fig. 4. The C-HTC-SAPO-11 displays a spheroidal crystal with a rough surface (Fig. 4A), and the margin of TEM image (Fig. 4E) shows that the primary C-HTC-SAPO-11 crystal was nano-cubic, having a morphology similar to that studied by Liu et al.14 On the other hand, the C-SAC-SAPO-11 displays a heterogeneous particle aggregation, as shown in Fig. 4B and F. The primary crystal size is smaller than that of C-HTC-SAPO-11 and no irregularly shaped amorphous phase is present. This phenomenon was attributed to the fast nucleation in the quasi-solid dry gel systems. As shown in Fig. 4C and D, the crystal of the SISAC-SAPO-11 becomes rougher and the surface comprises an abundant number of branches, which looks just like a dandelion.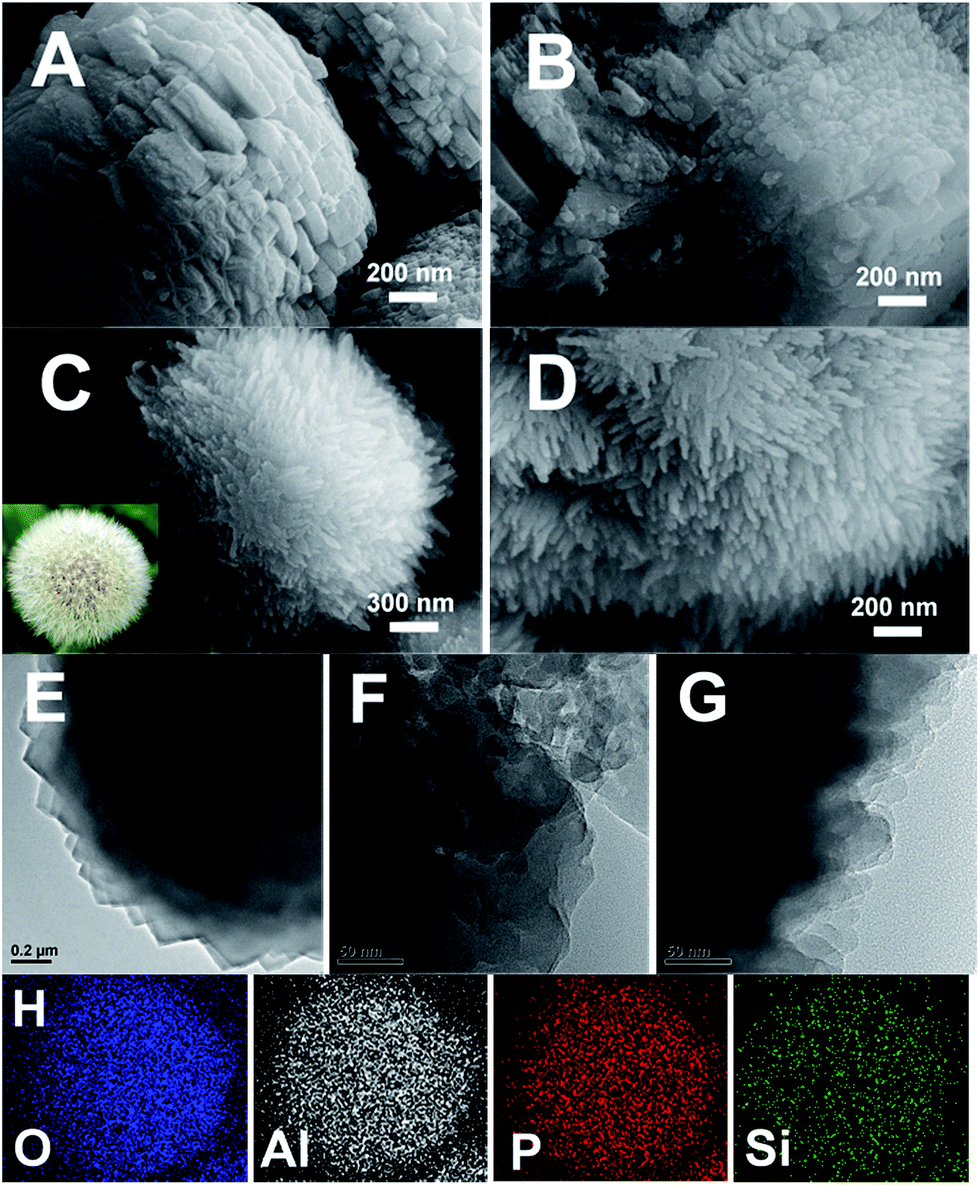 | ||
| Fig. 4 SEM images of C-HTC-SAPO-11 (A), C-SAC-SAPO-11 (B), and SISAC-SAPO-11 (C, D); TEM image of C-DTC-SAPO-11 (E), C-SAC-SAPO-11 (F), SISAC-SAPO-11 (G); EDX element mapping of dandelion-like SISAC-SAPO-11 (H). | ||
TEM and SEM images of the dandelion-like SAPO-11 show that the branches are uniform with dimensions of 200–250 nm in length and 20 - 30 nm in diameter. The elemental maps of O, Al, P, and Si (Fig. 4H) all show homogeneous distribution throughout the whole dandelion-like SAPO-11.
Compared to C-HTC-SAPO-11 and C-SAC-SAPO-11, the discrepancy in the crystallographic morphology of SISAC-SAPO-11 is probably due to the existence of a slight embryonic AEL structure in DG-160 and is the dominant difference in crystal growth orientation of SAPO-11, as is indicated by analyzing the difference between the two dry gels using XRD, 31P and 27Al solid-state MAS NMR. We also prepared seed-SAC-SAPO-11 as a reference material by directly adding 1 wt% C-HTC-SAPO-11 samples to the synthesis gel in a similar manner. As revealed by the SEM images (Fig. S4, ES4†), the ordinary foreign seed addition method causes a tremendous difference compared with SISAC-SAPO-11. Although the surfaces of the aggregates are composed of abundant nanosheets, the integral morphology is inhomogeneous, unlike the regular dandelion-like morphology obtained using the SISAC method. These results suggest that the interaction intensity between the seed structure and gel plays a pivotal role in the further crystallization process, and the formation of the dandelion-like SAPO-11 was contributed to by a nanoscale interaction between the seed structure and the gel.
The wide-angle range XRD patterns of three samples are plotted in Fig. 5A. All of the samples exhibit the high crystallinity of AEL-structured materials without traces of impure crystalline phases, reflecting the successful production of a SAPO-11 zeolite.15,54,55 The present results indicate that the SAC and SISAC processes do not affect the crystal structure of SAPO-11. Their relative crystallinities calculated by the ratio of the highest peak values are listed in Table 1. Depending on the C-SAC process, the RC value is 86.3% and SISAC-SAPO-11 is 96% of the C-HTC-SAPO-11 value. Moreover, it is noteworthy that the intensities decreased and the line widths broadened when the samples were treated using the SAC method, which is attributable to the decreased crystal size.5 This confirms that the SAC method is favorable for the formation of smaller crystals and as a result, a greater number of external active sites were exposed.
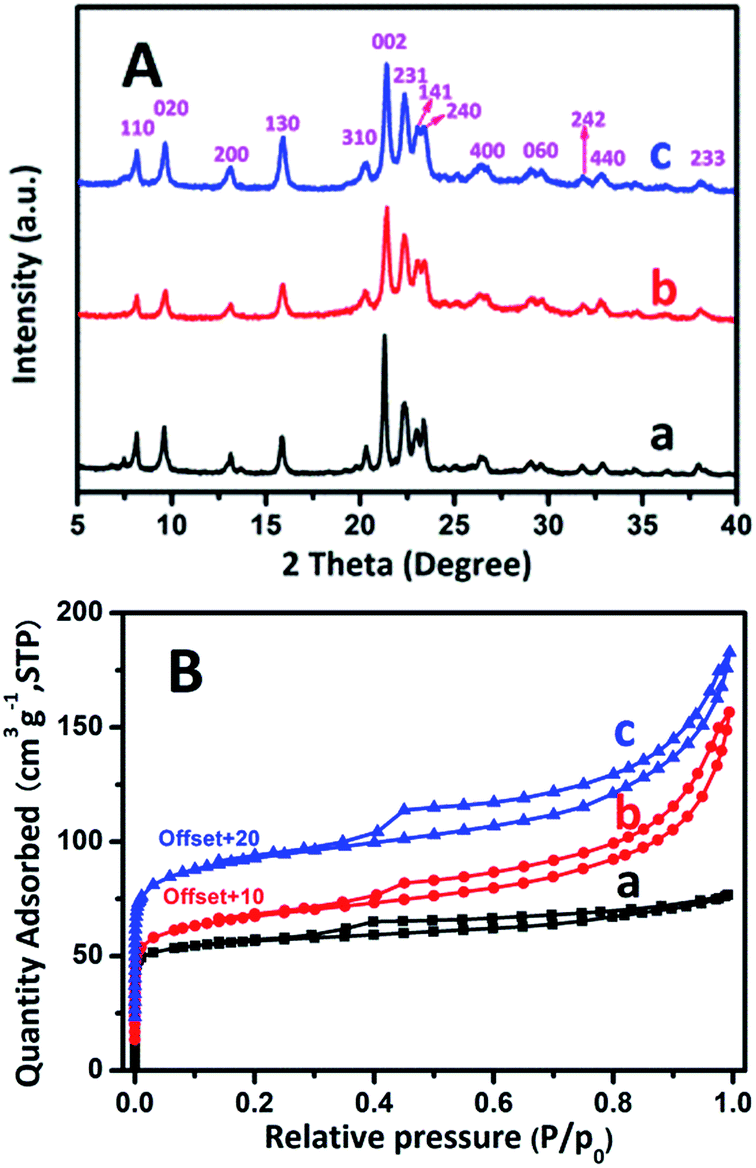 | ||
| Fig. 5 XRD patterns (A) and Ar adsorption/desorption isotherms (B) of the calcined (a) C-HTC-SAPO-11, (b) C-SAC-SAPO-11 and (c) SISAC-SAPO-11 samples. | ||
| Sample | SBET (m2 g−1) | Smic (m2 g−1) | Sext (m2 g−1) | Vmic (cm3 g−1) | Vmeso (cm3 g−1) | Bulk composition via XRF | Surface composition via XPS | RC (%) |
|---|---|---|---|---|---|---|---|---|
| C-HTC-SAPO-11 | 167.5 | 130.6 | 36.9 | 0.056 | 0.042 | (Si0.054Al0.495P0.456)O2 | (Si0.093Al0.460P0.447)O2 | 100 |
| C-SAC-SAPO-11 | 172.8 | 99.2 | 73.6 | 0.041 | 0.124 | (Si0.061Al0.486P0.452)O2 | (Si0.088Al0.448P0.464)O2 | 86.3 |
| SISAC-SAPO-11 | 200.3 | 118.3 | 82.0 | 0.056 | 0.125 | (Si0.107Al0.483P0.423)O2 | (Si0.174Al0.400P0.426)O2 | 96.0 |
The Ar adsorption/desorption isotherms of the calcined C-HTC-SAPO-11, C-SAC-SAPO-11 and SISAC-SAPO-11 samples are shown in Fig. 5B, and the corresponding textural properties are summarized in Table 1. The isotherm of C-HTC-SAPO-11 exhibits a typical type I profile with a very high uptake at low relative pressures (P/P0 < 0.02) and a plateau at high relative pressures (0.35 < P/P0 < 1.0), indicating that the C-HTC-SAPO-11 is a purely microporous phase with negligible mesoporosity. In addition to a high uptake at low relative pressure, it shows a typical type IV isotherm profile characterized by an H4-type hysteresis loop and an apparent uptake at high relative pressures (0.35 < P/P0 < 1.0), demonstrating the coexistence of microporosity and mesoporosity in them.
For C-SAC-SAPO-11, slightly lower Smicropore and Vmicropore values are exhibited in comparison with C-HTC-SAPO-11, consistent with its lower relative crystallinity (86.3%). In accord with this data, it possessed a larger external surface area and mesopore volume as compared with C-HTC-SAPO-11. It is notable that the SAPO-11 synthesized by the SISAC method not only retains its microporous properties (almost equal to the C-HTC-SAPO-11) but also exhibits the largest external surface area (Sext: 82.0 m2 g−1) and mesopore volume (Vmesopore: 0.125 cm3 g−1) among them. This indicates significantly decreased crystal thickness compared to C-HTC-SAPO-11 (Sext: 36.9 m2 g−1 and Vmesopore: 0.042 cm3 g−1, respectively). Recent studies24,26 suggested that the SAC systems further decrease the grain size of zeolite, and as a result, extend the external surface areas and mesopore volumes of the zeolite. This coincides with our study. In summary, the extension of such textural characteristics constitutes a major preponderance of the SISAC-SAPO-11 support, particularly when more acid sites (active sites) are exposed on the external surface.
The bulk and surface compositions of SAPO-11 synthesized by different methods are measured by XRF and XPS (Table 1).
The Si content using the SAC method is greater than that using the CHT method, especially for the SISAC method. Accordingly, the dry gel system24 (nonaqueous condition) might favor the incorporation of Si into the framework of AlPO-11, which determines the acidity of SAPO-11.16 Combining the results of XRF and XPS, one can conclude that the Si content on the surface is much higher than that in the bulk for all the samples, but the incorporation of the Si content and the distribution of Si atoms on the surface was facilitated by the SISAC crystallization method. These results indicate that the acidity and the catalytic behavior of SISAC-SAPO-11 may different when compared with C-HTC-SAPO-11 and C-SAC-SAPO-11, and the promotion of catalytic activity for heptane hydroisomerization originates from a much greater accessibility to the external acidity.
The NH3-TPD technique is useful for characterizing the acidic concentration and strength. Fig. S5† shows the NH3-TPD curves of the three samples. There are two desorption peaks marked in Fig. S5† corresponding to the weak and medium/strong acid sites of SAPO-11.14 We noted that the acid sites are mostly composed of weak acid sites. Obviously, the total ammonia desorption areas of SISAC-SAPO-11 implied that SISAC-SAPO-11 had the largest number of total acid sites.
To further understand the acidity type and spatial distribution of acid sites, FT-IR investigations were carried out using pyridine (Py, 0.57 nm in diameter) and 2,6-dimethylpyridine (2,6-DMPy, 0.67 nm in diameter, which is too large to enter the pore channel of SAPO-11) as the base probe molecules, to quantify the number of total and external acid sites, respectively. The Py-IR and 2,6-DMPy-IR spectra of the three SAPO-11 zeolites are shown in Fig. 6. As illustrated in Fig. 6A, the adsorption bands at 1450 cm−1 and 1540 cm−1 are assigned to the chemisorbed pyridine, which correspond to the pyridine interaction with both Lewis and Brønsted acid sites,12,13 respectively. For the 2,6-DMPy spectra (Fig. 6B), the bands located at 1630–1650 cm−1 can be assigned to 2,6-DMPy adsorbed on Brønsted acid sites only.36
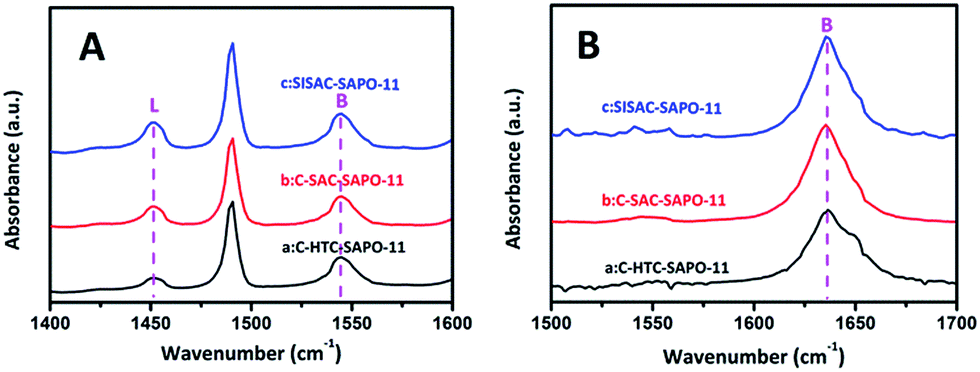 | ||
| Fig. 6 IR spectra of pyridine (A) and 2,6-dimethylpyridine (B) adsorption for (a) C-HTC-SAPO-11, (b) C-SAC-SAPO-11 and (c) SISAC-SAPO-11 samples at 423 K. | ||
The quantitative analyses of the numbers of total and external Brønsted acid sites are summarized in Table 2. Compared with C-HTC-SAPO-11 and C-SAC-SAPO-11, the SISAC-SAPO-11 shows a larger number of total Brønsted acid sites and total Lewis acid sites, indicating the promotion effect of the SISAC method on the incorporation of Si atoms. This is in good agreement with the NH3-TPD analysis. For external Brønsted acidity, the number of Brønsted acid sites at the pore mouths increased in the order of C-HTC-SAPO-11 < C-SAC-SAPO-11 < SISAC-SAPO-11. These results show that both SAC methods are encouraging for producing more detected Brønsted acid sites, especially the SISAC method, owing to its smaller nanocrystals (dandelion-like SAPO-11 composed of multiple branches) and the increased surface areas. Combining the acidity results and the above Ar adsorption–desorption and SEM results, it can be inferred that the smaller crystallites of the dandelion-like SAPO-11 with a greater external surface area produced a larger number of Brønsted acid sites at the pore mouths of SAPO-11. These results are in agreement with the bulk compositions of the three SAPO-11 measured by XRF, and a high Si content is found on the surface by XPS analysis. A number of papers12,13,15,16 describe an increase in catalytic activity and performance as a result of the presence of more acid sites, especially on the external surface of SAPO-11 material. These acid sites act as catalytic sites for hydroisomerization by providing protons that were generated by Si atoms incorporated into the neutral AlP0-11 framework via two different substitution mechanisms.
| Sample | Total Brønsted acidity (mmol g−1) | Total Lewis acidity (mmol g−1) | External Brønsted acidity (mmol g−1) |
|---|---|---|---|
| C-HTC-SAPO-11 | 1.85 | 0.296 | 0.080 |
| C-SAC-SAPO-11 | 1.69 | 0.608 | 0.091 |
| SISAC-SAPO-11 | 2.27 | 0.802 | 0.104 |
Catalytic performance for hydroisomerization of heptane
The C-HTC-SAPO-11, C-SAC-SAPO-11 and SISAC-SAPO-11 are selected as supports for 0.5 wt% Pt catalyst via impregnation method to test the hydroisomerization performance of heptane. H2-TPR profiles of C-HTC-SAPO-11, C-SAC-SAPO-11 and SISAC-SAPO-11 are shown in Fig. 7. Three catalysts showed the similar reduced profiles. The peak around 450 K was ascribed to the reduction of platinum species in the channel of SAPO-11 and the strong peak at 320 K in the profiles was attributed to the reduction of platinum particles on the external surface.56 The weak and broad peak around 750 K was contributed to the strong interaction between Pt species and SAPO-11 support. We can draw conclusion that the peak areas of the Pt existed in external surface was higher than the peak areas of Pt existed in channels. These results indicated that lager number of platinum existed in external surface and small part of platinum remained in channels. Moreover, the peak area at 320 K of three samples showed the sequence of 0.5% Pt/SISAC-SAPO-11 > 0.5% Pt/C-SAC-SAPO-11 > 0.5% Pt/C-HTC-SAPO-11, which means that the multibranched SAPO-11 supported catalyst has higher (de)hydrogenation function of Pt in the hydroisomerization of heptane.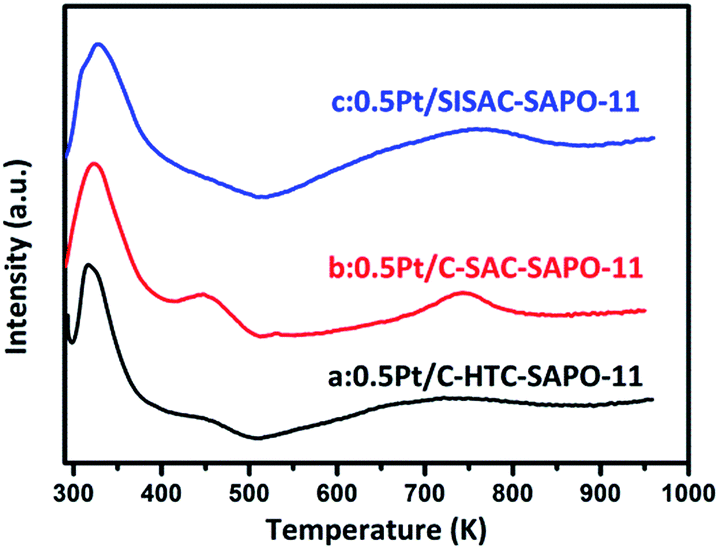 | ||
| Fig. 7 H2-TPR profiles of (a) C-HTC-SAPO-11, (b) C-SAC-SAPO-11 and (c) SISAC-SAPO-11. | ||
CO chemisorption on catalyst is carried out to determine the Pt dispersion (DPt) and particle sizes (dPt), and the data is given in Table 3. The CO chemisorption results show that 0.5Pt/SISAC-SAPO-11 catalyst exhibits the greatest DPt (51.1%) and smaller dPt (2.2 nm) compared to 0.5Pt/C-HTC-SAPO-11 and 0.5Pt/C-SAC-SAPO-11. These results indicated that the multibranched SAPO-11 support not only promote the distribution but also decrease the size of Pt nanoparticles.
| Sample | Pt/C-HTC-SAPO-11 | Pt/C-SAC-SAPO-11 | Pt/SISAC-SAPO-11 |
|---|---|---|---|
| a Reaction conditions: temperature 573 K, WHSV = 10 h−1, H2/heptane = 400, and hydrogen pressure 1.5 MPa.b MB: mono-branched iso-C7.c DB: di-branched and multibranched iso-C7.d Cracking products: C1–C6. | |||
| Method | C-HTC | C-SAC | SISAC |
| DPt (%) | 36.8 | 46.8 | 51.1 |
| dPt (nm) | 3.1 | 2.7 | 2.2 |
| Conversion (%) | 64.3 | 71.3 | 79.7 |
| Iso-C7 selectivity (%) | 97.0 | 95.1 | 93.7 |
| Iso-C7 yield (%) | 62.4 | 67.8 | 74.7 |
| MB yield (%)b | 61.1 | 65.8 | 69.5 |
| DB yield (%)c | 1.32 | 1.99 | 5.23 |
| Cracking yield (%)d | 1.73 | 3.26 | 4.14 |
Measures of the catalytic hydroisomerization reaction have been carried out at temperatures varying from 513 K to 673 K. Fig. 8 shows the temperature dependence of the conversion over 0.5 wt% Pt/SAPO-11 for the three samples synthesized by different methods. It shows that the conversion increased as the temperature of the reaction was increased. Previous studies52,57 have shown that under a loading of 0.5 wt% Pt, the (de)hydrogenation becomes kinetically unimportant, and thereby the catalytic performance is mainly governed by the properties of the SAPO-11 as well as the size and distribution of Pt. The conversion for Pt/SISAC-SAPO-11 is highest among them for the entire range of testing temperatures, and the activities of the three catalysts show the order 0.5% Pt/SISAC-SAPO-11 > 0.5% Pt/C-SAC-SAPO-11 > 0.5% Pt/C-HTC-SAPO-11, consistent with the H2-TPR results and the sequence of the numbers of external acid sites, the dispersity and the metal size of Pt.
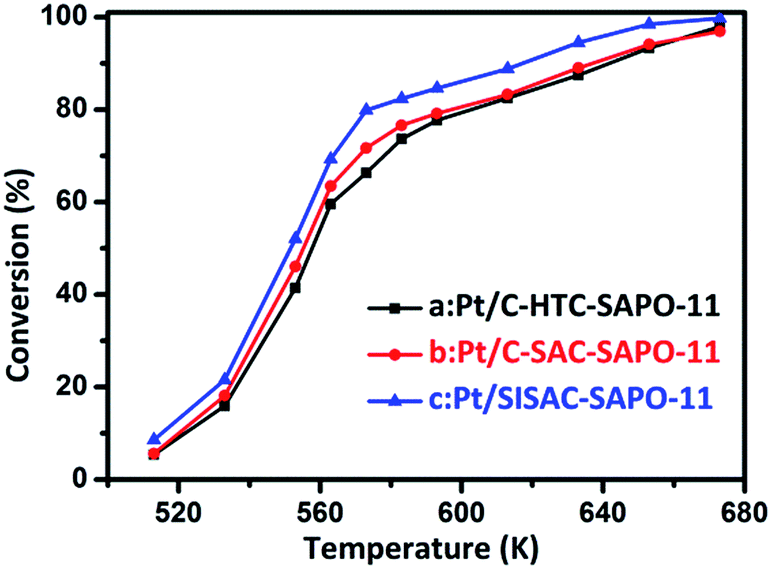 | ||
| Fig. 8 Conversion of n-heptane by (a) 0.5% Pt/C-HTC-SAPO-11, (b) 0.5% Pt/C-SAC-SAPO-11 and (c) 0.5% Pt/SISAC-SAPO-11 catalysts plotted as a function of temperature (reaction conditions: WHSV = 10 h−1, H2/heptane = 400, and hydrogen pressure = 1.5 MPa). | ||
In all cases, the major products were divided into cyclized, cracked, mono-branched, di-branched and multi-branched products. However, the cyclized product is negligible, and both the selectivity for DB (di-branched and multi-branched iso-C7) and cracked products over all catalysts increased with the increasing n-C7 conversion. This suggests that the increase in n-C7 conversion not only promotes the formation of DB but also enhances the undesirable cracking reactions. 2-Methylhexane (2-MC6) and 3-methylhexane (3-MC6) are the major mono-branched products over all the catalysts, in good agreement with the product shape selectivity in SAPO-11.13 The major di-branched product over all the catalysts is 2,4-dimethyl pentane. For a detailed comparison, a summary of the results for heptane hydroisomerization and hydrocracking product fractions is shown in Table 3. The 0.5% Pt/SISAC-SAPO-11 catalyst delivers a remarkably enhanced hydroisomerization activity (79.7%) than others under the same reaction condition, and the total yield of iso-C7 for 0.5% Pt/SISAC-SAPO-11 catalyst (72.2%) is the highest among them, whereas 0.5% Pt/C-HTC-SAPO-11 only shows a yield of 64.3%. These results indicate that the dandelion-like SAPO-11 promoted both catalytic activity and iso-C7 yield, implying that the activities of the catalysts depended on the crystallite size, the acid content and metal distribution. The smaller crystallite size, and much higher external acid sites of SAPO-11 cooperated with better metal distribution may cause easy accessibility for the heptane molecule, and then more intermediates can be generated and further transfer can take place. As a result, the catalyst 0.5% Pt/SISAC-SAPO-11 with its larger mesopore volume, external surface, higher external Brønsted acidity, smaller Pt size and a high degree of metal dispersion shows better activity than the other catalysts, as has been observed for other hierarchical SAPO-11 (ref. 15 and 16) derived from other methods.
Conclusions
In summary, a dandelion-like SAPO-11 has been successfully synthesized by a facile “SISAC” synthesis strategy, in which some of the design embryonic AEL structure seed was embedded into dry gel in situ by pre-crystallization process and further crystal growth by a steam-assisted conversion. In this strategy, artificially integrated the advantage of both low temperature and quasi-solid system promoted the nucleation of the zeolite, by which the smaller promoted the nucleation of the zeolite, by which the smaller particles to be readily formed. Particularly, a layered intermediate with some embryonic AEL structure was obtained at pre-crystallization process, as evidence by XRD, 31P, 27Al MAS NMR and Raman spectrum analysis. The nanoscale interaction between seed and amorphous structure play a vital role for the formation of multibranched dandelion-like SAPO-11. Besides the morphological advantage, the dandelion-like SAPO-11 not only provide an enlarged surface area and mesopore volume, but also form much external acid sites, where the interparticle voids lead to an improved accessibility and diffusion. After loading 0.5 wt% Pt nanoparticles, benefiting from the promoting effect of multibranched SAPO-11, the size and distribution of Pt was smaller and higher respectively. As a result, the 0.5% Pt/SISAC-SAPO-11 catalyst possess superior heptane hydroisomerization performance than 0.5% Pt/C-HTC-SAPO-11 and 0.5% Pt/C-SAC-SAPO-11, in which support was prepared by conventional hydrothermal conversion method and conventional steam-assisted method, respectively. The superior catalytic activities is owing to the effect of morphological modification (i.e., introducing more external surface reaction sites, facilitating the distribution of Pt nanoparticles). The work presented here not only for SAPO-11, such easy SISAC method create a new perspective as a viable and effective pathway for the synthesis of other zeolite framework types and related materials with different morphology and structural properties.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21373168, 21473143) and the Science and Technology Foundation of Guangxi (14125008-2-10).Notes and references
- V. M. Akhmedov and S. H. Al-Khowaiter, Catal. Rev., 2007, 49, 133–139 Search PubMed.
- N. Batalha, L. Pinard, C. Bouchy, E. Guillon and M. Guisnet, J. Catal., 2013, 307, 122–131 CrossRef CAS.
- N. Li, J. G. Wang, J. X. Xu, J. Y. Liu, H. J. Zhou, P. C. Sun and T. H. Chen, Nanoscale, 2012, 4, 2150–2156 RSC.
- N. Musselwhite, K. Na, K. Sabyrov, S. Alayoglu and G. A. Somorjai, J. Am. Chem. Soc., 2015, 137, 10231–10237 CrossRef CAS PubMed.
- J. Kim, W. Kim, Y. Seo, J. C. Kim and R. Ryoo, J. Catal., 2013, 301, 187–197 CrossRef CAS.
- Y. Wang, Z. Tao, B. Wu, J. Xu, C. Huo, K. Li, H. Chen, Y. Yang and Y. Li, J. Catal., 2015, 322, 1–13 CrossRef CAS.
- P. Liu, J. Ren and Y. Sun, Catal. Commun., 2008, 9, 1804–1809 CrossRef CAS.
- J. Walendziewski and B. Pniak, Appl. Catal., A, 2003, 250, 39–47 CrossRef CAS.
- D. P. Serrano, J. Aguado and J. M. Escola, ACS Catal., 2012, 2, 1924–1941 CrossRef CAS.
- G. Majano, K. Raltchev, A. Vicente and S. Mintova, Nanoscale, 2015, 7, 5787–5793 RSC.
- Y. Seo, S. Lee, C. Jo and R. Ryoo, J. Am. Chem. Soc., 2013, 135, 8806–8809 CrossRef CAS PubMed.
- Y. Fan, H. Xiao, G. Shi, H. Liu and X. Bao, J. Catal., 2012, 285, 251–259 CrossRef CAS.
- L. Guo, X. Bao, Y. Fan, G. Shi, H. Liu and D. Bai, J. Catal., 2012, 294, 161–170 CrossRef CAS.
- Y. Liu, W. Qu, W. Chang, S. Pan, Z. Tian, X. Meng, M. Rigutto, A. van der Made, L. Zhao, X. Zheng and F. S. Xiao, J. Colloid Interface Sci., 2014, 418, 193–199 CrossRef CAS PubMed.
- M. Y. Kim, K. Lee and M. Choi, J. Catal., 2014, 319, 232–238 CrossRef CAS.
- L. Guo, Y. Fan, X. Bao, G. Shi and H. Liu, J. Catal., 2013, 301, 162–173 CrossRef CAS.
- M. Choi, R. Srivastava and R. Ryoo, Chem. Commun., 2006, 4380–4382 RSC.
- Z. Liu, L. Liu, H. Song, C. Wang, W. Xing, S. Komarneni and Z. Yan, Mater. Lett., 2015, 154, 116–119 CrossRef CAS.
- K. Moller, B. Yilmaz, R. M. Jacubinas, U. Muller and T. Bein, J. Am. Chem. Soc., 2011, 133, 5284–5295 CrossRef CAS PubMed.
- H. Tao, H. Yang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Eng. J., 2013, 225, 686–694 CrossRef CAS.
- R.-F. Tsai, K.-J. Du, T.-Y. Cheng, G. H. Ho, P.-H. Wu, S.-B. Liu and T.-C. Tsai, Catal. Today, 2013, 204, 30–37 CrossRef CAS.
- Y. Liu, X. Cui, L. Han, Z. Yu and X. Liu, Microporous Mesoporous Mater., 2014, 198, 230–235 CrossRef CAS.
- L. Han, Y. Liu, F. Subhan, X. Liu and Z. Yan, Microporous Mesoporous Mater., 2014, 194, 90–96 CrossRef CAS.
- C.-M. Song, Y. Feng and L.-L. Ma, Microporous Mesoporous Mater., 2012, 147, 205–211 CrossRef.
- Z. Wang, C. Li, H. J. Cho, S. C. Kung, M. A. Snyder and W. Fan, J. Mater. Chem. A, 2015, 3, 1298–1305 CAS.
- K. Zhu, J. Sun, J. Liu, L. Wang, H. Wan, J. Hu, Y. Wang, C. H. F. Peden and Z. Nie, ACS Catal., 2011, 1, 682–690 CrossRef CAS.
- G. T. Neumann and J. C. Hicks, Cryst. Growth Des., 2013, 13, 1535–1542 CAS.
- Y. Zhang, K. Zhu, X. Zhou and W. Yuan, New J. Chem., 2014, 38, 5808–5816 RSC.
- D. Nandan, S. K. Saxena and N. Viswanadham, J. Mater. Chem. A, 2014, 2, 1054–1059 CAS.
- L. Gora, K. Streletzky, R. W. Thompson and G. D. Phillies, Zeolites, 1997, 19, 98–106 CrossRef CAS.
- X. Cheng, J. Mao, X. Lv, T. Hua, X. Cheng, Y. Long and Y. Tang, J. Mater. Chem. A, 2014, 2, 1247–1251 CAS.
- M. Niederberger and H. Colfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- Q. Zhang, S.-J. Liu and S. H. Yu, J. Mater. Chem., 2009, 19, 191–207 RSC.
- M. Kumar, H. Luo, Y. Roman-Leshkov and J. D. Rimer, J. Am. Chem. Soc., 2015, 137, 13007–13017 CrossRef CAS PubMed.
- S. Zeng, J. Blanchard, M. Breysse, Y. Shi, X. Shu, H. Nie and D. Li, Microporous Mesoporous Mater., 2005, 85, 297–304 CrossRef CAS.
- T. Onfroy, G. Clet and M. Houalla, Microporous Mesoporous Mater., 2005, 82, 99–104 CrossRef CAS.
- B. Chen and Y. Huang, J. Phys. Chem. C, 2007, 111, 15236–15243 CAS.
- B. Li, P. Tian, Y. Qi, L. Zhang, S. Xu, X. Su, D. Fan and Z. Liu, Chin. J. Catal., 2013, 34, 593–603 CrossRef CAS.
- B. Chen and Y. Huang, Microporous Mesoporous Mater., 2011, 143, 14–21 CrossRef CAS.
- K. Wang, J. Yu, P. Miao, Y. Song, J. Li, Z. Shi and R. Xu, J. Mater. Chem., 2001, 11, 1898–1902 RSC.
- S. Oliver, A. Kuperman, A. Lough and G. A. Ozin, Chem. Mater., 1996, 8, 2391–2398 CrossRef CAS.
- L. Zhang, J. Bates, D. Chen, H. Y. Nie and Y. Huang, J. Phys. Chem. C, 2011, 115, 22309–22319 CAS.
- T. Wang, L. Yu and W. Pang, J. Solid State Chem., 1990, 89, 392–395 CrossRef CAS.
- P. K. Dutta and J. Twu, J. Phys. Chem., 1991, 95, 2498–2501 CrossRef CAS.
- A. J. Holmes, S. J. Kirkby, G. A. Ozin and D. Young, J. Phys. Chem., 1994, 98, 4677–4682 CrossRef CAS.
- P. K. Dutta, D. Shieh and M. Puri, Zeolites, 1988, 8, 306–309 CrossRef CAS.
- Y. Zhang, Z. Deng, K. Zhu and X. Zhou, CrystEngComm, 2015, 17, 3214–3218 RSC.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- J. F. Banfield, S. A. Welch, H. Zhang, T. T. Ebert and R. L. Penn, Science., 2000, 289, 751–754 CrossRef CAS PubMed.
- J. Zhang, F. Huang and Z. Lin, Nanoscale, 2010, 2, 18–34 RSC.
- J. Chen, W. Hua, Y. Xiao, Q. Huo, K. Zhu and X. Zhou, Chemistry, 2014, 20, 14744–14755 CrossRef CAS PubMed.
- D. Jin, Z. Liu, J. Zheng, W. Hua, J. Chen, K. Zhu and X. Zhou, RSC Adv., 2016, 6, 32523–32533 RSC.
- B. Gao, P. Tian, M. Li, M. Yang, Y. Qiao, L. Wang, S. Xu and Z. Liu, J. Mater. Chem. A, 2015, 3, 7741–7749 CAS.
- T. Blasco, A. Chica, A. Corma, W. Murphy, J. Agundezrodriguez and J. Perezpariente, J. Catal., 2006, 242, 153–161 CrossRef CAS.
- P. Meriaudeau, V. Tuan, V. T. Nghiem, S. Lai, L. Hung and C. Naccache, J. Catal., 1997, 169, 55–66 CrossRef CAS.
- S. Yuvaraj, T. H. Chang and C. T. Yeh, J. Catal., 2004, 221, 466–473 CrossRef CAS.
- M. Höchtl, A. Jentys and H. Vinek, J. Catal., 2000, 190, 419–432 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26522g |
| This journal is © The Royal Society of Chemistry 2017 |