
Amine synthesis via transition metal homogeneous catalysed hydrosilylation
Bin
Li
ab,
Jean-Baptiste
Sortais
a and
Christophe
Darcel
*a
aUMR 6226 CNRS-Université de Rennes 1 “Institut des Sciences Chimiques de Rennes”, Team “Organometallics: Materials and Catalysis”, “Centre for Catalysis and Green Chemistry”, Campus de Beaulieu, 35042 Rennes Cedex, France. E-mail: christophe.darcel@univ-rennes1.fr
bSchool of Chemical & Environmental Engineering, Wuyi University, Jiangmen 529020, Guangdong Province, P. R. China
First published on 1st June 2016
Abstract
Amines are among the most important compounds due to their wide applications in several areas of importance. Among the numerous synthetic methodologies, this review provides a comprehensive summary of the preparation of amines involving homogeneous transition metal catalysed hydrosilylation methodologies including the reduction of imine, amide, nitro and nitrile derivatives. In addition, challenging tandem reactions such as reductive amination or the use of CO2 as a C1-building block in N-methylation of amines will also be discussed.
A. Introduction
Amines are one of the most important compounds due to their wide applications in agrochemical, natural products, pharmaceuticals, as well as biologically valuable fine chemicals.1 Among the numerous synthetic methodologies developed, alkylation using alcohols or halo alkanes is acknowledged to be a simple way to prepare the most industrially significant amines. However, the degree of alkylation is always difficult to control with alkyl iodides and bromides.2 An alternative route for the synthesis of amine derivatives is the reduction of the corresponding imines, amides, nitriles, etc. or the reductive amination of carbonyl derivatives. In this area, traditional methods using stoichiometric amount of reactive alkali hydrides, such as lithium aluminium hydride and boron hydrides, are still a challenging task in fine chemistry, because of the air and moisture sensitivity of the reducing agents, the potential metallic salt by-product formation and the lack of functional group tolerance.3 In this area, catalytic reduction reaction via transition metal catalysed hydrogenation, hydrogen transfer or hydrosilylation seems to be a simple way in molecular synthesis to obtain amines.4 However, even if hydrogen is an ideal reducing reagent, harsh reaction conditions including high temperature and pressure and the lack of chemoselectivity still make this method challenging for many applications.5 During the last decade, hydrosilanes have attracted attention as competitive alternative reducing reagents to the classical alkali hydrides and hydrogen, mainly because they are usually air and water stable, easy to use, and activated under mild conditions.6 Moreover, this methodology might improve the chemo- and regioselectivity towards to the target products in the reduction reactions. Since the first example of hydrosilylation of 1-octene with trichlorosilane in the presence of acetyl peroxide discovered by Sommer in 1947,7 reduction of carbonyl groups are well exemplified during the last 3 decades.8Within the last few years, the transition metal catalysed hydrosilylation processes have tremendously contributed to the amines synthesis. As several reviews have recently presented the reduction of carbonyl groups by Beller,9 Nolan,10 Nishiyama,11in this review, we will summarize the development on transition metal catalysed hydrosilylation of imines, amides, sequential reaction with aldehydes or alkynes towards to amines. The use of carbon dioxide as a C1source for the methylation of amines under hydrosilylation conditions will also be treated as it appears as a green alternative to the classical Eschweiler–Clarke methodology.
B. Hydrosilylation of imines leading to amines
Among the direct and efficient methodologies for the synthesis of amines, imine catalysed-reduction methodologies4 (such as hydrogenation,4 hydrogen transfer12 or hydrosilylation)13 were developed using transition metals such as ruthenium, iridium, and rhodium and are some of the most ubiquitous protocols in organic synthesis. In this paragraph, transition metal catalysed hydrosilylation of imines will be described.B.1. Group 3 (ytterbium) catalysts
Takaki et al. reported a catalytic hydrosilylation of imines using ytterbium–imine complex [Yb(η2-Ph2CNPh)(hmpa)6] C1 as the catalyst (hmpa = hexamethylphosphoramide).14 Aminosilanes and diaminosilanes were usually produced as a mixture by hydrosilylation of corresponding imines in the presence of 10 mol% of ytterbium–imine catalyst, 1 equiv. of PhSiH3 in THF at room temperature for 20 h (Scheme 1). Notably, ketimines were less reactive than aldimines, and both triphenylsilane and diphenylsilane were unreactive in this catalytic reaction.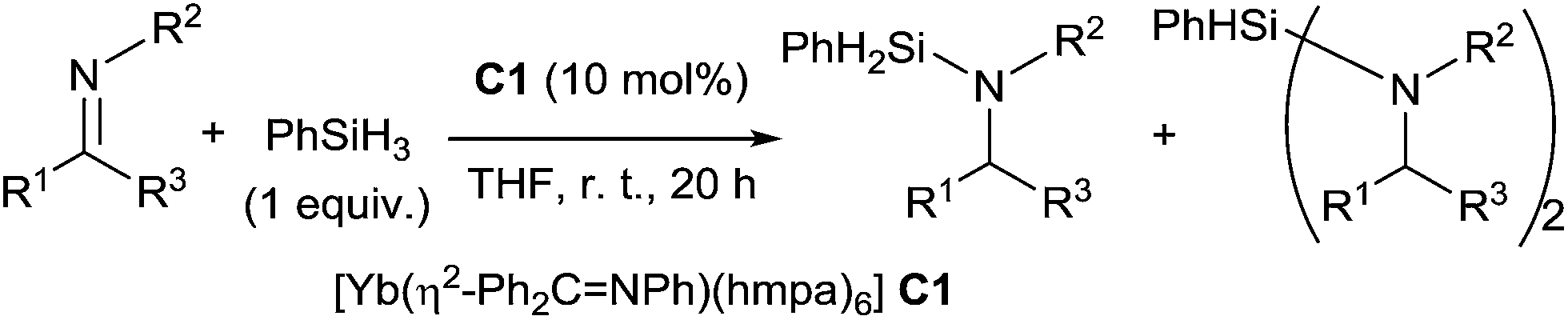 | ||
| Scheme 1 Ytterbium based catalyst for hydrosilylation of imines. | ||
B.2. Group 4 (titanium) catalysts
Since the pioneering report of Nagai in 1988,15 and the intense work of Buchwald in the 90s,16 the use of titanocene based complexes as catalysts for the reduction of carbonyl containing compounds such as aldehydes, ketones and esters is a well-established methodology to produce alcohols under mild conditions. Using a titanium complex bearing a m-tert-butyl-o-diphenylphosphinophenol ligand CpTiCl2(o-PPO) C2 (5 mol%) treated with n-BuLi (10 mol%) and phenylsilane (2.2 equiv.), Buchwald has shown that hydrosilylation of N-benzylidene-methylamine was achieved in full conversion at 65 °C in 54 h.17 Thus, titanocene based complexes were found to be catalytically active in hydrosilylation of imines.18 The first enantioselective catalytic hydrosilylation of N-alkylimines using titanium catalyst was reported by Buchwald et al.19 using the in situ prepared catalyst (0.02–2.5 mol%) (S,S)-ethylene-bis(η5-tetrahydroindenyl)titanium hydride (S,S)-[(EBTHI)TiH] C3b, by reaction of the difluoride (S,S)-[(EBTHI)TiF2] complex C3a with phenylsilane. The corresponding amines were obtained in good yields and enantiomeric excesses up to 99% (Scheme 2).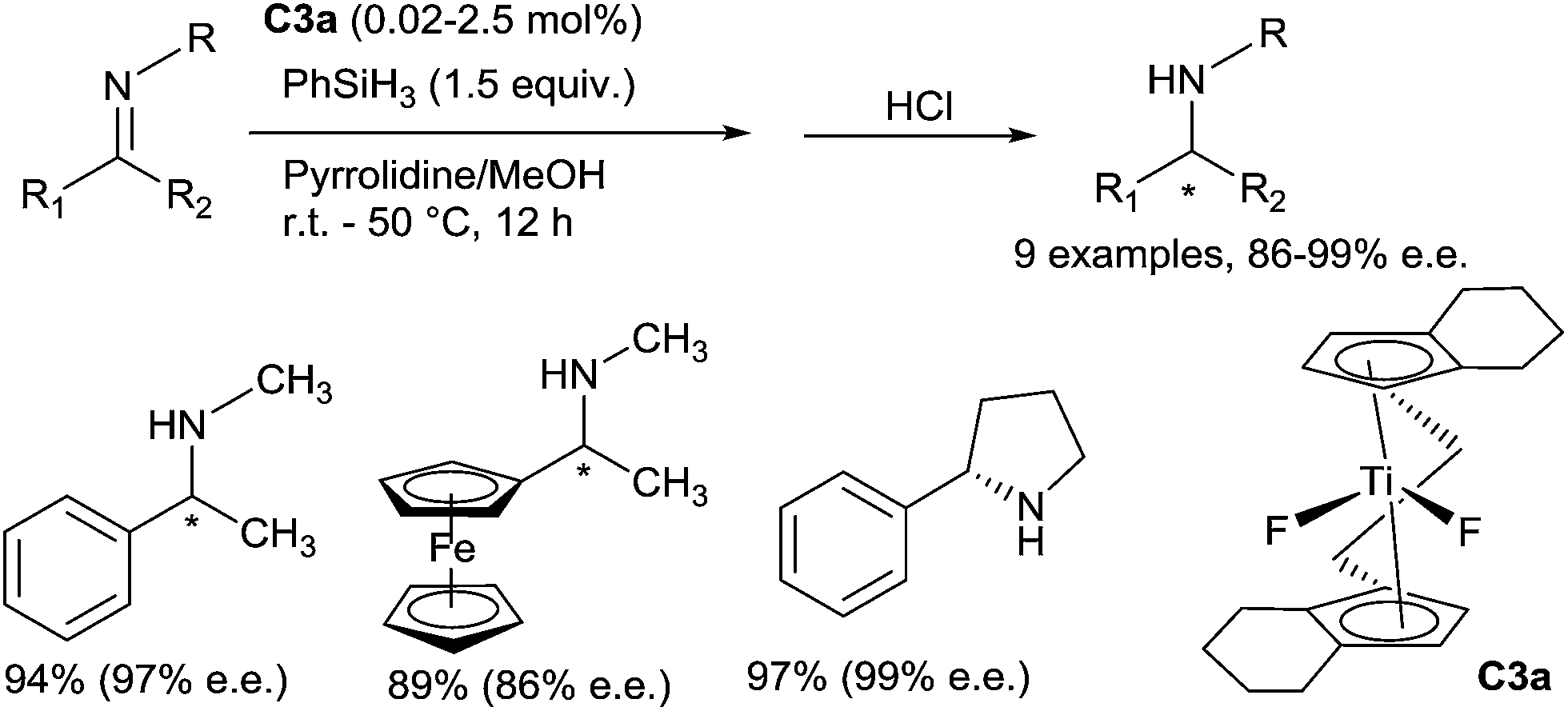 | ||
| Scheme 2 Titanium catalysed enantioselective hydrosilylation. | ||
Titanium-catalysed asymmetric hydrosilylation of N-alkyl imines was also developed using PMHS (polymethyl-hydrosiloxane) as the reducing reagent thanks to the use of primary alkylamines as additives at 60 °C20 (Scheme 3).
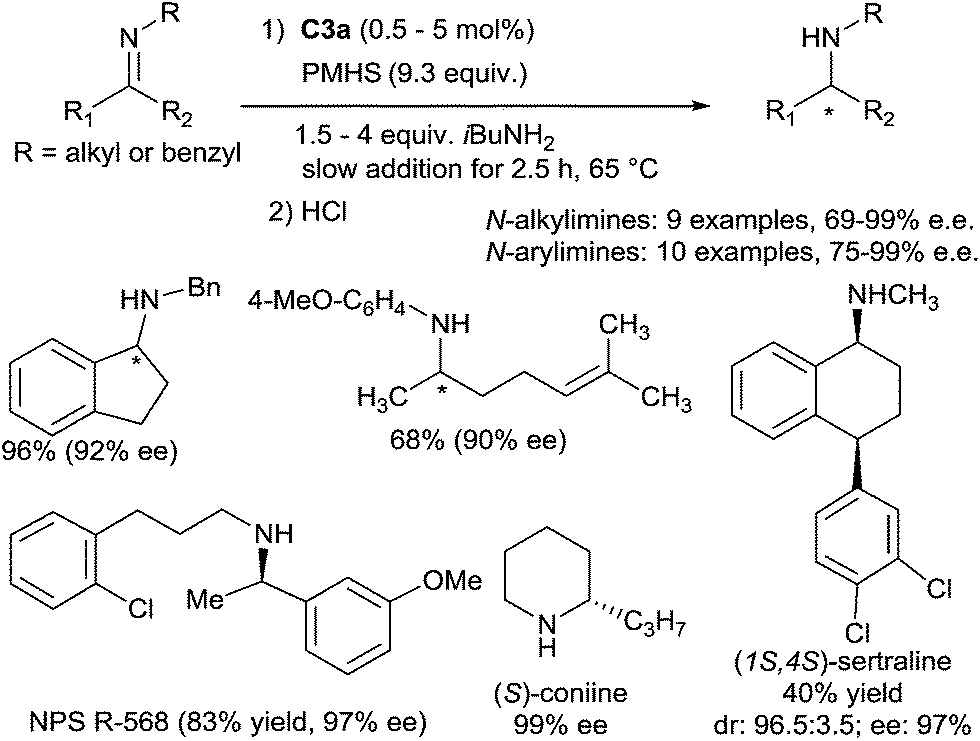 | ||
| Scheme 3 Titanium catalysed asymmetric hydrosilylation of imines using PMHS. | ||
It is noteworthy that PMHS, convenient and inexpensive hydride source, was used instead of PhSiH3.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 The titanocene hydride catalytic species was generated at 60 °C from the titanocene difluoride by reaction with phenylsilane, and PMHS was then used for the hydrosilylation of the imines. Notably, the obtained ee of amines (69–99%) were not correlated to the E:Z ratio of the starting imines.
21 The titanocene hydride catalytic species was generated at 60 °C from the titanocene difluoride by reaction with phenylsilane, and PMHS was then used for the hydrosilylation of the imines. Notably, the obtained ee of amines (69–99%) were not correlated to the E:Z ratio of the starting imines.
In the proposed mechanism, the insertion of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the imine into the Ti–H bond of the titanocene catalytic species C3b generated the chiral titanocene amido intermediate 4-I. The use of primary alkylamines, as an additive, permitted to release the chiral amine by reaction with the sterically encumbered titanium-amido intermediate 4-I and to form a new amido complex 4-II. Finally, the reaction of the less hindered species 4-II with the hydrosilane regenerates the active catalyst C3b (Scheme 4a). Moreover, the N-silylamine 4-III, which is silylated from the primary amine, could facilitate the cleavage through the silylation of Ti–N bond by coordination to the titanium centre followed by σ-bond metathesis, then regenerated the intermediate 4-II and desired product (Scheme 4b).20 Notably, the reaction of primary alkylamines with titanocene hydride C3b led to the intermediate titanocene amido 4-II with release one molecule of hydrogen, which permitted to explain the formation of the species 4-III in an initial step.
N bond of the imine into the Ti–H bond of the titanocene catalytic species C3b generated the chiral titanocene amido intermediate 4-I. The use of primary alkylamines, as an additive, permitted to release the chiral amine by reaction with the sterically encumbered titanium-amido intermediate 4-I and to form a new amido complex 4-II. Finally, the reaction of the less hindered species 4-II with the hydrosilane regenerates the active catalyst C3b (Scheme 4a). Moreover, the N-silylamine 4-III, which is silylated from the primary amine, could facilitate the cleavage through the silylation of Ti–N bond by coordination to the titanium centre followed by σ-bond metathesis, then regenerated the intermediate 4-II and desired product (Scheme 4b).20 Notably, the reaction of primary alkylamines with titanocene hydride C3b led to the intermediate titanocene amido 4-II with release one molecule of hydrogen, which permitted to explain the formation of the species 4-III in an initial step.
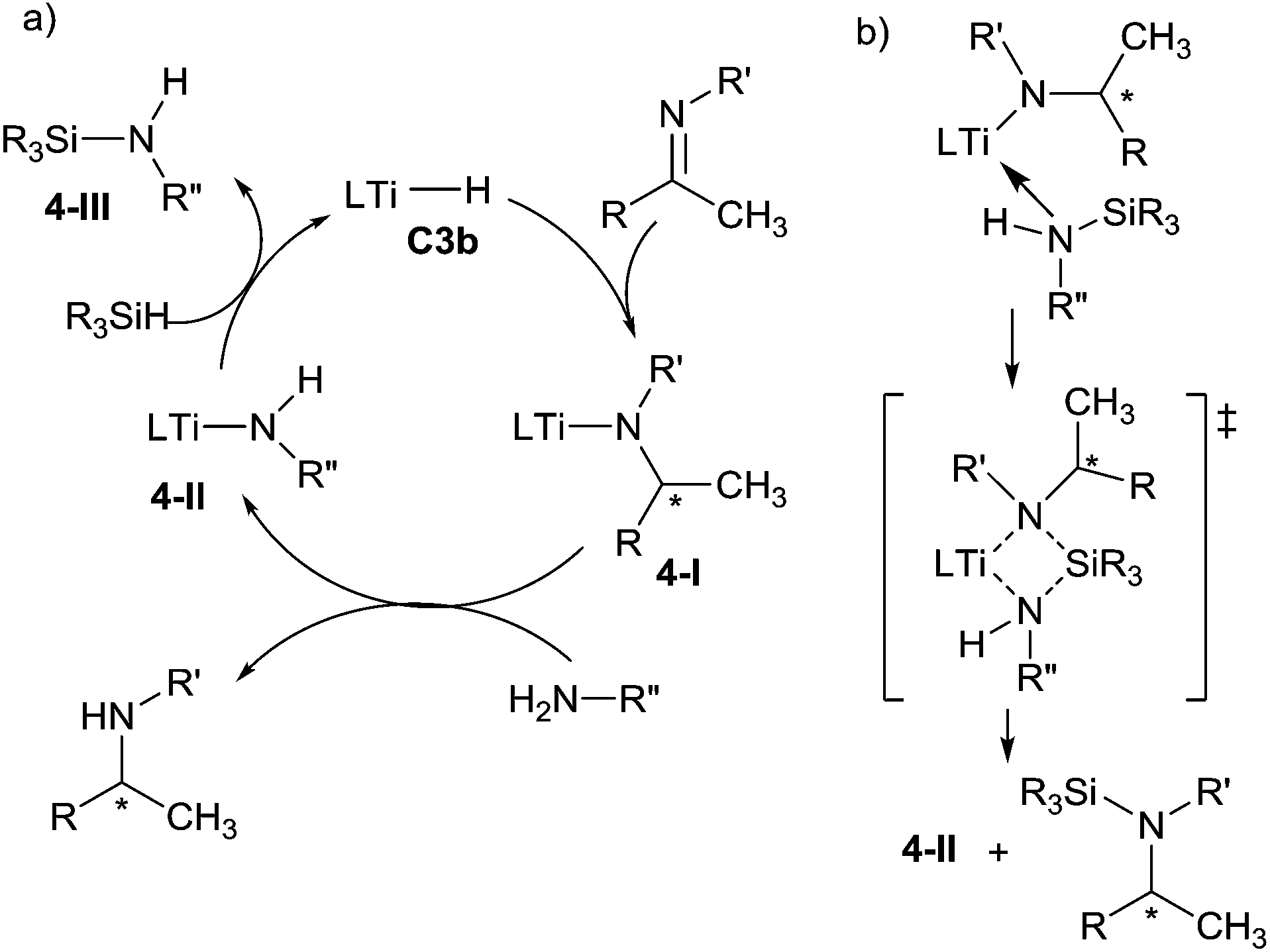 | ||
| Scheme 4 Mechanism of titanium catalysed hydrosilylation of imines. | ||
Similar conditions were applied to develop a Ti-catalysed asymmetric hydrosilylation of N-arylimines.22 A variety of secondary N-arylamines were obtained by reduction of the corresponding N-arylimines derived from nonaromatic ketones with moderate to good yields (63–90%) and ee's (88–99%). It must be underlined that N-arylimines derived from aromatic ketones such as acetophenone derivatives lead to the corresponding amines with low ee (6–13%), by contrast with N-arylimines derived from indanone furnishing the amine with 97% ee. Interestingly, this methodology was applied for the preparation of NPS R-568, an active compound for the treatment of hyperparathyroidism, which was obtained from the corresponding imine in 83% yield and 97% ee using 1 mol% of the catalyst C3a23a (Scheme 3). Piperidine alkaloids such as (S)-coniine and (2R,6R)-trans-solenopsin A can be also prepared using C3a as the catalyst from the corresponding imines.23b Notably, Buchwald described the kinetic resolution of imine derivatives of indanones and tetralones having a substituent at a position remote from the reaction center under hydrosilylation conditions in the presence of 2.5 mol% of C3a. Thus, in the presence of 2 equiv. of PhSiH3, (1S,4S)-sertraline, an antidepressant drug, was obtained with high diastereo- and enantio-selectivities (96.5:3.5 and 97% respectively) and 40% yield.23c
In 2009, Khinast et al. reported the hydrosilylation of imines using in situ catalyst precursor species derived from the reaction of (R,R)-ethylene-1,2-bis-(η5-4,5,6,7-tetrahydro-1-indenyl) titanium (R)-1,1′-binaphth-2-olate C4a or dichloride C4b with organolithium and silane.24 Even though the efficiency of this asymmetric catalytic hydrosilylation is lower (ee = 33–78%) than the one with similar titanium difluoride complex C3a, which generates a Ti(III)–H active species,17,18,21 the formation of a new Ti(IV) active species C6 was proposed through the reaction of phenylsilane with a dialkyltitanium species C5 (Scheme 5).24 The structure of this catalytic Ti(IV) active species C6 was confirmed by DFT calculations, IR, NMR, and EPR analysis. Noticeably, menthyl-substituted metallocene titanium Cp′2TiF2 (Cp′ = η5-menthyl-C5H4) C7 exhibited similar activities than C3a, but lower enantioselectivities (7–27% ee).25
 | ||
| Scheme 5 Titanium catalysts for asymmetric hydrosilylation of imines. | ||
The same group described in 2012 tethered titanocene complexes immobilized onto 3-mercaptopropyl-functionalized silica gel C8 and their activity for the hydrosilylation of 2-phenylpyrroline, after activation with BuLi and PhSiH3 (conversion up to 95%, TOF up to 20 h−1)26 (Scheme 6).
 | ||
| Scheme 6 Immobilized tethered titanocene for catalysed hydrosilylation of 2-phenylpyrroline. | ||
B.3. Group 7 (rhenium) catalysts
Toste et al. have first developed a rhenium based catalytic system for the hydrosilylation of carbonyl derivatives using a rhenium(V)-dioxo complex, (PPh3)2Re(O)2I C9, yielding silyl ethers.27 Using a chiral (CNbox)Re(V)-oxo complex C10b, synthesized from Re(O)(Cl)3(OPPh3)(SMe2) C10a (3 mol%), by reaction with 1.1 equiv. of the bidentate cyanobis(oxazoline) chiral ligand, enantioselective hydrosilylation of imines was reported in the presence of 2 equiv. of dimethylphenylsilane at room temperature.28 A wide range of phosphinylimines led to corresponding phosphinylamines in 51–89% yields with high ee up to 99% (Scheme 7). Interestingly, the Re(V)-catalysed hydrosilylation of α-imino esters and α,β-unsaturated imines led to the corresponding phenyl glycine derivatives and allylic amines with highly chemo- and enantioselectivity without reduction of the ester and alkene groups29 (Scheme 7).
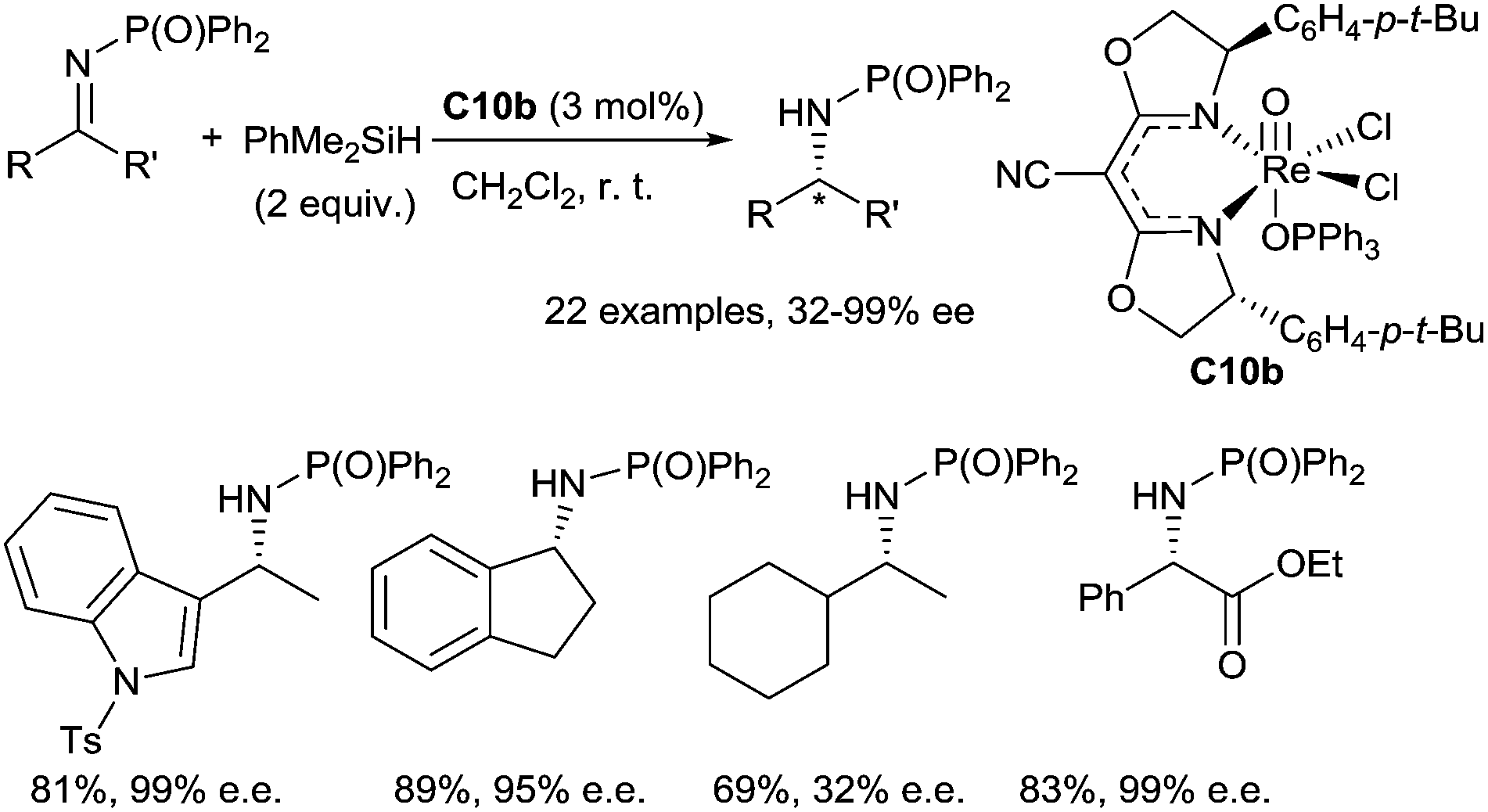 | ||
| Scheme 7 Rhenium catalysed hydrosilylation of imines. | ||
B.4. Group 8 (ruthenium and iron) catalysts
Ruthenium complexes were widely used in reduction area, more particularly in asymmetric hydrogenation and hydrogen transfer of imines.30 In comparison, ruthenium catalysed hydrosilylation of imines is still underdeveloped. The first catalytic asymmetric hydrosilylation of imines using the RuCl2(PPh3)(oxazolinyl-ferrocenylphosphine) complex C11 as the catalyst was described by Hidai on one imine, 2-phenyl-1-pyrroline, which led to the corresponding cyclic amine in 60% yield and 88% ee after 48 h at 0 °C using diphenylsilane as a reductant31 (Scheme 8).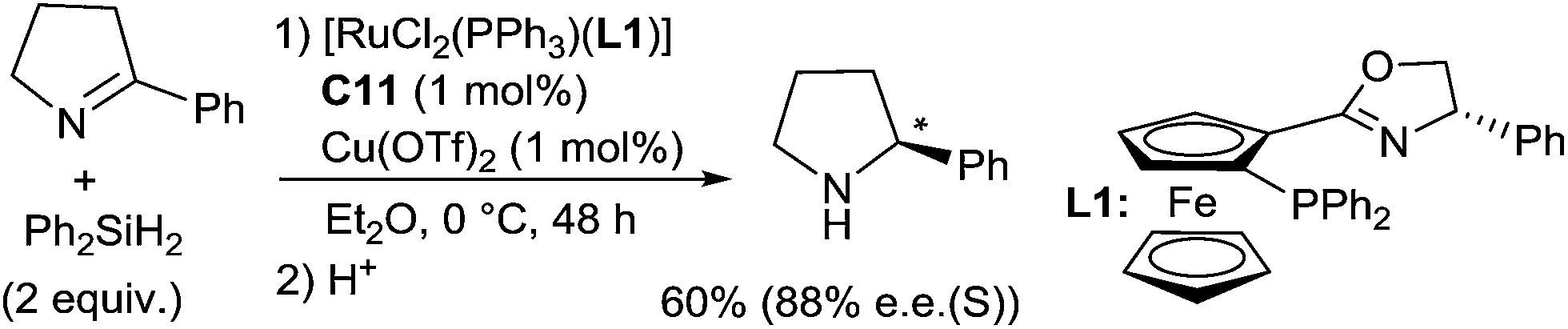 | ||
| Scheme 8 Asymmetric ruthenium catalysed hydrosilylation of 2-phenyl-1-pyrroline. | ||
Interestingly, under similar conditions, chiral primary amines were prepared by hydrosilylation of ketoximes using 2 mol% of [RuCl2(PPh3)(oxazolinylferrocenylphosphine)] complexes associated with 2 mol% of AgOTf, and 2 equiv. of Ph2SiH2 in THF or DME at room temperature (Scheme 9). Low to moderate isolated yields (5–62%) of chiral primary amines were obtained with ee up to 89%.32
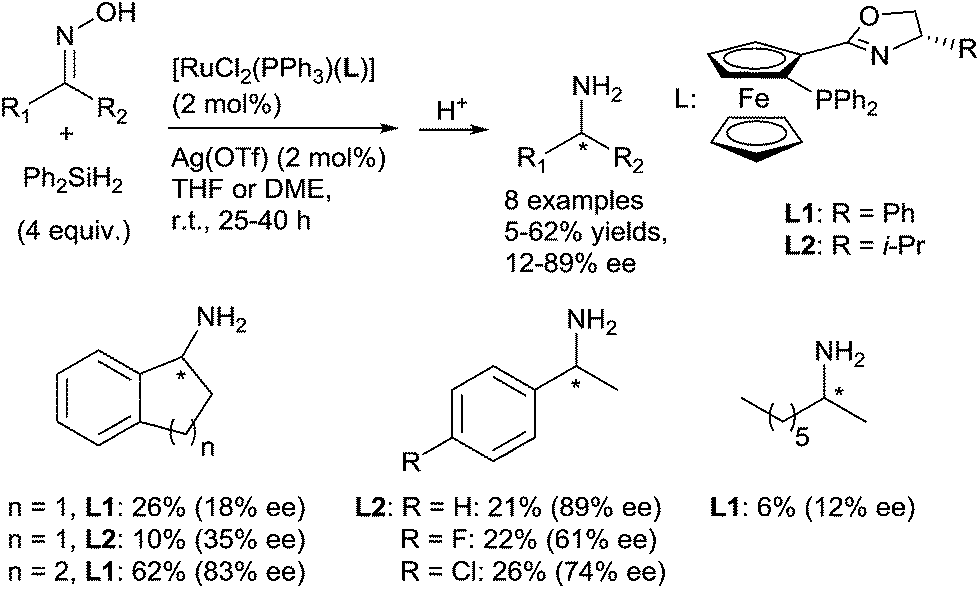 | ||
| Scheme 9 [RuCl2(PPh3)(L)] catalysed hydrosilylation of hydroxyimine. | ||
A binuclear μ-silane complex, [Ru(CO)2(SiHTol)]2(μ-dppm)(μ-η2:η2-H2SiTol2) C12 (dppm = Ph2PCH2PPh2, Tol = p-CH3–C6H4), can also be used to hydrosilylate with di-p-tolylsilane N-aryl imines leading to aminosilanes at 50 °C for 2.5–8 days.33 Notably, no hydrosilylation occurred when a monohydrosilane was used instead of a dihydrosilane (Scheme 10).
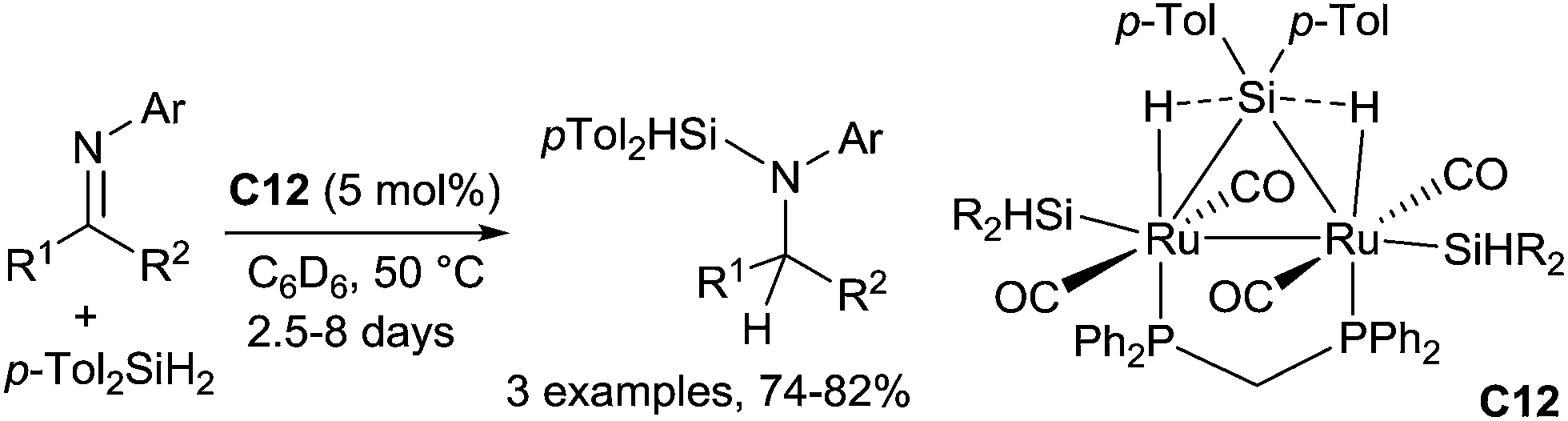 | ||
| Scheme 10 Binuclear μ-silane complex C12 catalysed hydrosilylation of N-arylimines. | ||
Non classical high valent nitridoruthenium(VI) complex, [RuN(saldach)(CH3OH)][ClO4] C13 can be an interesting alternative catalyst for hydrosilylation of aldimines34 (saldach = dianion of the racemic N,N′-cyclohexandi-yl-bis(salicyl-ideneimine)) (Scheme 11).
 | ||
| Scheme 11 Nitridoruthenium(VI) based catalyst for hydrosilylation. | ||
In a sustainable and economic point of view, PMHS is also an interesting alternative hydrosilane as it is an inexpensive residue from silicon industry. Our group has reported an efficient catalytic hydrosilylation of imines with simple [RuCl2(p-cymene)]2C14 catalyst in ethanol under air at room temperature for 2 h.35 A variety of aldimines were directly reduced to secondary amines in 69–97% yields in the presence of 1–2 mol% of [RuCl2(p-cymene)]2C14, with 2 equiv. of PMHS (Scheme 12). Interestingly, this hydrosilylation of imines offered a very good chemoselectivity to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, NO, CO and CC bonds.
N, NO, CO and CC bonds.
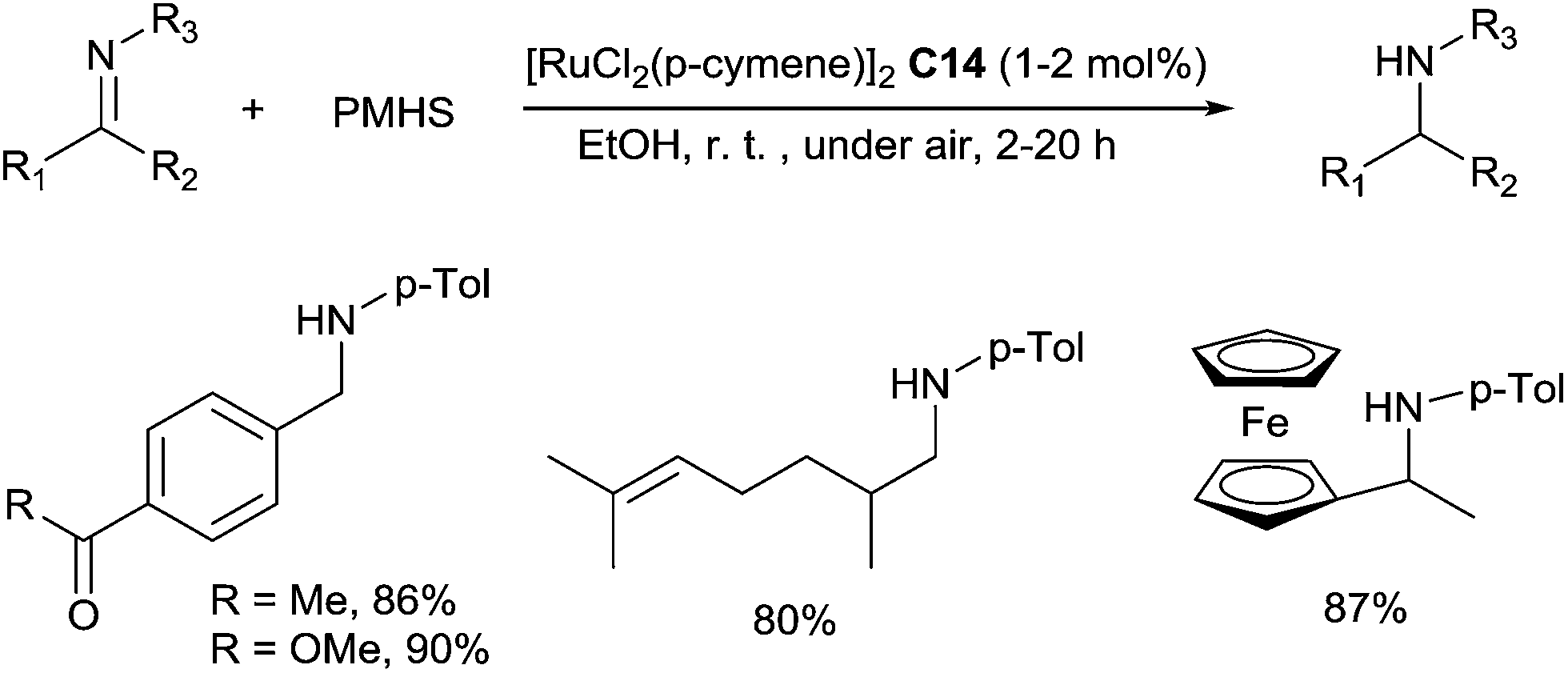 | ||
| Scheme 12 [RuCl2(p-cymene)]2C14 catalysed hydrosilylation of imines. | ||
Notably, the use of ethanol as the solvent avoids further desilylation step. These conditions were also successfully applied to the hydrosilylation of ketimines using 2 mol% of [RuCl2(p-cymene)]2C14 at rt for 4–20 h (Scheme 12).
The preparation of secondary bulky amines via a sequential ruthenium(II) catalysed C–H bond activation and a hydrosilylation of in situ formed imines were subsequently performed.36 It is noteworthy that the [RuCl2(p-cymene)]2 catalyst C14 was used in both C–H bond activation and hydrosilylation steps. The secondary bulky amines were then isolated in 70–86% yields based on initial imines (Scheme 13).
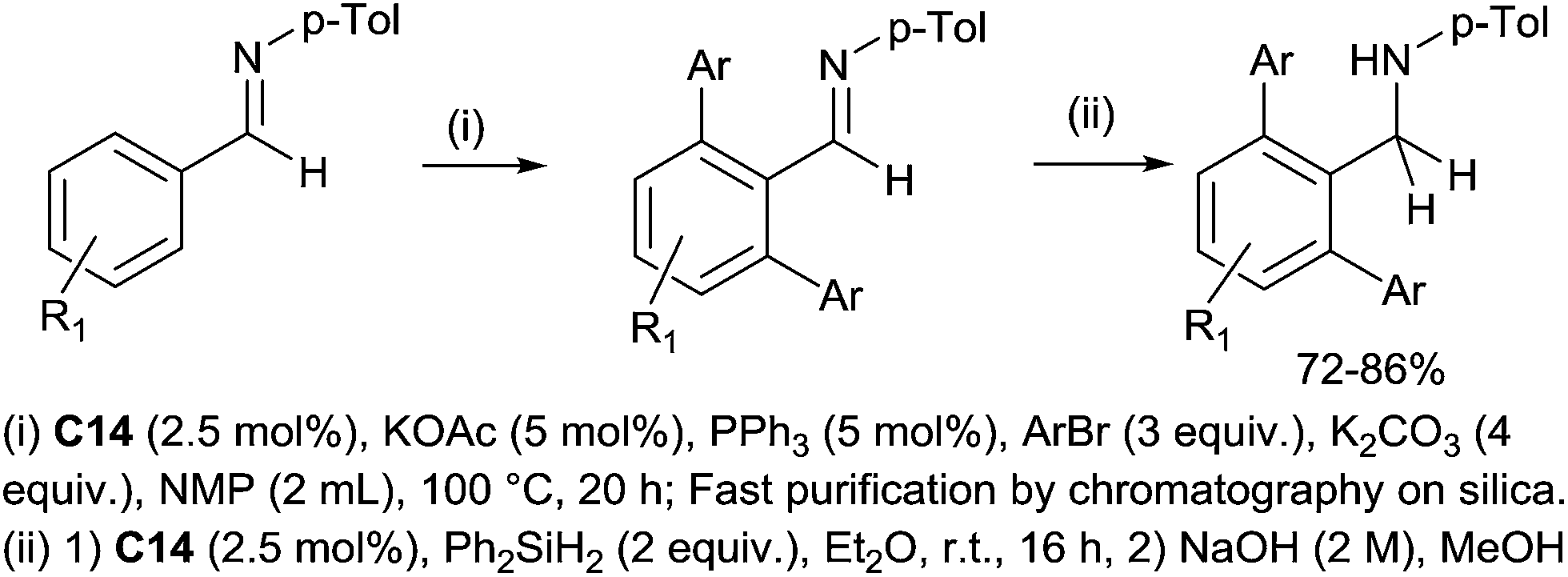 | ||
| Scheme 13 Synthesis of arylated amines via sequential Ru(II) catalysed arylation and hydrosilylation of imines. | ||
Interestingly, Oestreich has shown that modifying the complex architecture can have drastic effect on the chemoselectivity: using cationic tethered ruthenium(II)thiolate complexes such as C15a (1 mol%), in the presence of 1.0 equiv. of dimethylphenylsilane in C6D6 at rt, enolizable N-arylimines lead to the corresponding N-silylated enamines as the major products (28–99%) with C,N-disilylated enamines as by products (up to 36%). Noticeably, this reaction proceeded without addition of a base37 (Scheme 14).
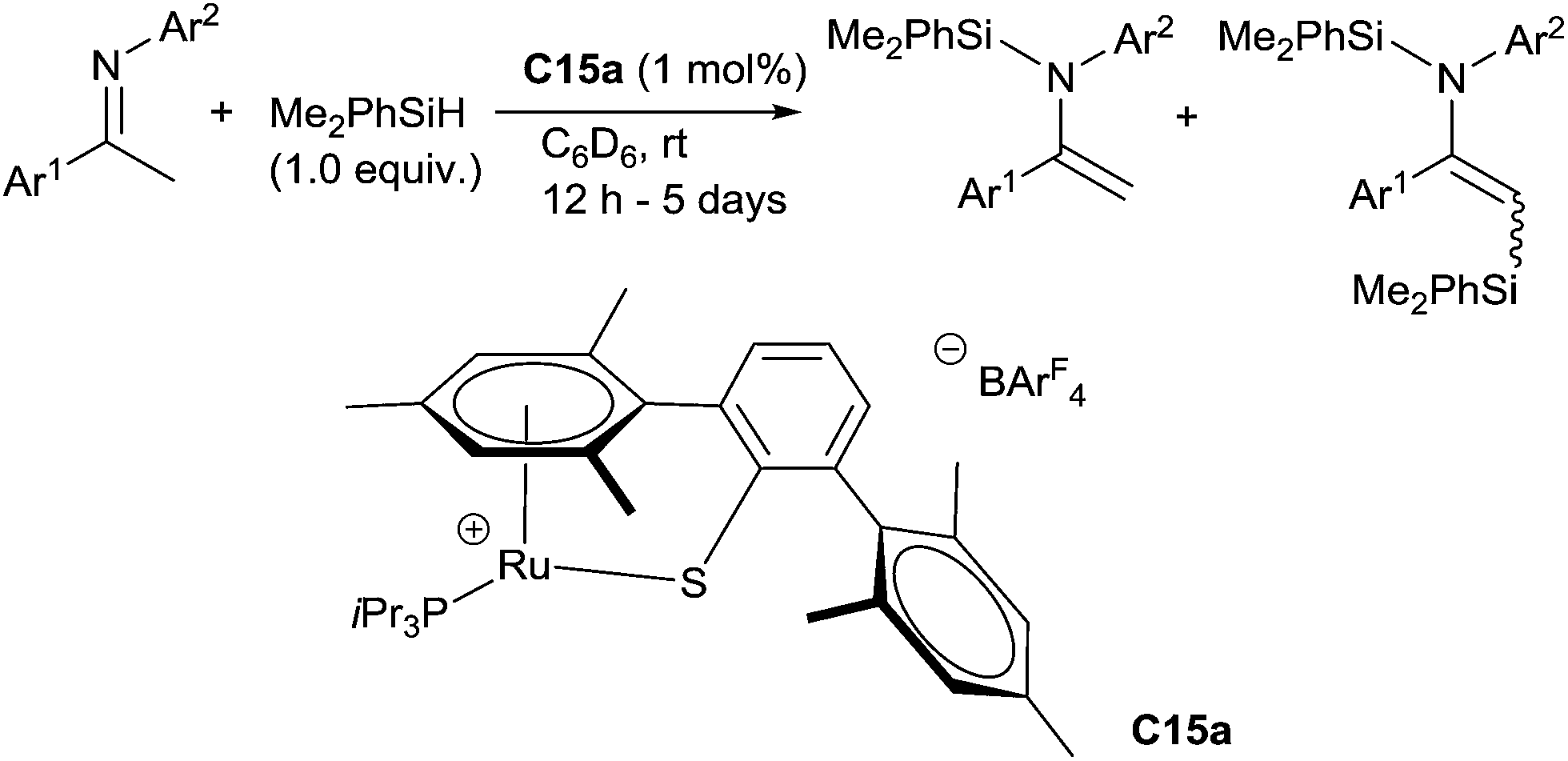 | ||
| Scheme 14 Ru(II)-thiolate complex C15a catalysed hydrosilylation. | ||
The first examples of iron-catalysed hydrosilylation of imines were reported by Beller38 and Nishiyama.39 Using 4 mol% of Fe3(CO)12, in the presence of 4.0 equiv. of PMHS at 100 °C in toluene for 24 h, PhCHN–Ph, was reduced in the corresponding amine in 96% yield.38 With the in situ generated catalytic system from Fe(OAc)2 (5 mol%) and TMEDA (tetramethyldiamine) (10 mol%), in the presence of 2 equiv. of (EtO)2MeSiH in THF at 65 °C for 24 h, PhCHNPh could be reduced in 75% yield in the corresponding amine.39
An efficient hydrosilylation of both aldimines and ketimines was also developed using a well-defined NHC carbene cyclopentadienyl iron complex C16.40 A wide range of aldimines were tested by using 2 mol% of C16, and 2 equiv. of PhSiH3 at 30 °C without solvent upon visible light irradiation for 30 h (Scheme 15). Interestingly, heteroaromatic imines (i.e.: furyl, pyridyl) gave the corresponding amines in good yields. Notably, interesting chemoselectivities can be reached under such mild conditions, as the reduction conditions were tolerant towards ketone and ester groups.
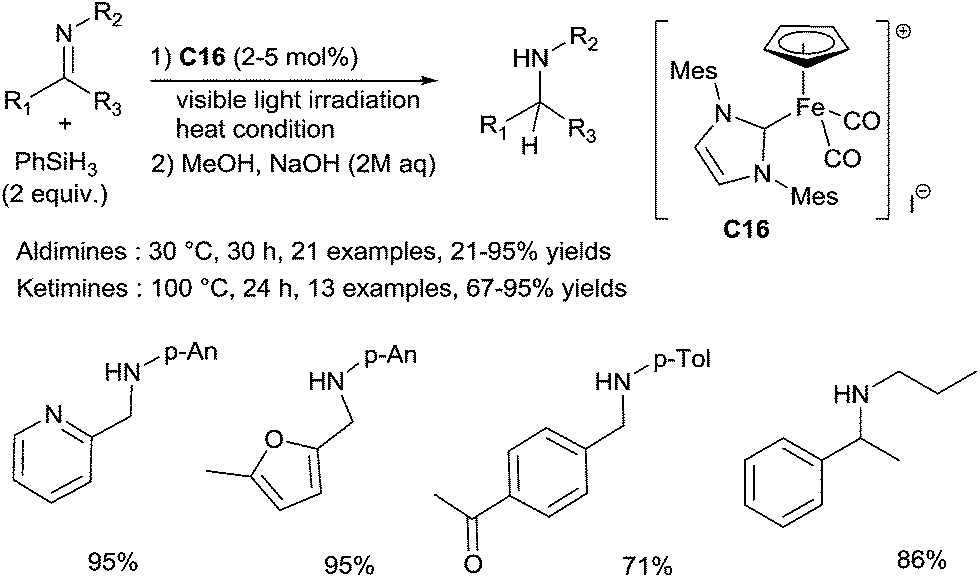 | ||
| Scheme 15 Catalysed hydrosilylation of aldimines by NHC iron complex C16. | ||
The similar conditions could be applied for the reduction of ketimines in presence of 5 mol% of C16 at 100 °C for 24 h. Various N-aryl or N-alkyl ketimines led to corresponding amines in 57–93% yields (Scheme 15).
B.5. Group 9 (iridium and rhodium) catalysts
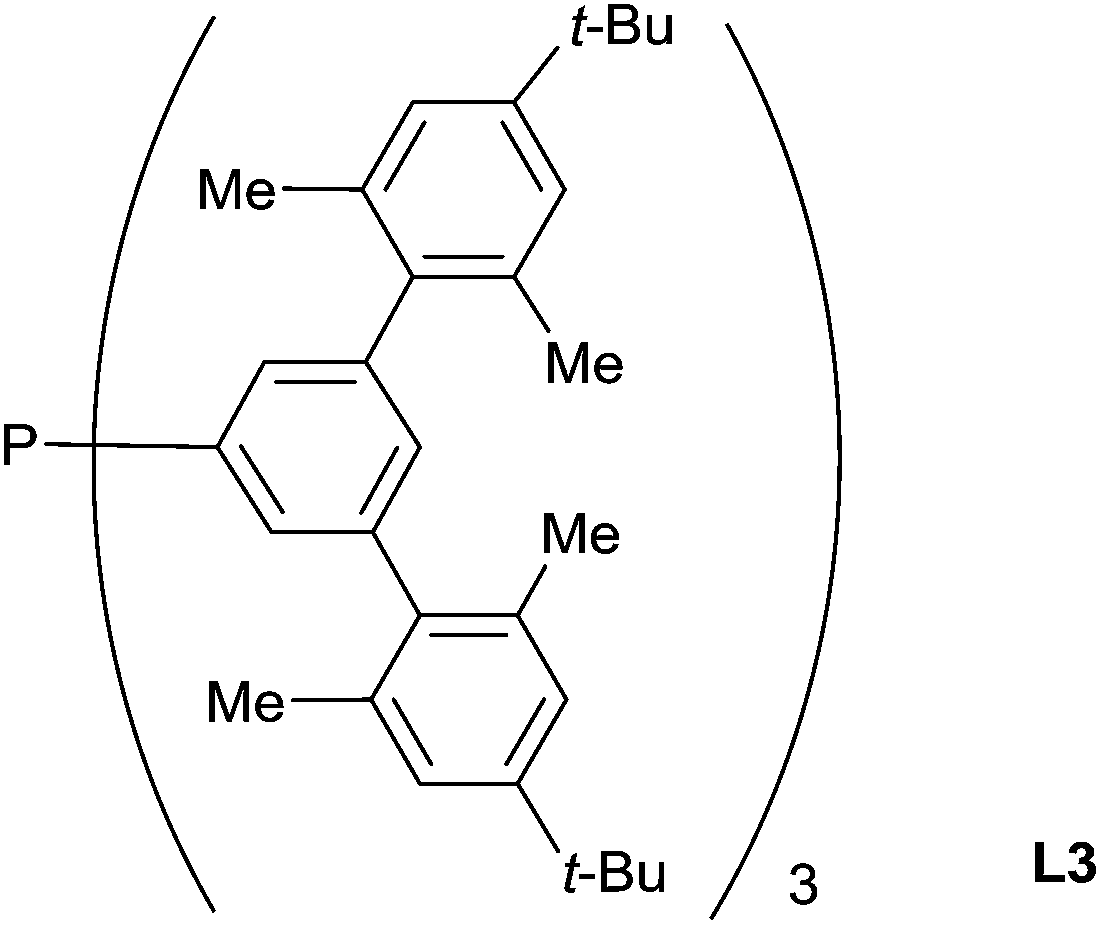
In pioneering contributions, in 1973, Kagan has reported the first asymmetric hydrosilylation of imines using a rhodium-(+)-diop complex as the catalyst (1 mol%): in presence of Ph2SiH2 (2 equiv.), N-(α-methylbenzylidene)benzylamine led to the corresponding amine in 98% yield and 50% ee at 24 °C.41Using a “bowl-shaped” phosphine L3 (3 mol%) associated to the precursor [RhCl(C2H4)2]2C17 (1.5 mol%), Tsuji reported the hydrosilylation of imines in the presence of PhMe2SiH at 60 °C for 8–16 h leading to the corresponding amines with 80–88% yields. These phosphines have a huge Tolman angles (>200°) and showed better activities than the conventional very bulky and basic monophosphines such as PCy3 and P(t-Bu)3 which showed only moderated activities42 (Fig. 1).
 | ||
| Fig. 1 Bowl-shaped monophosphine. | ||
In their continuous work on asymmetric hydrosilylation using chiral oxazolinylphosphine ligands, Hidai et al. developed iridium and rhodium-catalysed hydrosilylation of imines.43 For the hydrosilylation of 2-phenyl-1-pyrroline, the Ir-catalytic system showed a better catalytic activity and selectivity (95% yield, 85% ee) than the Rh-system (75% yield, 34% ee), similar to the Ru one. Notably, the size of the cyclic imine is crucial to obtain good enantioselectivities as the hydrosilylation of 6-phenyl-2,3,4,5-tetrahydropyridine to 2-phenylpiperidine gave lower ee. The enantioselectivity was also substrate dependant for acyclic imines: the Ir-catalyst was the most effective in terms of activity (56% yield) and enantioselectivity (89%) in the case of the hydrosilylation of (1-phenylethyl-1-ylidene)methanamine (Scheme 16).
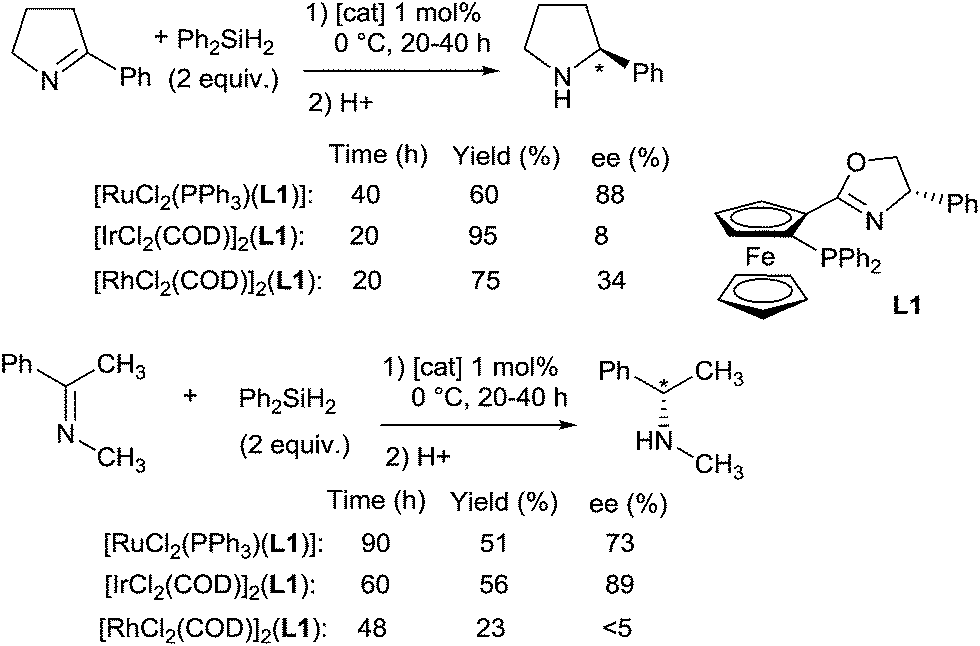 | ||
| Scheme 16 Ir and Rh catalysed hydrosilylation of imines. | ||
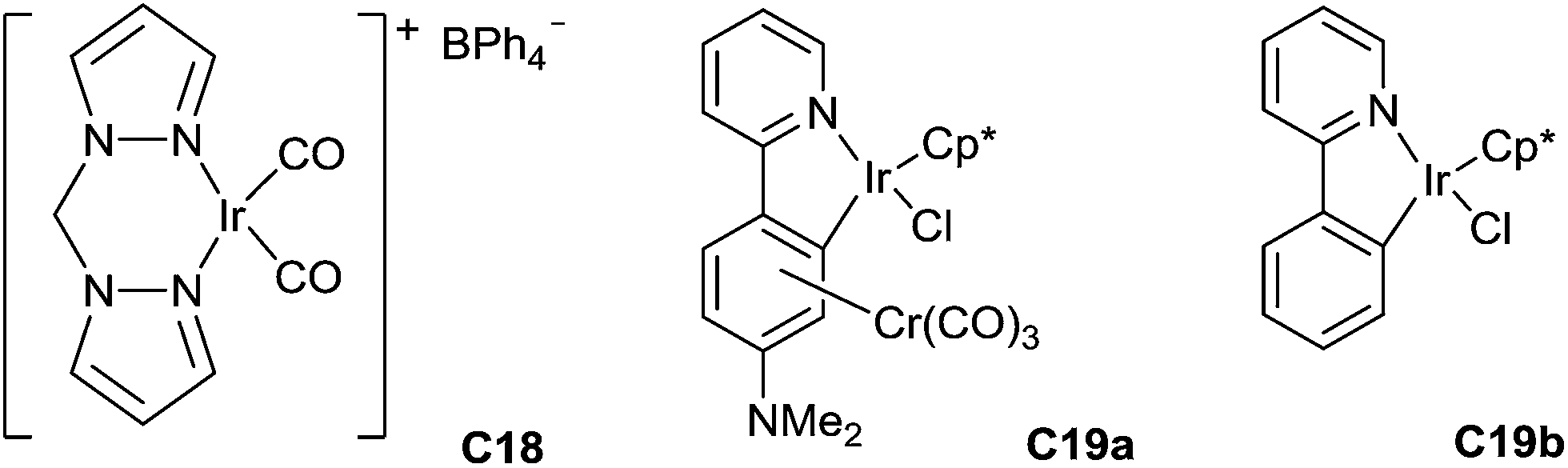
Messerle has also reported the catalysed hydrosilylation of N-alkyl- and N-aryl imines using a cationic Ir(I) complex [Ir(bpm)(CO)2][BPh4] C18 (bpm = bis(pyrazol-1-yl)methane) as the catalyst (Fig. 2). The reaction worked at room temperature in few minutes using 1 mol% of the complex in the presence of 2 equiv. of Et3SiH under air, N-phenyl imines giving higher conversions than the other imines studied. Using 0.5 mol% of C18, 80% conversion was obtained after 2 min for the hydrosilylation of N-arylketimines, with the turnover frequency up to 5000 h−1.44
 | ||
| Fig. 2 Iridium based complexes for hydrosilylation of imines. | ||
Similarly, Djukic et al. have described in 2012 the use of chromium tricarbonyl coordinated iridacycle C19a (2.5 mol%) for the reduction of aromatic imines using 1.3 equiv. of Et3SiH in methanol at 25 °C for 2–3 h leading quantitatively to the corresponding amines45 (Fig. 2). Recently, the iridacycle C19b (1 mol%) was found to be even more active when associated to sodium tetrakis[(3,5-trifluoromethyl)phenyl]borate as an additive (2 mol%) at 25 °C for 0.25–2 h in most of the examples.46
B.6. Group 10 (nickel, palladium) catalysts
In the area of nickel catalysis, even if the hydrosilylation of carbonyl derivatives is well-exemplified,47 only scarce examples of hydrosilylation of imines were reported. In 1995, Berkessel described the use of a nickel catalyst formed in situ from nickel(II)acetate and thiosemicarbazones L4a–c for the hydrosilylation of N-phenylimines and N-benzylphenylmethylimine in the presence of 2 equiv. of triethylsilane (Scheme 17) at 35 °C for 8 h.48a The secondary amines were then obtained after basic hydrolysis with moderate to high conversions (24–98%).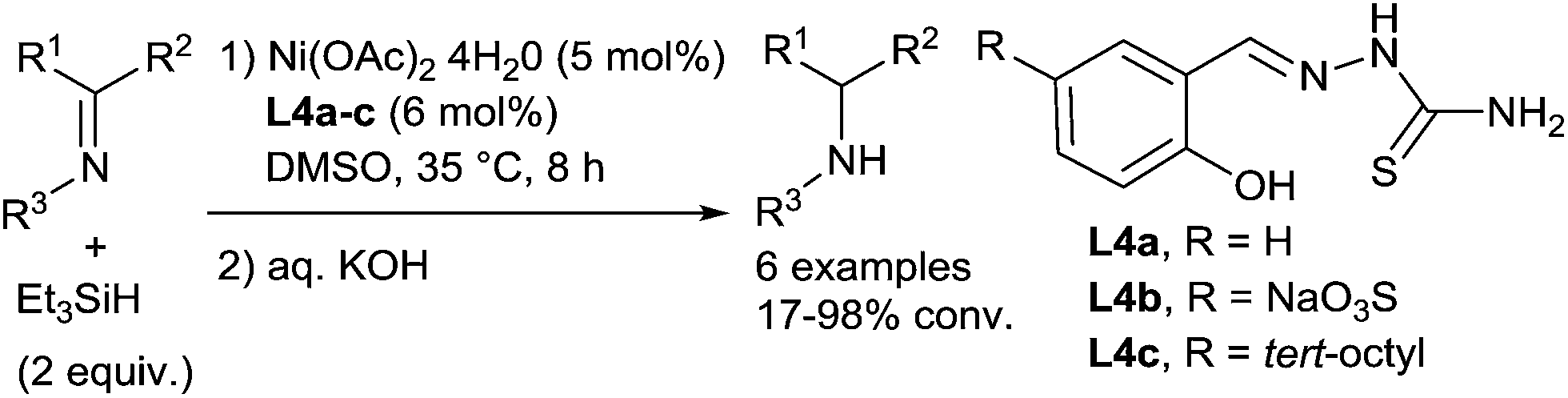 | ||
| Scheme 17 Nickel/thiosemicarbazones based catalysts for hydrosilylation of imines. | ||
More recently, our group have developed a general methodology for hydrosilylation of both aldimines and ketimines using half sandwich cyclopentadienyl NHC nickel complexes as the catalysts.
Using the nickel hydride complex, [Ni(Mes2NHC)HCp] (Mes2NHC = 1,3-dimesitylimidazol-2-ylidene, Cp = cyclopenta-dienyl) – generated in situ by reaction of [Ni(Mes2NHC)ClCp] C20a (1 mol%) and NaHBEt3 (2 mol%) – as the catalyst, aldimines can be hydrosilylated at rt for 17 h, and the corresponding amines were obtained after basic hydrolysis with 39–90% isolated yields. When using the cationic [Ni(Mes2NHC)(NCMe)Cp][PF6] complex C20b as the catalyst (1 mol%), similar results were obtained, but operating at 50 °C for 24 h. For the reduction of ketimines, more drastic conditions have to be used to reach full conversions: starting from C20a (1 mol%) and NaHBEt3 (2 mol%), the reaction has to be warmed at 50 °C for 17 h to be completed, whereas 5 mol% of C20b have to be used as the catalyst at 50 °C for 24 h (Scheme 18).48b
 | ||
| Scheme 18 Half sandwich cyclopentadienyl NHC nickel complexes for catalytic hydrosilylation of imines. | ||
Recently, using a (3-iminophosphine)palladium catalyst C21 (5 mol%), aromatic and alkyl N-allylimine derivatives were reduced to the corresponding N-allylamines in the presence of 1.2 equiv. of PhSiH3 in CDCl3 at 40 °C for 2–36 h (53–92% yields, 26 examples)49 (Fig. 3). Notably, cyano groups were tolerated.
 | ||
| Fig. 3 Palladium complex for hydrosilylation of N-allylimines. | ||
B.7. Group 11 (copper) catalysts
Copper based catalysts were widely described in the hydrosilylation of carbonyl derivatives, mainly using copper hydride complexes.50Lipshutz in 2004 described the use of an “in situ” generated copper hydride complex for the efficient catalytic asymmetric hydrosilylation of phosphinyl ketimines.51 Several phosphinyl amines were produced with excellent yields (90–99%) and high ee (94–99%) using 6 mol% of the [CuH] species generated in situ from CuCl, NaOMe, and (R)-(−)-DTBM-SEGPHOS L5 as the ligand, in the presence of 3 equiv. of TMDS (1,1,3,3-tetramethylsiloxane) and 3.3 equiv. of t-BuOH in toluene at rt for 17 h (Scheme 19). It must be underlined that imines activated by bis-arylphosphinyl moieties were required to obtain good activity. Notably, this copper catalytic system remains one of the most effective in asymmetric hydrosilylation of aryl and alkyl ketimines with 94–99% ee.
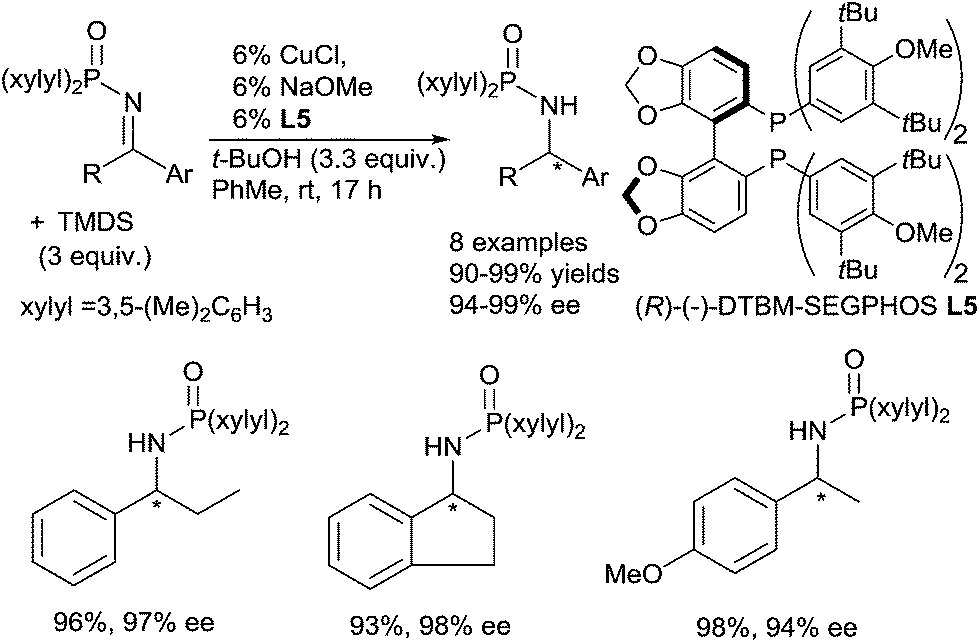 | ||
| Scheme 19 Copper catalysed hydrosilylation of imines. | ||
B.8. Group 12 (zinc) catalysts
One major breakthrough in the area of hydrosilylation of carbonyl compounds was reported in 1998 by Mimoun who has found zinc complexes as efficient precatalysts for the reduction of various chemical functionalities with PMHS.52 Thus, imines can be chemoselectively reduced at rt in 18–24 h in methanol leading to the corresponding amines in 65–99% GC-yields in a one-step procedure using polymethylhydrosiloxane (PMHS) (2–4 equiv.) and a simple zinc–diamine catalyst, [EtZnNBn(C2H4)NHBn]2, (2 mol%) which is formed in situ from a 1:1 combination of diethylzinc and N,N′-dibenzylethylenediamine (dbea).53
A highly enantioselective hydrosilylation of imines using a chiral Zn/diamine catalyst was then developed by Yun et al.54 Various N-phosphinyl amines were produced by reduction of the corresponding imines in the presence of 5 mol% ZnEt2, with 5 mol% of chiral diamine ligand L6, 3 equiv. of PMHS in MeOH/THF [2:8 (v/v)] at rt for 12 h, then followed by the desilylation with NaOH (1 N) (Scheme 20). It is noteworthy that MeOH plays an important role to enhanced reaction rate of the zinc-catalysed hydrosilylation of imines.
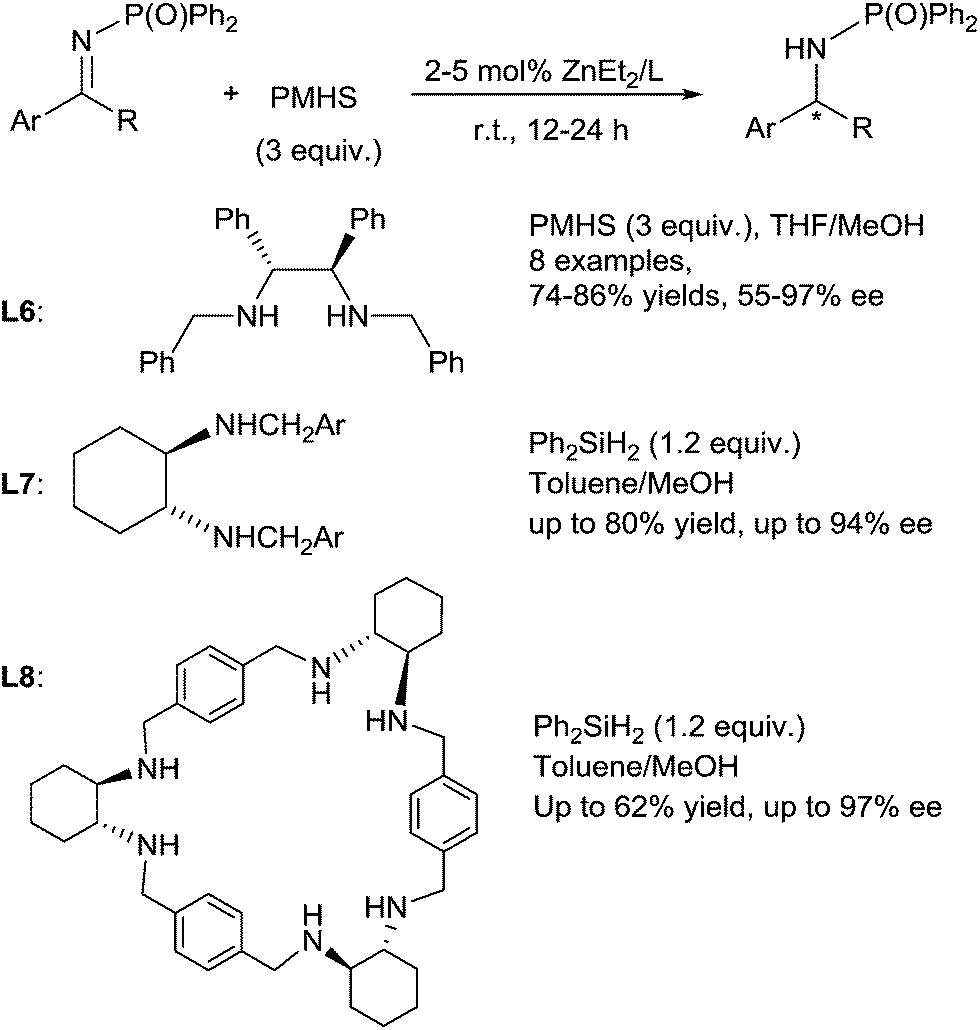 | ||
| Scheme 20 Asymmetric Zn-catalysed hydrosilylation of ketimines. | ||
In 2011, Kwit et al. reported zinc-trans-1,2-diaminocyclohexane derivatives L7 or zinc-chiral hexamine macrocycle L8 derived from trans-1,2-diaminocyclohexane catalysed enantioselective hydrosilylation of phosphinyl ketimines with Ph2SiH2 as the reducing agent in toluene–methanol at rt for 24 h.55 Notably, ketimines bearing an electron-withdrawing phosphinyl group have to be used as substrates for the reduction into their corresponding protected amines whereas imines bearing different N-substituents such as OH, OBn, or p-methoxyphenyl (PMP) were unreactive. Moreover, better results on hydrosilylation of N-diphenylphosphinyl ketimines were obtained with the ZnEt2-hexamine macrocycle catalyst in comparison to the trans-1,2-diaminocyclohexane ligand-ZnEt2 catalyst (Scheme 20).
In 2016, using the catalytic system from 5 mol% ZnEt2 and 5 mol% of L6, Mlynarski reported the diastereoselective hydrosilylation of N-(tert-butylsulfinyl)imines in the presence of 2 equiv. of (EtO)3SiH in THF leading to the corresponding amines in 54–85% yields and 75–98% de. Noticeably ester, nitro, bromo, styryl groups were tolerated.56
C. Hydrosilylation of amides to amines
An interesting route to amines is the reduction of carboxamides, which are the least reactive species among carbonyl derivatives toward hydride addition. An alternative method to the use of stoichiometric amount of metal hydride reducing agents such as LiAlH4 deals with transition metal catalytic reduction of amides which can be achieved using hydrosilanes as the reductants.C.1. Group 4 (titanium) catalysts
The first titanocene-catalyzed reduction of tertiary amides to tertiary amines was reported by Harrod et al.57 The tertiary amines were synthesized by reduction of the corresponding tertiary amides in moderate to very good yields (40–96%) using Cp2TiF2C22a or Cp2TiMe2C22b as the catalyst (10 mol%), with 2 equiv. of PhMeSiH2 in toluene at 80 °C for 30 minutes. Notably, when this reaction was carried out at room temperature using various tertiary acetamides, a mixture of tertiary and secondary amines were obtained, the latter produced via the hydrogenolysis of the C(O)–N bond (Scheme 21). It must be noticed that secondary amides were very slowly reduced under optimized conditions. | ||
| Scheme 21 Ti-catalysed hydrosilylation of amides. | ||
This selectivity is different than the one obtained by Buchwald when using a stoichiometric amount Ti(O–i-Pr)4 in the presence of Ph2SiH2 as aldehydes were obtained starting from acetamide derivatives.58 Furthermore, even if performed in a stoichiometry way, Lemaire et al. reported the more challenging reduction of primary amides to primary amines, purified as the corresponding amine hydrochloride salts, using 1 equiv. of Ti(OiPr)4 with 4 equiv. of PMHS in toluene at 100 °C. Aromatic and aliphatic primary amines were isolated in 87–95% yields. However, the tertiary and secondary amines were only obtained in 33–40% yields due to the reduction of tertiary and secondary amides into aldehydes after acidic treatment.59
C.2. Group 6 (molybdenum) catalysts
Both tertiary and secondary amides were reduced to the corresponding amines in moderate to good yields (40–87%) by using 10 mol% of MoO2Cl2C23 with 2 equiv. of PhSiH3 in refluxing toluene60 (Scheme 22). It must be underlined that this catalytic system is suitable for tertiary amides to tertiary amines. Using Mo(CO)6C24 (5 mol%) as the catalyst, Tinnis and Adolfsson et al. described a chemoselective hydrosilylation of α,β-unsaturated amides leading to allylamines in 51–95% yields using 1.5 equiv. of TMDS at 65 °C for 24 h.61 | ||
| Scheme 22 Molybdenum catalysed hydrosilylation of amides. | ||
On the other hand, Enthaler has developed a selective cleavage of the C–N bond of carboxamides leading to primary alcohols and amines using a well-defined bimetallic molybdenum complex C2562 (Scheme 23). The key intermediate for both C–O bond or C–N bond cleavage is O-silylated N,O-acetal 23-I, which is formed from the Mo–H species addition to the amide. In the C–O bond cleavage pathway, 23-I is transformed to the iminium ion species 23-IV by elimination of a silanolate and the transfer of a second equivalent of activated hydrosilane to 23-IV then led to the tertiary amine. By contrast, in the C–N bond cleavage pathway, the second equivalent of the Mo–H species transfers a hydride to the carbon of 23-I and cleaves the C–N bond to generate a silyl ether and a silyl amine, finally affords the corresponding alcohol and amine after work-up. Those two reports show that the nature of the molybdenum complexes and the hydrosilanes are crucial for the chemoselectivities of the reduction of amides.
 | ||
| Scheme 23 Mechanism of Mo-catalysed hydrosilylation of amides. | ||
C.3. Group 8 (ruthenium, osmium and iron) catalysts
 | ||
| Scheme 24 Ru- and Os-catalysed hydrosilylation of amides. | ||
In 2001, Nagashima reported a ruthenium cluster, (μ3,η2,η3,η5-acenaphthylene)-Ru3(CO)7C28 which was able to catalyse the reduction of tertiary carboxamides leading to the corresponding tertiary amines in the presence of 2.5 equiv. of EtMe2SiH in 1,4-dioxane at 20 °C for 0.5–2 h (3 examples, 90–99% NMR yields).64 With the same catalyst C28 (1 mol%), carboxamides can be also reduced using 4.4 equiv. of PMHS in tetrahydropyran (THP) at 30 °C for 15 h65 (Scheme 25). In this protocol, the ruthenium catalyst species is easily and effectively self-encapsulated into polymer gel which resulting from the active ruthenium species cross-link with silicone resin. It is noteworthy that the encapsulated catalyst species can be facilely recovered and reused as the catalyst for the reduction without losing the activity.
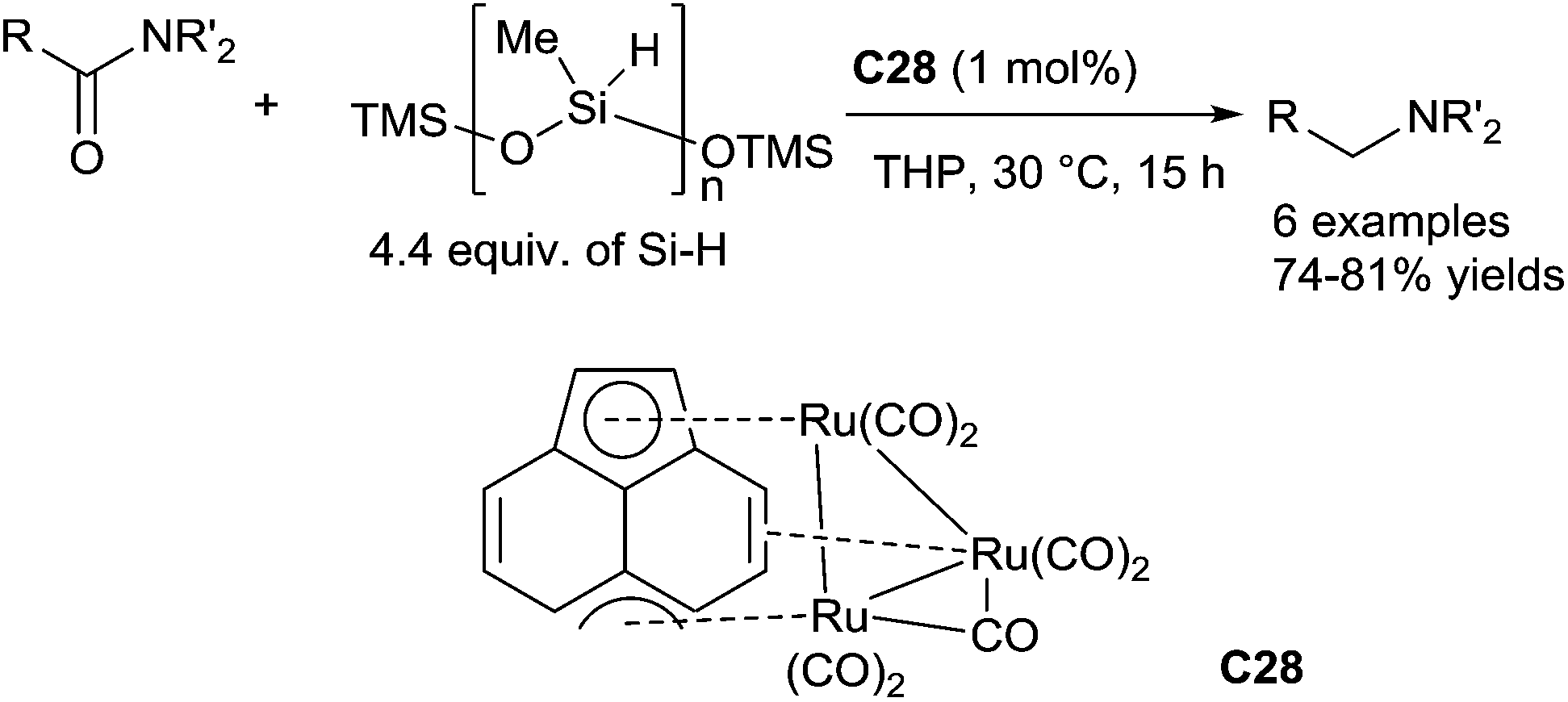 | ||
| Scheme 25 Ruthenium cluster for catalysed hydrosilylation of amides. | ||
Notably, this ruthenium cluster complex C28 could be applied to catalyse highly chemo-selective hydrosilylation of tertiary amides without reducing ketone or ester moieties, which is a hard task in molecular synthesis. This reaction was carried out with 1 mol% of ruthenium catalyst, 2.5–3.5 equiv. of PhMe2SiH in the presence or absence of 1 equiv. of Et3N at room temperature (Scheme 26).66
 | ||
| Scheme 26 Ruthenium cluster for catalysed chemoselective hydrosilylation of tertiary amides. | ||
Starting from aliphatic and aromatic secondary amides, using the ruthenium cluster C28, different chemoselectivities towards secondary amines or tertiary amines were obtained depending of the nature of the hydrosilane used.67
The secondary amines were produced from the corresponding secondary amides in the presence of 3 mol% of C28, with 3.2–4.5 equiv. of [H(Me)2Si]2O (TMDS) in DME at 40–60 °C for 6–12 h (Scheme 27a).
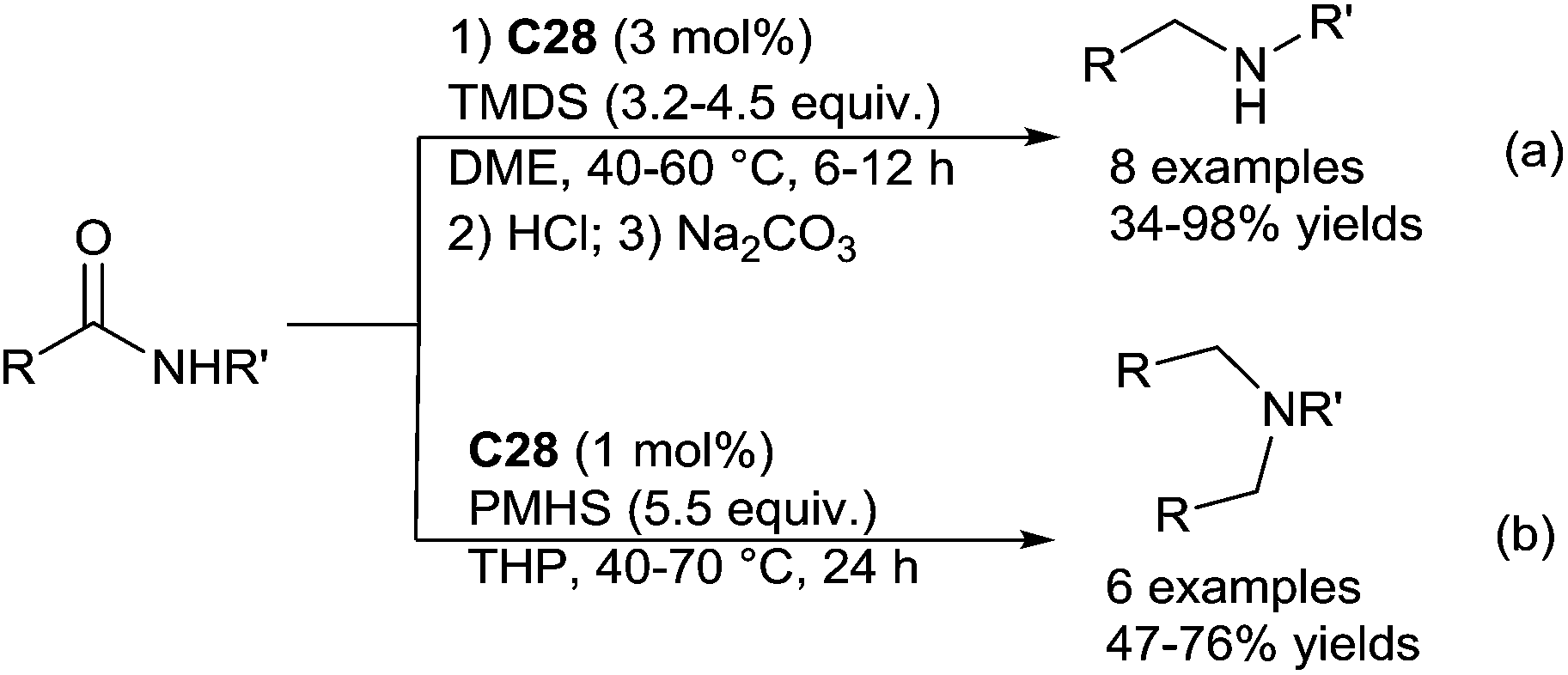 | ||
| Scheme 27 C28 catalysed hydrosilylation of secondary amides. | ||
By contrast, using PMHS (5.5 equiv.) as the hydride source, with secondary amides bearing bulky substituents on the nitrogen atom such as N-isopropyl or N-tert-butyl, the corresponding secondary amines were obtained, even if the reaction is slower (70 °C, 24 h). An interesting outcome was observed when using secondary amides with less bulky N-substituents, as tertiary amines were obtained when working in THP at 70 °C for 24 h (Scheme 27b). It must be noticed that such production of the tertiary amines from secondary amides involving a reductive N-alkylation reaction was previously reported in the presence of POCl3 and NaBH4.68
Similarly, using Ru3(CO)12C29 (1 mol%) in the presence of 4 equiv. of TMDS in toluene at 50 °C for 24 h, Senanayake succeeded to reduce tertiary, secondary and primary amides leading the corresponding amines in good yields (67–93% yields, 18 examples). Interestingly this methodology was applied to the synthesis of calcimimetic drug cinacalcet. It must be noted that this catalytic system can also reduce 4-bromobenzonitrile to 4-bromobenzylamine in 95% conversion after 76 h at 70 °C using 5 equiv. of TMDS.69
In 2013, we reported an unexpected selective ruthenium(II)-catalysed hydrosilylation of primary amides leading to symmetric secondary amines.70 This reaction was performed with 1–2 mol% of [RuCl2(mesitylene)]2C30, 3 equiv. of PhSiH3 under solvent-free condition at 100 °C (Scheme 28). Substituted benzamides, thiophenamides, benzylamides and alkylamides led successfully to respected secondary amines in moderate to good yields.
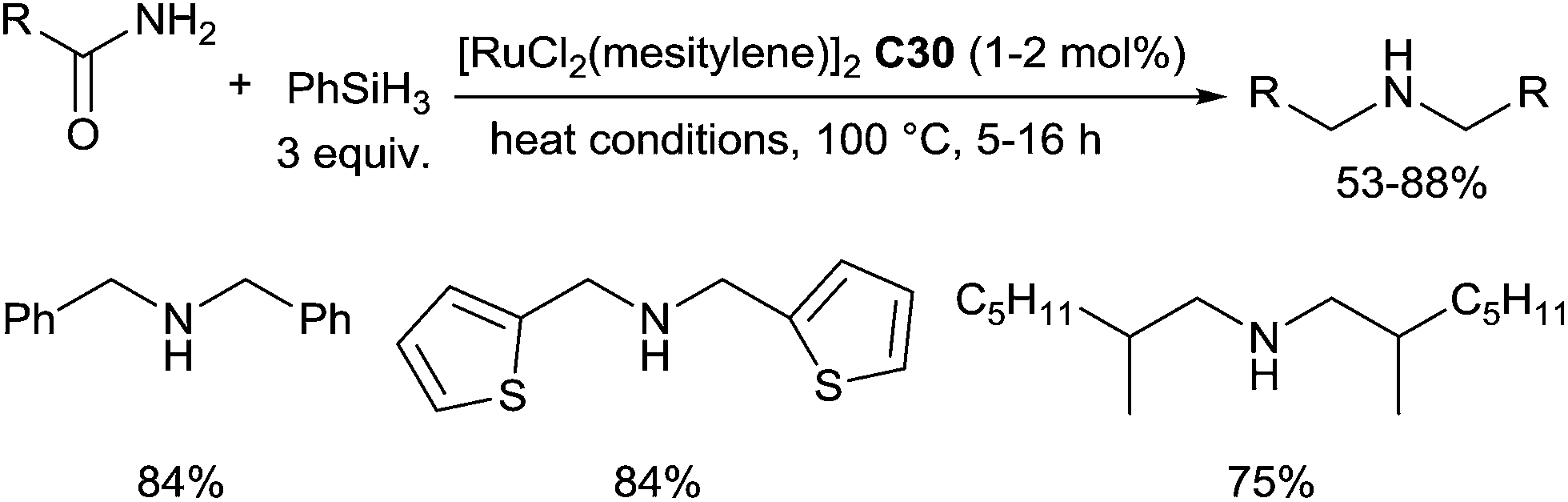 | ||
| Scheme 28 Ru(II) catalysed hydrosilylation of primary amides. | ||
 | ||
| Scheme 29 Fe3(CO)12 catalysed hydrosilylation of tertiary amides. | ||
The mechanism of Fe-catalysed reduction of amides is similar to the ruthenium catalysed procedure. The O-silylated N,O-acetal intermediate was firstly generated from the carbonyl group of the amide, then transformed into an iminium species which is finally reduced into the corresponding amine.
On the other hand, Nagashima reported similar hydrosilylation reaction of tertiary carboxamides using [Fe(CO)5] or [Fe3(CO)12] C31 as the catalyst, but with TMDS as the reducing agent.71 Similar results could be obtained in both thermal conditions at 100 °C for 24 h and photo-assisted conditions by irradiation with a 400 W high pressure mercury lamp for 9 h at rt (Scheme 30).
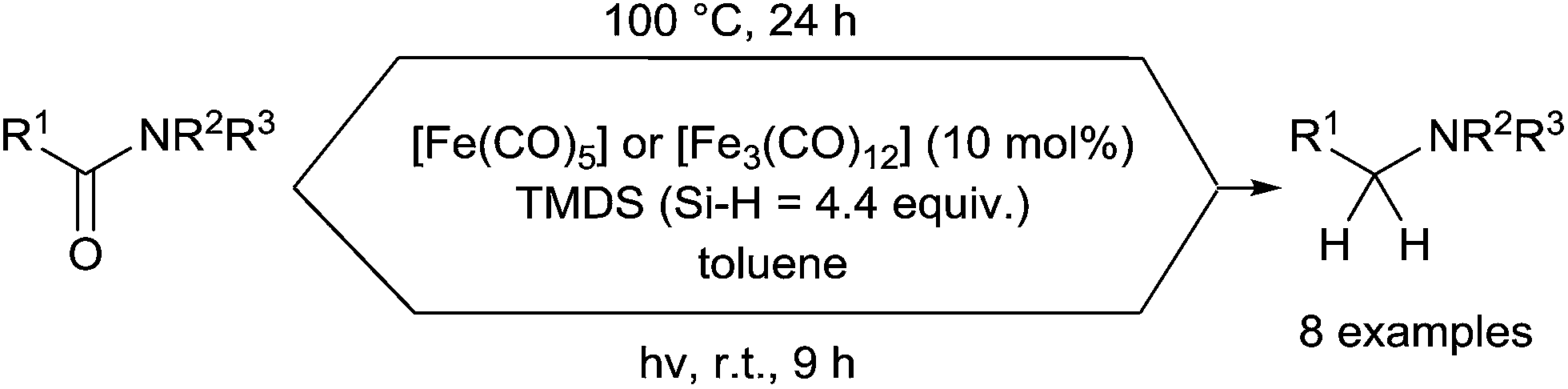 | ||
| Scheme 30 Thermal vs. photochemical Fe-catalysed hydrosilylation of tertiary amides. | ||
It must be underlined that the Fe-catalysed reduction of N,N-dimethyl-p-nitrobenzamide didn't give to the corresponding N,N-dimethyl-p-nitrobenzamine with respect to the ruthenium or platinum catalytic processes, but the aniline product resulting from the chemoselective reduction of the nitro group and which was isolated in 77% yield (Scheme 31).71
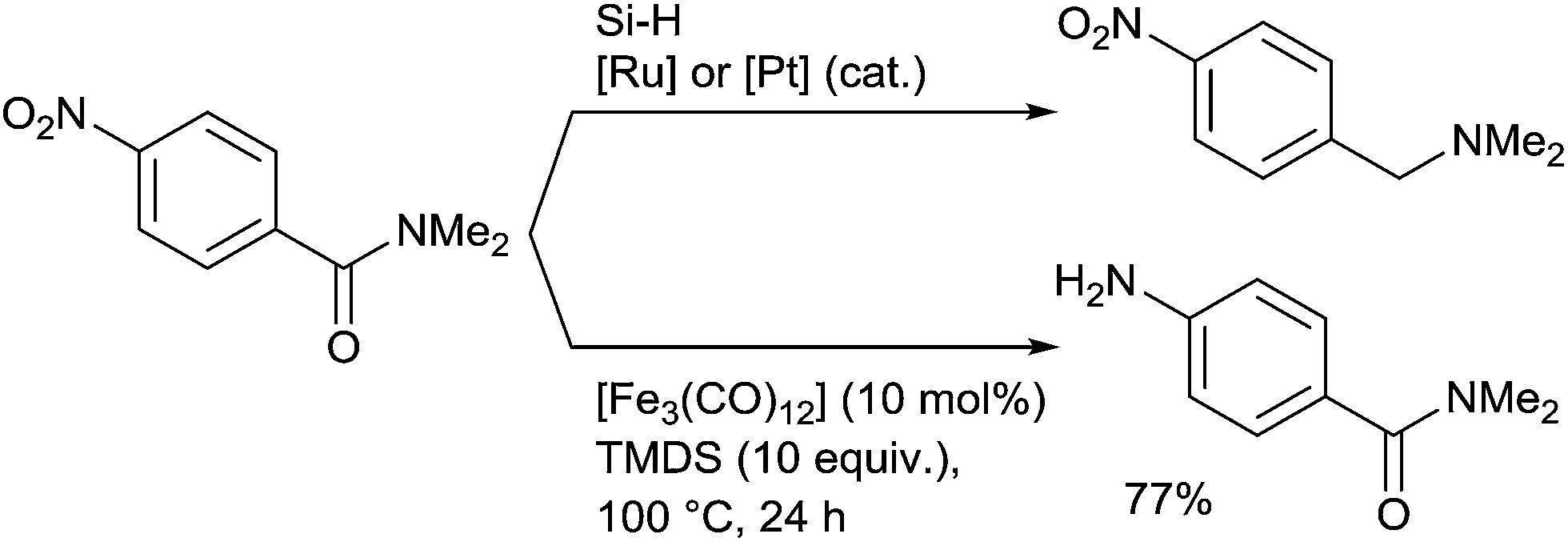 | ||
| Scheme 31 Chemoselectivity in Fe-catalysed hydrosilylation of tertiary amides. | ||
An original heptanuclear iron carbonyl cluster, [Fe3(CO)11(μ-H)]2-Fe(DMF)4C33 was found to be as an efficient catalyst for the reduction of carboxamides in the presence of 1,2-bis(dimethylsilyl)benzene (BDSB) as the reducing agent.73 Various amides were reduced to corresponding amines in the presence of only 0.5 mol% of Fe-cluster C33 in toluene at 100 °C for 3 h. C33 was found to be more active in comparison to the classical iron carbonyl complexes (Scheme 32).
 | ||
| Scheme 32 Iron catalysed hydrosilylation of carboxamides. | ||
Well-defined cyclopentadienyl NHC iron complexes can catalyze the hydrosilylation of amides to amines with PhSiH3 as the reducing agent (2 equiv.) under visible light irradiation at 100 °C for 24 h.74 Using the complex [CpFe(IMes)(CO)2][I] C16 (5 mol%), various tertiary and secondary amides led to the corresponding amines in good to excellent yields, and the CC bond can be tolerated. It must be noticed that using similar conditions with 4 equiv. of phenylsilane, primary amides gave selectively and quantitatively the corresponding nitrile derivatives (Scheme 33).
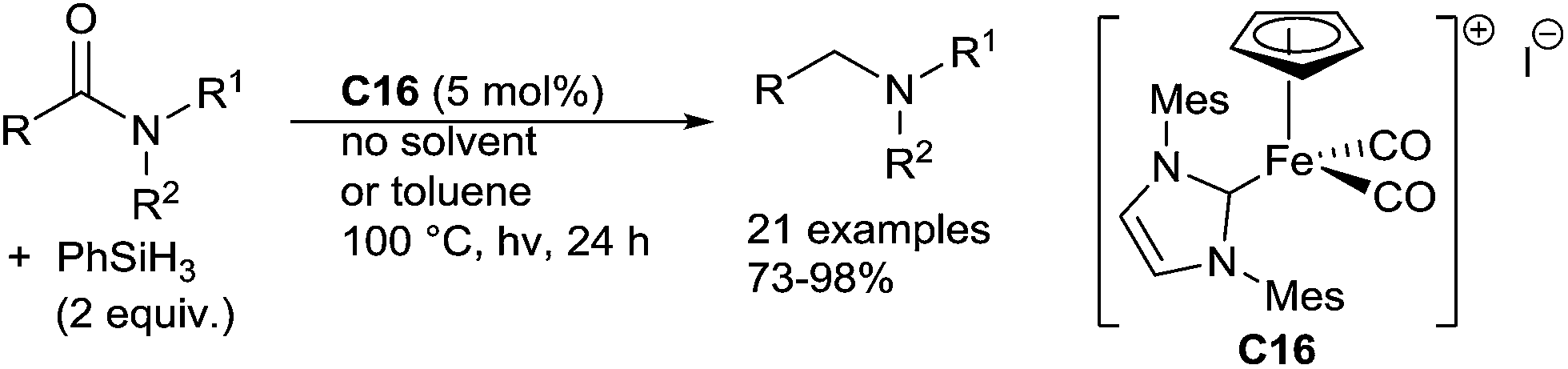 | ||
| Scheme 33 C16-catalyzed hydrosilylation of carboxamides. | ||
The in situ generated iron/NHC complex from 1-(2-hydroxyethyl)-3-methylimidazolium triflate ([HEMIM][OTF]) L9 and iron(II)acetate, was recently reported by Adolfsson and catalysed efficiently the hydrosilylation of tertiary amides to amines.75 The catalytic species was carefully prepared from 1 mol% of Fe(OAc)2, 1.1 mol% of L9, 1 mol% of LiCl which were treated under vacuum for 10 min, followed by the successive addition of THF and 2.2 mol% of n-BuLi. Notably, the reaction works with 3 equiv. of PMHS in THF at 65 °C for 5–14 h, but was not suitable for the reduction of primary and secondary amides (Scheme 34). LiCl seems to play an important role for the improvement of the selectivity of the reaction.
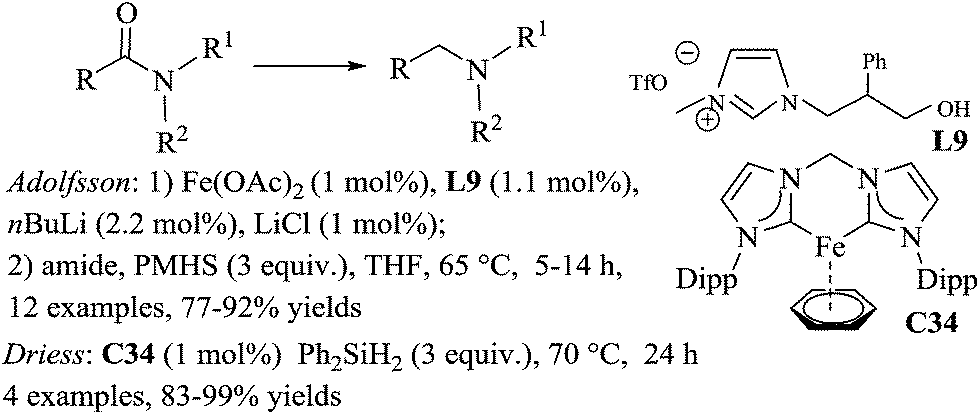 | ||
| Scheme 34 Fe-catalysed hydrosilylation of carboxamides. | ||
The reduction of tertiary amides can also be performed using 1 mol% of the well-defined NHC–Fe(0) complex bis-(N-Dipp-imidazole-2-ylidene)methylene-Fe(η6-benzene) C34 in the presence of 3 equiv. of diphenylsilane at 70 °C in THF for 24 h.76
While secondary and tertiary amides were successfully reduced to corresponding amines, the reduction of primary amides to primary amines is still a challenging task in iron catalysis field. To overcome this goal, Beller et al. reported the first reduction of primary amides to primary amines using two iron catalysts ([Et3NH][HFe3(CO)11]) C32 and Fe(OAc)2, in association with phenanthroline type ligand L10 and (EtO)2MeSiH as the reducing agent.77 This catalytic system includes two steps: (i) the dehydration of carboxamides to the corresponding nitriles using C32 as the catalyst in the presence of (EtO)2MeSiH; (ii) the hydrosilylation of nitrile to the corresponding primary amines in the presence of the in situ prepared catalyst from Fe(OAc)2 and L10 with (EtO)2MeSiH. Various aromatic and aliphatic amines were obtained in moderate to good yields (Scheme 35).
 | ||
| Scheme 35 Fe-catalysed hydrosilylation of primary carboxamides. | ||
C.4. Group 9 (cobalt, rhodium and iridium) catalysts
 | ||
| Scheme 36 Cobalt catalysed hydrosilylation of amides. | ||
The secondary amide is not favorable with this catalytic system. It is noteworthy that excellent chemoselectivities were found in the presence of ester and alkene groups.
C bond and CC bonds were not tolerated.
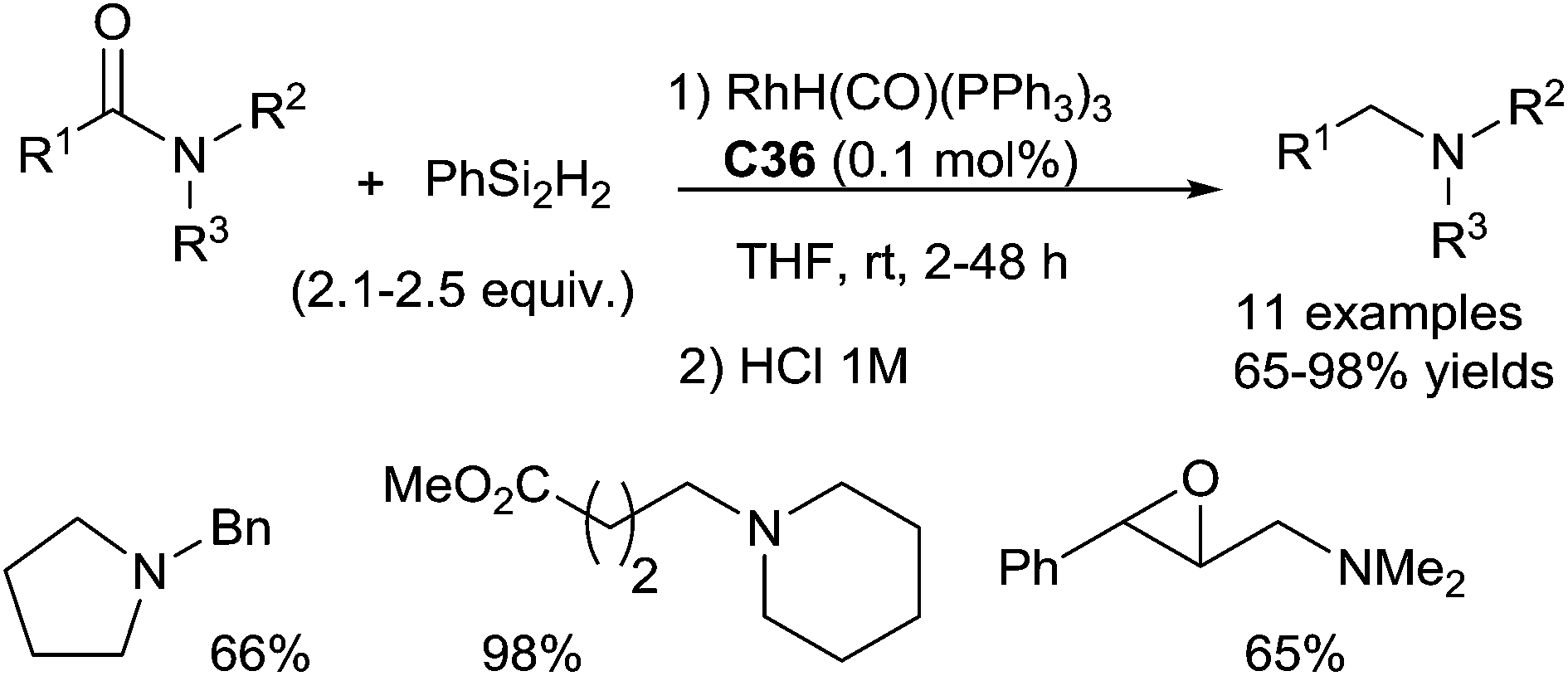 | ||
| Scheme 37 Rhodium catalysed hydrosilylation of amides. | ||
In 2015, Beller et al. reported a selective rhodium-catalyzed hydrosilylation of tertiary β-lactams giving the corresponding azetidines in 35–99% yields under mild conditions using phenylsilane as the reducing agent and the combination of [Rh(cod)2][BF4] C37 (2.5 mol%) and dppp (bis(diphenylphos-phino)propane) (5 mol%). Notably, starting with optically active compounds, no erosion of the stereochemistry was observed81 (Scheme 38).
 | ||
| Scheme 38 Rhodium-catalysed hydrosilylation of tertiary β-lactams. | ||
The same group also reported the efficient reduction of the tertiary amide bond in amino acid derivatives and peptides using the same catalytic system C37 (1 mol%) and dppp (2 mol%) in the presence of 2 equiv. of phenylsilane as a reductant at rt – 45 °C. This methodology can be applied for the chemospecific reduction leading to biologically interesting peptides. Noticeably, the catalytic system tolerates numerous functional groups such as secondary amides, ester, nitrile, etc.82
Using a zwitterionic N-heterocyclic silylene–rhodium and iridium complexes C38a–b as the catalysts, Driess et al. reported the hydrosilylation of N-acyl dibenzoazepine. The rhodium complex C38a (2.5 mol%) gave specifically the N-ethyl dibenzoazepine, resulting from the C–O cleavage in 68% GC-yield by reaction with 2.5 equiv. of PhSiH3 at rt for 24 h. In similar conditions, even if more active, the iridium complex C38b (2.5 mol%) led to a mixture of the N-ethyl dibenzoazepine (78%) and dibenzoazepine (9%) resulting from the C–N cleavage83 (Scheme 39). Interestingly the beneficial role of the N-heterocyclic silylene have been demonstrated as with [Rh(cod)Cl]2 and [Ir(cod)Cl]2, 53 and 30%, respectively, of N-ethyl dibenzoazepine were obtained.
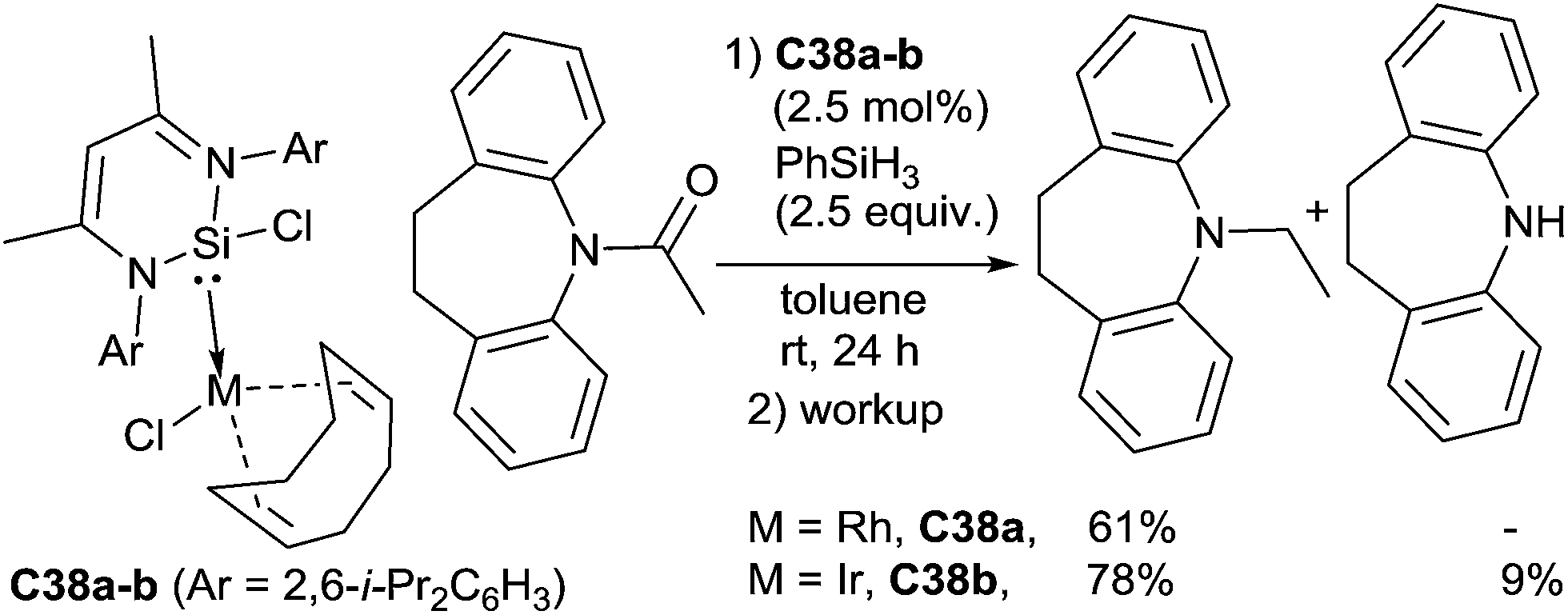 | ||
| Scheme 39 N-Heterocyclic silylene rhodium and iridium complexes for hydrosilylation of amides. | ||
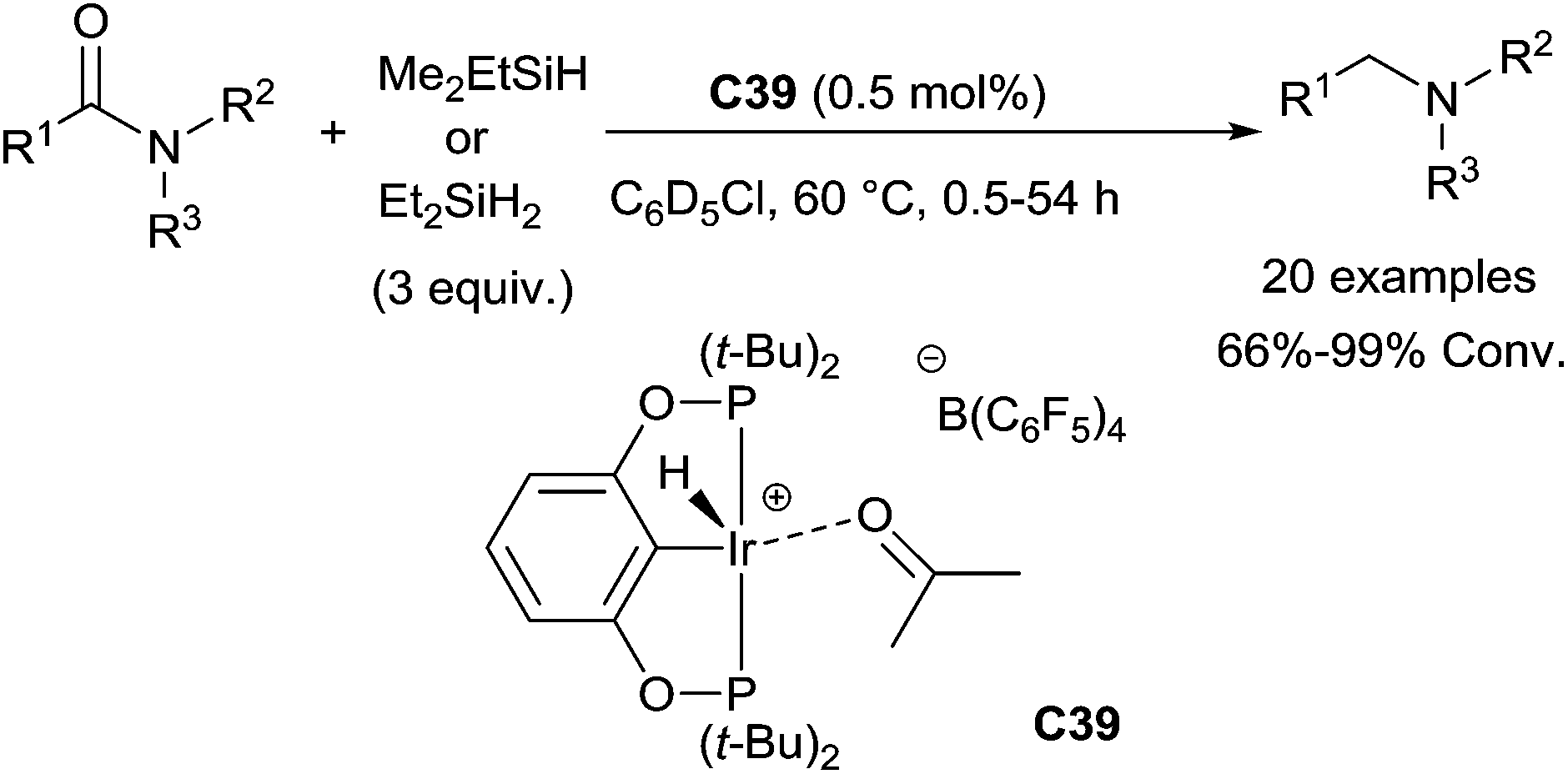 | ||
| Scheme 40 Iridium catalysed hydrosilylation of amides. | ||
Notably, under such mild conditions, functional or sensitive groups such as cyclopropyl, furanyl, phenyldiazo, terminal alkenes, internal alkynes, cyclic ethers and nitriles were tolerated, whereas ketone and ester were hydrosilylated in competition with the amide reduction. It is noteworthy that the presence of 1 atm H2 accelerated the reduction and the turnover numbers is up to 10000. An in-depth mechanism study, the active species Ir(IV)(POCOP)Ir(H)3(SiHEt2) generated in situ from the cationic Ir(III)acetone complex has been identified. The presence of 1 atm H2 could accelerate the reduction by shifting the equilibrium from Ir(III)silyl hydride to the active species Ir(IV).
The same year, Brookhart developed [Ir(COE)2Cl]2C40 catalysed selective reduction of secondary amides to secondary amines or imines using diethylsilane as the reductant. Secondary amines were synthesized using 0.5 mol% of [Ir(COE)2Cl]2C40 with 4 equiv. of Et2SiH2 at rt or 80 °C, followed by an acidic workup.85 Interestingly, when the catalyst loading decreased to 0.1 mol% with only 2 equiv. of Et2SiH2 at rt for short reaction times (0.2–1 h), imines were obtained and isolated in good yields (Scheme 41).
 | ||
| Scheme 41 Iridium catalysed selective hydrosilylation of amides. | ||
In 2009, Nagashima described an original selectivity towards aldenamines when performing the hydrosilylation of tertiary amides using 5 mol% of IrCl(CO)(PPh3)2C41 in the presence of 2 equiv. of TMDS in toluene at room temperature for 30 min or 1 mol% of C41 with 4 equiv. of PMHS86 (Scheme 42). It must be underlined the advantage to use PMHS for the purification of the aldenamines as the insoluble silicone resin containing the iridium residue was simply filtrated.
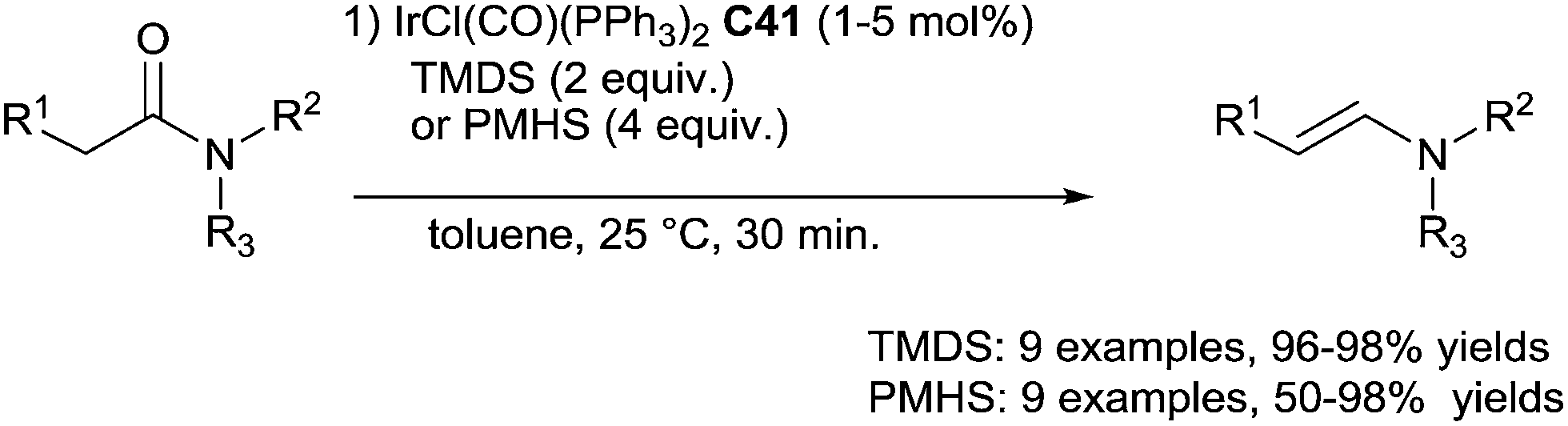 | ||
| Scheme 42 Iridium catalysed hydrosilylation of amides to enamines. | ||
Recently, the same group has shown that using IrCl(CO)(PR3)2 bearing electron-withdrawing phosphorus ligands as catalysts, higher catalytic activity were observed. Thus, high catalytic efficiency (TON > 10000) was achieved with the iridium catalyst bearing a tri(pentafluoroaryl)phosphite P(OC6F5)3, (catalyst loading 0.001 mol%).87
C.5. Group 10 (nickel, platinum) catalysts
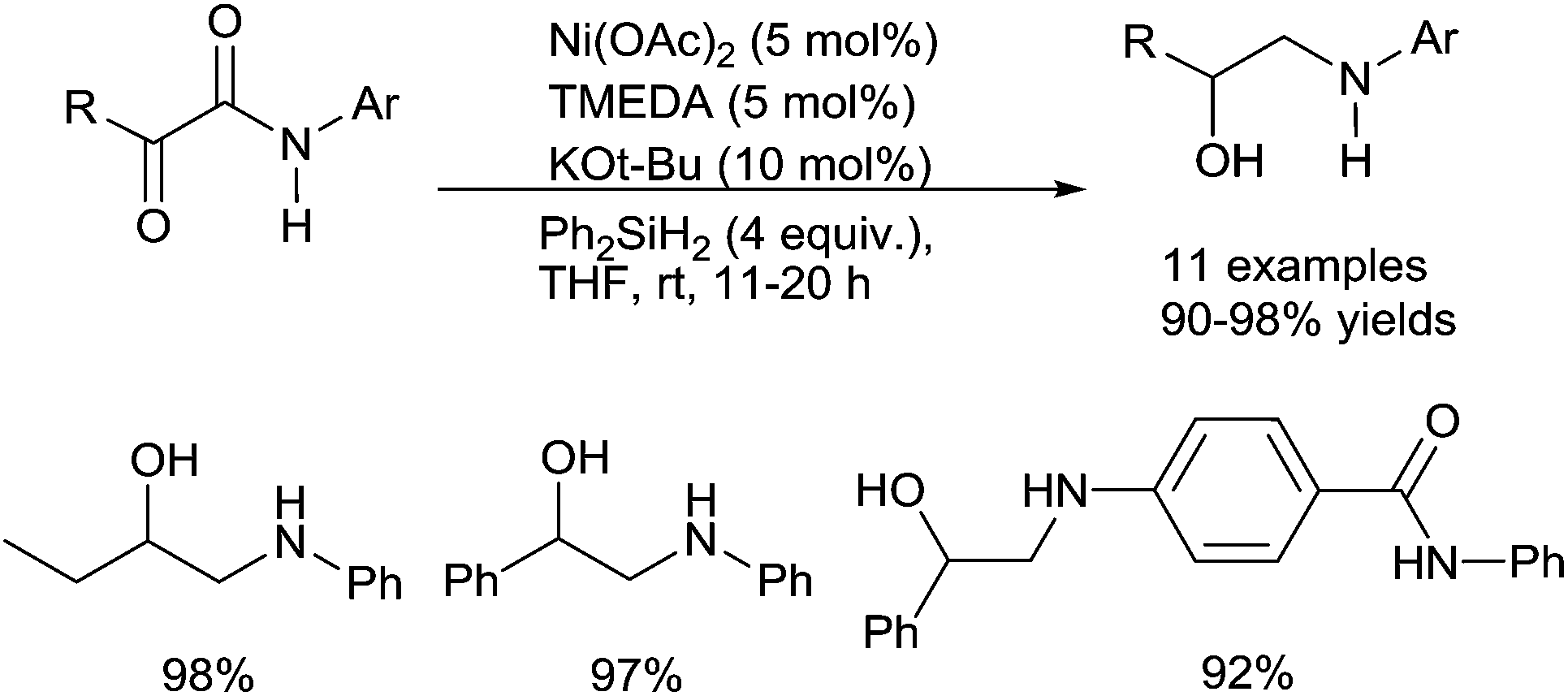 | ||
| Scheme 43 Nickel catalysed hydrosilylation of α-keto amides. | ||
This reaction corresponds to the concomitant reduction of the amido moiety in amines and the reduction of the keto part in alcohol. Furthermore, it must be underlined that the reduction is highly chemoselective as isolated amido groups were not reduced under such conditions, and only α-ketoamido moiety are involved in the reduction process showing the importance of either the keto- and/or the hydroxyl group on the efficiency of the reduction of the amido part. Notably, when using PMHS as the reductant (4 equiv.) and NaOAc (10 mol%) as the base, only the keto group was reduced to lead to the α-hydroxy amides.
 | ||
| Scheme 44 Platinum catalysed hydrosilylation of amides. | ||
Beller reported also a Pt–NHC based complexes C43 as catalysts (1 mol%) for the hydrosilylation of tertiary amides to the corresponding tertiary amines in the presence of 2 equiv. of Ph2SiH2 in THF at 40 °C for 1 h (Scheme 45). Notably, with secondary amides, drastic conditions (100 °C) were used and led the amines with moderate yields (5–30%).90
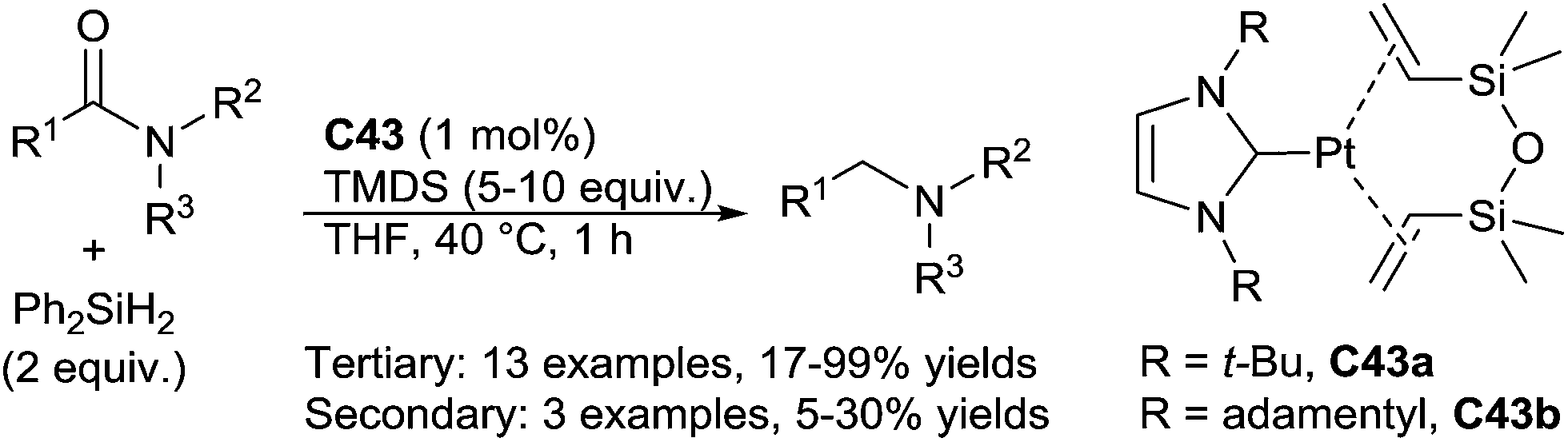 | ||
| Scheme 45 Pt–NHC catalysed hydrosilylation of amides. | ||
C.6. Group 11 (copper) catalysts
Beller reported a hydrosilylation of amides leading to amines using in situ generated cationic copper catalysts from Cu(OTf)2 (10 mol%) and the ligand L11 (1.25–3 mol%), in the presence of 3 equiv. of TMDS in toluene (3 mL), at 65–100 °C. This methodology is particularly suitable for the reduction of secondary amides including aromatic, aliphatic, and heterocyclic amides to secondary amines in 70–90% yields91 (Scheme 46). Notably, ester functional groups were tolerated under such conditions whereas aldehyde and ketone were preferentially reduced.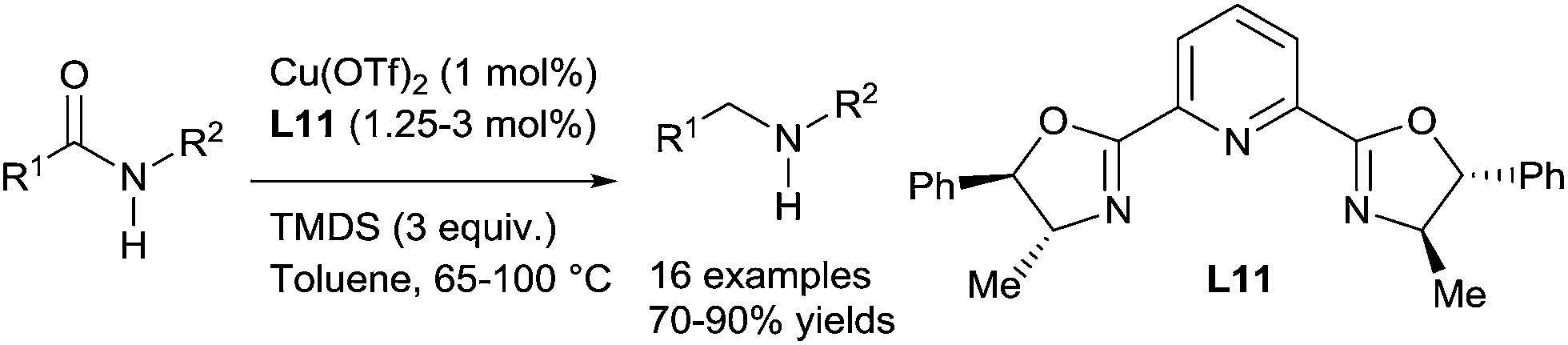 | ||
| Scheme 46 Copper catalysed hydrosilylation of secondary amides. | ||
C.7. Group 12 (zinc) catalysts
Using zinc as an inexpensive catalyst, the pioneering report was made in 1962 by Calas who described the ZnCl2-catalyzed reduction of tertiary amides to tertiary amines in the presence of several trialkylsilanes at 140–155 °C.92 Only four decades later, Beller described a general and mild methodology for the zinc-catalysed reduction of amides to amines with hydrosilanes.93 Using 10 mol% of Zn(OAc)2 in the presence of 3 equiv. of (EtO)3SiH at rt or 40 °C for 22 h, a wide range of tertiary amides, including aromatic, aliphatic, alicyclic and heterocyclic amides were successfully reduced to corresponding amines in 65–99% yields. Interestingly, good chemoselectivity was found in the presence of ester, ketone, ether, nitro, cyano, and phenylazo substituents, even with isolated and conjugated CC bonds, as the corresponding amines were synthesized without reducing the functional groups (Scheme 47).
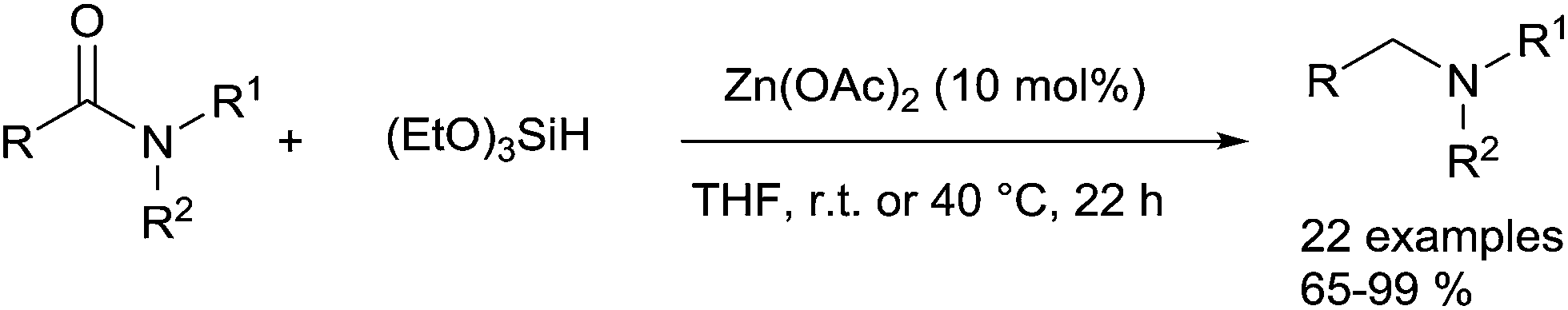 | ||
| Scheme 47 Zn(OAc)2-catalysed hydrosilylation of amides. | ||
Interestingly, the mechanistic studies have shown that zinc acetate didn't activate the carbonyl group of amide, but the triethoxysilane generating the active species 48-I. The latter then reacted with the amide to form the intermediate N,O-acetal 48-III by reduction of 48-II. The product was finally obtained via the reduction of the iminium species 48-V, which was formed from the intermediate 48-III with release of an anionic zinc ether 48-IV (Scheme 48).
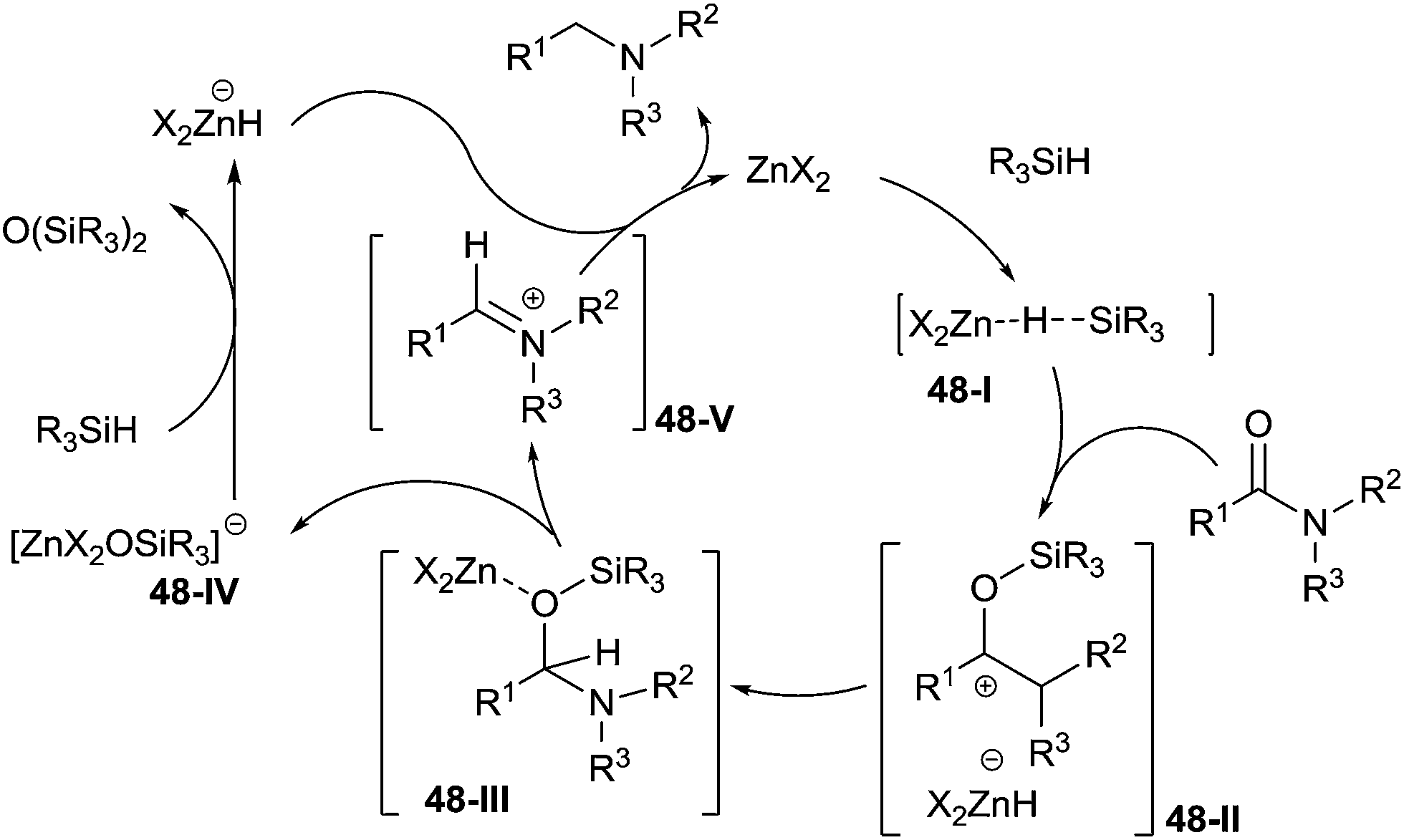 | ||
| Scheme 48 Proposed mechanism of zinc-catalysed hydrosilylation of amides. | ||
In 2011, the same group expand the reduction of tertiary amides using 3 equiv. of methyldiethoxysilane in the presence of 10 mol% of Zn(OAc)2 in THF at 65 °C for 20–30 h.94 Moreover, the more challenging reduction of secondary amides to secondary amines was achieved using 3 equiv. of TMDS with 20 mol% of Zn(OTf)2 in toluene at 100 °C for 28–72 h (Scheme 49). It must be underlined that various functional groups, such as halogeno, ketone, ester, ether, alkenes, nitro, cyano, and phenylazo bond were tolerated in both reduction of tertiary amides and secondary amides and that aniline or phenol moieties inhibited the reactivity.
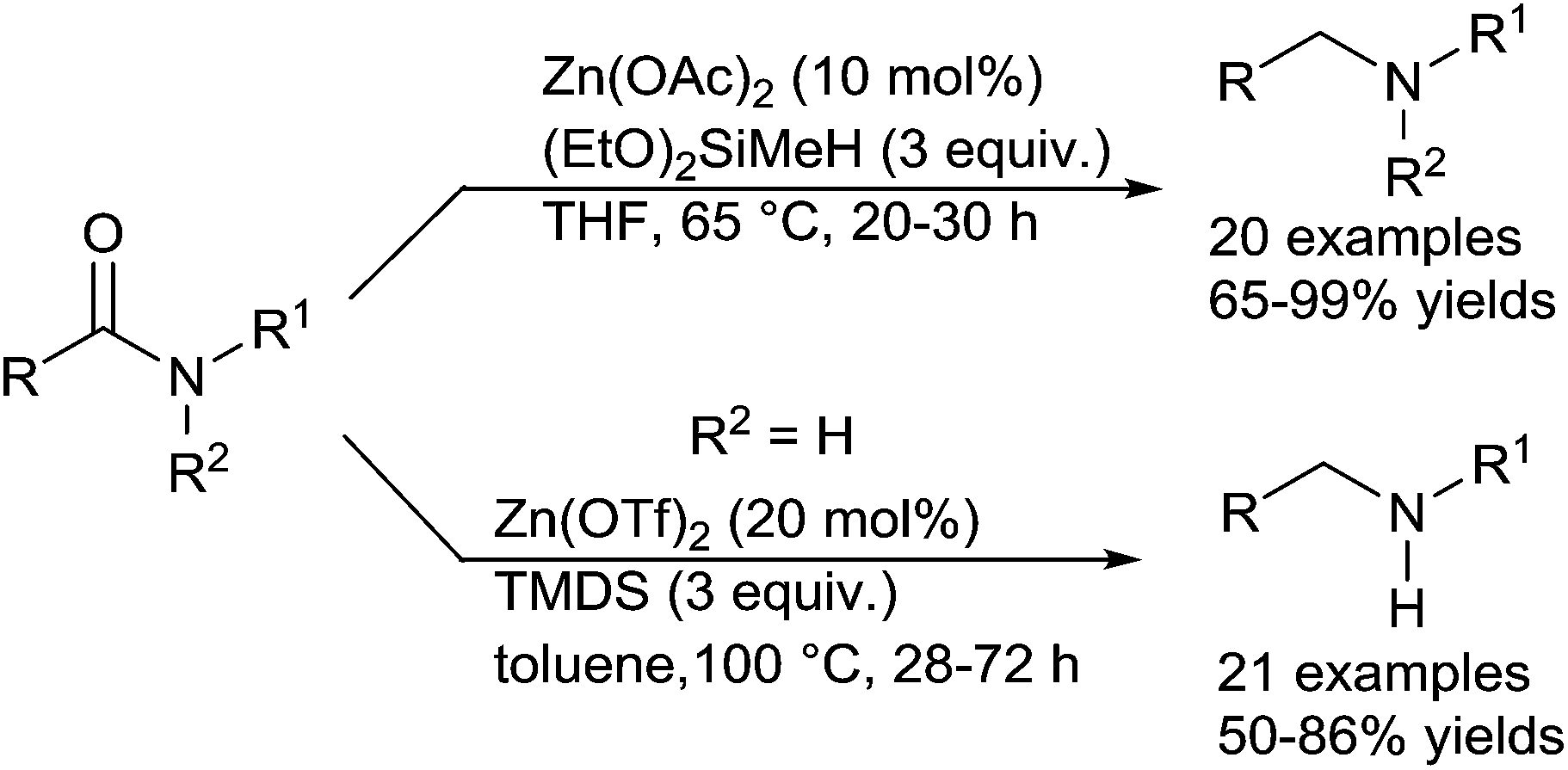 | ||
| Scheme 49 Zinc-catalysed hydrosilylation of amides. | ||
Recently, Adolfsson reported the use of Et2Zn as a catalyst for the hydrosilylation of tertiary amides leading to the corresponding amines in 52–98% yields.95 Indeed, Et2Zn (5 mol%) in the presence of 10 mol% of LiCl and 3 equiv. of PMHS in THF (2 mL) catalysed the reduction of amide (1.0 mmol), at rt for 24 h. Functional groups such as cyano, nitro, cinnamyl can be tolerated, but the reaction did not proceed with primary and secondary amides. The presence of LiCl is described to be crucial to increase the rate and the selectivity of the reaction.
D. Hydrosilylation of nitriles to amines
By contrast with transition-metal-catalyzed hydrosilylations of amides and imines, very little is known about the hydrosilylation of nitrile derivatives. After a pioneering work in the early 1960 on the use of ZnCl2 as the catalyst for the hydrosilylation of organonitriles in the presence of Et3SiH at 140–150 °C for 36 h leading mixture of compounds including triethylsilylamine,96 in 1982, Corriu described the hydrosilylation of nitriles using the Wilkinson catalyst RhCl(PPh3)3 (1 mol%) in the presence of 1 equiv. of 1,2-bis(dimethylsilyl)benzene in refluxing toluene: a mixture of trans-N,N-disilylated enamines (major compounds) and N,N-disilylated amines (minor compounds) was obtained.97 Using catalytic amount of [Co2(CO)8] C35 (8 mol%), Murai et al. reported the hydrosilylation of aromatic nitriles in the presence of 10 equiv. of trimethylsilane at 60 °C for 20 h under an atmosphere of CO to give N,N-disilyl amines in 36–91% yields (Scheme 50).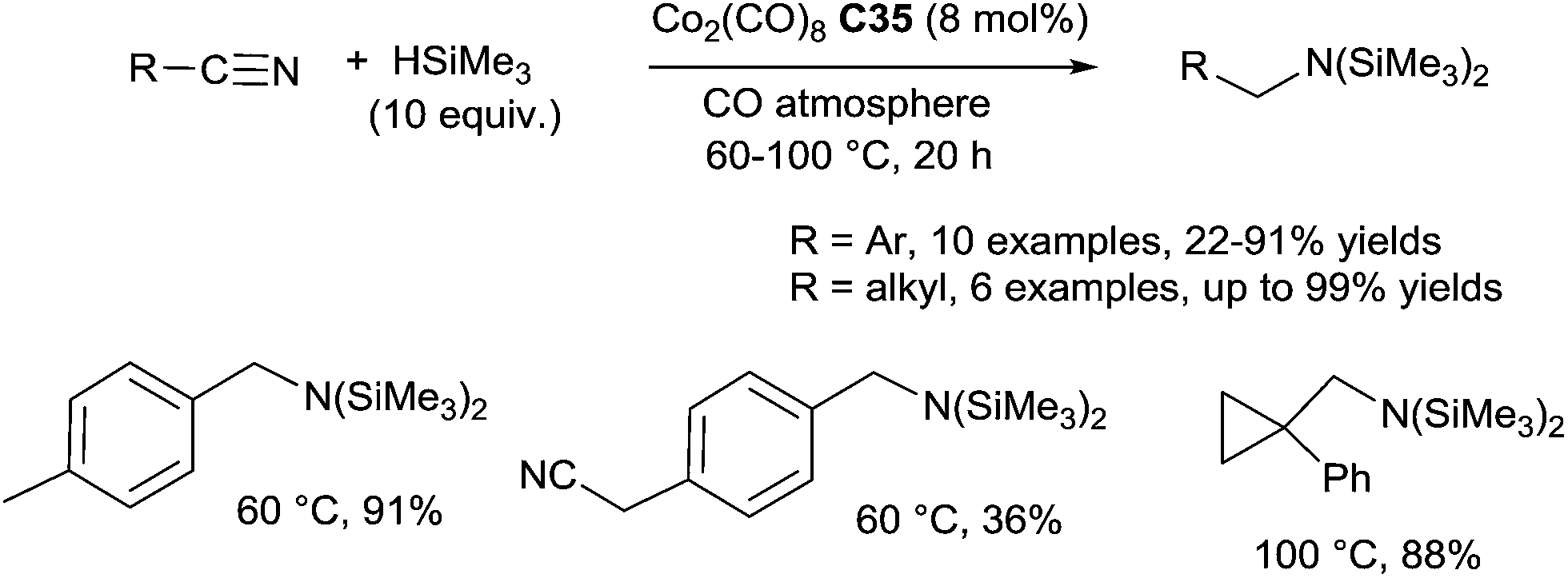 | ||
| Scheme 50 Cobalt-catalysed hydrosilylation of nitriles. | ||
Interestingly, electron donating groups on the aromatic nitriles facilitated the reaction, whereas electron-withdrawing groups decreased the reaction rate.98 For aliphatic nitriles, a slight modification of the catalytic species generated from 8 mol% of CO2(CO)8C35 and 16 mol% of PPh3 and a higher reaction temperature (100 °C) were required. When they performed the reaction with the most challenging α,β-unsaturated nitriles, a mixture of four products were obtained.99
Using rhodium metal particles, Caporusso developed a rhodium-catalyzed hydrosilylation of nitriles: in the presence of 1 mol% of nanoparticles and 5 equiv. of trimethylsilane at 100 °C for 15 h, aromatic nitriles were reduced to the corresponding disilylated amine derivatives in 65–95% yields, showing the functional group tolerance with ester.100 With homobimetallic rhodium NHC complex C44, nitrile compounds can be also hydrosilylated at 100 °C for 4–6 h leading the corresponding disilylated amine in moderate yields (42–77%)101 (Fig. 4).
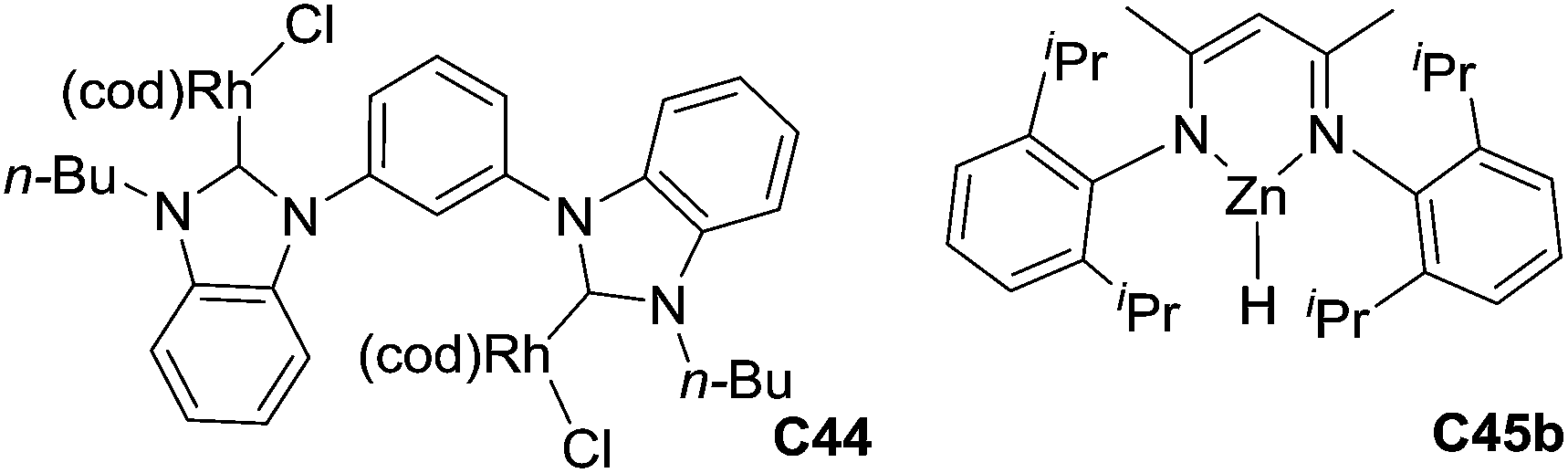 | ||
| Fig. 4 Rhodium and zinc complexes for catalysed hydrosilylation of nitriles. | ||
In 2010, Nikonov reported an elegant and efficient ruthenium-catalyzed hydrosilylation of nitriles to silylimines with a broad chemoselectivity as most common functional groups were tolerated using the air-stable catalyst [Cp(iPr3P)Ru(NCCH3)2][B(C6F5)4] C45a (4–5 mol%) and 1.1 equiv. of Me2PhSiH at rt. Using 2 equiv. of silane, the disilylated amines could be obtained specifically with electron rich aromatic nitriles, whereas with electron poor ones, the reaction stopped at the imine stage.102 Similarly, zinc hydride complex, DippNacNacZnH C45b (3 mol%), catalyzed the chemoselective reduction of nitriles to N-silyl imines in the presence of HSi(OEt)3 in C6D6 at rt for 7–24 h, but with a limited functional group tolerance (Fig. 4).103
The reduction of nitriles to the corresponding primary amines via hydrosilylation can be also performed using oxo-rhenium complexes such as ReIO2(PPh3)2C9 (10 mol%) as the catalyst.104 In the presence of 3 equiv. of PhSiH3 in refluxing toluene under air, a series of aromatic nitriles bearing functional groups such as halides, CF3, SO2CH3 and NHTs were efficiently reduced in 51–92% yields.
E. Hydrosilylation of nitro to amines
Reduction of nitroarenes leading to aniline derivatives is one of the standard reactions in aromatic chemistry. By comparison with hydrogenation mainly promoted by heterogeneous catalysts, hydrosilylation is still largely underrepresented.In the early 1970's, one example of reduction of nitroarenes, namely nitrobenzene, using 3.3 equiv. of PMHS as the hydrosilane and Pd/C (5 mol%) as the catalyst in ethanol at 60 °C for 1 h was reported by Lipowitz.105 Using 5 mol% of Pd(OAc)2 in the presence of 3–6 equiv. of PMHS and 2 equiv. of aqueous KF in THF at RT for 30 min, nitroarenes were reduced leading the corresponding amines in high yields, with wide functional group tolerance such as esters, carboxylic acid, aldehydes, amides and nitriles. With nitroarenes bearing alkenyl or alkynyl substituents, both nitro and unsaturated CC were reduced106 (Scheme 51). By contrast, using similar conditions with Pd(OAc)2 and triethylsilane in THF/water solution, aliphatic nitro compounds were reduced to the corresponding hydroxylamines.
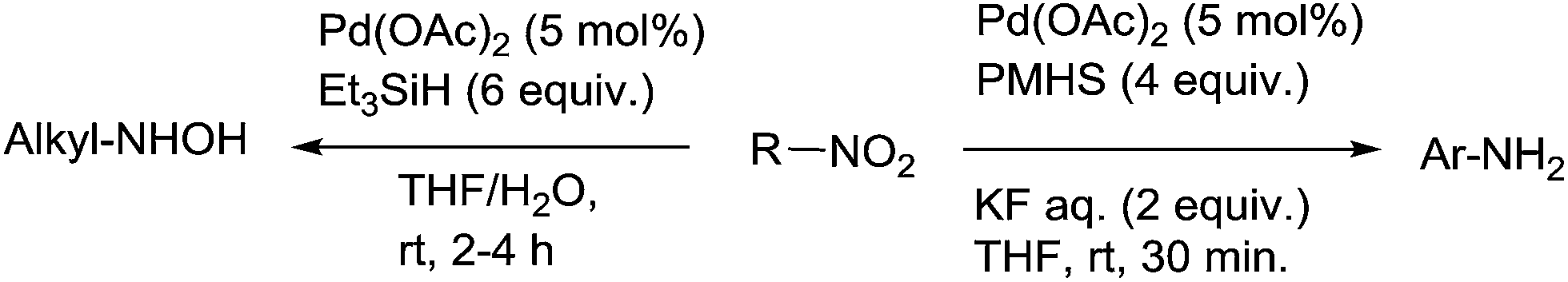 | ||
| Scheme 51 Palladium-catalysed hydrosilylation of nitro derivatives. | ||
Ni(acac)2 was found to be an efficient catalyst (10 mol%) for the chemoselective hydrosilylation of nitroarenes in the presence of 1.2–4.0 equiv. of PMHS in 1,4-dioxane at 80 °C for 3–5 h. A high functional group tolerance were observed as thiocarbonyl, cyano, ester, carboxylic acid, aldehyde, alkenyl, primary amide, ether, were compatible with the reaction conditions.107
Wilkinson catalyst RhCl(PPh3)3 in the presence of triethylsilane in refluxing toluene can also be an efficient catalyst to reduce nitroarenes to aniline derivatives after acidic treatment.108 Esters can be tolerated whereas aldehydes and ketones were simultaneously reduced.
Such reduction can be also performed using high-valent oxo-rhenium such as ReIO2(PPh3)2C9 or ReOCl3(PPh3)2 (5 mol%) in the presence of 3.6 mol% of PhMe2SiH in refluxing toluene for 1–48 h. Anilines derivatives was then isolated in moderate to good yields (31–96%). It is noticeable that this catalytic transformation tolerated a huge variety of functional groups such as halides, esters, amides, sulfones, and nitriles. By contrast, the hydrosilylation of nitroalkanes such as 2-nitroethylbenzene led to the corresponding nitrile in 38%.109
During their studies on hydrosilylation of amides to amines, Nagashima et al. discovered the reduction of nitroarenes to anilines as the Fe-catalysed reduction of N,N-dimethyl-p-nitrobenzamide didn't give to the corresponding N,N-dimethyl-p-nitrobenzamine but the aniline product resulting from the chemoselective reduction of the nitro group and which was isolated in 77% yield (see Scheme 31).71 Thus using 10 mol% of [Fe3(CO)12] C31 as the catalyst in the presence of 5 equiv. of TMDS in toluene at 100 °C for 24 h, 4 examples of nitroarenes were reduced in the corresponding anilines in 73–93% yields. Using an in situ generated catalyst from FeBr2 (10 mol%) and PPh3 (12 mol%) in the presence of 2.5 equiv. of phenylsilane, Beller et al. developed another efficient chemoselective methodology for the preparation of functional anilines starting from the corresponding nitroarenes in refluxing toluene for 16 h. Indeed, functional groups such as halides, esters, hydroxyl, nitriles, and conjugated internal CC bonds are tolerated.110 Simultaneously, Lemaire et al. reported an alternative catalytic system with 10 mol% of Fe(acac)3 in the presence of 4 equiv. of TMDS in THF at 60 °C for 24–48 h with similar group tolerance.111
In 2015, Baran et al. described an interesting sequence involving a nitroarene reduction following by a hydroamination of alkenes leading to the corresponding secondary amines.112a Indeed, using 30 mol% of Fe(acac)3 as the catalyst in the presence of 3 equiv. of olefins, 1 equiv. of nitro(hetero)arenes and 2 equiv. of phenylsilane, in ethanol at 60 °C for 1 h followed by the addition of 20 equiv. of zinc and aq. HCl, >110 examples of secondary amines were prepared in moderate yields (24–79% yields). Impressively, ketone, acetal, benzyloxy, amides, primary alkylamines, secondary anilines, alkylalcohols, boronic acids, triflates, and halides were tolerated. Furthermore, this methodology was applied to prepare pharmaceutical targets in a reduced number of steps.
Recently, Thomas reported the use of an amine-bis(phenolate) iron(III) complex C46 (2 mol%) for the efficient and chemoselective reduction of nitroarenes to aniline derivatives in the presence of 4 equiv. of (EtO)3SiH in acetonitrile at 80 °C for 3–8 h. Interestingly reducible functional groups such as esters, nitriles, sulfones and tertiary amides were tolerated (Scheme 52).
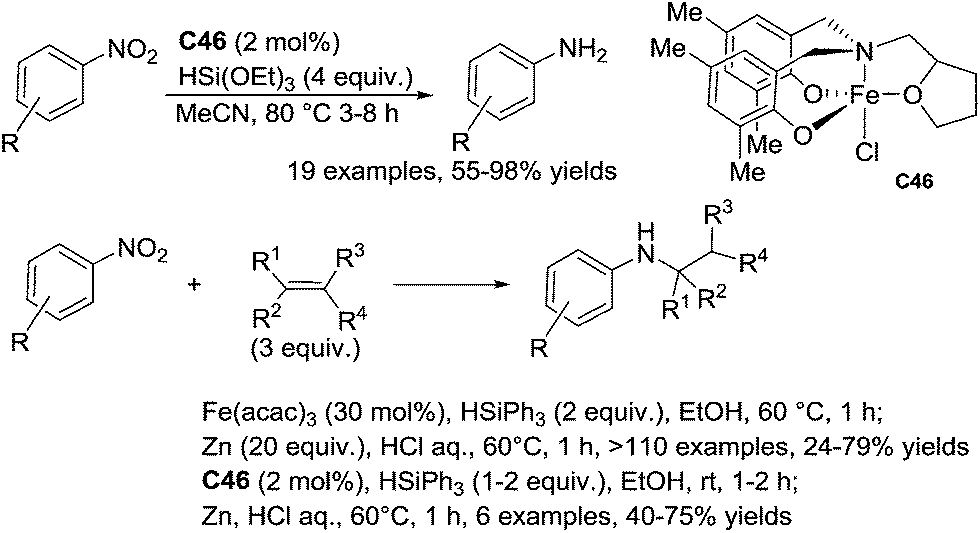 | ||
| Scheme 52 Iron-catalyzed nitroarenes reduction and reduction/hydroamination. | ||
Similarly to Baran, he used C46 (2 mol%) to catalyze the sequence nitroarene reduction/hydroamination of alkenes leading to the corresponding secondary amines in the presence of 1–2 equiv. of phenylsilane and 3 equiv. of alkenes in ethanol at rt for 1–2 h (40–75% yields).112b
F. Hydroamination/hydrosilylation of alkynes with aniline
One-pot tandem hydroamination and hydrosilylation using transition metals has been recently developed to synthesize secondary amines. The tandem reaction including two steps: (i) the synthesis of imines through the addition of N–H bond of the carbon–carbon triple bond, and (ii) the reduction of the intermediate imines by hydrosilanes.In 2003, Messerle et al. reported the first cyclic amine synthesis via one-pot two step tandem hydroamination/hydrosilylation procedure using iridium based catalyst.113 Indeed, the cyclisation of 4-pentyn-1-amine was carried out in the presence of 2 mol% of Ir(I)dicarbonyl complexes [{Ir(bpm)(CO2)BPh4}] C18 (bpm = bis(pyrazol-1-yl)methane) or [{Ir(bim)(CO2)BPh4}] C47 (bim = bis(1-methylimidazol-2-yl)methane) in THF at 60 °C, leading to the in situ generated 2-methyl-1-pyrroline, which was then treated with triethylsilane affording to 1-(triethylsilyl)-2-methylpyrrolidine (Scheme 53). The complex C18 was more efficient than the complex C47 in the hydroamination step (3 h for complex C18vs. 21 h for complex C47), but both catalysts have similar catalytic efficiency in the hydrosilylation step.
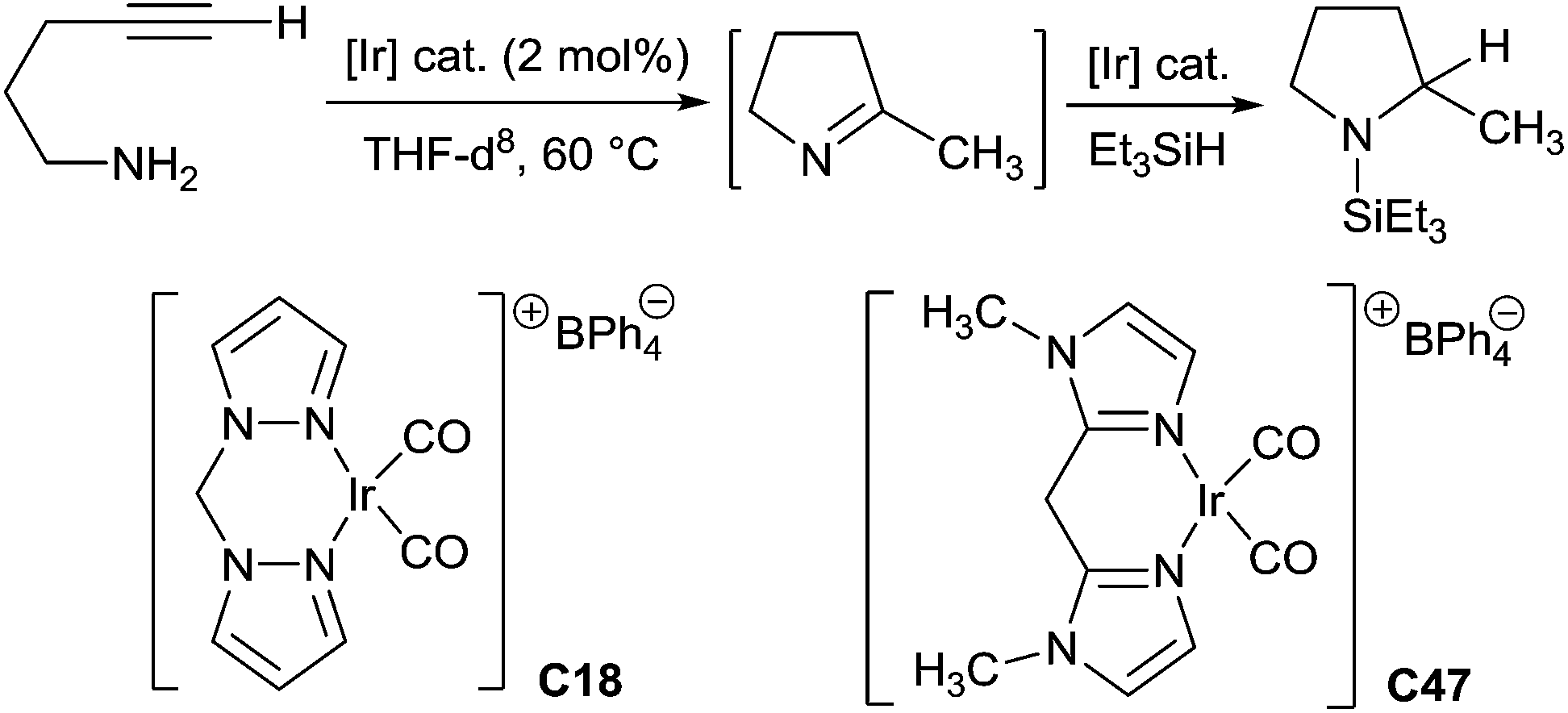 | ||
| Scheme 53 Ir-catalysed intramolecular hydroamination/hydrosilylation of terminal alkynes. | ||
An intermolecular version can also be performed using the metalated PNC–pincer iridium complex C48 as the catalyst to perform the one pot two step tandem hydroamination/hydrosilylation of arylethynes with aniline derivatives. Arylethynes and anilines (1.2 equiv.) in the presence of 1 mol% of iridium complex C48 and 10.5 mol% of NaBArF4 (sodium tetrakis(3,5-trifluoromethylphenyl)borate) as the catalytic system was refluxed in toluene for 12–20 h. Et3SiH (1.2 equiv.) was then added and stirred for 2 additional hours (Scheme 54).114
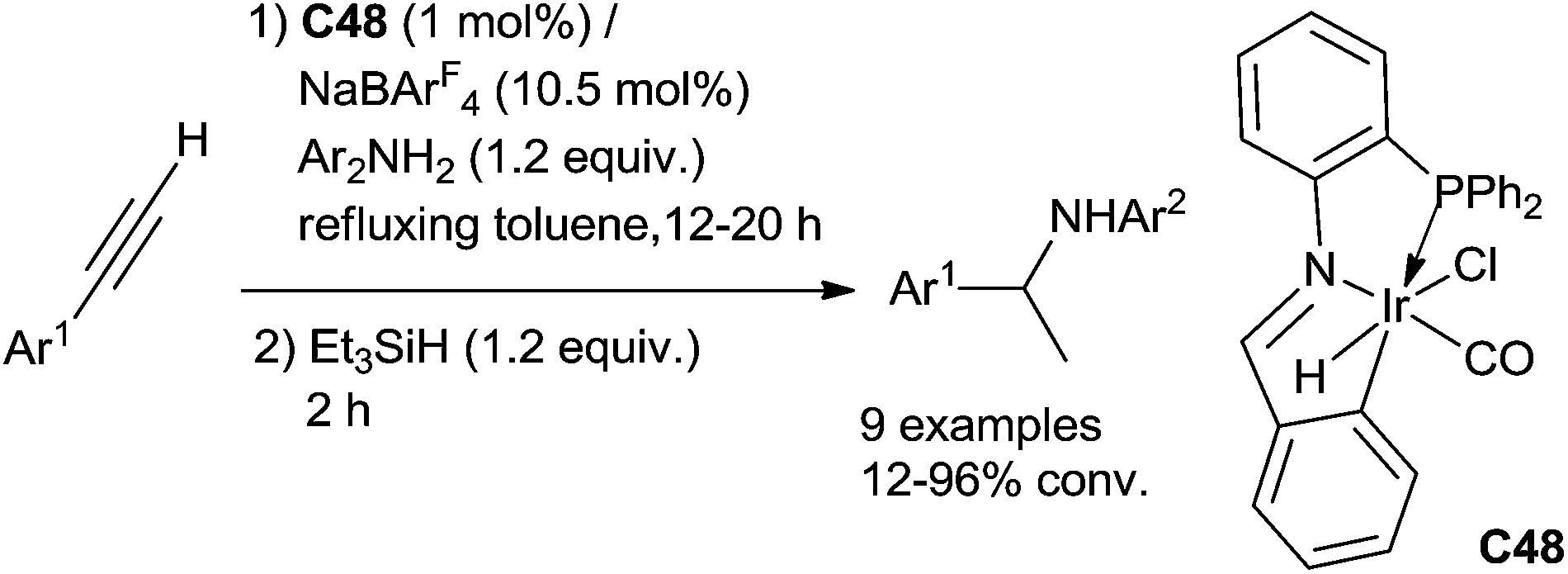 | ||
| Scheme 54 Ir-catalysed intermolecular hydroamination/hydrosilylation of terminal alkynes. | ||
Unfortunately, internal alkynes could not undergo such a reaction under this catalytic condition. The role of the NaBArF4 is to assist the dissociation of the chloride to generate a Ir(III)-hydride species able to act as a Lewis acid to coordinate the CC bond.
Using a tricarbonyl chromium-bound iridacycle C19a as the catalyst (2.5 mol%), an one pot sequential intermolecular hydroamination–hydrosilylation of terminal arylethynes with aniline leading to corresponding secondary amines was performed at rt when working in methanol in the presence of 1.3 equiv. of Et3SiH45 (Scheme 55). Interestingly, a hydrido-Ir intermediate was observed at −15.1 ppm in the 1H NMR by the addition of N,N-dimethylaminopyridine to a mixture of tricarbonylchromium-bound iridacycle with Et3SiH, which initially release H2.
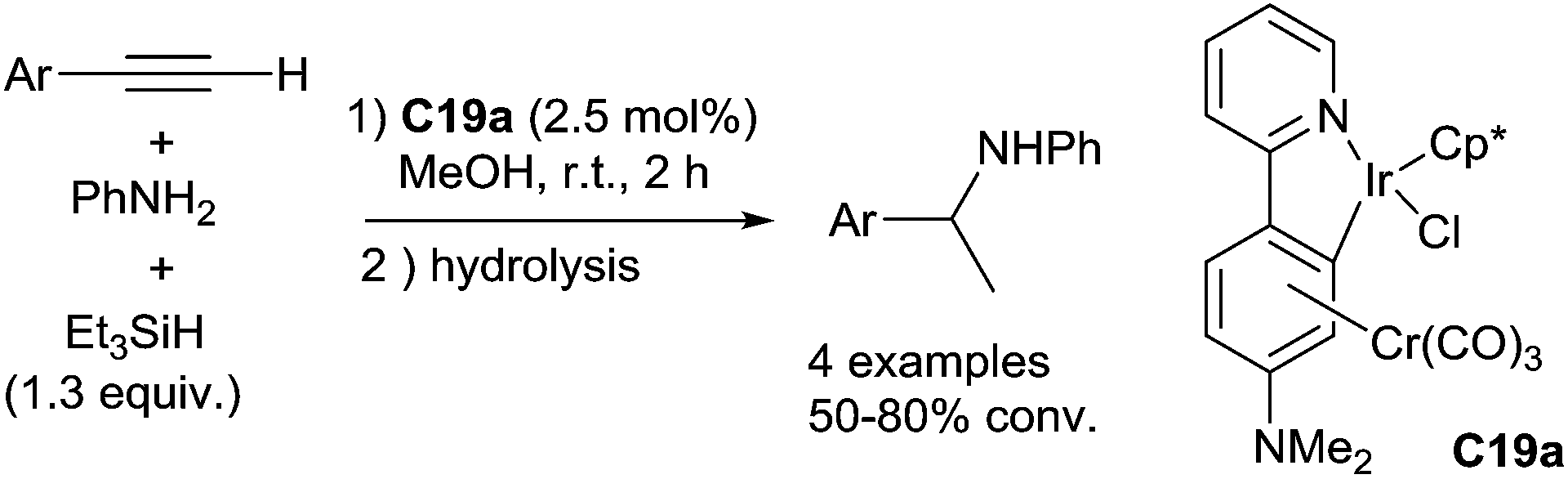 | ||
| Scheme 55 Ir-catalysed intermolecular hydroamination/hydrosilylation of terminal alkynes. | ||
In titanium series, Doye et al. reported a catalysed sequential one pot two step reaction of terminal and internal alkynes with amines.115 Starting from aniline derivatives and primary alkylamines by reaction with 1-phenylprop-1-yne, various secondary amines were synthesized in poor (6%) to excellent (99%) yields. In a first step, the alkyne, primary amines and Cp2TiMe2C22b (10 mol%) or the complex C49 (10 mol%) are warmed in toluene at 105 °C for 24 h. Then 3 equiv. of phenylsilane, 40 mol% of piperidine and 40 mol% of methanol are added to the mixture and warmed 24 additional hours at 105 °C (Scheme 56).
 | ||
| Scheme 56 Titanium catalysed hydroamination/hydrosilylation of alkynes with amines. | ||
It must be underlined the outstanding regioselectivity observed as mainly one isomer was obtained when starting from internal alkynes. Furthermore, alkylamines were less reactive than anilines. With terminal alkynes, even if the corresponding secondary amines can be obtained in good yields, the observed regioselectivity was lower (31:69 to 72:24). Interestingly, this one-pot protocol could be applied to synthesize cyclic amines.
G. Reductive amination with amines or anilines
Direct Reductive Amination (DRA) of secondary or primary amines with aldehydes or ketones is one of the powerful methodologies to synthesize tertiary or secondary amines through the reduction of iminium intermediate using reducing agents. In hydrosilylation area, only scarce examples were reported using transition metals as the catalysts.In 2005, Ishii reported an iridium catalysed reductive alkylation of secondary amines in the presence of triethylsilane. Typically, by reaction of secondary alkylamines with alkanals with 0.5–2 mol% of [IrCl(cod)2] C50 in the presence 1 equiv. of triethylsilane in 1,4-dioxane at 75 °C for 8 h, the corresponding tertiary amines were obtained in good yields (71–90%). Notably IrCl3 can be also used as an alternative catalyst, and PMHS as a silane substitute.116 It was shown that a [Ir–H] species generated from [IrCl(cod)]2 and organosilanes was a catalytic active species for the reductive amination.
Using oxo-rhenium ReIO2(PPh3)2 complex C9 as a catalyst (2.5 mol%), Fernandes have developed an efficient system to promoted the DRA of benzaldehyde derivatives with primary and secondary anilines in refluxing THF for 5 min to 7 h leading to the corresponding amines with 72–90% yields. Noticeably, nitro, sulfone, ester, nitrile, amide, halides including iodide can be tolerated.117 The same group have also developed a series of oxo-rhenium complexes bearing heterocyclic ligands to perform this transformation: the oxo-rhenium complex [ReOBr2(L12)(PPh3) C51 was used as a catalyst (2.5 mol%) with the same efficiency and chemoselectivity in refluxing THF118 (Scheme 57).
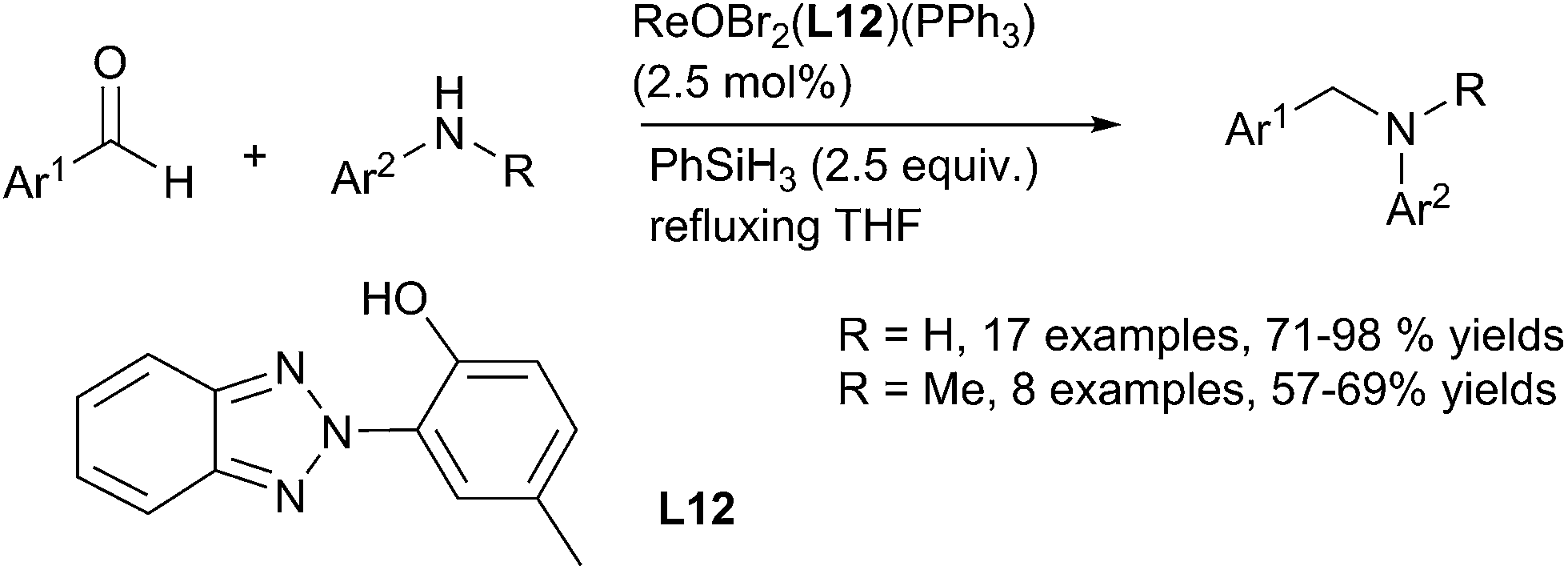 | ||
| Scheme 57 Oxo-rhenium catalysed DRA reaction. | ||
Furthermore, Ghorai reported a direct reductive amination of ketones such as alkanones and cycloalkanones with electron-deficient amines using Re2O7/NaPF6 as the catalytic system and triethylsilane as the reductant in dichloromethane at 50 °C for 12–48 h (Scheme 58).119
 | ||
| Scheme 58 Oxo-rhenium catalysed DRA reaction of ketones with electron deficient amines. | ||
Among the inexpensive and eco-friendly transition metals, zinc is an interesting candidate, and is also able to catalyse the reductive amination of aldehydes and primary amines to produce secondary amines using PMHS as the hydrosilane as shown by Enthaler.120 This reaction was carried out with 1 equiv. of aldehyde, 1 equiv. of aniline, 5 mol% of Zn(OTf)2, 0.5 mL of PMHS in THF at 60 °C for 24 h. A wide range of aldehydes and amines were used to synthetize the corresponding secondary amines in good yields (57–95%). Excellent chemoselectivity was observed in the presence of ketones, esters, CC bonds, nitro, hydroxyl and epoxide groups. Zn(OTf)2(thf)4 complex has been isolated and could be seen as intermediate or precatalyst. The water generated from the condensation of aldehyde and amine plays a key role for the desilylation of N-silylated intermediate and led to free secondary amine.
Another cheap and low toxic transition metal, iron, also catalysed reductive amination of aldehydes. Indeed, Enthaler has developed an efficient DRA reaction of aromatic and aliphatic aldehydes with primary aniline derivatives yielding the corresponding secondary anilines in 23–99% yields using FeCl3 as a catalyst in the presence of PMHS in THF at 60 °C for 24 h.121 Our group has also developed a series of well-defined piano-stool iron phosphane–pyridine iron complexes such as C52 which were efficient catalysts for the DRA of aromatic aldehydes with secondary amines under mild conditions (30–40 °C), using PMHS as a reductant in dimethylcarbonate as the solvent122 (Scheme 59). Various tertiary amines were produced in moderate to good yields. It is noteworthy that P–N bidentate iron complexes are more efficient than the monodentate phosphine complexes in this catalytic system.
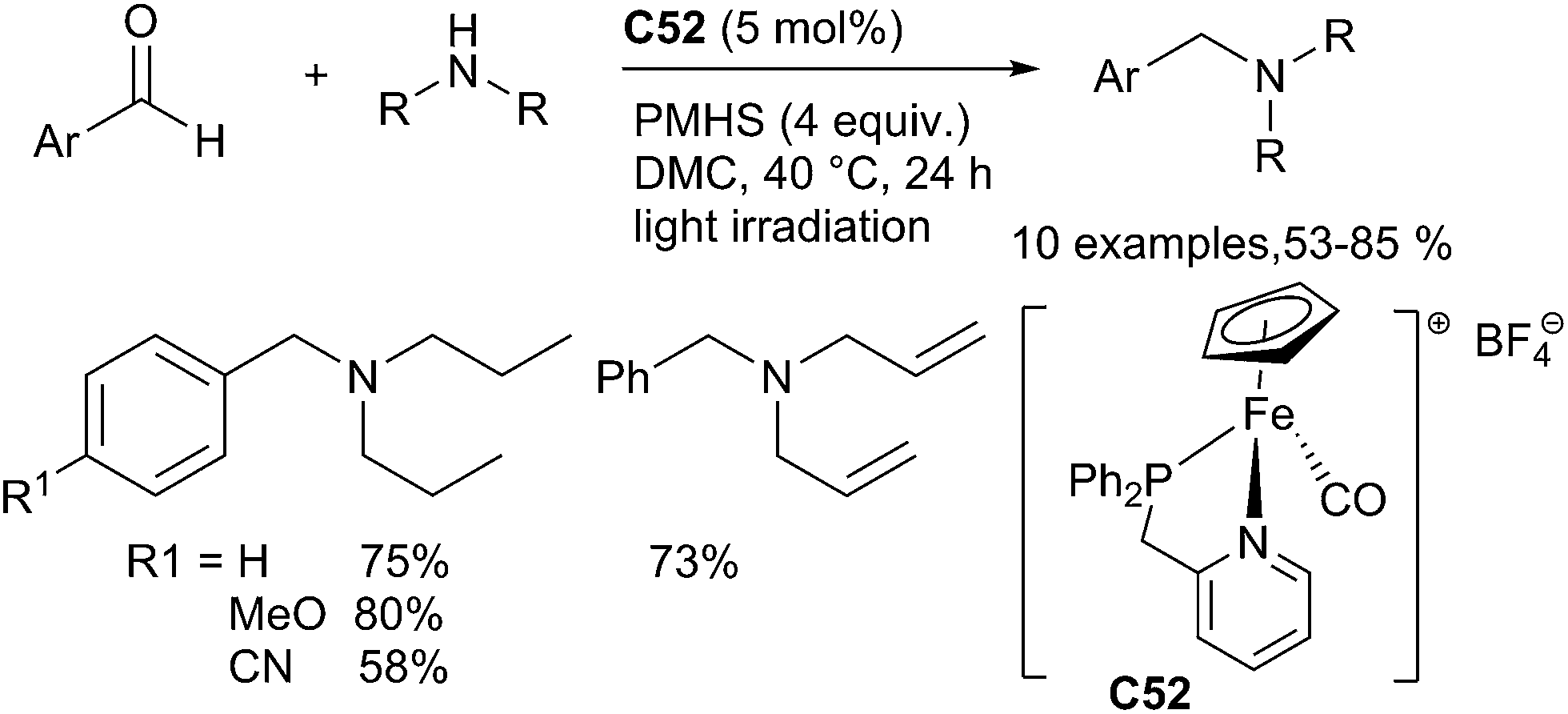 | ||
| Scheme 59 Iron catalysed DRA under hydrosilylation conditions. | ||
The nickel version of reductive amination reaction of aldehydes with primary amines was also reported. The catalytic system was prepared in situ starting from Ni(OAc)2 (5 mol%) and tricyclohexylphosphine (10 mol%) and the reaction was performed in THF at 70 °C, and 1.5 equiv. of TMDS as the hydrosiloxane was added after 8 h of reaction and the methanolysis (MeOH, NaOH) was perform after 16 additional hours at 70 °C. Up to 21 examples of amines was reported with moderate to good yields (46–97%).123
One interesting alternative is the use of readily available and stable carboxylic acid derivatives instead of sensitive aldehydes to perform such reaction. In 2012, Nagashima reported a ruthenium-catalyzed N-alkylation of N-tosylamides with esters under hydrosilylation conditions.124 Using 1 mol% of the ruthenium complex C28 in the presence of 3 equiv. of TMDS in CH2Cl2 at rt for 6–20 h, secondary tosylamide derivatives were N-alkylated with 2–2.5 equiv. of esters leading to tertiary tosylamides with good yields (32–99%). Interestingly, azacycloalkanes and azaspirocycles can be obtained by intramolecular reductive N-alkylation of amide–ester under similar conditions (48–99% yields). In particular, the reductive alkylation of optically active N-tosylated dimethylglutamate led to the corresponding methyl ester of the N-tosylated proline, without racemization (Scheme 60).
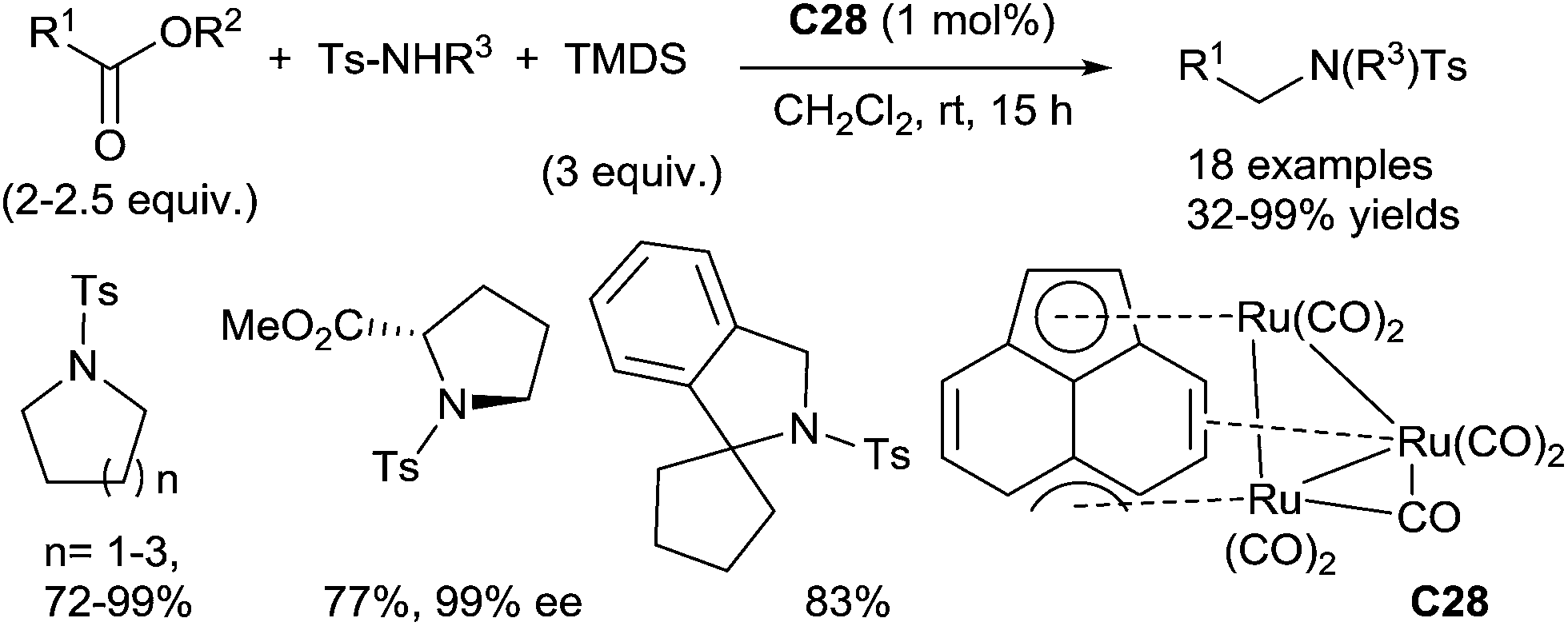 | ||
| Scheme 60 Ruthenium-catalysed RDA of carboxylic esters with tosylamines. | ||
Beller succeeded to perform similar reaction starting from carboxylic acids using an in situ prepared catalyst from Karstedt complex Pt[(CH2CHSiMe2)2O] C53 (0.5 mol%) and dppe (0.5 mol%). By reaction at rt – 120 °C for 18 h in Bu2O in the presence of phenylsilane (4 equiv.), carboxylic acids (2.2 equiv.) reacted with primary and secondary amines leading to the corresponding amines in 48–99% yields.
The synthetic utility of this efficiency methodology was highlighted by the preparation of cinacalcet hydrochloride, a selective calcimimetic agent used for the treatment of secondary hyperparathyroidism125 (Scheme 61).
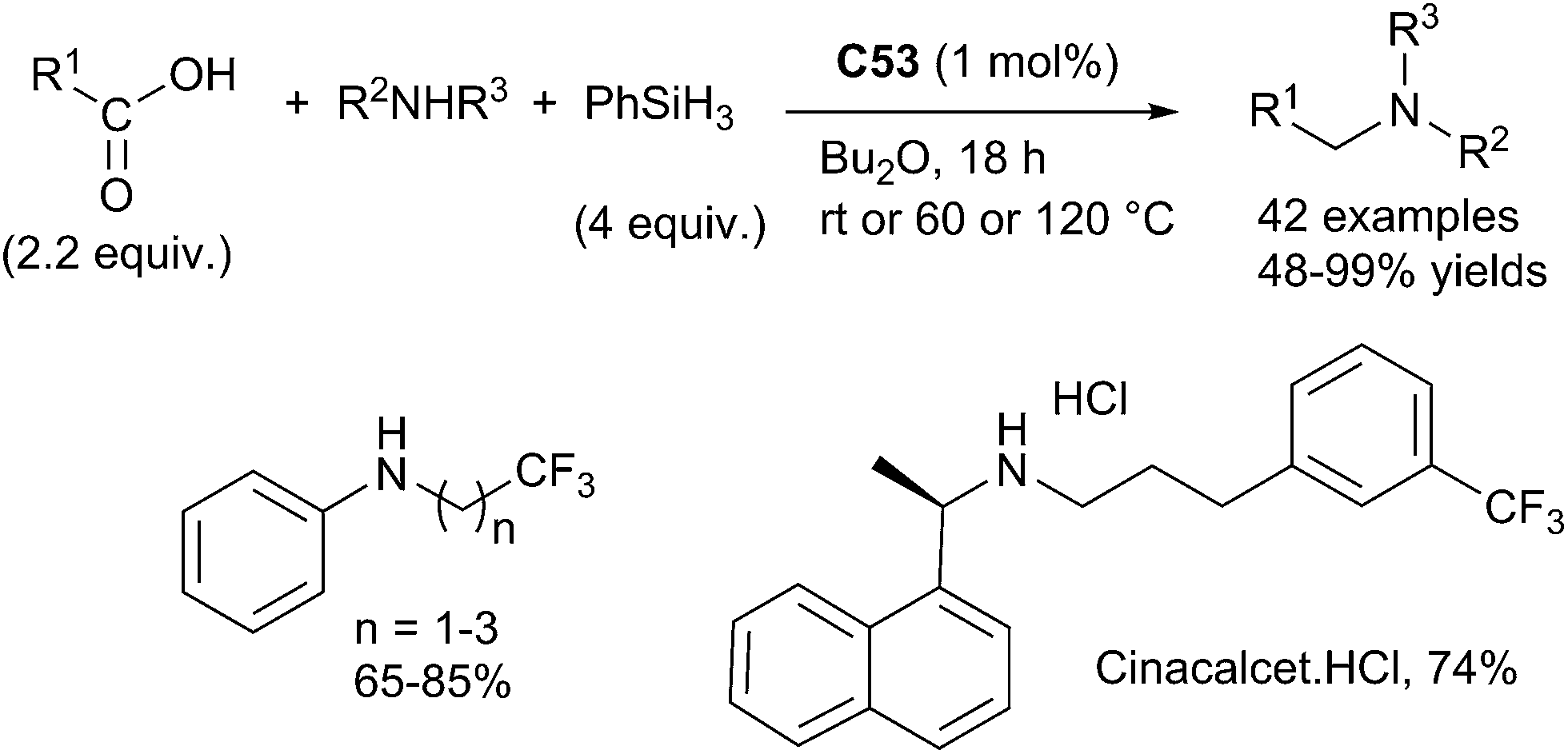 | ||
| Scheme 61 Platinum-catalysed RDA of carboxylic acids with primary and secondary amines. | ||
Other interesting readily available and stable starting materials are alcohols. Starting from allylic or homoallylic alcohols and secondary and primary anilines using 5 mol% of Fe(cod)(CO)3 (cod = cycloocta-1,5-diene) C54 in the presence of 3 equiv. of PMHS in ethanol at 50–70 °C under light irradiation, in a cascade pathway, DRA was performed. This transformation corresponds to a formal DRA of (homo)allylic alcohols via a tandem isomerization/condensation/hydrosilylation reaction (Scheme 62).126 Interestingly anilines featuring a hydroxylated N-substituent can be prepared with moderate yields (61–92%) by reaction of (Z)-but-2-ene-1,4-diol with various substituted primary aniline compounds.
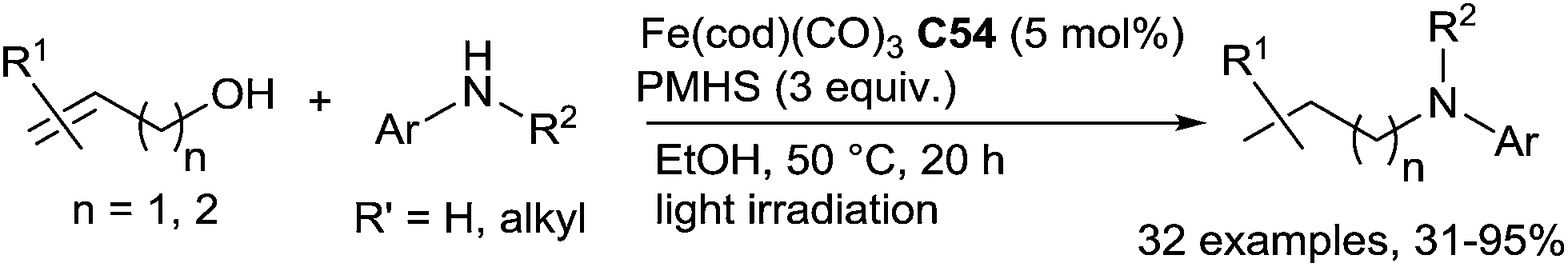 | ||
| Scheme 62 Cascade iron-catalysed isomerization/RDA reaction. | ||
Using a copper N-heterocyclic carbene complex C55 (2 mol%) in the presence of 2 equiv. of KOH in CH2Cl2 at rt under oxygen atmosphere, primary alcohols were efficiently oxidized leading to aldehydes. Using the same conditions, in the presence of 1.1 equiv. of primary amines, imines were selectively produced via a dehydrogenative condensation. A one pot process can then be applied for the preparation of amines from benzylic alcohol derivatives and amines via a copper-catalyzed dehydrogenative condensation/hydrosilylation: using the former conditions up to the full consumption of alcohols, 4 mmol of PMHS and additional 2 mol% of C55 were added leading to the amines in 75–97% yields127 (Scheme 63).
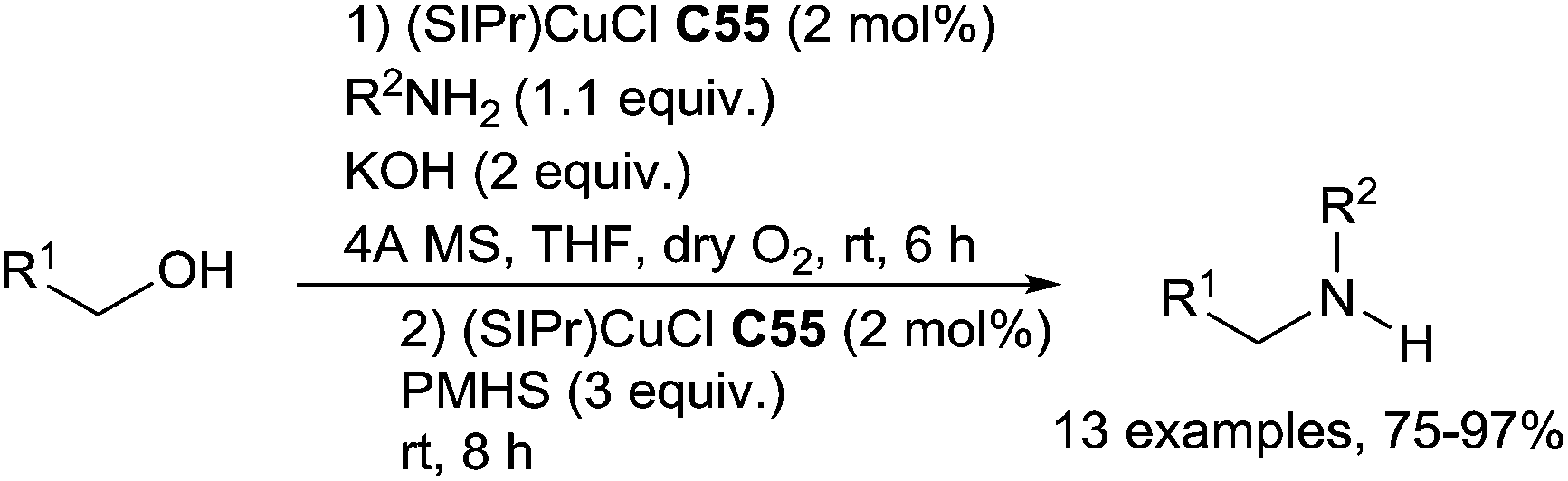 | ||
| Scheme 63 Cascade iron-catalysed oxidation/RDA reaction. | ||
H. N-Methylation of amines using carbon dioxide as C1 source
N-Methylation of amines is an important chemical modification which classical approaches involve the nucleophilic substitution with hazardous electrophilic methylating agents, such as methyl iodide or dimethyl sulfate, the use of formaldehyde in the presence of a reductant, and in an industrial scale production, the use of heterogeneous dehydration catalysts for the reaction ammonia and methanol.128Carbon dioxide is the most abundant carbon source in nature, and it can be used as a C1 building block in organic synthesis. Thus, its utilization for N-methylation of amines is obviously highly attractive.129 In a pioneering report, in 2013, Cantat et al. reported the catalytic methylation of aliphatic and aromatic, primary and secondary amines using CO2 (1–5 bar) and phenylsilane (2 equiv.) as the reductant at 100 °C for 20 h using a zinc complex bearing a N-heterocyclic carbene ligand, IPrZnCl2C56, as the in situ generated catalyst130 (Scheme 64). Notably, formamides, R2N–CHO, were detected in trace amounts in the reaction indicating that they are intermediate of this reaction.
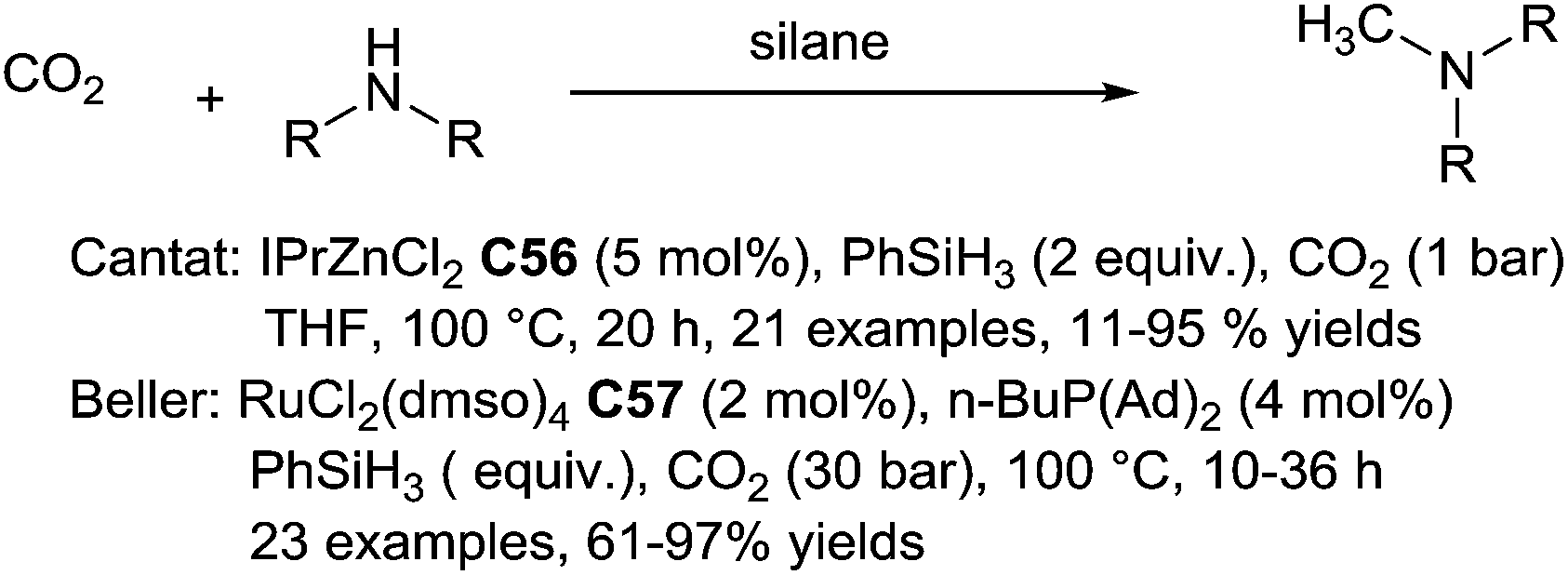 | ||
| Scheme 64 Zinc and ruthenium-catalysed N-methylation of amines using CO2 and hydrosilanes. | ||
Soon afterwards, Beller reported a ruthenium version using an in situ generated catalyst from RuCl2(dmso)4C57 (2 mol%) and n-BuP(Ad)2 (4 mol%) under 30 bar of carbon dioxide in the presence of 4 equiv. of phenylsilane at 100 °C for 10–36 h131 (Scheme 64). It was also shown that the selective methylation of the amine moiety when amino alcohols or aminoesters were used. Indeed, ephedrine led to N-methylephedrine in good yields (73%) and selectivity (>7:1).
Using inexpensive transition metals, this transformation is also possible. With nickel–phosphine complexes such as [(dippe)Ni(μ-H)]2C58 as a catalyst, [dippe = bis-(diisopropylphosphino)ethane] selective N-methylation of aliphatic primary amine was performed at 100 °C in the presence of 4 equiv. of phenylsilane and 1 bar of CO2 in toluene at 100 °C for 20 h.132 Indeed, starting from benzylamine, 69% of N-methylbenzylamine was obtained with N-methyl-N,N′-dibenzylurea being the major by-product (25%). Notably using an in situ generated catalyst from Ni(cod)2 and dcype [dcype = bis-(dicyclohexylphosphino)ethane], similar result was obtained. Using these catalytic systems, aliphatic amines can be methylated in 43–69% yields. By contrast, aniline gave lower activities with 33–37% yields (Scheme 65).
 | ||
| Scheme 65 Nickel catalysed N-methylation of amines using CO2 and hydrosilanes. | ||
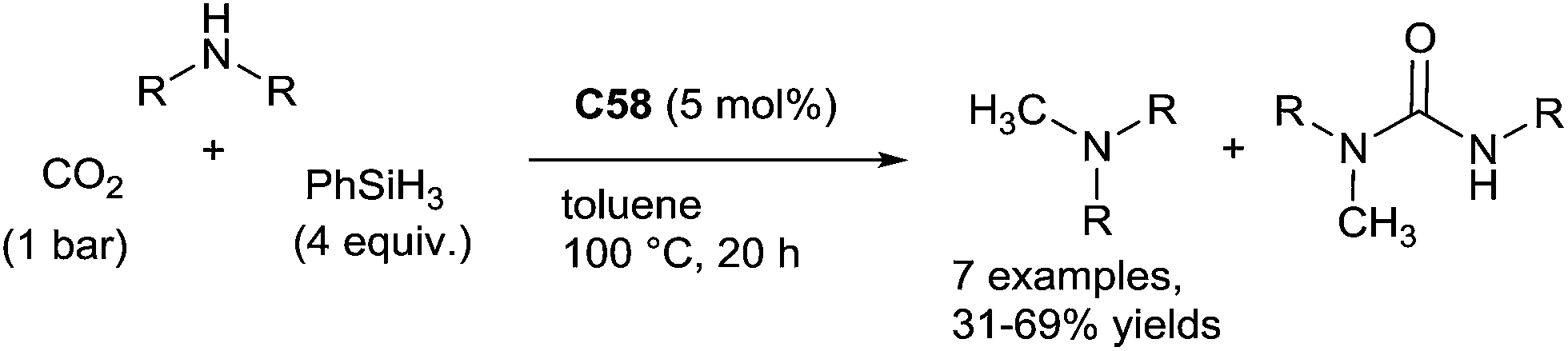
Using an in situ generated iron catalyst from Fe(acac)2/P(CH2CH2PPh2)3 (L13, 5 mol%), the hydrosilylation of carbon dioxide in the presence of amines can lead selectively to formamide and methylamine derivatives. By reaction of primary and secondary amines with 1 equiv. of phenylsilane in THF at room temperature for 18 h, CO2 (1 bar) was transformed to the corresponding formamides in 24–95% GC-yields. It must be underlined that using 4 equiv. of phenylsilane in THF at 100 °C for 18 h under 1 bar of CO2, the catalytic system (10 mol%) was able to perform the methylation of methylarylamines to give a mixture of formamides and tertiary methylated amines when starting from secondary anilines133 (Scheme 66).
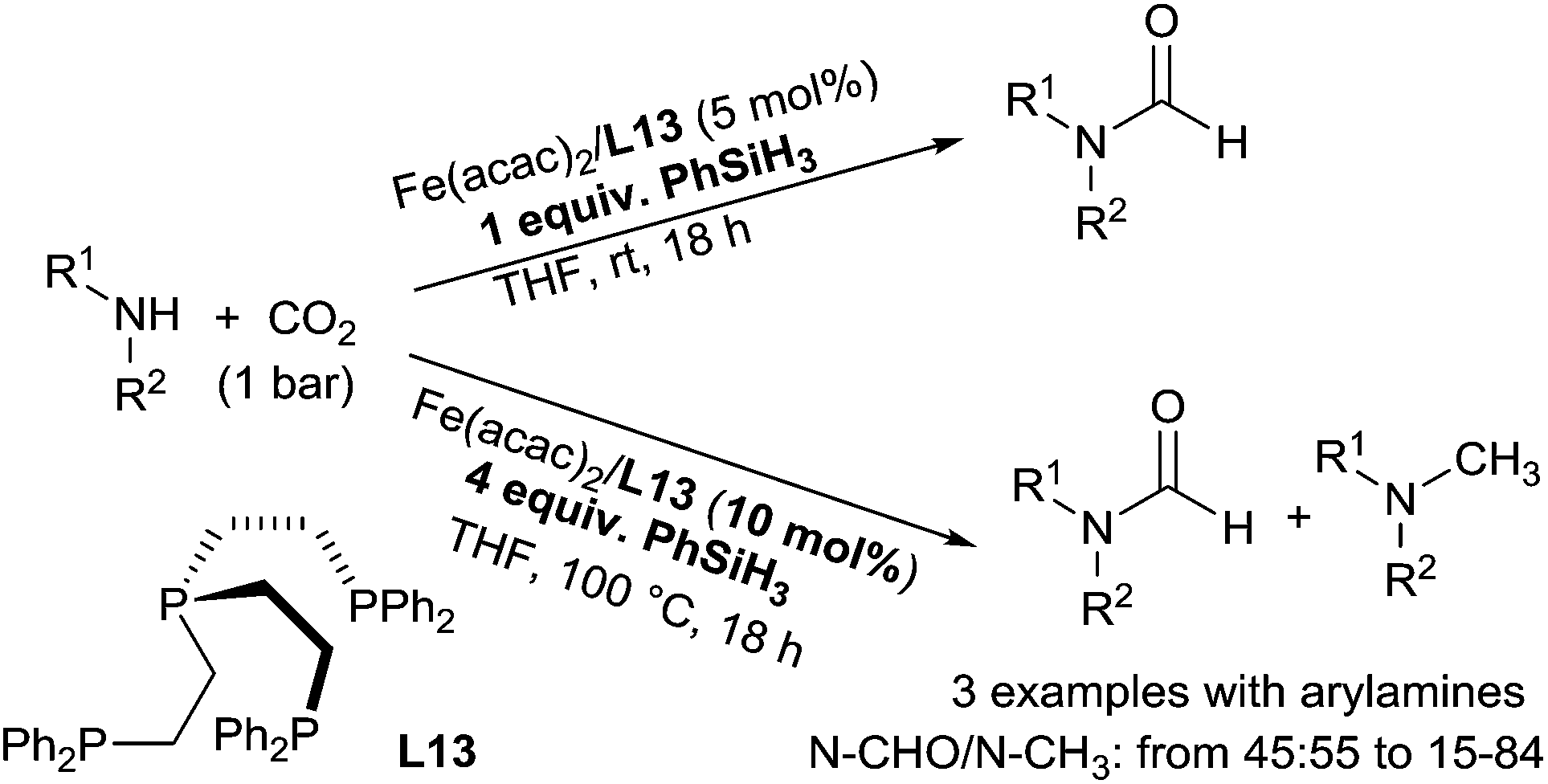 | ||
| Scheme 66 Iron catalysed reductive functionalization of CO2via hydrosilylation. | ||
Chelating bis(1,2,3-triazol-5-ylidene)-rhodium complexes C59 were also reported for the reductive methylation of amines: using carbon dioxide (25 atm) in the presence of diphenylsilane (2.5 equiv.) in CH2Cl2 at 25 °C for 4 h, amines were fully formylated, and after removal of CO2 and addition of 2 equiv. of Ph2SiH2 led to the methylated amines in 45–88% yields (Scheme 67). Remarkably, this catalyst was also able to reduce amides leading to amines (0.25 mol% C59, 1.6 equiv. of PhSiH3, 30 °C, 20 h) and carboxylic acids giving alcohols (0.5 mol% C59, 3 equiv. of PhSiH3, 30 °C, 20 h). Furthermore, the reductive alkylation of amines using carboxylic acids (1.75 equiv.) have been achieved (0.25 mol% C59, 4 equiv. of PhSiH3, 30 °C, 20 h).134
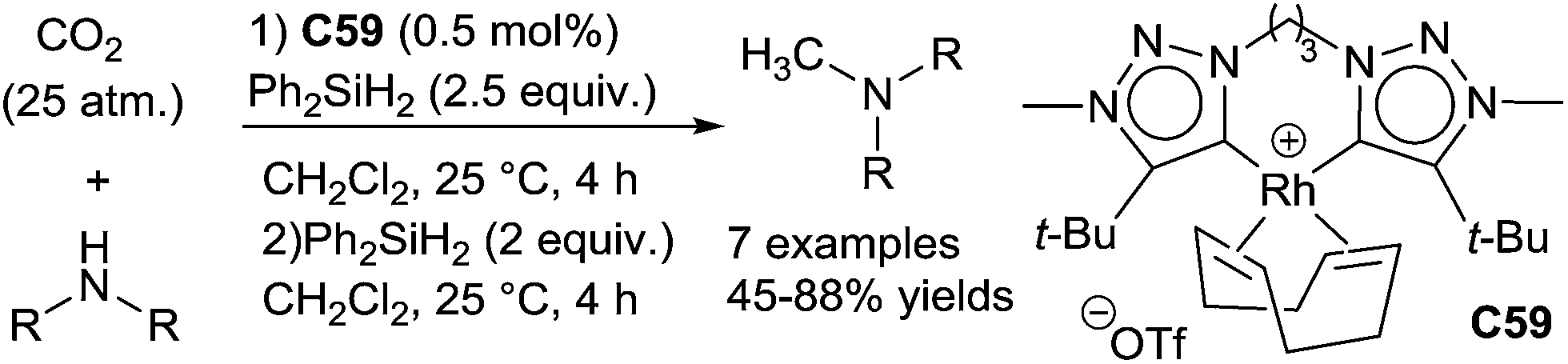 | ||
| Scheme 67 Rhodium catalysed reductive functionalization of CO2via hydrosilylation. | ||
The methylation of secondary amines was also achieved using dimethylcarbonate (DMC) or diethylcarbonate as the C1 source: using the NHC–Fe complex [CpFe(IMes)(CO)2][I] C16 as a catalyst (5 mol%) in the presence of 5 equiv. of phenylsilane in DMC as the solvent, at 100 °C under light irradiation for 24 h, reductant, methylated amines were obtained in 54–98% isolated yields.135
I. Hydrosilylation of pyridines
In 1998, Harrod et al. reported the first results dealing with homogeneous hydrosilylation of pyridines using a titanocene complex, Cp2TiMe2C22b as the catalyst (10 mol%).136 Thus, pyridine derivatives in the presence of 2 equiv. of PhMeSiH2 in neat conditions at 80 °C led to the silylated dihydropyridine compounds in 70–99% NMR-yields. In 2008, Crabtree developed the hydrosilylation of quinoline derivatives in the presence of [Rh(nbd)(PPh3)2][PF6] C60 (nbd = norbornadiene) as the catalyst (3 mol%) and freshly distilled silane (4 equiv.) in toluene at rt leading to 1,2-dihydroquinoline derivatives in good yields. By contrast, isoquinoline gave the tetrahydro derivative as the major product.137In 2010, Nikonov et al. did a significant advance in this area developing a hydrosilylation of pyridine derivatives using cationic ruthenium(II) complexes [Cp(iPr3P)Ru(MeCN)2][X] C61 [X = PF6 or B(C6F5)4; Cp = cyclopentadienyl] (5 mol%).138
Pyridines reacted with HSiMe2Ph in CH2Cl2 at rt to give selectively the product of 1,4-addition. It must be noted that substitution in the 3- and 5-positions of the pyridine was tolerated, whereas substitution in the 2-, 4-, and 6-positions inhibited the reaction. Furthermore, sensitive groups such as cyano, aldehyde, ketone and ester were not tolerated (Scheme 68).
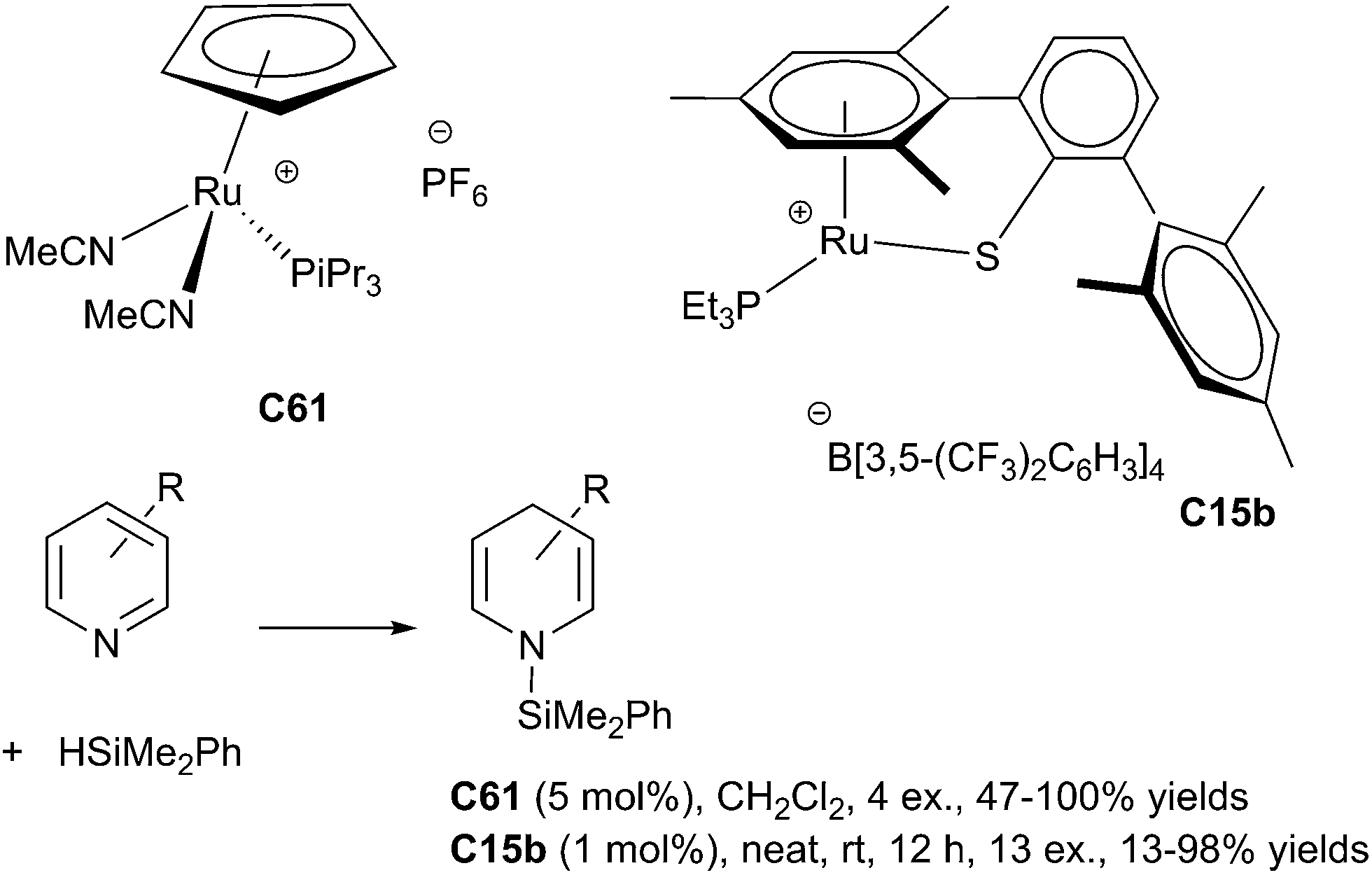 | ||
| Scheme 68 Ruthenium catalysed hydrosilylation of pyridines. | ||
Interestingly, the complex [Cp*(phen)Ru(CH3CN)][B(C6F5)4] C62 [phen = phenanthroline, Cp* = pentamethylcyclopentadienyl] catalyzed the regioselective hydrosilylation of ring-fused N-heteroaromatics, such as quinoline, isoquinoline and acridine.139
In 2013, Oestreich et al. reported a general hydrosilylation of pyridines and its benzannulated congeners using a tethered cationic ruthenium(II) complex C15b as the catalyst and a equimolar amount of PhMe2SiH in neat conditions at rt for 12 h leading the silylated 1,4-dihydropyridine resulting from the 1,4-addition (Scheme 68). Notably, using the same catalyst, 1,4-selective hydrosilylation of quinolines/acridine and 1,2-selective hydrosilylation of isoquinolines/phenanthridine were achieved in 71–98% yields.140
J. Conclusions
During the last decades, transition metal hydrosilylation reactions have made huge progresses, particularly for the reduction of CX (X = O, N, S) bond containing compounds. Thus the preparation of amines via the hydrosilylation of imines, amides, nitro and nitrile compounds via transition metal catalysis is nowadays an efficient alternative methodology to produce amines. It must be pointed out the remarkable observed chemoselectivities as with most of the systems, numerous reducible functional groups were tolerated. Another interesting point is that non precious, earth abundant and eco-friendly transition metals can be used with high efficiencies. Inexpensive silane such as PMHS can be also advantageously used. Furthermore, the topical reduction of carbon dioxide is also an exalting area of research, and pioneering exciting results on the reductive amination of CO2 is an interesting and eco-friendly alternative.
Acknowledgements
L. B., J.-B. S. and C. D. acknowledge the University of Rennes 1 and the CNRS for financial support. L. B. also thanks the Support of the Science Foundation for Young Teachers of Wuyi University (No. 2014zk04), the Natural Science Foundation of Guangdong Province (No. 2014A030310211), and the Foundation of Department of Education of Guangdong Province (No. 2014KQNCX156).Notes and references
- Selected representative books: (a) R. C. Larock, in Comprehensive Organic Transformations: a Guide to Functional Group Preparation, Wiley-VHC, New York, 1989 Search PubMed; (b) A. Ricci, Modern Amination Method, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, New York, 2000 CrossRef; (c) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (d) A. Ricci, in Amino Group Chemistry. From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- K. Eller, E. Henkes, R. Rossbacher and H. Höke, Amines, Aliphatic, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
- J. Seyden-Penne, in Reductions by the Alumino- and Borohydrides in Organic Synthesis, Wiley, New York, 2nd edn, 1997 Search PubMed.
- (a) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS PubMed; (b) N. Fleury-Brégeot, V. de la Fuente, S. Castillon and C. Claver, ChemCatChem, 2010, 2, 1346 CrossRef; (c) J. G. de Vries and C. J. Elservier, Handbook of Homogeneous Hydrogenation, Wiley-VCH, Weinheim, 2007 Search PubMed; (d) M. Beller and C. Bolm, Transition Metals for Organic Synthesis, Wiley-VCH, Weinheim, 2nd edn, 2004 CrossRef; (e) A. Fabrello, A. Bachelier, M. Urrutigoïty and P. Kalck, Coord. Chem. Rev., 2010, 254, 273 CrossRef CAS.
- Representative examples: (a) B. Wojcik and H. Adkins, J. Am. Chem. Soc., 1934, 56, 2419 CrossRef CAS; (b) A. A. Núňez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154 RSC; (c) G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, Adv. Synth. Catal., 2010, 352, 869 CrossRef CAS; (d) G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2010, 269, 93 CrossRef CAS; (e) M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231 CrossRef CAS.
- (a) I. Ojima, in The Chemistry of Organic Silicon Compounds, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1989, vol. 1 Search PubMed; (b) B. Marciniec, J. Gulinsky, W. Urbaniak and Z. Kornetka, in Comprehensive Handbook on Hydrosilylation, ed. B. Marciniec, Pergamon, Oxford, 1992 Search PubMed; (c) V. B. Pukhnarevich, E. Lukevics, L. T. Kopylova and M. G. Voronkov, in Perspectives of Hydrosilylation, ed. E. Lukevics, Institute of Organic Synthesis, Riga, 1992 Search PubMed; (d) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000 Search PubMed; (e) H. Nishiyama, in Transition Metals For Organic Synthesis, ed. M. Beller and C. Bolm, 2nd edn, 2004, vol. 2, p. 182 Search PubMed; (f) B. Marciniec, Coord. Chem. Rev., 2005, 249, 2374 CrossRef CAS; (g) O. Riant, in Modern Reduction Methods, ed. P. G. Anderson and I. J. Munslow, 2008, p. 321 Search PubMed; (h) C. G. Arena, Minireviews in Organic Chemistry, 2009, vol. 6, p. 159 Search PubMed; (i) H. Nishiyama, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, 2012, vol. 5, p. 31 Search PubMed.
- F. C. Whitmore, E. W. Pietrusza and L. H. Sommer, J. Am. Chem. Soc., 1947, 69, 2108 CrossRef CAS.
- (a) R. J. P. Corriu and J. J. E. Moreau, J. Organomet. Chem., 1975, 85, 19 CrossRef CAS; (b) J. Boyer, R. J. P. Corriu, R. Perz and C. Réyé, J. Organomet. Chem., 1979, 172, 143 CrossRef CAS; (c) J. Boyer, R. J. P. Corriu, R. Perz and C. Réyé, Tetrahedron, 1981, 37, 2165 CrossRef CAS; (d) G. L. Larson and J. L. Fry, Ionic and Organometallic-Catalyzed Organosilane Reductions, John Wiley & Sons Inc, 2010 Search PubMed.
- D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS PubMed.
- (a) S. Diez-Gonzalez and S. P. Nolan, Org. Prep. Proced. Int., 2007, 3, 523 CrossRef; (b) S. Diez-Gonzalez and S. P. Nolan, Acc. Chem. Res., 2008, 41, 349 CrossRef CAS.
- H. Nishiyama, Hydrosilylation of Carbonyl and Imino Groups, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, vol. 1, ch. 6.3 Search PubMed.
- (a) S. Gladiali and E. Alberico, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2004, vol. 2, p. 145 Search PubMed; (b) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201 CrossRef CAS.
- (a) O. Riant, N. Mostefaï and J. Courmarcel, Synthesis, 2004, 2943 CrossRef CAS; (b) J.-F. Carpentier and V. Bette, Curr. Org. Chem., 2002, 6, 913 CrossRef CAS.
- K. Takaki, T. Kamata, Y. Miura, T. Shishido and K. Takehira, J. Org. Chem., 1999, 64, 3891 CrossRef CAS.
- T. Nakano and Y. Nagai, Chem. Lett., 1988, 481 CrossRef CAS.
- (a) S. C. Berk, K. A. Kreutzer and S. L. Buchwald, J. Am. Chem. Soc., 1991, 113, 5093 CrossRef CAS; (b) K. J. Barr, S. C. Berk and S. L. Buchwald, J. Org. Chem., 1994, 59, 4323 CrossRef CAS; (c) M. B. Carter, B. Schiøtt, A. Gutierrez and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 11667 CrossRef CAS.
- C. A. Willoughby, R. R. Duff Jr, W. M. Davies and S. L. Buchwald, Organometallics, 1996, 15, 472 CrossRef CAS.
- A. Tillack, C. Lefeber, N. Peulecke, D. Thomas and U. Rosenthal, Tetrahedron Lett., 1997, 38, 1533 CrossRef CAS.
- X. Verdaguer, U. E. W. Lange, M. T. Reding and S. L. Buchwald, J. Am. Chem. Soc., 1996, 118, 6784 CrossRef CAS.
- X. Verdaguer, U. E. W. Lange and S. L. Buchwald, Angew. Chem., Int. Ed., 1998, 37, 1103 CrossRef CAS.
- (a) M. T. Reding and S. L. Buchwald, J. Org. Chem., 1995, 60, 7884 CrossRef CAS; (b) S. W. Breeden and N. J. Lawrence, Synlett, 1994, 833 CrossRef CAS.
- M. C. Hansen and S. L. Buchwald, Org. Lett., 2000, 2, 713 CrossRef CAS.
- (a) M. C. Hansen and S. L. Buchwald, Tetrahedron Lett., 1999, 40, 2033 CrossRef CAS; (b) M. T. Reeding and S. L. Buchwald, J. Org. Chem., 1998, 63, 6344 CrossRef; (c) J. Yun and S. L. Buchwald, J. Org. Chem., 2000, 65, 767 CrossRef CAS.
- H. Gruber-Woelfler, J. G. Khinast, M. Flock, R. C. Fischer, J. Sassmannshausen, T. Stanoeva and G. Gescheidt, Organometallics, 2009, 28, 2546 CrossRef CAS.
- M. Klahn, P. Arndt, A. Spannenberg, A. Gansäuer and U. Rosenthal, Organometallics, 2008, 27, 5846 CrossRef CAS.
- H. Gruber-Woelfler, G. J. Lichtenegger, C. Neubauer, E. Polo and J. G. Khinast, Dalton Trans., 2012, 41, 12711 RSC.
- J. J. Kennedy-Smith, K. A. Nolin, H. P. Gunterman and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 4056 CrossRef CAS.
- K. A. Nolin, R. W. Ahn and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 12462 CrossRef CAS PubMed.
- K. A. Nolin, R. W. Ahn, Y. Kobayashi, J. J. Kennedy-Smith and F. D. Toste, Chem.–Eur. J., 2010, 16, 9555 CrossRef CAS PubMed.
- Selected examples: (a) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (b) T. Ohkuma, H. Doucet, T. Pham, K. Mikami, T. Korenaga, M. Terada and R. Noyori, J. Am. Chem. Soc., 1998, 120, 1086 CrossRef CAS; (c) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- Y. Nishibayashi, I. Takei, S. Uemura and M. Hidai, Organometallics, 1998, 17, 3420 CrossRef CAS.
- I. Takei, Y. Nishibayashi, Y. Ishii, Y. Mizobe, S. Uemura and M. Hidai, Chem. Commun., 2001, 2360 RSC.
- H. Hashimoto, I. Aratani, C. Kabuto and M. Kira, Organometallics, 2003, 22, 2199 CrossRef CAS.
- S. Abbina, S. Bian, C. Oian and G. Du, ACS Catal., 2013, 3, 678 CrossRef CAS.
- B. Li, J.-B. Sortais, C. Darcel and P. H. Dixneuf, ChemSusChem, 2012, 5, 396 CrossRef CAS.
- (a) B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221 CrossRef CAS; (b) B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, Top. Catal., 2014, 57, 833 CrossRef CAS.
- J. Hermeke, H. F. T. Klare and M. Oestreich, Chem.–Eur. J., 2014, 20, 9250 CrossRef CAS PubMed.
- S. Zhou, K. Junge, D. Addis, S. Das and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9507 CrossRef CAS PubMed.
- H. Nishiyama and A. Furuta, Chem. Commun., 2007, 760 RSC.
- L. C. Misal Castro, J.-B. Sortais and C. Darcel, Chem. Commun., 2012, 48, 151 RSC.
- (a) N. Langlois, D. Tang Phat and H. B. Kagan, Tetrahedron Lett., 1973, 49, 4865 CrossRef; (b) H. B. Kagan, N. Langlois and D. Tang Phat, J. Organomet. Chem., 1975, 90, 353 CrossRef CAS.
- O. Niyomura, T. Iwasawa, N. Sawada, M. Tokunaga, Y. Obora and Y. Tsuji, Organometallics, 2005, 24, 3468 CrossRef CAS.
- I. Takei, Y. Nishibayashi, Y. Arikawa, S. Uemura and M. Hidai, Organometallics, 1999, 18, 2271 CrossRef CAS.
- L. D. Field, B. A. Messerle and S. L. Rumble, Eur. J. Org. Chem., 2005, 2881 CrossRef CAS.
- W. Iali, F. La Paglia, X.-F. Le Goff, D. Sredojevic, M. Pfeffer and J.-P. Djukic, Chem. Commun., 2012, 48, 10310 RSC.
- Y. Corre, W. Iali, M. Hamdaoui, X. Trivelli, J.-P. Djukic, F. Agbossou-Niedercorn and C. Michon, Catal. Sci. Technol., 2015, 5, 1452 RSC.
- For representative examples of Ni-catalyzed hydrosilylation of carbonyl derivatives, see: (a) F.-G. Fontaine, R.-V. Nguyen and D. Zargarian, Can. J. Chem., 2003, 81, 1299 CrossRef CAS; (b) S. Chakraborty, J. A. Krause and H. Guan, Organometallics, 2009, 28, 582 CrossRef CAS; (c) B. L. Tran, M. Pink and D. J. Mindiola, Organometallics, 2009, 28, 2234 CrossRef CAS; (d) S. Kundu, W. W. Brennessel and W. D. Jones, Inorg. Chem., 2011, 50, 9443 CrossRef CAS; (e) J. Zheng, C. Darcel and J.-B. Sortais, Catal. Sci. Technol., 2013, 3, 81 RSC; (f) L. P. Bheeter, M. Henrion, L. Brelot, C. Darcel, M. J. Chetcuti, J.-B. Sortais and V. Ritleng, Adv. Synth. Catal., 2012, 354, 2619 CrossRef CAS; (g) L. Postigo and B. Royo, Adv. Synth. Catal., 2012, 354, 2613 CrossRef CAS.
- (a) A. H. Vetter and A. Berkessel, Synthesis, 1995, 419 CrossRef CAS; (b) L. P. Bheeter, M. Henrion, M. J. Chetcuti, C. Darcel, V. Ritleng and J.-B. Sortais, Catal. Sci. Technol., 2013, 3, 3111 RSC.
- H. Tofazolian and J. A. R. Schmidt, Catal. Sci. Technol., 2016, 6, 685 RSC.
- For representative reviews, see: (a) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498 CrossRef CAS; (b) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916 CrossRef CAS.
- B. H. Lipshutz and H. Shimizu, Angew. Chem., Int. Ed., 2004, 43, 2228 CrossRef CAS.
- As representative examples, see: (a) H. Mimoun, J. Org. Chem., 1999, 64, 2582 CrossRef CAS; (b) H. Mimoun, J. Y. de Saint Laumer, L. Giannini, R. Scopelliti and C. Floriani, J. Am. Chem. Soc., 1999, 121, 6158 CrossRef CAS; (c) V. Bette, A. Mortreux, D. Savoia and J.-F. Carpentier, Tetrahedron, 2004, 60, 2837 CrossRef CAS; (d) V. Bette, A. Mortreux, F. Ferioli, G. Martelli, D. Savoia and J.-F. Carpentier, Eur. J. Org. Chem., 2004, 3040 CrossRef CAS.
- V. Bette, A. Mortreux, C. W. Lehmann and J.-F. Carpentier, Chem. Commun., 2003, 332 RSC.
- (a) B.-M. Park, S. Mun and J. Yun, Adv. Synth. Catal., 2006, 348, 1029 CrossRef CAS; (b) B.-M. Park, X. Feng and J. Yun, Bull. Korean Chem. Soc., 2011, 32, 2960 CrossRef CAS; (c) A. Bezlada, M. Szewczyk and J. Mlynarski, J. Org. Chem., 2016, 81, 336 CrossRef CAS.
- J. Gajewy, J. Gawronski and M. Kwit, Org. Biomol. Chem., 2011, 9, 3863 RSC.
- A. Adamkiewicz and J. Mlynarski, Eur. J. Org. Chem., 2016, 1060 CrossRef CAS.
- K. Selvakumar, K. Rangareddy and J. F. Harrod, Can. J. Chem., 2004, 82, 1244 CrossRef CAS.
- S. Bower, K. A. Kreutzer and S. L. Buchwald, Angew. Chem., Int. Ed., 1996, 35, 1515 CrossRef CAS.
- S. Laval, W. Dayoub, L. Pehlivan, E. Métay, A. Favre-Réguillon, D. Delbrayelle, G. Mignani and M. Lemaire, Tetrahedron Lett., 2011, 52, 4072 CrossRef CAS.
- A. C. Fernandes and C. C. Romao, J. Mol. Catal. A: Chem., 2007, 272, 60 CrossRef CAS.
- A. Volkov, F. Tinnis, T. Slagbrand, I. Pershagen and H. Adolfsson, Chem. Commun., 2014, 50, 14508 RSC.
- S. Krackl, C. I. Someya and S. Enthaler, Chem.–Eur. J., 2012, 18, 15267 CrossRef CAS.
- M. Igarashi and T. Fuchikami, Tetrahedron Lett., 2001, 42, 1945 CrossRef CAS.
- K. Matsubara, T. Iura, T. Maki and H. Nagashima, J. Org. Chem., 2001, 67, 4985 CrossRef.
- Y. Motoyama, K. Mitsui, T. Ishida and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150 CrossRef CAS.
- H. Sasakuma, Y. Motoyama and H. Nagashima, Chem. Commun., 2007, 4916 RSC.
- (a) S. Hanada, T. Ishida, Y. Motoyama and H. Nagashima, J. Org. Chem., 2007, 72, 7551 CrossRef CAS; (b) Y. Motoyama, C. Itonaga, T. Ishida, M. Takasaki and H. Nagashima, Org. Synth., 2005, 82, 188 CrossRef CAS.
- M. E. Kuehne and P. J. Shannon, J. Org. Chem., 1977, 42, 2082 CrossRef CAS.
- J. T. Reeves, Z. Tan, M. A. Marsini, Z. S. Han, Y. Xu, D. C. Reeves, H. Lee, B. Z. Lu and C. H. Senanayake, Adv. Synth. Catal., 2013, 355, 47 CrossRef CAS.
- B. Li, J.-B. Sortais and C. Darcel, Chem. Commun., 2013, 49, 3691 RSC.
- Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511 CrossRef CAS.
- S. Zhou, D. Addis, S. Das, K. Junge and M. Beller, Chem. Commun., 2009, 4883 RSC.
- H. Tsutsumi, Y. Sunada and H. Nagashima, Chem. Commun., 2011, 47, 6581 RSC.
- D. Bézier, G. T. Venkanna, J.-B. Sortais and C. Darcel, ChemCatChem, 2011, 3, 1747 CrossRef.
- A. Volkov, E. Buitrago and H. Adolfsson, Eur. J. Org. Chem., 2013, 2066 CrossRef CAS.
- B. Blom, G. Tan, S. Enthaler, S. Inoue, J. D. Epping and M. Driess, J. Am. Chem. Soc., 2013, 135, 18108 CrossRef CAS.
- S. Das, B. Wendt, K. Möller, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 1662 CrossRef CAS.
- T. Dombray, C. Helleu, C. Darcel and J.-B. Sortais, Adv. Synth. Catal., 2013, 355, 3358 CrossRef CAS.
- Recent review: K. Riener, M. P. Högerl, P. Gigler and F. E. Kühn, ACS Catal., 2012, 2, 613 CrossRef CAS.
- R. Kuwano, M. Takahashi and Y. Ito, Tetrahedron Lett., 1998, 39, 1017 CrossRef CAS.
- C. Bornschein, A. J. J. Lennox, S. Werkmeister, K. Junge and M. Beller, Eur. J. Org. Chem., 2015, 1915 CrossRef CAS.
- S. Das, Y. Li, C. Bornschein, S. Pisiewicz, K. Kiersch, D. Michalik, F. Gallou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 12389 CrossRef CAS.
- M. Stoelzel, C. Präsang, B. Blom and M. Driess, Aust. J. Chem., 2013, 66, 1163 CrossRef CAS.
- S. Park and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 640 CrossRef CAS.
- C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304 CrossRef CAS PubMed.
- Y. Motoyama, M. Aoki, N. Takaoka, R. Aoto and H. Nagashima, Chem. Commun., 2009, 1574 RSC.
- A. Tahara, Y. Miyamoto, R. Aoto, K. Shigeta, Y. Une, Y. Sunada, Y. Motoyama and H. Nagashima, Organometallics, 2015, 34, 4895 CrossRef CAS.
- N. C. Mamillapalli and G. Sekar, Chem. Commun., 2014, 50, 7881 RSC.
- (a) S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032 CrossRef CAS; (b) S. Hanada, Y. Motoyama and H. Nagashima, Tetrahedron Lett., 2006, 47, 6173 CrossRef CAS.
- S. Pisiewicz, K. Junge and M. Beller, Eur. J. Inorg. Chem., 2014, 2345 CrossRef CAS.
- S. Das, B. Join, K. Junge and M. Beller, Chem. Commun., 2012, 48, 2683 RSC.
- (a) R. Calas, E. Frainnet and A. Bazouin, C. R. Chim., 1962, 254, 2357 CAS; (b) E. Frainnet, A. Bazouin and R. Calas, C. R. Chim., 1963, 257, 1304 Search PubMed; (c) R. Calas, Pure Appl. Chem., 1966, 13, 61 CAS; (d) R. Calas, J. Organomet. Chem., 1980, 200, 11 CrossRef CAS.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS PubMed.
- S. Das, D. Addis, K. Junge and M. Beller, Chem.–Eur. J., 2011, 17, 12186 CrossRef CAS.
- O. O. Kovalenko, A. Volkov and H. Adolfsson, Org. Lett., 2015, 17, 446 CrossRef CAS.
- R. Calas, E. Frainnet and A. Bazouin, C. R. Chim., 1961, 252, 420 CAS.
- R. J. P. Corriu, J. J. E. Moreau and M. Pataud-Sat, J. Organomet. Chem., 1982, 228, 301 CrossRef CAS.
- T. Murai, T. Sakane and S. Kato, Tetrahedron Lett., 1985, 26, 5145 CrossRef CAS.
- T. Murai, T. Sakane and S. Kato, J. Org. Chem., 1990, 55, 449 CrossRef CAS.
- A. M. Caporusso, N. Panziera, P. Pertici, E. Pitzalis, P. Salvadori, G. Vitulli and G. Martra, J. Mol. Catal. A: Chem., 1999, 137, 275 CrossRef.
- A. J. Huckaba, T. K. Hollis and S. W. Reilly, Organometallics, 2013, 32, 6248 CrossRef CAS.
- D. V. Gutsulyak and G. I. Nikonov, Angew. Chem., Int. Ed., 2010, 49, 7553 CrossRef CAS.
- C. Boone, I. Korobkov and G. I. Nikonov, ACS Catal., 2013, 3, 2336 CrossRef CAS.
- I. Cabrita and A. C. Fernandes, Tetrahedron, 2011, 67, 8183 CrossRef CAS.
- J. Lipowitz and S. A. Bowman, J. Org. Chem., 1972, 48, 162 Search PubMed.
- (a) R. J. Rahaim Jr and R. E. Maleczka Jr, Org. Lett., 2005, 7, 5087 CrossRef; (b) R. J. Rahaim Jr and R. E. Maleczka Jr, Synthesis, 2006, 3316 Search PubMed.
- S. Sun, Z. Quan and X. Wang, RSC Adv., 2015, 5, 84574 RSC.
- H. R. Brinkman, W. H. Miles, M. D. Hilborn and M. C. Smith, Synth. Commun., 1996, 26, 973 CrossRef CAS.
- R. G. de Noronha, C. C. Romão and A. C. Fernandes, J. Org. Chem., 2009, 74, 6960 CrossRef CAS.
- K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC.
- L. Pehlivan, E. Métay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2010, 51, 1939 CrossRef CAS.
- (a) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Scmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886 CrossRef CAS; (b) K. Zhu, M. P. Shaver and S. P. Thomas, Chem. Sci., 2016, 7, 3031 RSC.
- L. D. Field, B. A. Messerle and S. L. Wren, Organometallics, 2003, 22, 4393 CrossRef CAS.
- R.-Y. Lai, K. Surekha, A. Hayashi, F. Ozawa, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Organometallics, 2007, 26, 1062 CrossRef CAS.
- A. Heutling, F. Pohlki, I. Bytschkov and S. Doye, Angew. Chem., Int. Ed., 2005, 44, 2951 CrossRef CAS.
- T. Mizuta, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2005, 70, 2195 CrossRef CAS.
- S. C. A. Sousa and A. C. Fernandes, Adv. Synth. Catal., 2009, 352, 2218 CrossRef.
- J. R. Bernardo, S. C. A. Sousa, P. R. Florindo, M. Wolff, B. Machura and A. C. Fernandes, Tetrahedron, 2013, 69, 9145 CrossRef CAS.
- B. G. Das and P. Ghorai, Org. Biomol. Chem., 2013, 11, 4379 RSC.
- S. Enthaler, Catal. Lett., 2011, 141, 55 CrossRef CAS.
- S. Enthaler, ChemCatChem, 2010, 2, 1411 CrossRef CAS.
- H. Jaafar, H. Li, L. C. Misal Castro, J. Zheng, T. Roisnel, V. Dorcet, J.-B. Sortais and C. Darcel, Eur. J. Inorg. Chem., 2012, 3546 CrossRef CAS.
- J. Zheng, T. Roisnel, C. Darcel and J.-B. Sortais, ChemCatChem, 2013, 5, 2729 CrossRef.
- T. Nishikata and H. Nagashima, Angew. Chem., Int. Ed., 2012, 51, 5363 CrossRef CAS.
- I. Sorribes, K. Junge and M. Beller, J. Am. Chem. Soc., 2014, 136, 14314 CrossRef CAS.
- H. Li, M. Achard, C. Bruneau, J.-B. Sortais and C. Darcel, RSC Adv., 2014, 4, 25892 RSC.
- L.-W. Zhan, L. Han, P. Xing and B. Jiang, Org. Lett., 2015, 17, 5990 CrossRef CAS.
- M. F. Ali, B. M. El Ali and J. G. Speight, Handbook of Industrial Chemistry – Organic Chemicals, McGraw-Hill, New York, 2005 Search PubMed.
- For a representative review on reductive functionalization of CO2 with amines, see: A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157 RSC.
- O. Jacquet, X. Frogneux, C. Das Neves Gomes and T. Cantat, Chem. Sci., 2013, 4, 2127 RSC.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568 CrossRef CAS.
- L. Gonzalez-Sebastian, M. Flores-Alamo and J. J. Garcia, Organometallics, 2015, 34, 763 CrossRef CAS.
- X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529 RSC.
- T. V. Q. Nguyen, W.-J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2016, 358, 452 CrossRef CAS.
- J. Zheng, C. Darcel and J.-B. Sortais, Chem. Commun., 2014, 50, 14229 RSC.
- L. Hao, J. F. Harrod, A.-M. Lebuis, Y. Mu, R. Shu, E. Samuel and H.-G. Woo, Angew. Chem., Int. Ed., 1998, 37, 3126 CrossRef CAS.
- A. M. Voutchkova, D. Gnanamgari, C. E. Jakobsche, C. Butler, S. J. Miller, J. Parr and R. H. Crabtree, J. Organomet. Chem., 2008, 693, 1815 CrossRef CAS.
- D. V. Gutsulyak, A. van der Est and G. I. Nikonov, Angew. Chem., Int. Ed., 2011, 50, 1384 CrossRef CAS.
- S.-H. Lee, D. V. Gutsulyak and G. I. Nikonov, Organometallics, 2013, 32, 4457 CrossRef CAS.
- C. D. F. Königs, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 10076 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |