Magnetic iron oxide–fluorescent carbon dots integrated nanoparticles for dual-modal imaging, near-infrared light-responsive drug carrier and photothermal therapy†
Hui
Wang
a,
Jing
Shen
a,
Yingyu
Li
a,
Zengyan
Wei
b,
Guixin
Cao
c,
Zheng
Gai
c,
Kunlun
Hong
c,
Probal
Banerjee
a and
Shuiqin
Zhou
*a
aDepartment of Chemistry, The College of Staten Island, and The Graduate Center, The City University of New York, Staten Island, NY 10314, USA. E-mail: shuiqin.zhou@csi.cuny.edu; Fax: +1 718 982 3910; Tel: +1 718 982 3897
bDepartment of Chemistry and Biochemistry, Hunter College, The City University of New York, New York, NY 10065, USA
cCenter for Nanophase Materials Sciences and Chemical Science Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 13th February 2014
Abstract
Multifunctional hybrid nanoparticles (NPs, ∼100 nm) that combine magnetic Fe3O4 nanocrystals and fluorescent carbon dots (CDs) in porous carbon (C) were successfully synthesized using a one-pot solvothermal method by simply increasing the H2O2 concentration. The resultant Fe3O4@C-CDs hybrid NPs not only demonstrate excellent magnetic responsive properties (Ms = 32.5 emu g−1) and magnetic resonance imaging ability (r*2 = 674.4 mM−1 s−1) from the Fe3O4 nanocrystal core, but also exhibit intriguing photoluminescent (quantum yield ∼6.8%) properties including upconversion fluorescence and excellent photostability from the CDs produced in the porous carbon. The hybrid NPs can enter the intracellular region and illuminate mouse melanoma B16F10 cells under different excitation wavelengths. Meanwhile, the mesoporous carbon shell and hydrophilic surface functional groups endow the hybrid NPs with high loading capacity (835 mg g−1) for the anti-cancer drug doxorubicin and excellent stability in aqueous solutions. More importantly, the hybrid NPs can absorb and convert near-infrared (NIR) light to heat due to the existence of CDs, and thus, can realise NIR-controlled drug release and combined photothermo/chemotherapy for high therapeutic efficacy. Such nanostructured Fe3O4@C-CDs hybrid NPs demonstrate great promise towards advanced nanoplatforms for simultaneous imaging diagnostics and high efficacy therapy.
1. Introduction
Multifunctional nanoparticles (NPs) that can combine magnetic and optical properties have attracted a great deal of attention due to their increasing and important applications in the biomedical field.1–5 While magnetic NPs can be used as contrast agents in magnetic resonance imaging (MRI) and facilitate magnetic responsive separation, detection, targeting, and hyperthermia, luminescent or plasmonic NPs enable optical bioimaging and photothermal therapy.1–9 Many systems and methods have been developed to combine the magnetic and optical materials into single nanohybrids to achieve the aforementioned multiple functions. Typically, magnetic iron oxide NPs have been combined with organic dyes or polymers,10–14 semiconducting quantum dots (QDs),15–17 noble metal NPs,18–23 and rare earth metal NPs.24–28 However, these materials have certain limitations for practical applications. Organic dyes show poor photostability and potential toxicity, QDs have problems associated with the inherent heavy metal toxicity, and noble metal/rare earth metal materials involve high cost. In addition, the preparation of these magnetic–fluorescent hybrid NPs involves complicated multi-step synthetic procedures.Recently explored fluorescent carbon dots (CDs) or graphene nanosheets with tunable emission as a new type of fluorescence marker in nanobiotechnology are widely considered to be advantageous over semiconductor QDs concerning material toxicity and over conventional organic dyes in terms of optical brightness and photostability.29–38 Furthermore, CDs can be produced at relatively low cost on a large scale by several approaches.35 On the other hand, nanomaterial-based photothermal therapy (PTT) for treating solid tumors has also attracted considerable interest because these nanostructured materials, including gold, carbon nanotubes, and graphenes, can strongly absorb near-infrared (NIR) light and locally produce cytotoxic heat to kill cancer cells.39–46 The big challenge is that it is still very hard to achieve complete tumor ablation, because the local photothermal cytotoxicity from these NPs is not sufficient to eliminate the diffused tumor cells, and heat transfer in organisms is complicated.47,48 In order to enhance the therapeutic efficacy, the combination of PTT with chemotherapy is a desirable method because chemotherapeutic agents exhibit an enhanced cytotoxicity at elevated temperatures.49,50 In addition, combined photothermal/chemotherapy can lower the drug dosage requirements but still achieve a comparable cytotoxicity to the corresponding unheated drug dose, and thus minimize systemic side-effects of chemotherapeutic agents.51,52 However, to the best of our knowledge, neither have nanosystems been reported to integrate magnetic nanocrystals with CDs for dual-modal imaging, nor have CD-based drug carriers been synthesized to combine the photothermal therapy and chemotherapy functions. Therefore, the purpose of this work is to develop a simple one-pot method to synthesize a new type of highly integrated hybrid NPs that can combine magnetic iron oxide nanocrystals and fluorescent CDs into a single mesoporous carbon nanosystem, which will not only show MRI and fluorescent dual-modal imaging ability, but also manifest ability as a drug carrier for combined photothermal/chemotherapy.
Herein, we report a new type of highly integrated hybrid NPs that can combine the magnetic iron oxide nanocrystals and fluorescent CDs into a single carbon-based nanosystem, with the Fe3O4 nanocrystals clustered in the core and the CDs implanted in the porous carbon (C) shell. Such nanostructured Fe3O4@C-CDs hybrid NPs were synthesized through an improved solvothermal method.53,54 Our synthetic strategy is to ensure sufficient oxidizing agent, H2O2, is present in the reaction medium, such that abundant small-sized fluorescent CDs can be formed in situ in the porous carbon shell from the oxidation and deep decomposition of the precursor ferrocene. In such a design, NPs with combined magnetic–fluorescent features can be synthesized in one-step in a high yield. As shown in Fig. 1, we expect that the resultant Fe3O4@C-CDs hybrid NPs will not only demonstrate the excellent weak ferromagnetic properties of the Fe3O4 nanocrystals for MRI contrast, but also manifest the intriguing photoluminescent (PL) properties of the CDs, including excitation-tunable emission, upconversion PL, and excellent photostability for fluorescent imaging. Furthermore, the mesoporous structure of the carbon matrix and the photothermal conversion ability of the CDs should provide the hybrid NPs with a high drug loading capacity for chemotherapy and a high efficiency for photothermal therapy, respectively. Therefore, these newly developed Fe3O4@C-CDs hybrid NPs, which are non-toxic and low cost, should be ideal candidates for combined dual-modal imaging diagnosis, near-infrared (NIR) photothermal therapy, and chemotherapy.
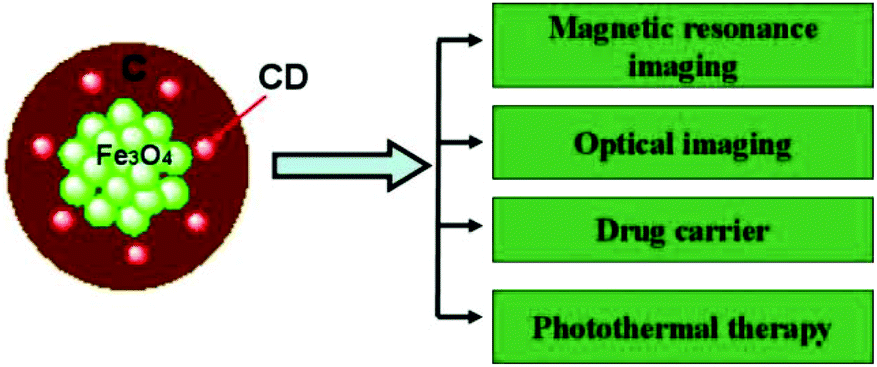 | ||
| Fig. 1 Schematic illustration of multifunctional Fe3O4@C-CDs hybrid NPs. The magnetic Fe3O4 nanocrystals and CDs can be used for MRI and fluorescent imaging contrast, respectively. The porous carbon shell embedded with CDs can not only provide high loading capacity for an anti-cancer drug, but also convert the NIR light to heat for combined photothermal/chemotherapy to provide high therapeutic efficacy. | ||
2. Materials and methods
2.1 Materials
All chemicals were purchased from Aldrich. Ferrocene (Fe(C5H5)2, ≥98%), hydrogen peroxide (H2O2, 30%) and acetone (C3H6O) were used as received without further purification. The water used in all experiments was of Millipore Milli-Q grade.2.2 Synthesis of Fe3O4@C-CDs hybrid NPs
In a typical synthesis, ferrocene (0.10 g) was dissolved in acetone (30 mL). After intense sonication for 30 min, 2.5 mL of 30% H2O2 solution was slowly added into the above solution, which was then vigorously stirred for 30 min with magnetic stirring apparatus. The precursor solution was transferred to a 50.0 mL Teflon-lined stainless steel autoclave. After sealing, the autoclave was heated to and maintained at 200 °C for 48 h. The autoclave was then cooled naturally to room temperature. After intense sonication for 15 min, the products from the Teflon-lined stainless steel autoclave were magnetized for 10 min by a magnet of 0.20 T. The supernatant was discarded under a magnetic field. The precipitates were then washed with acetone three times to remove excess ferrocene. Finally, the black products were dried at room temperature in a vacuum oven.2.3 Internalization of Fe3O4@C-CDs hybrid NPs into mouse melanoma cells B16F10
Round glass coverslips were placed in the wells of a 24-well plate and treated with 0.1% poly-L-lysine in 100 mM phosphate buffered saline (PBS) for 40 min. Following the treatment, the solution was aspirated and the wells were washed with PBS three times each. Next, B16F10 cells (2 × 104 cells per well) were plated on the glass coverslips at 80% confluence in DMEM containing 10% FBS and 1% penicillin–streptomycin. After 24 h, 500 μL dispersions of Fe3O4@C-CDs NPs in serum-free DMEM (50 mg mL−1) were respectively added to the marked wells. In a control well, 500 μL of serum-free DMEM was added. The plate was incubated at 37 °C for 2 h. The medium was then aspirated and fresh serum-free DMEM was added to each well. Finally, the coverslips with cells were removed from the wells and mounted onto slides for confocal microscopy studies.2.4 Drug loading and release
Doxorubicin (DOX) drug loading was performed by adding the Fe3O4@C-CDs NPs (1 mg) into a phosphate buffered saline (PBS) solution (10 mL, 0.005 M, pH = 7.4) containing DOX (1 mg) under magnetic stirring at 37 °C for 24 h. Then the mixture was separated by a magnet of 0.20 T. In order to remove unloaded DOX, the precipitate was redispersed in 10 mL PBS solution of pH = 7.4, and further purified and washed five times until the separated solution was clear. All the supernatant was collected for measurements of the drug loading content. The unloaded DOX present in the supernatant was quantified by a UV-Vis spectrophotometer at 480 nm. The DOX drug loading content of the hybrid NPs was calculated by (M0 − Mt)/MN × 100%, where M0 and Mt are the total mass of DOX dissolved in the initial solution and remaining in the supernatant solution, respectively. MN is the mass of the Fe3O4@C-CDs NPs used in the loading process.The in vitro release test of DOX from the Fe3O4@C-CDs NPs was evaluated by the dialysis method. The DOX-loaded Fe3O4@C-CDs NPs were redispersed in 10 mL PBS solution (0.005 M, pH = 7.4). Two dialysis bags filled with 5 mL diluted DOX-loaded Fe3O4@C-CDs NPs were immersed in 50 mL 0.005 M buffer solutions of pH = 7.4 at 37 °C, with one of them exposed to a NIR lamp with an output power of 1.5 W cm−2 for 5 min. The released DOX outside of the dialysis bag was sampled at defined time periods and assayed by UV-vis spectrometry at 480 nm. Cumulative release is expressed as the total percentage of drug released through the dialysis membrane over time.
2.5 In vitro cytotoxicity of the unloaded and DOX-loaded Fe3O4@C-CDs NPs with or without NIR irradiation
The B16F10 cells were cultured in a 96-well microplate in 100 μL medium containing about 2000 cells seeded into each well. After an overnight attachment, the medium was removed and another 100 μL medium containing the Fe3O4@C-CDs NPs unloaded or loaded with DOX was added so as the final exact concentrations of NPs were 200, 150, 100, and 50 μg mL−1. The hybrid NPs were well-dispersed in the culture medium and demonstrated excellent stability (see Fig. S1, ESI†). Wells containing the normal medium without NPs were used as a control. The cells were irradiated with 1.5 W cm−2 NIR light for 5 min in photothermal and photothermal–chemo combined treatments, whereas in chemotherapy alone, the cells were not exposed to NIR light. After incubation at 37 °C for 24 h, 10 μL of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg mL−1 in PBS) was added into the wells and the plate was incubated in a humidified environment of 5% CO2 and 37 °C. The medium was removed after 2 h and 100 μL of DMSO solution was added to each well. The plates were gently agitated until the formazan precipitate was dissolved. Cell viability was measured using a microplate reader at 570 nm. Positive controls contained no NPs, and negative controls contained MTT.2.6 Sample characterization
The powder X-ray diffraction (XRD) patterns were collected on a Japan Rigaku D/MAX-γA X-ray diffractometer equipped with Cu Kα radiation (λ = 1.54178 Å) over the 2θ range of 10–70°. Transmission electron microscopy images were obtained on a Hitachi H-800 transmission electron microscope, using an accelerating voltage of 100 kV. High-resolution transmission electron microscopy images were characterized by a JEM 2100 instrument with an acceleration voltage of 200 kV. Energy-dispersive X-ray analysis was obtained with an EDAX detector installed on the JEM 2100. The Raman spectrum was obtained on a LABRAM-HR Confocal Laser Micro-Raman spectrometer using an Ar+ laser at 514.5 nm at room temperature. The FTIR spectrum of the sample was recorded with a Nicolet Instrument Co. MAGNA-IR 750 Fourier transform infrared spectrometer. The UV-Vis absorption spectra were obtained on a Thermo Electron Co. Helios β UV-vis Spectrometer. The surface area and pore volume data were collected by accelerated surface area and porosimetry analysis. The photoluminescence spectra of the hybrid NPs were obtained on a JOBIN YVON Co. FluoroMax®-3 Spectrofluorometer equipped with a Hamamatsu R928P photomultiplier tube, calibrated photodiode for excitation reference correction from 200 to 980 nm, and an integration time of 1 s. The surface potential of the hybrid NPs was measured using a seveneasy™ pH meter (Mettler Toledo). The photothermal experiments on the hybrid NPs were conducted using a Philips infrared reflector lamp with a power density of 1.5 W cm−2 and a filter to block ultraviolet light. The B16F10 cells with incorporated hybrid NPs were imaged using a confocal laser scanning microscope (LEICA TCS SP2 AOBSTM) equipped with a HC PL APO CS 20 × 0.7 DRY lens. A superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS XL-7) was used to measure the magnetic properties of the hybrid NPs. Relaxation properties of hybrid NPs in aqueous solution were measured at 25 °C with a small animal 7.0 T MR scanner (Varian, America) with a 63/95 mm quad birdcage coil and gradient strength of up to 400 mT m−1. Dynamic light scattering (DLS) measurements were performed on a standard laser light scattering spectrometer (BI-200SM) equipped with a BI-9000 AT digital time correlator (BIC) to determine the hydrodynamic diameter (Dh) distributions of the hybrid NPs dispersed in water at a scattering angle of 60° and temperature of 22 °C. A He–Ne laser (35 mW, 633 nm) was used as the light source.3. Results and discussion
The composition of the Fe3O4@C-CDs hybrid NPs was firstly analyzed by X-ray diffraction (XRD, Fig. S2†). The observed reflections of 220, 311, 222, 400, 422, 511, 440 in the typical XRD pattern can be indexed to the magnetic Fe3O4 nanocrystals with a size of around 9 nm calculated from the Debye–Scherrer formula. The small reflection peak of carbon found at 2θ = 25° can be indexed well as the (002) reflection of graphite.29Fig. 2a shows the typical transmission electron microscopy (TEM) images of the Fe3O4@C-CDs NPs, which demonstrate a sphere-like morphology with an average size of 100 nm. This size was obtained from the number average of the measured diameters of 270 dried hybrid NPs in the TEM images (see size distribution in Fig. S3a†). To further characterize the size/size distribution and stability of the hybrid NPs dispersed in water, DLS was used to determine the hydrodynamic size of the NPs. Fig. S3b† shows the hydrodynamic diameter (Dh) distribution of the hybrid NPs in water, which indicates that the hybrid NPs have a narrow size distribution with polydispersity index μ2/<Γ>2 = 0.053 and average Dh = 124.5 nm. The DLS result also indicates that the hybrid NPs are very stable in water and do not form aggregates. The slightly larger diameter of the hybrid NPs dispersed in water compared with those in the dried state in the TEM images is understandable and is due to the hydrated surface layer from the hydroxyl/carboxyl groups of the hydrophilic surface (see discussion of Fig. 3a). When the aqueous dispersion of the hybrid NPs was exposed to UV light (365 nm), blue light was obviously emitted (inset in Fig. 2a). The TEM image of a single hybrid NP (Fig. 2b) demonstrates a core–shell structure with a carbon shell thickness of about 6 nm. Fig. 2c shows the high-resolution TEM image of the carbon shell. Some nanocrystals with a size of about 3 nm can be easily found in the carbon shell. The 2D lattice fringes of these small nanocrystals (inset of Fig. 2c) demonstrate an interplanar distance of about 0.206 nm, which corresponds to the (102) lattice planes of graphitic (sp2) carbon.55,56 The unsaturated carbon bonds were confirmed by the Raman spectrum of the hybrid NPs (Fig. S4†). The peak at 1603 cm−1 (G band) is associated with the E2g mode of the graphite and is related to the vibration of sp2-hybridized carbon atoms in a two-dimensional hexagonal lattice.57 The D band around 1350 cm−1 is ascribed to the vibrations of carbon atoms with dangling bonds in the termination plane of disordered graphite or glassy carbon.58 The low intensity ratio of the D-band to that of the G-band (ID/IG) suggests that the hybrid NPs contain less amorphous carbon and more crystalline graphitic structures. To further confirm the composition of the Fe3O4@C-CDs NPs and the nanocrystals embedded in the carbon shell, energy dispersive analysis of X-rays were conducted. The energy dispersive spectrum (EDS) of the single Fe3O4@C-CDs hybrid NP (Fig. 2d) shows the existence of Fe, O, C, and Cu elements. While the Cu is from the copper grid for TEM sample preparation, the Fe, O, and C should be attributed to the magnetic iron oxide nanocrystals in the core and carbon shell. In contrast, the EDS of the single small-sized nanocrystal embedded in the shell (Fig. 2e) reveals only the presence of C and Cu elements, which indicates that the nanocrystals in the shell only contain carbon. Combining all these analytical results, it is reasonable to conclude that the Fe3O4@C-CDs NPs are composed of iron oxide nanocrystals in the core and fluorescent CDs embedded in the carbon network shell.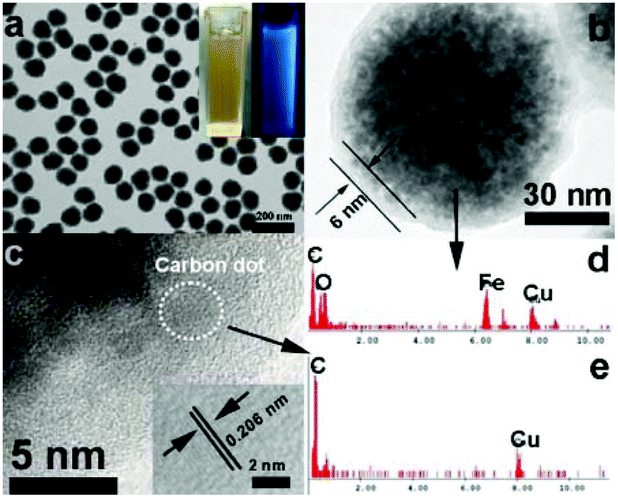 | ||
| Fig. 2 (a and b) TEM images of the Fe3O4@C-CDs hybrid NPs. The inset in (a) is the photographs of aqueous dispersions of the hybrid NPs under visible light and UV light (365 nm); (c) High resolution TEM image of the carbon shell. The inset is the lattice fringe of CDs; (d) and (e) Energy dispersive spectrum of the single Fe3O4@C-CDs NP and single CD, respectively. | ||
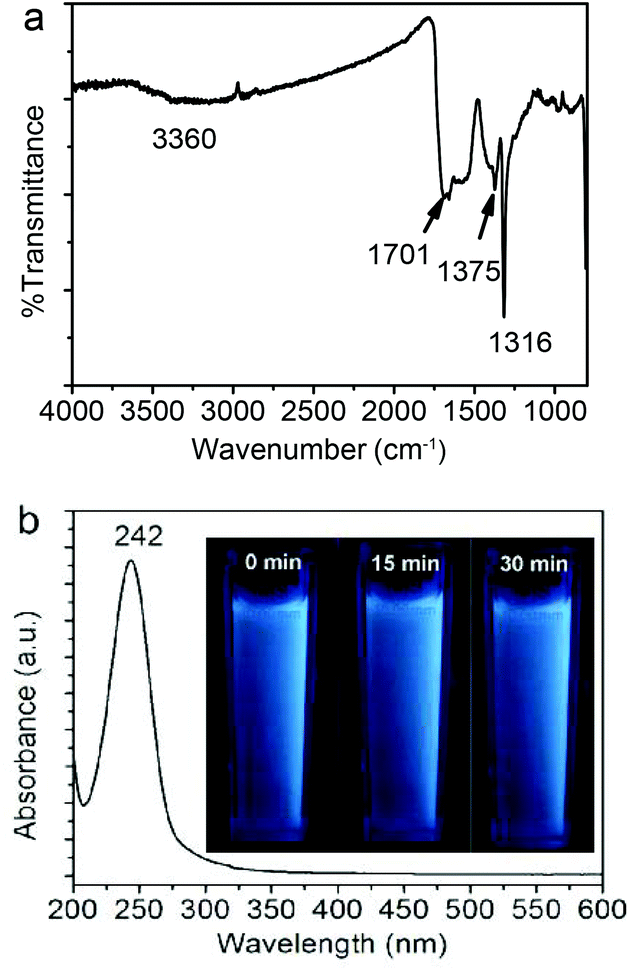 | ||
| Fig. 3 (a) Typical FT-IR spectrum and (b) UV-Vis spectrum of the as-obtained Fe3O4@C-CDs NPs. The inset in (b) shows photographs of the aqueous dispersions of the Fe3O4@C-CDs NPs under UV light (365 nm) with different exposure times from 0 min to 30 min. | ||
The formation of the Fe3O4@C-CDs hybrid NPs in the reaction medium must involve very complicated chemical processes. A potential mechanism should involve two stages. In stage I, in the sealed reactor containing ferrocene and H2O2 dissolved in acetone at 200 °C, the ferrocene quickly decomposes to form iron and cyclopentadiene. The C–H chemical bonds in the cyclopenta-1,3-diene are ruptured, leading to the formation of carbon-based free radicals. These small carbon free radicals can form relatively large carbon-based fragments or NPs with disordered (amorphous carbon) or ordered (CD) structures under the high temperature and high pressure from the gasification of the solvent acetone. Meanwhile, the O2 resulting from decomposition of the H2O2 can oxidize the Fe atoms to form Fe2+ and iron oxide. The presence of Fe2+ can in turn catalyze the decomposition of H2O2 to form highly reactive free radicals such as hydroxyl HO˙ and hydroperoxyl HOO˙ and form more gaseous O2 and H2O. These highly reactive free radicals can speed up the oxidation of Fe to form iron oxide nanocrystals and rapidly react with the carbon fragment free radicals to form the hydrophilic hydroxyl and carboxyl groups on the surface of carbon materials. All these reactions are very fast at stage I in the gaseous reaction medium at high temperature and pressure. In stage II, with the increase in concentration, the as-obtained iron oxide NPs in the supersaturated solution will aggregate into larger secondary clusters.59 Then the carbon-based fragments or NPs with conjugated double bonds and surface hydroxyl/carboxyl groups can be adsorbed on the surface of iron oxide NPs by chemical bonding.60 The carbon atoms deposited on the surface of iron oxide NPs are highly mobile and thus form the coating layer on the cluster of iron oxide NPs.61 With the continuous extension of the carbon-based NPs, carbon-encapsulated magnetic NPs are formed. Fig. S5† shows a schematic illustration of this potential mechanism for the formation of Fe3O4@C-CDs hybrid NPs.
Fig. 3a shows the FT-IR spectrum of the Fe3O4@C-CDs NPs. An apparent absorption peak of the –OH group at 3360 cm−1 and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode at 1701 cm−1 of the carboxylic acid groups conjugated with condensed aromatic carbons were respectively observed.62 The presence of the carboxyl groups on the surface of Fe3O4@C-CDs NPs can be further supported by the negative zeta potential of −40.13 mV of the hybrid NPs at neutral pH, at which the carboxylic acid groups dissociate to form negatively charged carboxylate groups. These data indicate that the Fe3O4@C-CDs NPs are surrounded by hydrophilic groups, thus can disperse well in aqueous media. The UV-Vis absorption spectrum of the Fe3O4@C-CDs NPs (Fig. 3b) shows a sharp absorption at about 242 nm, which likely originates from the formation of multiple polyaromatic chromophores.63,64 Interestingly, the increase in the feeding ratio of oxidizing agent H2O2 to the ferrocene precursor (γH2O2/ferrocene) in the solvothermal synthesis induced a red shift of the UV band (Fig. S6†), which may result from the increased degree of graphitization and π conjugation in the structure of Fe3O4@C-CDs NPs.29 Nevertheless, the hybrid NPs synthesized at a broad range of γH2O2/ferrocene can emit blue light under UV irradiation (Fig. S7†). In addition, the photographs (inset in Fig. 3b) of the aqueous dispersion of the Fe3O4@C-CDs NPs under UV light did not show an observable color change after continuous exposure to UV light of 365 nm for 30 min, which indicates their excellent photostability.
O stretching mode at 1701 cm−1 of the carboxylic acid groups conjugated with condensed aromatic carbons were respectively observed.62 The presence of the carboxyl groups on the surface of Fe3O4@C-CDs NPs can be further supported by the negative zeta potential of −40.13 mV of the hybrid NPs at neutral pH, at which the carboxylic acid groups dissociate to form negatively charged carboxylate groups. These data indicate that the Fe3O4@C-CDs NPs are surrounded by hydrophilic groups, thus can disperse well in aqueous media. The UV-Vis absorption spectrum of the Fe3O4@C-CDs NPs (Fig. 3b) shows a sharp absorption at about 242 nm, which likely originates from the formation of multiple polyaromatic chromophores.63,64 Interestingly, the increase in the feeding ratio of oxidizing agent H2O2 to the ferrocene precursor (γH2O2/ferrocene) in the solvothermal synthesis induced a red shift of the UV band (Fig. S6†), which may result from the increased degree of graphitization and π conjugation in the structure of Fe3O4@C-CDs NPs.29 Nevertheless, the hybrid NPs synthesized at a broad range of γH2O2/ferrocene can emit blue light under UV irradiation (Fig. S7†). In addition, the photographs (inset in Fig. 3b) of the aqueous dispersion of the Fe3O4@C-CDs NPs under UV light did not show an observable color change after continuous exposure to UV light of 365 nm for 30 min, which indicates their excellent photostability.
To further explore the optical properties of the Fe3O4@C-CDs NPs, a detailed PL study was carried out under different excitation wavelengths (λex). As shown in Fig. 4a, when the λex increases from 260 nm to 540 nm, the emission peak red-shifts to longer wavelength and the PL intensity gradually increases, which indicates a distribution of the different surface energy traps of the CDs.65 The maximum PL emission locates at 520 nm obtained with λex = 440 nm. Using rhodamine B as a standard, the PL quantum yield of the Fe3O4@C-CDs NPs was found to be 6.8%. Fig. 4b shows the PL spectra of the Fe3O4@C-CDs NPs excited by long-wavelength light from 660 to 460 nm, which clearly demonstrate the upconverted emissions from 470 to 370 nm. The upconversion PL property of the Fe3O4@C-CDs NPs can be attributed to the multiphoton active process similar to the previously reported CDs.38 Importantly, the Fe3O4@C-CDs NPs can emit light in the NIR range under the excitation of NIR light (Fig. S8†), which is highly desirable for biological applications. Furthermore, the Fe3O4@C-CDs hybrid NPs exhibit excellent photostability. The maximum PL intensity of the hybrid NPs at 520 nm (Fig. S9†) only changes slightly (∼2.1%) after 2 h of continuous exposure to the excitation of λex = 440 nm in a fluorospectrometer. These results indicate that the Fe3O4@C-CDs hybrid NPs not only demonstrate tunable excitation and upconversion PL properties, but also show excellent photostability against light illumination.
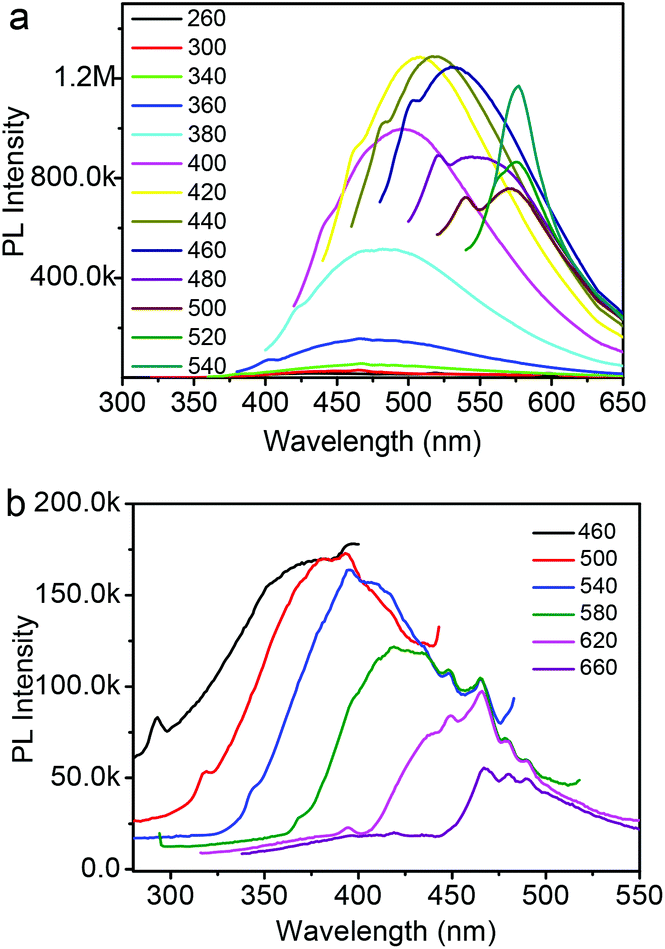 | ||
| Fig. 4 (a) PL spectra of Fe3O4@C-CDs NPs obtained with different excitation wavelengths; (b) Upconversion PL spectra of Fe3O4@C-CDs NPs obtained with different excitation wavelength. | ||
The magnetic hysteresis loop (Fig. 5a) of the Fe3O4@C-CDs NPs was measured at 300 K in an applied magnetic field of up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe, which indicates that the saturation magnetization (Ms) of the hybrid NPs is 32.5 emu g−1. The expanded low-field plot in the inset reveals the weakly ferromagnetic nature of the hybrid NPs at room temperature. The temperature dependent zero-field-cooled (ZFC) and field-cooled (FC) magnetization curves of the hybrid NPs (Fig. S10†) separate from each other from 5 K to 325 K and no transition from ferromagnetism to superparamagnetism can be observed, which further confirms the weak ferromagnetism of the hybrid NPs at room temperature.66 When the aqueous dispersion of the hybrid NPs is subjected to a magnetic field (0.30 T), the NPs can be attracted toward the magnet within minutes (inset of Fig. S10†). Slight agitation can bring the hybrid NPs back into the uniform dispersion after the magnet is removed. The quick magnetic response and excellent redispersibility of the Fe3O4@C-CDs NPs in water is undoubtedly very important for future applications in bioseparation, storage, and magnetic targeting drug delivery. In addition, the Fe3O4@C-CDs NPs could be used as a T2-weighted MRI contrast agent. Fig. 5b shows the inverse relaxation times 1/T*2 of the Fe3O4@C-CDs hybrid NPs at different concentrations. The specific relaxivity (r*2) of the NPs was determined to be 674.4 mM−1 s−1. Fig. 5c reveals that the MRI signal intensity declines with the increase in hybrid NP concentration (corresponding to an increase in iron concentration), due to the fact that the magnetic Fe3O4 nanocrystals shorten the T*2 by dephasing the spins of neighboring water protons.67 The incremental decrease in signal intensity is indicated by the enhanced darkness at increased Fe concentration from 0 to 0.128 mM. These results indicate that the Fe3O4@C-CDs NPs have high potential as T*2 contrast agents for MR imaging diagnosis.
000 Oe, which indicates that the saturation magnetization (Ms) of the hybrid NPs is 32.5 emu g−1. The expanded low-field plot in the inset reveals the weakly ferromagnetic nature of the hybrid NPs at room temperature. The temperature dependent zero-field-cooled (ZFC) and field-cooled (FC) magnetization curves of the hybrid NPs (Fig. S10†) separate from each other from 5 K to 325 K and no transition from ferromagnetism to superparamagnetism can be observed, which further confirms the weak ferromagnetism of the hybrid NPs at room temperature.66 When the aqueous dispersion of the hybrid NPs is subjected to a magnetic field (0.30 T), the NPs can be attracted toward the magnet within minutes (inset of Fig. S10†). Slight agitation can bring the hybrid NPs back into the uniform dispersion after the magnet is removed. The quick magnetic response and excellent redispersibility of the Fe3O4@C-CDs NPs in water is undoubtedly very important for future applications in bioseparation, storage, and magnetic targeting drug delivery. In addition, the Fe3O4@C-CDs NPs could be used as a T2-weighted MRI contrast agent. Fig. 5b shows the inverse relaxation times 1/T*2 of the Fe3O4@C-CDs hybrid NPs at different concentrations. The specific relaxivity (r*2) of the NPs was determined to be 674.4 mM−1 s−1. Fig. 5c reveals that the MRI signal intensity declines with the increase in hybrid NP concentration (corresponding to an increase in iron concentration), due to the fact that the magnetic Fe3O4 nanocrystals shorten the T*2 by dephasing the spins of neighboring water protons.67 The incremental decrease in signal intensity is indicated by the enhanced darkness at increased Fe concentration from 0 to 0.128 mM. These results indicate that the Fe3O4@C-CDs NPs have high potential as T*2 contrast agents for MR imaging diagnosis.
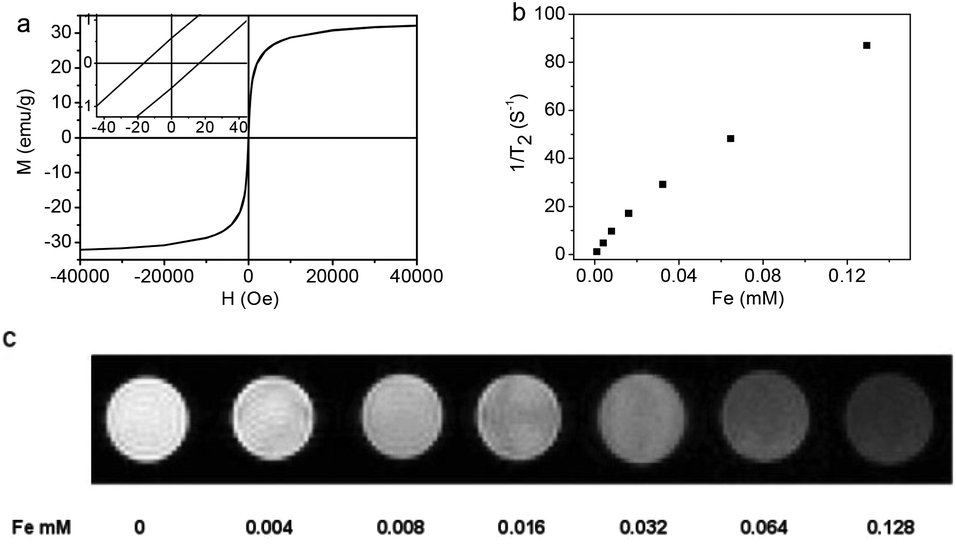 | ||
| Fig. 5 (a) The hysteresis loop of Fe3O4@C-CDs NPs measured at room temperature. The left inset shows the respective expanded plots for fields between −40 and 40 Oe; (b) T*2 relaxation rates as a function of iron concentration (mM) for Fe3O4@C-CDs NPs; (c) T*2-weighted MR images of Fe3O4@C-CDs NPs at different concentrations, corresponding to different amount of Fe. | ||
It is expected that the developed Fe3O4@C-CDs NPs containing both Fe3O4 and CD nanocrystals should have a dual-modal imaging ability for both MRI and fluorescent imaging. After confirming the strong fluorescence of the Fe3O4@C-CDs hybrid NPs in Fig. 4, mouse melanoma cells B16F10 were selected as a model to evaluate the optical cellular imaging function of these hybrid NPs as a fluorescent marker. Fig. 6 shows the laser scanning confocal images of the B16F10 cells incubated with the Fe3O4@C-CDs hybrid NPs under laser irradiation at wavelengths of 405, 488, and 546 nm, respectively. Obviously, the Fe3O4@C-CDs hybrid NPs containing fluorescent CDs produced a bright fluorescence and can illuminate the B16F10 cells in multicolor forms, which probably originates from the different sizes of the fluorescent CDs embedded in the carbon matrix. The Z-scanning confocal fluorescence images of the B16F10 cells incubated with the Fe3O4@C-CDs NPs (Fig. S11†) confirm that the hybrid NPs can overcome the cellular barriers to enter the intracellular region. The confocal images of B16F10 cells did not show a fluorescent signal change after continuous irradiation with the excitation laser at 488 nm for 30 min (Fig. 7), which further indicates that the Fe3O4@C-CDs NPs have excellent photostability and can be used for long-term cellular imaging. This stable multicolor fluorescent imaging ability of the CDs, together with the demonstrated MRI ability of the Fe3O4 nanocrystals, provides the Fe3O4@C-CDs hybrid NPs with a great potential for multimodal bioimaging function.
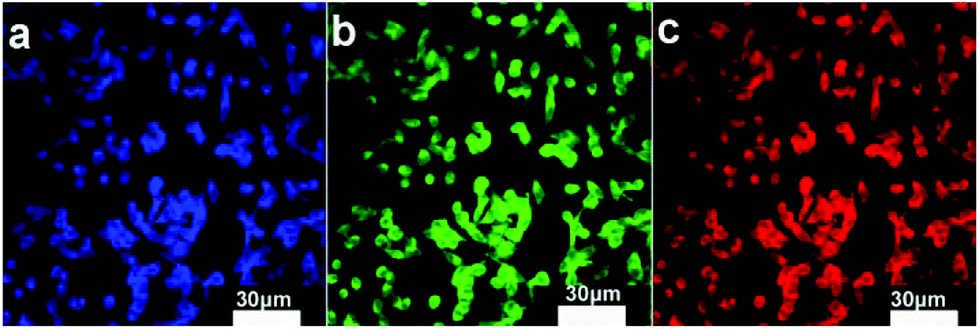 | ||
| Fig. 6 Laser scanning confocal microscopy images of B16F10 cells incubated with Fe3O4@C-CDs NPs under different excitation wavelengths: (a) 405 nm; (b) 488 nm; (c) 546 nm. | ||
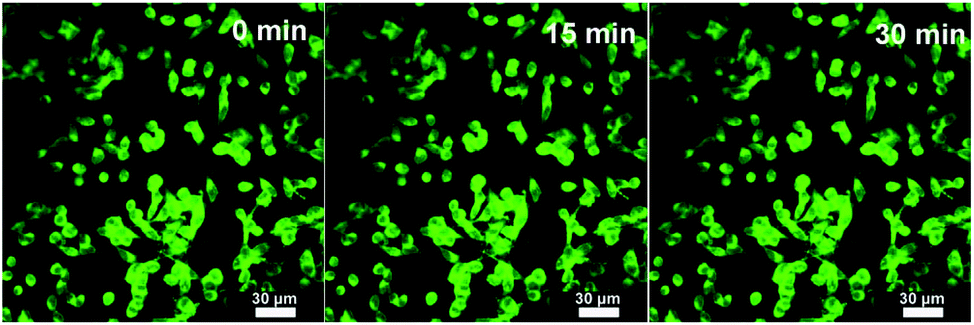 | ||
| Fig. 7 Laser scanning confocal microscopy images of B16F10 cells incubated with the Fe3O4@C-CDs NPs under different excitation times. Excitation wavelength = 488 nm. | ||
Fig. 8 shows the representative nitrogen adsorption/desorption isotherms and the corresponding pore size distribution of the Fe3O4@C-CDs NPs. The determined Brunauer–Emmett–Teller (BET) surface area and total pore volume of the hybrid NPs are 93.6 m2 g−1 and 0.29 cm3 g−1, respectively, which are relatively large as the magnetic nanocrystal core has been included in the calculations. The average Barrett–Joyner–Halenda (BJH) pore diameters of the hybrid NPs calculated from the desorption branch of the isotherm are 3.5 nm and 6 nm. The porous structure and large surface area of the Fe3O4@C-CDs NPs are highly desirable for drug carrier applications. Here, we have selected doxorubicin (DOX) as a model anti-cancer drug to test the drug loading capacity of the hybrid NPs. The drug loading capacity of the hybrid NPs reaches 835 mg g−1. This result shows that the DOX drug molecules can be readily loaded into the mesoporous carbon network of the Fe3O4@C-CDs hybrid NPs simply by mixing the NPs into the PBS solution of DOX. The high drug loading capacity of the hybrid NPs can be attributed to both the porous structure of the carbon matrix and the interactions between DOX molecules and the surface carboxyl/hydroxyl groups of the hybrid NPs.68
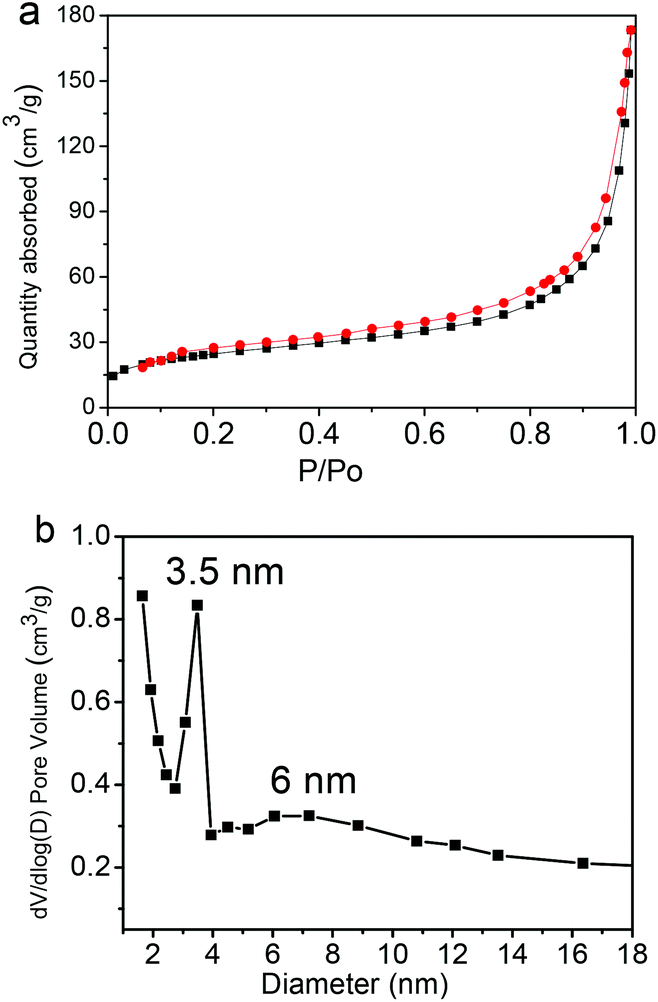 | ||
| Fig. 8 (a) N2 adsorption/desorption isotherms and (b) the pore size distribution of the Fe3O4@C-CDs hybrid NPs. | ||
Fig. 9a shows the photothermal effect of the Fe3O4@C-CDs hybrid NPs. When exposed to NIR light at a power density of 1.5 W cm−2, the Fe3O4@C-CDs NPs and their corresponding hollow C-CDs carbon shell particles (obtained by dissolving iron oxide nanocrystals using acid solution) showed a temperature increase of almost 25 °C within 5 min. In contrast, the temperature change of water (control) was much less significant under the same irradiation conditions. This result indicates that the CDs embedded in the carbon shell possess an excellent photothermal effect in response to NIR light irradiation. Fig. 9b compares the DOX release behavior from the Fe3O4@C-CDs hybrid NPs dispersed in PBS solution at 37 °C, with and without NIR light irradiation, respectively. Without NIR irradiation, the release of DOX molecules from the hybrid NPs is slow at an almost steady rate. The drug molecules are nearly completely released from the NPs after 80 h. In contrast, the short time (5 min) of irradiation with NIR light can speed up the release of DOX drug molecules from the hybrid NPs. When the radiation was turned off, heating ceased and the drug release returned to its regular slow rate. Such a significantly enhanced drug release rate under NIR light exposure is attributed to the local heat produced by the efficient photothermal conversion of the fluorescent CDs, which could weaken the drug–host interactions of DOX molecules with the surface carboxyl/hydroxyl groups of porous carbon and increase the mobility of DOX molecules at elevated temperatures. We expect that the combination of high drug loading capacity and NIR photothermally triggerable drug release behavior of the Fe3O4@C-CDs NPs will provide high therapeutic efficacy. While the porous Fe3O4@C-CDs hybrid NPs can act as a regular drug carrier for basal chemotherapy, they can also offer fast-acting dosage under NIR photoactivation when necessary.
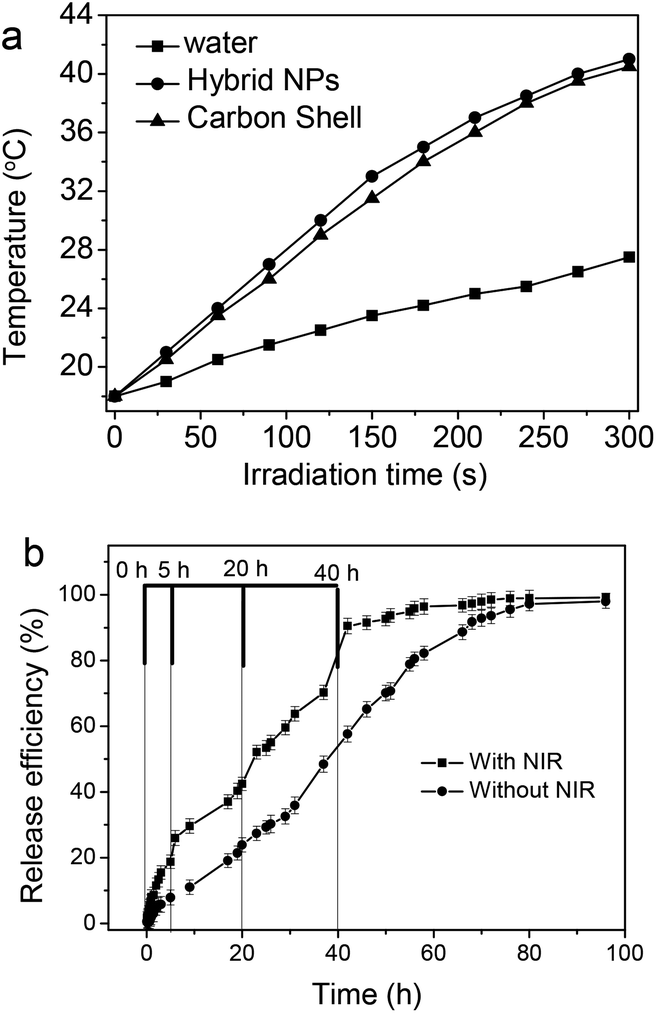 | ||
| Fig. 9 (a) The photothermal curves of water, Fe3O4@C-CDs hybrid NPs in water (100 mg L−1), and corresponding hollow C-CDs carbon shell particles in water under NIR irradiation for 5 min. (b) Release profiles of DOX from the Fe3O4@C-CDs NPs in PBS at 37 °C, irradiated with/without 1.5 W cm−2 NIR for 5 min at certain time points. | ||
Fig. 10 shows the in vitro cytotoxicity against B16F10 cells of the free DOX as a control and the corresponding drug-free and DOX-loaded Fe3O4@C-CDs hybrid NPs at different concentrations, without and with the 5 min treatment of 1.5 W cm−2 NIR light, respectively. The results indicate that the drug-free Fe3O4@C-CDs NPs are non-toxic to B16F10 cells after 24 h incubation at concentrations of up to 200 μg mL−1. The non-toxicity of the hybrid NPs is a crucial characteristic for future biological applications. In contrast, the cell viability dramatically decreased when the cells were incubated with the DOX-loaded hybrid NPs under the same conditions, indicating that the DOX-loaded hybrid NPs can demonstrate high anti-cancer activity. Compared to the control group of free DOX at the same concentrations as those loaded into hybrid NPs, the cytotoxicity of DOX-loaded hybrid NPs is lower than that of free DOX solution at all the studied concentrations. This can be attributed to the sustained-release property of the DOX-loaded hybrid NPs. Considering that only less than 60% of the loaded DOX was released in 24 h under physiological conditions (Fig. 9b), the lower cytotoxicity of the DOX-loaded hybrid NPs compared with the free DOX solutions is understandable. To investigate the photothermal effect, the B16F10 cells were irradiated with 1.5 W cm−2 NIR light for 5 min at a certain time point during the incubation process with the drug-free or drug-loaded hybrid NPs. The cytotoxicity results show that the Fe3O4@C-CDs hybrid NPs can significantly kill the tumor cells with the assistance of 5 min NIR irradiation, thus can serve as an effective photothermal therapeutic agent. In contrast, the 5 min NIR irradiation has a negligible effect on the viability of B16F10 cells in normal culture medium without hybrid NPs (Fig. S12†), which indicates that the photothermal effect can only be observed in the presence of Fe3O4@C-CDs hybrid NPs. The combination of the 5 min NIR irradiation with the released DOX treatment could kill the tumor cells even more effectively in comparison with the treatment by either DOX-loaded hybrid NPs alone (chemotherapy) or 5 min NIR irradiation alone (photothermal therapy using drug-free NPs) under the same conditions. However, the 5 min NIR irradiation treatment has little influence on the cytotoxicity of the free DOX solutions, which further supports the observation that the photothermal effect can only be observed in the presence of Fe3O4@C-CDs hybrid NPs. The IC50 values of free DOX with/without 5 min NIR treatment are 18.0 and 20.0 μg mL−1, respectively. While the drug-free hybrid NPs without NIR treatment are non-toxic to the cells, the IC50 of the hybrid NPs with 5 min NIR treatment is 150.6 μg mL−1. These results demonstrate that the Fe3O4@C-CDs hybrid NPs as drug carriers can facilitate combined photothermal/chemotherapy to provide high therapeutic efficacy.
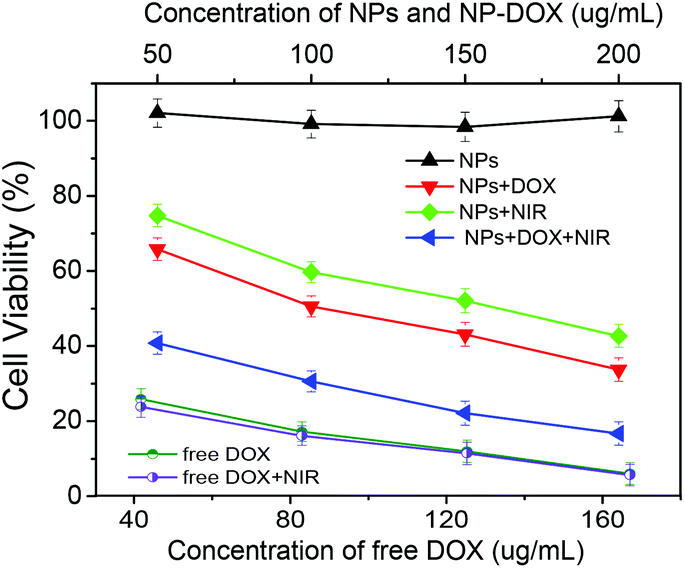 | ||
| Fig. 10 Comparison of B16F10 cell viability following treatment with drug-free Fe3O4@C-CDs hybrid NPs, DOX-loaded hybrid NPs, and free DOX at different concentrations with/without NIR treatment, respectively. | ||
4. Conclusions
In summary, we have prepared a new type of hybrid NPs integrated with magnetic Fe3O4 nanocrystals and fluorescent CDs in porous carbon using a one-pot solvothermal synthesis method. In addition to the magnetic responsive properties and MRI ability of the Fe3O4 nanocrystal core, the newly developed Fe3O4@C-CDs hybrid NPs also exhibit attractive optical properties from CDs with strong and upconversion fluorescence, excellent photostability, and an NIR photothermal effect, thus providing additional ability for fluorescent imaging and photothermal treatment. The mesoporous structures and hydrophilic surface functional groups endow the hybrid NPs with high drug loading capacity and excellent dispersibility in aqueous solutions. Combining the non-toxicity and low cost of the carbon materials, the as-prepared Fe3O4@C-CDs hybrid NPs demonstrate great promise towards advanced nanoplatforms for simultaneous imaging diagnostics and high efficacy therapy.Acknowledgements
We gratefully acknowledge the financial support from the American Diabetes Association (Basic Science Award 1-12-BS-243) and PSC-CUNY Research Award (66076-00 44). A portion of this research was conducted at the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy.Notes and references
- C. Xu and S. Sun, Adv. Drug Delivery Rev., 2013, 65, 732 CrossRef CAS PubMed.
- S. P. Foy, R. L. Manthe, S. T. Foy, S. Dimitrijevic, N. Krishnamurthy and V. Labhasetwar, ACS Nano, 2010, 4, 5217 CrossRef CAS PubMed.
- M. J. Sailor and J.-H. Park, Adv. Mater., 2012, 24, 3779 CrossRef CAS PubMed.
- A. Fu, R. J. Wilson, B. R. Smith, J. Mullenix, C. Earhart, D. Akin, S. Guccione, S. X. Wang and S. S. Gambhir, ACS Nano, 2012, 6, 6862 CrossRef CAS PubMed.
- J. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18 CrossRef CAS PubMed.
- H. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133 CrossRef CAS.
- M. Arruebo, R. Fernández-Pacheco, M. Ibarra and J. Santamaría, Nano Today, 2007, 2, 22 CrossRef.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed.
- M. M. Yallapu, S. F. Othman, E. T. Curtis, B. K. Gupta, M. Jaggi and S. C. Chauhan, Biomaterials, 2011, 32, 1890 CrossRef CAS PubMed.
- J. Gao, L. Li, P.-L. Ho, G. C. Mak, H. Gu and B. Xu, Adv. Mater., 2006, 18, 3145 CrossRef CAS.
- A. Quarta, R. D. Corato, L. Manna, S. Argentiere, R. Cingolani, G. Barbarella and T. Pellegrino, J. Am. Chem. Soc., 2008, 130, 10545 CrossRef CAS PubMed.
- P. Chang, K. C. Hwang and C.-S. Chiang, J. Am. Chem. Soc., 2008, 130, 15476 CrossRef PubMed.
- E.-K. Lim, J. Yang, C. P. N. Dinney, J.-S. Suh, Y.-M. Huh and S. Haam, Biomaterials, 2010, 31, 9310 CrossRef CAS PubMed.
- J. Liu, W. He, L. Zhang, Z. Zhang, J. Zhu, L. Yuan, H. Chen, Z. Cheng and X. Zhu, Langmuir, 2011, 27, 12684 CrossRef CAS PubMed.
- B. Sun, Y. Zhang, K.-J. Gu, Q.-D. Shen, Y. Yang and H. Song, Langmuir, 2009, 25, 5969 CrossRef CAS PubMed.
- P. Howes, M. Green, A. Bowers, D. Parker, G. Varma, M. Kallumadil, M. Hughes, A. Warley, A. Brain and R. Botnar, J. Am. Chem. Soc., 2010, 132, 9833 CrossRef CAS PubMed.
- Y. Xu, A. Karmakar, D. Wang, M. W. Mahmood, F. Watanabe, Y. Zhang, A. Fejleh, P. Fejleh, Z. Li, G. Kannarpady, S. Ali, A. R. Biris and A. S. Biris, J. Phys. Chem. C, 2010, 114, 5020 CAS.
- J. Gao, W. Zhang, P. Huang, B. Zhang, X. Zhang and B. Xu, J. Am. Chem. Soc., 2008, 130, 3710 CrossRef CAS PubMed.
- C. Y. Ang, L. Giam, Z. M. Chan, A. W. H. Lin, H. Gu, E. Devlin, G. C. Papaefthymiou, S. T. Selvan and J. Y. Ying, Adv. Mater., 2009, 21, 869 CrossRef CAS.
- R. Kas, E. Sevinc, U. Topal and H. Y. Acar, J. Phys. Chem. C, 2010, 114, 7758 CAS.
- Z. Wang, L. Wu, M. Chen and S. Zhou, J. Am. Chem. Soc., 2009, 131, 11276 CrossRef CAS PubMed.
- C. Xu, J. Xie, D. Ho, C. Wang, N. Kohler, E. G. Walsh, J. R. Morgan, Y. E. Chin and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 173 CrossRef CAS PubMed.
- H. Wang, J. Shen, G. Cao, Z. Gai, K. L. Hong, P. R. Debata, P. Banerjee and S. Zhou, J. Mater. Chem. B, 2013, 1, 6225 RSC.
- N. Hassan, V. Cabuil and A. Abou-Hassan, Angew. Chem., Int. Ed., 2013, 52, 1994 CrossRef CAS PubMed.
- C. Xu, B. Wang and S. Sun, J. Am. Chem. Soc., 2009, 131, 4216 CrossRef CAS PubMed.
- S. Peng, C. Lei, Y. Ren, R. E. Cook and Y. Sun, Angew. Chem., Int. Ed., 2011, 50, 3158 CrossRef CAS PubMed.
- L. Wang, H.-Y. Park, S. I. I. Lim, M. J. Schadt, D. Mott, J. Luo, X. Wang and C.-J. Zhong, J. Mater. Chem., 2008, 18, 2629 RSC.
- Z. Xu, Y. Hou and S. Sun, J. Am. Chem. Soc., 2007, 129, 8698 CrossRef CAS PubMed.
- H.-Y. Park, M. J. Schadt, L. Wang, I. I. S. Lim, P. N. Njoki, S. H. Kim, M.-Y. Jang, J. Luo and C.-J. Zhong, Langmuir, 2007, 23, 9050 CrossRef CAS PubMed.
- J. Jiang, H. Gu, H. Shao, E. Devlin, G. C. Papaefthymiou and J. Y. Ying, Adv. Mater., 2008, 20, 4403 CrossRef CAS.
- J. Chen, Z. Guo, H. Wang, M. Gong, X. Kong, P. Xia and Q. Chen, Biomaterials, 2013, 34, 571 CrossRef CAS PubMed.
- Z. Y. Ma, D. Dosev, M. Nichkova, S. J. Gee, B. D. Hammock and I. M. Kennedy, J. Mater. Chem., 2009, 19, 4695 RSC.
- S. Gai, P. Yang, C. Li, W. Wang, Y. Dai, N. Niu and J. Lin, Adv. Funct. Mater., 2010, 20, 1166 CrossRef CAS.
- H. Liu, T. Wang, L. Zhang, L. Li, Y. A. Wang, C. Wang and Z. Su, Chem.–Eur. J., 2012, 18, 3745 CrossRef CAS PubMed.
- L. Cheng, K. Yang, Y. Li, X. Zeng, M. Shao, S.-T. Lee and Z. Liu, Biomaterials, 2012, 33, 2215 CrossRef CAS PubMed.
- H. Zhu, J. Tao, W. Wang, Y. Zhou, P. Li, Z. Li, K. Yan, S. Wu, K. W. K. Yeung, Z. Xu, H. Xu and P. K. Chu, Biomaterials, 2013, 34, 2296 CrossRef CAS PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, B. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756 CrossRef CAS PubMed.
- X. Wang, L. Cao, S. Yang, F. Lu, M. Meziani, L. Tian, K. Sun, M. Bloodgood and Y. Sun, Angew. Chem., Int. Ed., 2010, 49, 5310 CrossRef CAS PubMed.
- R. Liu, D. Wu, S. Liu, K. Koynov, W. Knoll and Q. Li, Angew. Chem., Int. Ed., 2009, 48, 4598 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC.
- L. Cao, X. Wang, M. Meziani, F. Lu, H. Wang, P. Luo, Y. Lin, B. Harruff, L. Veca, D. Murray, S. Xie and Y. Sun, J. Am. Chem. Soc., 2007, 129, 11318 CrossRef CAS PubMed.
- S. Yang, L. Cao, P. Luo, F. Lu, X. Wang, H. Wang, M. Meziani, Y. Liu, G. Qi and Y. Sun, J. Am. Chem. Soc., 2009, 131, 11308 CrossRef CAS PubMed.
- M. A. Patel, H. Yang, P. L. Chiu, D. D. T. Mastrogiovanni, C. R. Flach, K. Savaram, L. Gomez, A. Hemnarine, R. Mendelsohn, E. Garfunkel, H. Jiang and H. He, ACS Nano, 2013, 7, 8147 CrossRef CAS PubMed.
- A. Zhu, Q. Qu, X. Shao, B. Kong and Y. Tian, Angew. Chem., Int. Ed., 2012, 51, 7185 CrossRef CAS PubMed.
- X. Huang, F. Zhang, L. Zhu, K. Y. Choi, N. Guo, J. Guo, K. Tackett, P. Anilkumar, G. Liu, M. Quan, H. S. Choi, G. Niu, Y. Sun, S. Lee and X. Chen, ACS Nano, 2013, 7, 5684 CrossRef CAS PubMed.
- J. Shen, Y. Zhu, X. Yang and C. Li, Chem. Commun., 2012, 48, 3686 RSC.
- Y. Fang, S. Guo, D. Li, C. Zhu, W. Ren, S. Dong and E. Wang, ACS Nano, 2012, 6, 400 CrossRef CAS PubMed.
- S. K. Bhunia, A. Saha, A. R. Maity, S. C. Ray and N. R. Jana, Sci. Rep., 2013, 3, 1473 CAS.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953 CrossRef CAS PubMed.
- P. G. Luo, S. Sahu, S.-T. Yang, S. K. Sonkar, J. Wang, H. Wang, G. E. LeCroy, L. Cao and Y.-P. Sun, J. Mater. Chem. B, 2013, 1, 2116 RSC.
- H. Wang, Q. W. Chen, Y. F. Yu, K. Cheng and Y. B. Sun, J. Phys. Chem. C, 2011, 115, 11427 CAS.
- H. Wang, J. Shen, Y. Y. Li, Z. Wei, G. Cao, Z. Gai, K. L. Hong, P. Banerjee and S. Q. Zhou, ACS Appl. Mater. Interfaces, 2013, 5, 9446 CAS.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803 CrossRef CAS.
- S. L. Hu, K. Y. Niu, J. Sun, J. Yang, N. Q. Zhao and X. W. Du, J. Mater. Chem., 2009, 19, 484 RSC.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2000, 61, 14095 CrossRef CAS.
- J. Ristein, R. T. Stief, L. Ley and W. Beyer, J. Appl. Phys., 1998, 84, 3836 CrossRef CAS PubMed.
- J. P. Ge, Y. X. Hu, M. Biasini, W. P. Beyermann and Y. D. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342 CrossRef CAS PubMed.
- H. Wang, Q. W. Chen, L. X. Sun, H. P. Qi, X. Yang, S. Zhou and J. Xiong, Langmuir, 2009, 25, 7135 CrossRef CAS PubMed.
- H. Hou, A. K. Schaper, F. Weller and A. Greiner, Chem. Mater., 2002, 14, 3990 CrossRef CAS.
- P. Duffy, L. Magno, R. Yadav, S. K. Roberts, A. D. Ward, S. W. Botchway, P. E. Colavita and S. J. Quinn, J. Mater. Chem., 2012, 22, 432 RSC.
- S. J. Yu, M. W. Kang, H. C. Chang, K. M. Chen and Y. C. Yu, J. Am. Chem. Soc., 2005, 127, 17604 CrossRef CAS PubMed.
- Y. R. Chang, H. Y. Lee, K. Chen, C. C. Chang, D. S. Tsai, C. C. Fu, T. S. Lim, Y. K. Tzeng, C. Y. Fang, C. C. Han, H. Chang and W. Fan, Nat. Nanotechnol., 2008, 3, 284 CrossRef CAS PubMed.
- F. Wang, S. P. Pang, L. Wang, Q. Li, M. Kreiter and C. Y. Liu, Chem. Mater., 2010, 22, 4528 CrossRef CAS.
- F. Zhang and C. Wang, J. Phys. Chem. C, 2008, 112, 15151 CAS.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427 CrossRef CAS PubMed.
- D. L. Hughes and R. A. Reamer, J. Org. Chem., 1996, 61, 2967 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S12. See DOI: 10.1039/c3bm60297d |
| This journal is © The Royal Society of Chemistry 2014 |