
Expeditious synthesis of CF3-phenanthridones through a base-mediated cross-conjugated vinylogous benzannulation (VBA)†
Madhu
Desagoni
ab,
Chavakula
Nagababu
ab and
Nagender
Punna
 *ab
*ab
aFluoro-Agro chemicals Division, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad 500007, India. E-mail: nagenderpunna@iict.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 8th November 2024
Abstract
Herein, we report a mild, efficient, and rapid approach for the preparation of CF3-phenanthridones through a cross-conjugated vinylogous [4 + 2] benzannulation of easily accessible 4-methyl-3-trifluoroacetylquinolones and nitro-olefins. The present transformation is superior to previous approaches for obtaining CF3-phenanthridones, in that it proceeds exclusively with the assistance of a simple base, eliminating the need for transition metal catalysts or oxidants. The strong electron-withdrawing nature of the CF3-group present in the quinolone moiety promotes the formation of a reactive cross-conjugated vinylogous enolate.
Phenanthridone derivatives are privileged tricyclic N-heterocyclic compounds frequently encountered in several biologically active molecules and natural products.1 As shown in Fig. 1, prastosine, N-methylcrinasiadine, and phenaglydon are naturally occurring alkaloids with anti-cancer and anti-inflammatory activities.1 PJ34 and PJ38 are lead molecules having the phenanthridone scaffold and they exhibit potent PARP inhibition activity.2 In addition, ARC-111 exhibits potent cytotoxic activity against multiple human cancer cell lines (Fig. 1).3 Over the past decade, significant advancements have been made in constructing phenanthridones.4–9 Suzuki couplings followed by condensations,5 intramolecular C–H-arylations,6 carbonylations/carboxylations of o-aryle anilines,7 and direct C–H aminations8 are some of the well-established metal-catalyzed protocols (Fig. 2a). Recently, the Lautens group demonstrated palladium/norbornene cooperative catalysis to furnish amino-substituted phenanthridones (Fig. 2a).9 Despite considerable progress, these reactions still depend on expensive transition metal catalysts and oxidants.
 | ||
| Fig. 1 Biologically active phenanthridones. | ||
 | ||
| Fig. 2 Approaches for the synthesis of phenanthridones: (a) TM catalysed approaches; (b) TM-free approaches; and (c) present work. | ||
Therefore, there is a persistent need for metal-free strategies for the synthesis of biologically active molecules such as phenanthridones to avoid potential contamination from toxic metals; yet these approaches are currently scarce. However, the first metal-free approach was disclosed through a radical C–C coupling of 2-halobenzamides using 1,10-phenanthroline/AIBN.10 Later in 2015, the Zang group disclosed an efficient radical decarboxylative cyclization of biaryl-2-oxamic acids using Na2S2O8 as an oxidant.11 However, many of these reactions require high temperatures, potent oxidants, and extended reaction times, substantially restricting their scope and functional group tolerance. Hence, the pursuit of mild, efficient, and metal-free synthetic strategies for obtaining high-value phenanthridone architectures has remained an area of universal interest. In this context, the vinylogous benzannulation approach (VBA) is the most efficient protocol for constructing benzo-fused architectures.12 Therefore, diverse vinylogous donors such as α,α-dicyanoolefins,13 alkylidene azlactones14 and β-methyl enone esters15 have been developed for the rapid construction of benzannulated products. The captivating features of the VBA approach have sparked continuous interest due to its ability to generate innovative variations toward high molecular diversity. For example, a base-mediated rapid benzannulation of α-cyanocrotonates with ynones was disclosed to access the corresponding aryl nitriles.16 Thus, the development of novel substrates for VBA reactions continuously expands the range of functionalized scaffolds. The present work, as shown in Fig. 2c, reports a novel VBA reaction of a trifluoromethyl substituted cross-conjugated vinylogous enolate generated in situ from the 4-methyl-3-(2,2,2-trifluoroacetyl)quinolin-2(1H)-one (1) and nitro-olefins to access CF3-phenanthridones. We predicted that the engineered 4-methyl-3-(2,2,2-trifluoroacetyl)quinolin-2(1H)-one would act as a novel vinylogous donor and participate in a VBA reaction through a cascade of Michael addition followed by intramolecular Henry reactions. Here, the –CF3 group potentially facilitates the activation of the γ-C–H site in compound 1 to form the cross-conjugated vinylogous enolate in situ in the presence of a suitable base. Hereof, aligning with our efforts to develop advanced and efficient strategies for the construction of fluorinated architectures,17 a rapid cross-conjugated vinylogous anionic annulation was developed under mild reaction conditions to synthesize high-value trifluoromethyl substituted phenanthridones.18
To study the vinylogous benzannulation cascade reaction, 1,4-dimethyl-3-(2,2,2-trifluoroacetyl) quinoline-2(1H)-one (1a) was selected as the model substrate to react with trans-β-nitrostyrene (2a) in acetonitrile solvent at room temperature using 2 equivalents of KOtBu (Table 1, entry 1). As expected, the desired VBA product CF3-phenanthridone 3a was formed in 32% yield. After that, a variety of solvents such as toluene, THF, DCM and 1,4-dioxane were tested in this VBA reaction; encouragingly, the reaction was successful with THF as the solvent, affording 85% product yield (Table 1, entries 2–5) in just 15 minutes. Next, the effect of bases in the VBA reaction was studied. The obtained results revealed that inorganic bases such as K2CO3, Cs2CO3, and NaOMe and organic bases such as DBU and DMAP were not efficient in improving the yield of compound 3a (Table 1, entries 6–10). Here, the high basicity of KOtBu and its greater solubility in THF solvent enable effective deprotonation, especially of weakly acidic protons, making it particularly suitable in reactions such as those used for the generation of vinylogous anions and enolates. Finally, the VBA reaction in the absence of a base failed to produce the desired CF3-phenanthridone (3a) (Table 1, entry 11).
|
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yieldb (%) |
| a Experiments were carried out using 1a (0.1 mmol) and 2a (0.15 mmol) in 2.0 mL of solvent at rt for 24 h. b Isolated yields. c Reaction completed in 15 minutes. | |||
| 1 | KOtBu | CH3CN | 32 |
| 2 | KOtBu | Toluene | 17 |
| 3c | KOtBu | THF | 85 |
| 4 | KOtBu | DCM | — |
| 5 | KOtBu | 1,4-Dioxane | 27 |
| 6 | K2CO3 | THF | — |
| 7 | Cs2CO3 | THF | 15 |
| 8 | NaOMe | THF | — |
| 9 | DBU | THF | — |
| 10 | DMAP | THF | — |
| 11 | — | THF | — |

With the optimized reaction conditions in hand, the generality of the VBA reaction was studied using nitro-styrenes (2), and the results revealed minimal substituent effects, delivering the desired CF3-phenanthridones (3) in good yields (Table 2). β-Nitro-styrenes having electron-donating substituents at the para-position of their phenyl rings such as methyl (2b) and methoxy (2c) were well tolerated in this VBA reaction and delivered the corresponding products 3b and 3c in 84% and 75% yields, respectively. Halogen substituents on β-nitrostyrenes such as 4-Br (2d) and 4-F (2e) also participated well in the VBA reaction to give CF3-phenanthridones 3d and 3e in good yields. The current protocol was successful with electron-withdrawing group containing substrates such as 4-CN (2f) and 4-CF3 (2g), affording the desired VBA products in excellent yields. Notably, the use of heteroaryl substituted β-nitrostyrenes such as furyl (2h) and thiophenyl (2i) was also effective in furnishing products 3h and 3i in 76% and 78% yields, respectively. Moreover, the bicyclic aromatic β-naphthyl nitroolefin (2j), featuring an extended π-system, was well tolerated under the standard reaction conditions, furnishing the requisite analogue 3j in a yield of 79%. Remarkably, the reaction proved viable with β-styryl nitroolefin (2k), sourced from cinnamaldehyde, and 3,4-di-OMe β-nitrostyrene (2l), derived from vanillin, yielding 3k and 3l in good yields, showcasing the universality of the methodology.
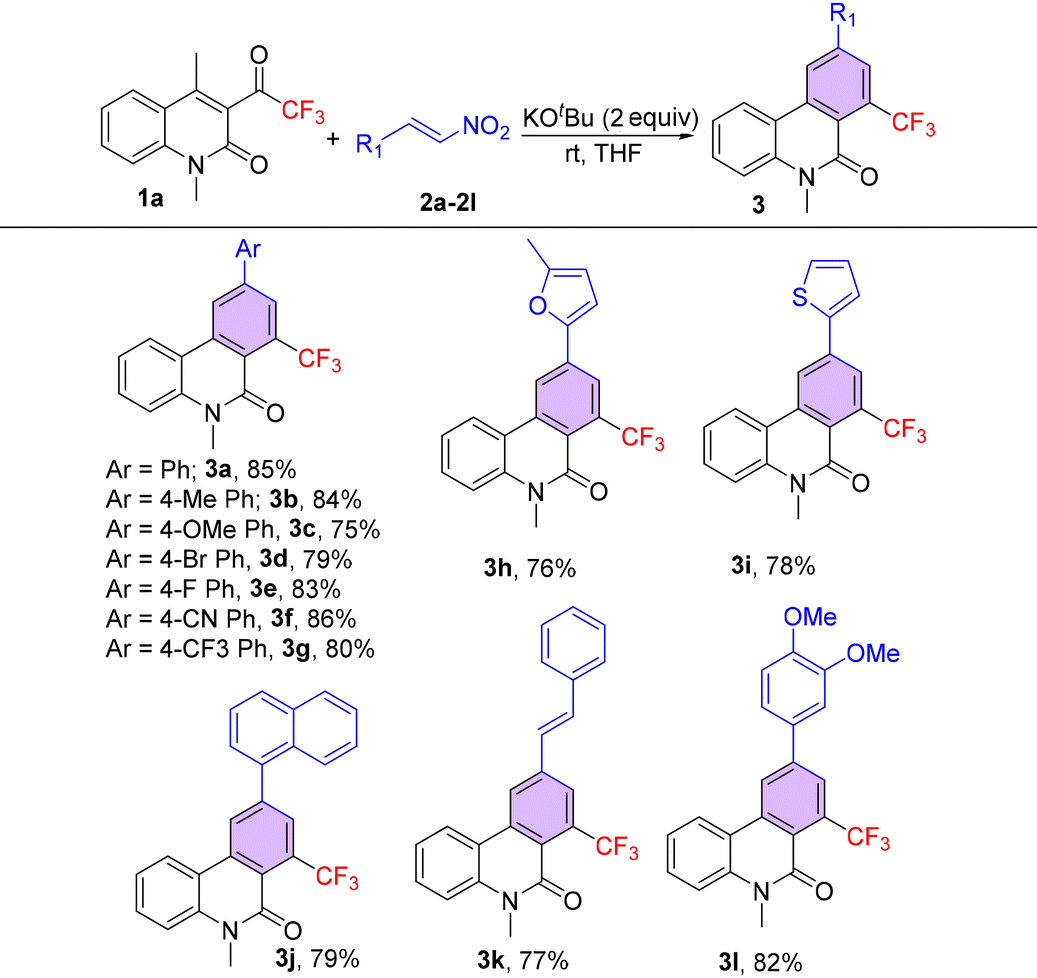
Utilizing conventional reaction conditions (Table 1, entry 3), the vinylogous benzannulation cascade was expanded to encompass a variety of substituted 2-quinolinones (1) (Table 3). Smooth reactions were observed with electron-donating groups on the quinolone substrates, exemplified by 1b (6-Me) and 1c (6-OMe), yielding products 3m and 3n in excellent yields. Introduction of halogen substituents on the phenyl ring of quinolinone substrates, such as 1d (6-Cl) and 1e (6-Br), resulted in good yields of phenanthridinones 3o and 3p. In addition, substrate 1f (6-ph) bearing an aryl group reacted favourably, providing product 3q in 80% yield. Notably, the electron-rich heterocycle bearing substrate 1g (6-thiophenyl) also delivered the desired product 3r in 71% yield. Furthermore, testing the reaction scope with substrates 1h (N-ethyl) and 1i (N-benzyl) was successful, furnishing the corresponding phenanthridinones 3s and 3t in 73% and 79% yields, respectively. However, the substrates with extended substitution at the 4-position of quinolone, 1j (4-Et) and 1k (4-benzyl), failed to produce the desired products.

Next, a gram-scale reaction was attempted to determine the efficiency of the VBA reaction under standard reaction conditions (Table 1, entry 3). To our delight, the reaction proceeded smoothly to afford the corresponding CF3-phenanthridone 3a in 82% yield (Scheme 1a). Next, to understand the influence of the CF3 group on the transformation, several control experiments were performed under optimized reaction conditions (Scheme 1b & c). Quinolone substrates having diverse substitutions such as methyl (1l) and phenyl (1m) instead of the CF3 group failed to furnish the desired products (Scheme 1b & c). In order to gain insight into the reaction mechanism, the standard reaction was attempted at a lower temperature with different β-nitrostyrenes such as 2a & 2b using 1 equivalent of the base. Notably, we observed the formation of densely substituted trifluoromethylated oxa-quaternary centered tetrahydro-phenanthridinones 4a and 4b with poor yield and diastereoselectivity (Scheme 1d). Here, the possibility of more number of diastereomers might be controlled by the high steric hindrance of the CF3 group, which further implies that the intramolecular Henry reaction proceeded in a highly selective manner. Next, the compatibility of aliphatic nitro-olefins was tested with (E)-1-nitropentene using 2 equivalents of the base, but the reaction yielded the non-aromatized tetrahydro-phenanthridone 4c in 51% yield (Scheme 1e). To obtain the desired aromatized compound, we attempted several experiments for the synthesis of alkyl-substituted CF3-phenanthridones from compound 4c. The reaction, under different sets of reaction conditions, failed to give the required aromatized product (ESI, Table S1†). Here, the less acidic nature of the carbon attached to the alkyl group in comparison to the carbon attached to the phenyl group may account for this outcome.
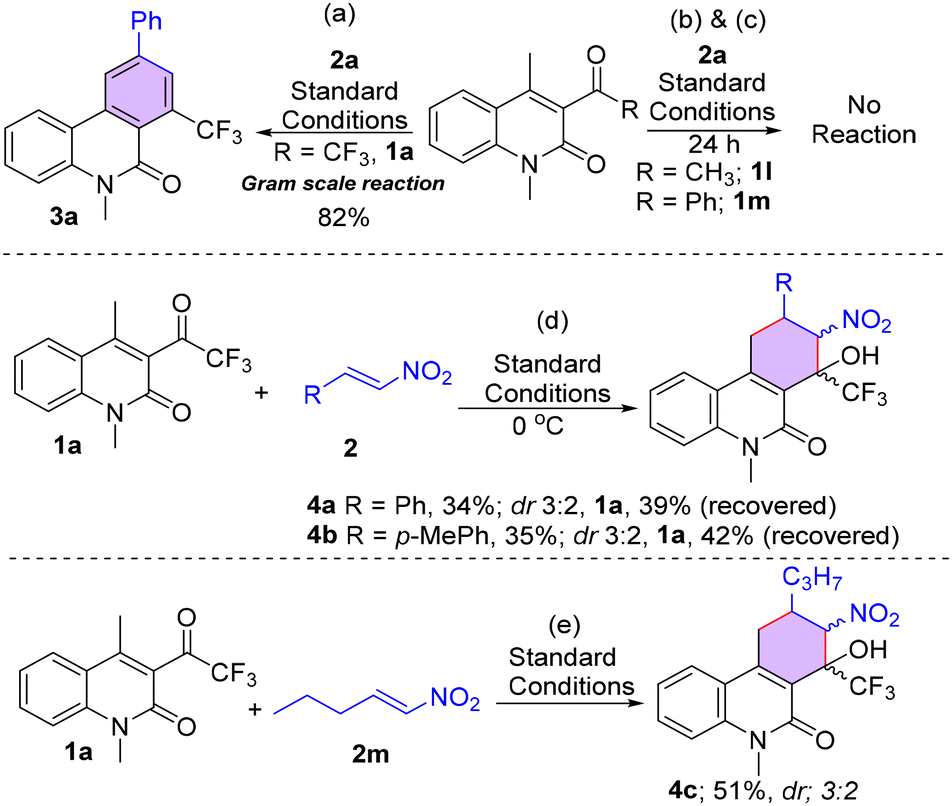 | ||
| Scheme 1 (a) Gram scale synthesis. (b&c) Control experiments. (d) Reactions at 0 °C. (e) Reaction with an aliphatic nitro olefin. | ||
Based on previous reports and control experiments, the plausible reaction mechanism is shown in Fig. 3. The synthesis of CF3-phenanthridones via a base-mediated cross-conjugated vinylogous benzannulation (VBA) starts with the deprotonation of 4-methyl-3-trifluoroacetylquinolone (1a) by a suitable base, such as potassium tert-butoxide (KOtBu), forming a cross-conjugated vinylogous enolate (A). This enolate undergoes a Michael addition with a nitro-olefin, facilitating a new carbon–carbon bond (intermediate B), followed by an intramolecular Henry (nitroaldol) reaction, leading to a tetrahydrophenanthridinone scaffold (C). The final steps involve the elimination of nitrous acid and aromatization, yielding the CF3-phenanthridone product 3a.
 | ||
| Fig. 3 Plausible reaction mechanism. | ||
In summary, we have disclosed a mild, rapid, and metal-free approach for the synthesis of CF3-phenanthridones using customized 1,4-dimethyl-3-(trifluoroacetyl) quinolin-2(1H)-one as a novel vinylogous pronucleophile and easily accessible nitro-alkenes. This protocol proceeds via a cross-conjugated vinylogous enolate, involving a cascade of Michael addition and intramolecular Henry reactions. This vinylogous benzannulation reaction has a wide substrate scope under optimized mild reaction conditions. A simple base effectively promotes the VBA reaction to construct structurally diverse and pharmaceutically valuable CF3-phenanthridinone scaffolds in a very short time.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Council of Scientific and Industrial Research (CSIR), Ministry of Science and Technology, Government of India. CSIR-IICT manuscript communication no: IICT/Pubs./2024/274.References
- (a) Z. Jin, Nat. Prod. Rep., 2009, 26, 363–381 RSC; (b) M. He, C. Qu, O. Gao, X. Hu and X. Hong, RSC Adv., 2015, 5, 16562–16574 RSC; (c) D. Weltin, V. Picard, K. Aupeix, M. Varin, D. Oth, J. Marchal, P. Dufour and P. Bischoff, Int. J. Immunopharmacol., 1995, 17, 265–271 CrossRef CAS PubMed; (d) A. L. Ruchelman, P. J. Houghton, N. Zhou, A. Liu, L. F. Liu and E. J. LaVoie, J. Med. Chem., 2005, 48, 792–804 CrossRef CAS.
- (a) S. Diani-Moore, J. Shoots, R. Singh, J. B. Zuk and A. B. Rifkind, Sci. Rep., 2017, 7, 2268 CrossRef PubMed; (b) M. Haddad, H. Rhinn, C. Bloquel, B. Coqueran, C. Szabó, M. Plotkine, D. Scherman and I. Margaill, Br. J. Pharmacol., 2006, 149, 23–30 CrossRef CAS; (c) S. H. Huang, Oncol. Rep., 2008, 20, 567–572 CAS; (d) T. Xiong, H. Wei, X. Chen and H. Xiao, Int. J. Oncol., 2015, 46, 223–232 CrossRef CAS; (e) C. Szabo, P. Jagtap, G. Southan and A. Salzman, US 6531464 B1, 2003.
- (a) T.-K. Li, P. J. Houghton, S. D. Desai, P. Daroui, A. A. Liu, E. S. Hars, A. L. Ruchelman, E. J. LaVoie and L. F. Liu, Cancer Res., 2003, 63, 8400–8407 CAS; (b) A. L. Ruchelman, P. J. Houghton, N. Zhou, A. Liu, L. F. Liu and E. J. LaVoie, J. Med. Chem., 2005, 48, 792–804 CrossRef CAS PubMed.
- For selected reviews, see: (a) L.-M. Tumir, M. R. Stojković and I. Piantanida, Beilstein J. Org. Chem., 2014, 10, 2930–2954 CrossRef PubMed; (b) A. Del Tito, H. O. Abdulla, D. Ravelli, S. Protti and M. Fagnoni, Beilstein J. Org. Chem., 2020, 16, 1476–1488 CrossRef CAS PubMed; (c) R. R. Aleti, A. A. Festa, L. G. Voskressensky and E. V. Van Der Eycken, Molecules, 2021, 26, 5560 CrossRef CAS; (d) V. Talukdar, A. Vijayan, N. K. Katari, K. V. Radhakrishnan and P. Das, Adv. Synth. Catal., 2021, 363, 1202–1245 CrossRef CAS.
- (a) Y. Kuwata, M. Sonoda and S. Tanimori, J. Heterocycl. Chem., 2017, 54, 1645–1651 CrossRef CAS; (b) K. Tanimoto, N. Nakagawa, K. Takeda, M. Kirihata and S. Tanimori, Tetrahedron Lett., 2013, 54, 3712–3714 CrossRef CAS.
- C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331–336 RSC.
- (a) S.-M. Lu and H. Alper, J. Am. Chem. Soc., 2005, 127, 14776–14784 CrossRef CAS PubMed; (b) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468–1471 CrossRef CAS PubMed; (c) D. Liang, Z. Hu, J. Peng, J. Huang and Q. Zhu, Chem. Commun., 2013, 49, 173–175 RSC; (d) M. Feng, B. Tang, N. Wang, H. Xu and X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 14960–14964 CrossRef CAS.
- (a) G. Wang, T. Yuan and D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380–1383 CrossRef CAS; (b) J. Karthikeyan and C. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880–9883 CrossRef CAS; (c) J. Karthikeyan, R. Haridharan and C. Cheng, Angew. Chem., 2012, 124, 12509–12513 CrossRef; (d) S. Pimparkar and M. Jeganmohan, Chem. Commun., 2014, 50, 12116–12119 RSC; (e) C. Lu, A. V. Dubrovskiy and R. C. Larock, J. Org. Chem., 2012, 77, 8648–8656 CrossRef CAS; (f) N. Senthilkumar, K. Parthasarathy, P. Gandeepan and C. Cheng, Chem. – Asian J., 2013, 8, 2175–2181 CrossRef CAS PubMed; (g) S. L. Yedage and B. M. Bhanage, J. Org. Chem., 2016, 81, 4103–4111 CrossRef CAS PubMed.
- X. Abel-Snape, A. Whyte and M. Lautens, Org. Lett., 2020, 22, 7920–7925 CrossRef CAS.
- B. S. Bhakuni, A. Kumar, S. J. Balkrishna, J. A. Sheikh, S. Konar and S. Kumar, Org. Lett., 2012, 14, 2838–2841 CrossRef CAS PubMed.
- M. Yuan, L. Chen, J. Wang, S. Chen, K. Wang, Y. Xue, G. Yao, Z. Luo and Y. Zhang, Org. Lett., 2015, 17, 346–349 CrossRef CAS PubMed.
- (a) B. Farajpour and A. Alizadeh, Org. Biomol. Chem., 2022, 20, 8366–8394 RSC; (b) C. Curti, L. Battistini, A. Sartori and F. Zanardi, Chem. Rev., 2020, 120, 2448–2612 CrossRef CAS; (c) Q. Zhao, C. Peng, G. Zhan and B. Han, RSC Adv., 2020, 10, 40983–41003 RSC; (d) E. Marcantonio, C. Curti, L. Battistini, A. Sartori, L. Cardinale, G. Pelosi and F. Zanardi, Adv. Synth. Catal., 2021, 363, 2625–2633 CrossRef CAS; (e) A. Ghosh, S. Shee and A. T. Biju, Org. Lett., 2022, 24, 2772–2777 CrossRef CAS.
- (a) V. Bhajammanavar, S. Mallik and M. Baidya, Adv. Synth. Catal., 2019, 361, 5472–5477 CrossRef CAS; (b) J.-R. Zhuo, B.-X. Quan, J.-Q. Zhao, M.-L. Zhang, Y.-Z. Chen, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2020, 76, 131115 CrossRef CAS; (c) C.-X. Hu, L. Chen, D. Hu, X. Song, Z.-C. Chen, W. Du and Y.-C. Chen, Org. Lett., 2020, 22, 8973–8977 CrossRef CAS.
- D. Cao, G. Chen, D. Chen, Z. Xia, Z. Li, Y. Wang, D. Xu and J. Yang, ACS Omega, 2021, 6, 16969–16979 CrossRef CAS PubMed.
- J. Huang and K. Shia, Angew. Chem., Int. Ed., 2020, 59, 6540–6545 CrossRef CAS.
- M. K. R. Singam, A. Nagireddy and S. R. Maddi, Green Chem., 2020, 22, 2370–2374 RSC.
- Some recent examples: (a) B. K. Jha, J. Prudhviraj, P. S. Mainkar, N. Punna and S. Chandrasekhar, RSC Adv., 2020, 10, 38588–38591 RSC; (b) C. Priyanka, M. Subbarao and N. Punna, Org. Biomol. Chem., 2021, 19, 8241–8245 RSC; (c) D. Madhu, V. R. Jetti, B. Narsaiah and N. Punna, J. Org. Chem., 2022, 87, 2301–2231 CrossRef CAS PubMed; (d) C. Nagababu, J. Prudhviraj and N. Punna, Adv. Synth. Catal., 2023, 365, 8–12 CrossRef CAS; (e) C. Nagababu, S. Sainadh, M. Desagoni, J. B. Nanubolu and N. Punna, Adv. Synth. Catal., 2024, 366, 3438–3442 CrossRef CAS.
- C. Priyanka, D. Madhu, P. S. Gangadhar, L. Giribabu and N. Punna, Chem. Commun., 2024, 60, 12233–12236 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01480d |
| This journal is © The Royal Society of Chemistry 2025 |