
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-meso-Fused pyrene–porphyrin scaffolds with panchromatic absorption features†
Christoph
Oleszak
a,
Christian L.
Ritterhoff
b,
Bernd
Meyer
 *b and
Norbert
Jux
*a
*b and
Norbert
Jux
*a
aDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials (ICMM), Chair of Organic Chemistry II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nikolaus-Fiebiger-Str. 10, 91058 Erlangen, Germany. E-mail: norbert.jux@fau.de
bInterdisciplinary Center for Molecular Materials (ICMM) & Computer Chemistry Center (CCC), Friedrich-Alexander-Universität Erlangen-Nürnberg, Nägelsbachstr. 25, 91052 Erlangen, Germany. E-mail: bernd.meyer@fau.de
First published on 15th October 2024
Abstract
The π-extension of porphyrins with pyrenes through the β-meso-fusion of five-membered rings is demonstrated. Three architectures resulting from combining up to two porphyrins and pyrenes were obtained straightforwardly in good overall yields. Although significantly planarized, the molecules retain excellent solubility and processability. Spectroscopic characterization and density-functional theory calculations reveal intriguing absorption features.
Introduction
In a fast-moving scientific world, porphyrin managed to keep its relevancy over the course of many decades. Of course, the main reason for this everlasting pertinence of the 18π-electron macrocycle is its standout photophysical and electronic properties, high chemical stability, and structural modifiability.1,2 The combination of these perks made it an invaluable component in the development of organic photovoltaics,3–6 optical transistors,7,8 light-emitting diodes,9,10 and other challenges of new-age synthetic organic chemistry.However, a key factor limiting the application of porphyrins in the mentioned fields is the relatively narrow width of their absorption curves and missing absorption in the near-infrared area of the solar spectrum.1 One of the most popular established approaches to circumvent these shortfalls is the peripheral extension of the porphyrins aromatic system. π-Extended porphyrin-based molecules usually show significant bathochromically shifted absorptions9–13 and other exciting properties like increases in two-photon absorption cross-section14–16 and changes in their aromaticity.17–19 In the literature, a plethora of methods and strategies employed in this context can be found.20 Notable examples from recent years include the triple β-meso-β-fusion of porphyrins to anthracenes, as demonstrated by the group of Anderson21–25 and the probing of varying fusing motifs by Osuka et al.26–31 The size and shape of the aromatic systems connected to the porphyrin periphery cover a wide range, reaching from simple phenyl rings to large nanoribbon-like architectures.9,13,32–34 In this work, we focus on pyrene, one of the most well-known and investigated polycyclic aromatic hydrocarbons (PAHs).35–37 The aromatic building block, consisting of four fused benzene rings, possesses a rigid and stable chemical structure, a high fluorescence quantum yield, and a long-living excited state.38–40 β-meso-Fused porphyrin–pyrene conjugates were primarily reported on by forming a six-ring between the fragments.41–43 Our herein presented approach relies on a different, five-ring fusion motif, which we already employed successfully, not only for the fusion of phenyl substituents32 but also for more immense aromatics like the hexa-peri-hexabenzocoronene13,44–50 (Fig. 1). In contrast to the examples from the literature, performing their experiments mostly on pyren-1-yl-porphyrin derivatives,42,43,51,52 we employ pyren-2-yl conjugates53 to achieve a selective β-meso five-ring instead of a six-ring formation.
 | ||
| Fig. 1 Transfer of the β-meso five-ring fusion concept demonstrated on A4-phenyl-porphyrins in 2020 to meso-pyrene-substituted conjugates.32,53 | ||
Results and discussion
We report a novel bottom-up approach toward synthesizing pyrene-fused porphyrins. We make use of our earlier developed protocols for the β-meso-fusion of phenyl32 and HBC substituents13 to porphyrins. The resulting conjugates show distinct optical and electronic properties, which were probed by means of absorption spectroscopy and density-functional theory (DFT) calculations.Initially, three different pyrene-nickel-porphyrin conjugates were synthesized. To this end, the borylated pyrenes 6 and 7 were obtained by functionalizing pyrene under Hartwig–Miyaura conditions (for experimental details, see Scheme S1†). Brominated nickel-trimesitylporphyrin 2 was synthesized starting from small molecular building blocks and attached to 6 and 7via palladium-catalyzed Suzuki cross-coupling (for experimental details, see ESI†). The nickel ion inside the porphyrin is not only necessary to run the subsequent coupling reaction successfully. It is also essential for the final cyclodehydrogenation step, which does not work with the free-base porphyrin.13 This way, conjugates 4 and 5 were formed in 86% and 77% yield, respectively. Furthermore, di-brominated dimesitylporphyrin 1 was prepared following published protocols (for experimental details, see ESI†) and used in a twofold Suzuki cross-coupling reaction with 6, yielding bis-pyrene-porphyrin 3 in 77% yield. Conjugates 3–5 were subjected to oxidative Scholl conditions with 16 equivalents of FeCl3 in a mixture of CHCl3 and nitromethane to achieve the β-meso five-ring fusion between the two meso-coupled fragments. A controlled connection between porphyrin and pyrene can be achieved due to the specific choice of mesityl groups in the periphery of the porphyrins. The mesityl groups enhance the solubility of precursor and target molecules and prevent unwanted fusion reactions with the phenyl rings. For the precursor bearing a single pyrene moiety 5, a much more rapid transformation was observed compared to the results from our group's earlier experiments on the fusion of phenyl and HBC substituents.13,32 In detail, this meant stirring the reaction at 0 °C under slow warming to rt for 1 h, which led to the complete consumption of the starting material.
After quenching the reaction by adding MeOH and NEt3, followed by a chromatographic work-up, PyrPor was obtained in an excellent yield of 93%. Identical reaction conditions were applied to the porphyrin bearing two pyrene groups 3. Hereby, slightly longer reaction times of 4 h were required. Furthermore, chromatographic separation from the mono-fused side product was necessary to obtain double-fused PyrPorPyr in 40% yield. The last conjugate of the series, PorPyrPor, was successfully isolated after 24 h of reaction time in 36% yield starting from precursor molecule 4. However, the reaction conditions for this last reaction had to be minorly modified due to the poor solubility of 4 in CH2Cl2. To maintain the concentration ratio of porphyrin and oxidant, a solvent mixture of CH2Cl2/CS2/CH3NO2 was employed, successfully circumventing the solubility issues without hampering the reaction (Scheme 1). Unfortunately, when comparing the yields of the double-fused conjugates PyrPorPyr and PorPyrPor with the one of mono-fused PyrPor, a significant drop-off in yield can be observed, which is not only due to the aforementioned incompletely fused side products but also due to an increased degree of susceptibility towards chlorination as soon as the second additional bond is formed in the molecules. Even though the chlorinated species can be removed conveniently by column chromatography, this is an interesting note regarding the reactivity difference between mono- and bis-fused macrocycles.
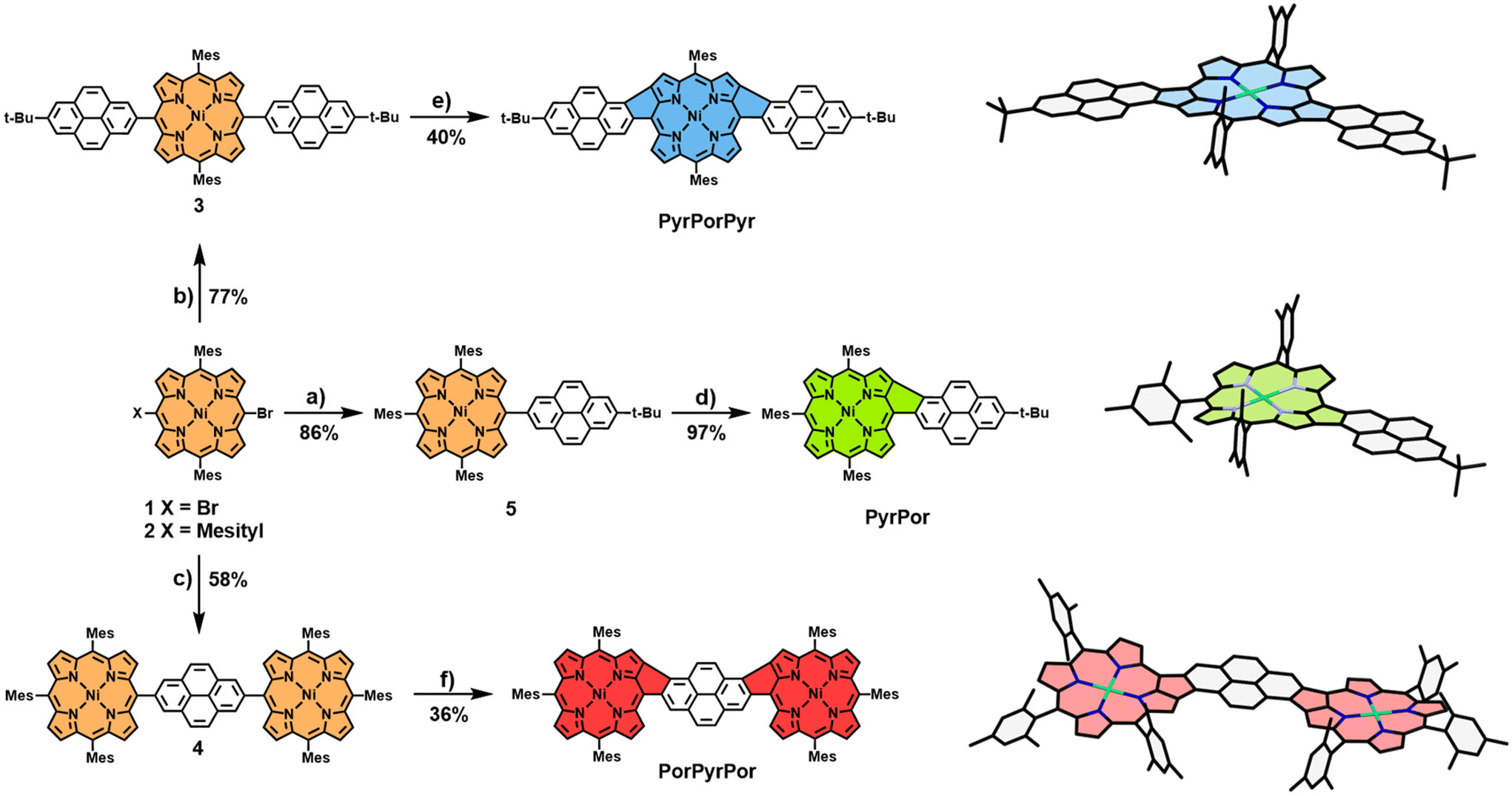 | ||
| Scheme 1 Synthesis of fused pyrene–porphyrins PyrPor, PyrPorPyr, and PorPyrPor. Reagents and conditions: (a) and (b) 2-(7-(tert-butyl)pyren-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane 6; Pd(PPh3)4, Cs2CO3, toluene, DMF, 18 h, 80 °C; (c) 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrene 7; Pd(PPh3)4, Cs2CO3, toluene, DMF, 18 h, 80 °C; (d) FeCl3 (16 equiv.), CH3NO2, CH2Cl2, 1 h, 0 °C → rt; (e) FeCl3 (16 equiv.), CH3NO2, CH2Cl2, 4 h, 0 °C → rt; (f) FeCl3 (16 equiv.), CH3NO2, CH2Cl2, CS2, 24 h, 0 °C → rt. DFT-optimized structures of PyrPor, PyrPorPyr, and PorPyrPor are displayed on the right side. | ||
Unambiguous structural characterization of PyrPor, PyrPorPyr, and PorPyrPor was conducted via NMR spectroscopic and mass spectrometric techniques. In the 1H and 13C NMR spectra of all three fused conjugates, an increase in the number of distinct signals caused by the loss of symmetry in the molecule can be observed (for precursor spectra, see ESI†). For the more extensively fused molecules PyrPorPyr and PorPyrPor, heating of the sample to 80 °C was necessary to obtain sharp signals in the area from 10 to 7.5 ppm in the 1H NMR (Fig. 2). However, by combining the obtained, well-resolved spectra with geometrical considerations, the definite position of the newly formed bonds could be determined successfully. In detail, for PyrPorPyr, the signal splitting of the aromatic mesitylene protons is strongly indicative of the bond geometry, as the number of distinct signals would differ between the two regioisomers due to the difference in molecular symmetry. In the case of PorPyrPor, the magnetic coupling between the pyrene-bound protons provides sufficient evidence by contemplating the symmetry-related changes in the magnetic equivalency of these hydrogens (for visualization of the assignments, see Fig. S38 and S39†). By taking this into account, a “cis” alignment of the two bonds for PyrPorPyr and PorPyrPor is unambiguously proven. This regioselectivity, according to findings in recent literature, is most likely caused by a biradical-cation-mediated reaction pathway.11 The results are also in line with the observations made for molecules we obtained in our earlier works on β-meso-fused congeners.13,32
 | ||
| Fig. 2 Aromatic region of the 1H NMR spectra of PyrPor (CD2Cl2, rt), PyrPorPyr (C2D2Cl4, 80 °C), and PorPyrPor (C2D2Cl4, 80 °C). | ||
The UV/Vis spectra show some general trends, like the significant flattening and red-shift of the absorption curves, which align with our previous studies on β-meso five-ring fused porphyrin conjugates (Fig. 3).13,32 Taking a closer look at the individual molecules, PyrPor, the molecule with the smallest π-system, as expected, shows the least bathochromically shifted absorption maxima at 500 nm, as well as the smallest integrated area in the range of 350–800 nm (green). For double-fused PyrPorPyr, a completely different spectral shape emerges, with a very intense maximum at 560 nm and strong concomitant absorption in the vicinity. Also, a strongly reduced absorption between 400 and 500 nm compared to PyrPor is observed (blue). For PorPyrPor, a curve reminiscent of PyrPorPyr but with a less intense absorption maximum at 554 nm is obtained (red). However, the integrated area is the largest of the three conjugates, which can be partially attributed to pronounced absorptions above 600 nm, which are only miniscule for both PyrPor and PyrPorPyr.
 | ||
| Fig. 3 UV/Vis absorptions of PyrPor, PyrPorPyr, and PorPyrPor (CH2Cl2). | ||
DFT calculations were performed to gain deeper insights into the electronic structure of the three fused conjugates. The mono-fused molecule PyrPor possesses the largest energy gap (2.28 eV) with HOMO and LUMO energy levels of −5.12 eV and −2.84 eV, respectively. Both frontier orbitals are delocalized over the whole aromatic system, however, with a less pronounced weight on the pyrene for the LUMO. A comparison of the orbital contour plots of PyrPor with those of its constituent building blocks, Por (Ni-tetramesitylporphyrin) and Pyr (2,7-di-tert-butylpyrene, see Fig. S40–S42 and Table S1 in the ESI†), provides an explanation for this. Both HOMO and LUMO of all fused molecules are linear combinations of the HOMO/HOMO−1 and LUMO of the building motifs. In the case of the HOMO, the orbital energies of Por and Pyr match closely. Therefore, they contribute equally to the resulting linear combination. However, the LUMO of Por is considerably lower in energy than the LUMO of Pyr, resulting in a higher weight of Por to the LUMO of the fused PyrPor.
The double-fused PyrPorPyr shows the same trends, with a further significant decrease in gap energy caused by an upward shift of the HOMO energy to −5.00 eV as well as a decrease in LUMO energy to −3.08 eV. The resulting energy gap of 1.92 eV is the smallest of the three investigated molecules. PorPyrPor has an energy gap of 2.15 eV. Its HOMO remains at a similar energy of −5.05 eV, while the lowering of the LUMO level (−2.90 eV) is much less pronounced than in PyrPorPyr. The reason is that the fusion of two porphyrins increases the weight of the central pyrene moiety to the LUMO of the PorPyrPor conjugate (Fig. 4), which is energetically unfavorable as discussed above, resulting in a higher energy of the LUMO level and, consequently, a larger than expected energy gap.
 | ||
| Fig. 4 Orbital contours and eigenvalues of the HOMO and LUMO of PyrPor, PyrPorPyr, and PorPyrPor in the gas phase, calculated by DFT at the B3LYP/def2-TZVPP level of theory including an implicit solvation model. | ||
Additional time-dependent-DFT (TD-DFT) calculations reveal the same trends as observed for the HOMO–LUMO gaps. The energy of the first optically active excitation, consisting of HOMO/HOMO−1 to LUMO/LUMO+1 transitions for all three conjugates, decreases from 2.0 to 1.96 and 1.78 eV for PyrPor, PorPyrPor, and PyrPorPyr, respectively (see Tables S2–S7 in the ESI†). The TD-DFT absorption spectra also reproduce perfectly the experimentally observed bathochromic shift from the mono- to the double-fused compounds (Fig. 5).
 | ||
| Fig. 5 Comparison of calculated (TD-DFT, B3LYP, implicit solvation model) and experimental absorption spectra of PyrPor, PyrPorPyr, and PorPyrPor (CH2Cl2). The theoretical transitions are broadened by a Gaussian function with a width of 10 nm and shifted by 80 nm to higher wavelengths to facilitate comparison. | ||
Conclusions
In summary, we could demonstrate the β-meso connection of nickel-porphyrins and pyrene in a straightforward fashion. The laid-out approach relies on creating architectures specifically tailored to form a five-ring in the final reaction step by fusing the two meso-coupled aromatic systems under oxidative cyclodehydrogenation conditions. Following this strategy, fusing one pyrene unit to a porphyrin was possible in excellent yields. Furthermore, it was also feasible to link two pyrenes to one porphyrin and vice versa in the same way. The solubility and processability of all three conjugates remained superb in common organic solvents. UV/Vis measurements unveiled the intriguing changes in absorption properties arising from the π-extension of the molecules. (TD)-DFT calculations provided deeper insight into the electronic structure of the molecules. The obtained HOMO–LUMO gaps, optical excitation energies, and absorption spectra are all in excellent agreement with the trends we observed in our earlier experiments13,32 with other β-meso five-ring fused conjugates. The progressive decrease of the gap energy for every additional bond formed between the porphyrin core and a meso-coupled substituent is splendidly demonstrated, and the experimentally seen bathochromic shift is replicated. To conclude, our herein laid-out synthetic approach also acts as a benchmark strategy for the design of five-ring fused conjugates. The step-wise approach based on the introduction of aromatic fragments in the porphyrins’ meso-positions via cross-coupling reactions and subsequent cyclodehydrogenation leading to planarization can potentially be applied by choosing from a plethora of well-defined PAHs. Investigations in this context, the more elaborate photophysical characterization of the presented conjugates, as well as further theoretical considerations are currently ongoing in our laboratories.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funded by the Deutsche Forschungsgemeinschaft (DFG) through SFB 953 (project number 182849149) and GRK 2423 (project number 377472739). C. O. and C. L. R. thank the Graduate School Molecular Science (GSMS) for financial support. Computational resources were provided by the Erlangen National High Performance Computing Center NHR@FAU.References
- K. Kadish, K. Smith and R. Guilard, Handbook of Porphyrin Science, World Scientific Publishing Company, 2010 Search PubMed.
- J. Yang, M.-C. Yoon, H. Yoo, P. Kim and D. Kim, Chem. Soc. Rev., 2012, 41, 4808 RSC.
- Y.-H. Chao, J.-F. Jheng, J.-S. Wu, K.-Y. Wu, H.-H. Peng, M.-C. Tsai, C.-L. Wang, Y.-N. Hsiao, C.-L. Wang and C.-Y. Lin, et al. , Adv. Mater., 2014, 26, 5205 CrossRef CAS PubMed.
- C.-H. Li, C.-C. Chang, Y.-H. Hsiao, S.-H. Peng, Y.-J. Su, S.-W. Heo, K. Tajima, M.-C. Tsai, C.-Y. Lin and C.-S. Hsu, ACS Appl. Mater. Interfaces, 2019, 11, 1156 CrossRef CAS.
- C.-L. Wang, Y.-C. Chang, C.-M. Lan, C.-F. Lo, E. Wei-Guang Diau and C.-Y. Lin, Energy Environ. Sci., 2011, 4, 1788 RSC.
- C.-H. Wu, M.-C. Chen, P.-C. Su, H.-H. Kuo, C.-L. Wang, C.-Y. Lu, C.-H. Tsai, C.-C. Wu and C.-Y. Lin, J. Mater. Chem. A, 2014, 2, 991 RSC.
- M. H. Hoang, Y. Kim, M. Kim, K. H. Kim, T. W. Lee, D. N. Nguyen, S.-J. Kim, K. Lee, S. J. Lee and D. H. Choi, Adv. Mater., 2012, 24, 5363 CrossRef CAS.
- X. Wang, Y. Lu, J. Zhang, S. Zhang, T. Chen, Q. Ou and J. Huang, Small, 2021, 17, 2005491 CrossRef CAS.
- Q. Chen, L. Brambilla, L. Daukiya, K. S. Mali, S. de Feyter, M. Tommasini, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2018, 57, 11233 CrossRef CAS PubMed.
- N. Fukui, W.-Y. Cha, S. Lee, S. Tokuji, D. Kim, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 9728 CrossRef CAS PubMed.
- N. Fukui, S.-K. Lee, K. Kato, D. Shimizu, T. Tanaka, S. Lee, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2016, 7, 4059 RSC.
- H. Mori, T. Tanaka, S. Lee, J. M. Lim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2015, 137, 2097 CrossRef CAS PubMed.
- C. Oleszak, P. R. Schol, C. L. Ritterhoff, M. Krug, M. M. Martin, Y. Bo, B. Meyer, T. Clark, D. M. Guldi and N. Jux, Angew. Chem., Int. Ed., 2024, e202409363 CAS.
- A. S. Bulbul, N. Chaudhri, M. Shanu, J. N. Acharyya, G. Vijaya Prakash and M. Sankar, Inorg. Chem., 2022, 61, 9968 CrossRef CAS PubMed.
- K. S. Kim, J. M. Lim, A. Osuka and D. Kim, J. Photochem. Photobiol., C, 2008, 9, 13 CrossRef CAS.
- S. Pascal, S. David, C. Andraud and O. Maury, Chem. Soc. Rev., 2021, 50, 6613 RSC.
- H. D. Root, D. N. Mangel, J. T. Brewster, H. Zafar, A. Samia, G. Henkelman and J. L. Sessler, Chem. Commun., 2020, 56, 9994 RSC.
- M.-C. Yoon, S. Cho, M. Suzuki, A. Osuka and D. Kim, J. Am. Chem. Soc., 2009, 131, 7360 CrossRef CAS PubMed.
- Z. S. Yoon, D.-G. Cho, K. S. Kim, J. L. Sessler and D. Kim, J. Am. Chem. Soc., 2008, 130, 6930 CrossRef CAS PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565 CrossRef CAS PubMed.
- J. M. Ball, N. K. S. Davis, J. D. Wilkinson, J. Kirkpatrick, J. Teuscher, R. Gunning, H. L. Anderson and H. J. Snaith, RSC Adv., 2012, 2, 6846 RSC.
- N. K. S. Davis, M. Pawlicki and H. L. Anderson, Org. Lett., 2008, 10, 3945 CrossRef CAS PubMed.
- N. K. S. Davis, A. L. Thompson and H. L. Anderson, Org. Lett., 2010, 12, 2124 CrossRef CAS PubMed.
- P. N. Taylor, A. P. Wylie, J. Huuskonen and H. L. Anderson, Angew. Chem., Int. Ed., 1998, 37, 986 CrossRef CAS.
- H. Zhu, Q. Chen, I. Rončević, K. E. Christensen and H. L. Anderson, Angew. Chem., Int. Ed., 2023, 62, e202307035 CrossRef PubMed.
- H. Furuta, T. Ishizuka, A. Osuka and T. Ogawa, J. Am. Chem. Soc., 2000, 122, 5748 CrossRef CAS.
- K. Fujimoto, J. Oh, H. Yorimitsu, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2016, 55, 3196 CrossRef CAS.
- H. Mori, T. Tanaka and A. Osuka, J. Mater. Chem. C, 2013, 1, 2500 RSC.
- Y. Nakamura, N. Aratani, H. Shinokubo, A. Takagi, T. Kawai, T. Matsumoto, Z. S. Yoon, D. Y. Kim, T. K. Ahn and D. Kim, et al. , J. Am. Chem. Soc., 2006, 128, 4119 CrossRef CAS.
- T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943 RSC.
- T. Tanaka and A. Osuka, Chem. – Eur. J., 2018, 24, 17188 CrossRef CAS PubMed.
- M. M. Martin, C. Oleszak, F. Hampel and N. Jux, Eur. J. Org. Chem., 2020, 2020, 6758 CrossRef CAS.
- Q. Chen, A. Lodi, H. Zhang, A. Gee, H. I. Wang, F. Kong, M. Clarke, M. Edmondson, J. Hart and J. N. O'Shea, et al. , Nat. Chem., 2024, 16, 1133 CrossRef CAS.
- K. R. Graham, Y. Yang, J. R. Sommer, A. H. Shelton, K. S. Schanze, J. Xue and J. R. Reynolds, Chem. Mater., 2011, 23, 5305 CrossRef CAS.
- Y. Gong, X. Zhan, Q. Li and Z. Li, Sci. China: Chem., 2016, 59, 1623 CrossRef CAS.
- X. Feng, J.-Y. Hu, C. Redshaw and T. Yamato, Chem. – Eur. J., 2016, 22, 11898 CrossRef CAS PubMed.
- J. M. Casas-Solvas, J. D. Howgego and A. P. Davis, Org. Biomol. Chem., 2014, 12, 212 RSC.
- F. Dumur, Eur. Polym. J., 2020, 126, 109564 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039 CrossRef CAS.
- J. Qiu, A. Hameau, X. Shi, S. Mignani, J.-P. Majoral and A.-M. Caminade, ChemPlusChem, 2019, 84, 1070 CrossRef CAS.
- Q. Zhong, V. V. Diev, S. T. Roberts, P. D. Antunez, R. L. Brutchey, S. E. Bradforth and M. E. Thompson, ACS Nano, 2013, 7, 3466 CrossRef CAS PubMed.
- V. V. Diev, K. Hanson, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, Angew. Chem., Int. Ed., 2010, 49, 5523 CrossRef CAS PubMed.
- V. V. Diev, C. W. Schlenker, K. Hanson, Q. Zhong, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, J. Org. Chem., 2012, 77, 143 CrossRef CAS PubMed.
- D. Lungerich, J. F. Hitzenberger, M. Marcia, F. Hampel, T. Drewello and N. Jux, Angew. Chem., Int. Ed., 2014, 53, 12231 CrossRef CAS PubMed.
- M. M. Martin, M. Dill, J. Langer and N. Jux, J. Org. Chem., 2019, 84, 1489 CrossRef CAS PubMed.
- M. M. Martin, C. Dusold, A. Hirsch and N. Jux, J. Porphyrins Phthalocyanines, 2020, 24, 268 CrossRef CAS.
- M. M. Martin and N. Jux, J. Porphyrins Phthalocyanines, 2018, 22, 454 CrossRef CAS.
- M. M. Martin, D. Lungerich, P. Haines, F. Hampel and N. Jux, Angew. Chem., Int. Ed., 2019, 58, 8932 CrossRef CAS.
- M. M. Martin, D. Lungerich, F. Hampel, J. Langer, T. K. Ronson and N. Jux, Chem. – Eur. J., 2019, 25, 15083 CrossRef CAS PubMed.
- J. M. Englert, J. Malig, V. A. Zamolo, A. Hirsch and N. Jux, Chem. Commun., 2013, 49, 4827 RSC.
- J. Pijeat, L. Chaussy, R. Simoës, J. Isopi, J.-S. Lauret, F. Paolucci, M. Marcaccio and S. Campidelli, ChemistryOpen, 2021, 10, 997 CrossRef CAS PubMed.
- V. V. Diev, K. Hanson, J. D. Zimmerman, S. R. Forrest and M. E. Thompson, Angew. Chem., Int. Ed., 2010, 122, 5655 CrossRef.
- S. Tomita, K. Hirabayashi, T. Shimizu, K. Goto and K. Sugiura, Synthesis, 2017, 49, 2182 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01447b |
| This journal is © The Royal Society of Chemistry 2025 |