
Facile synthesis of functionalized quinolinones in a green reaction medium and their photophysical properties†
Pari
Keerthana
 ,
Sundararajan
Suresh
and
Fazlur Rahman
Nawaz Khan
*
,
Sundararajan
Suresh
and
Fazlur Rahman
Nawaz Khan
*
Organic and Medicinal Chemistry Research Laboratory, School of Advanced Sciences, Vellore Institute of Technology, Vellore-632 014, Tamil Nadu, India. E-mail: nawaz_f@yahoo.co.in
First published on 30th October 2024
Abstract
A facile and green chemical approach was successfully developed to construct functionalized quinolinones utilizing substituted alcohols, alkyl acetoacetate, and α-bromo ketones. Various quinolinones bearing either electron-rich or electron-deficient groups at different positions were synthesized in moderate to good yields under mild reaction conditions. The plausible mechanistic pathway for this transformation is supported by experimental evidence and control experiments. This simple approach for synthesizing quinolinones could open new avenues for discovering novel biological and pharmaceutical compounds. The use of affordable nickel catalysts, mild reaction conditions, operational simplicity, and high atom economy are attractive features of this method. Furthermore, the synthetic efficiency has been demonstrated through gram-scale experiments. Our research also provides valuable insights into the photophysical properties of the synthesized derivatives. Notably, compound 6n exhibited the highest Stokes shift (216 nm) in DCM solvent. Furthermore, compounds 5d and 6j showed positive solvatochromism, displaying a stronger emission as the solvent polarity increased. Additionally, compound 6j displayed aggregation-induced emission (AIE) properties in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water mixture, making it suitable for use as a security ink, highlighting its potential applications in various fields.
:water mixture, making it suitable for use as a security ink, highlighting its potential applications in various fields.
Introduction
Quinolinone (quinolone) structural motifs1–7 are essential building blocks found widely across natural products, pharmaceuticals, and functional materials.8–11 In the domain of pharmaceutical science, quinolinone derivatives demonstrate a broad spectrum of biological activities including anti-bacterial,12 anti-acanthamoeba,13 antiulcer,14 antipsychotic and antidepressant activities.14,15In addition, they also serve as useful building blocks and synthetic intermediates in the synthesis of natural products and biologically active compounds.16,17 Notably, functionalized quinolinones are widely present in biologically active compounds, as shown in Fig. 1.18–23 Besides their phenomenal bioactivities, they have been extensively studied for their potential uses in industrial dyes, organic luminescent materials, and agrochemical compounds.24–26 Owing to their significance and utility, a straightforward synthesis of quinolinone scaffolds has become highly desirable recently. As a result, various synthetic approaches have been developed for the synthesis of quinolinone scaffolds owing to their crucial role in biological and medicinal chemistry. For example, in 2018, the Wang group reported a hypervalent iodine(III)-mediated synthesis of quinolinones from 2-vinyl-phenyl oxamic acids through an intramolecular decarboxylative Heck-type reaction.27 Later in 2021, Nan and co-workers documented the synthesis of quinolinone derivatives via C–H [5 + 1] carbonylation of alkenyl anilines and dioxazolones.28 Following this, in 2022, Hu and co-workers disclosed the dimethylamino pyridine (DMAP)-catalyzed synthesis of quinolinones through the Curtius rearrangement/intramolecular cyclisation of 2-alkenyl substituted benzoic acids with organic azides.29 Recently in 2024, Wang and co-workers reported the NaCl-catalysed synthesis of quinolinone using EtOS2K as a C-1 source (Scheme 1a).30 Despite their widespread use, these conventional methods face several drawbacks, including the generation of stoichiometric amounts of waste, low product yields due to limited selectivity, the formation of undesirable by-products, and the use of hazardous reagents. For these reasons, researchers are looking to synthesize quinolinone-based heterocycles using green solvents.31–33 The synthesis of quinolinone-based heterocycles using green solvents, although highly challenging, is highly appealing. In accordance with this, deep eutectic solvents (DES) have emerged as promising and sustainable alternatives for organic reaction media and other applications due to their easy availability and eco-friendliness.34,35 Despite being closely related to ionic liquids (ILs), DES offer more advantages due to their smaller size, higher polarity, lower toxicity, operational simplicity, and cost-effectiveness, and the biodegradability of their cations and anions.36–39 Notably, a low-melting mixture can serve as a reaction medium, enabling organic reactions to occur smoothly without the need for any catalysts or additives.40,41 In line with this, our research group has recently devised an efficient and environmentally sustainable method for synthesizing N-heterocyclic compounds.42–48 Building on this study, we present a nickel-catalyzed one-pot sequential strategy to synthesize mono-alkenylated and bi-alkenylated quinolinones from substituted alcohols, alkyl acetoacetate and α-bromo ketones in a DES reaction medium (Scheme 1b).
 | ||
| Fig. 1 Representation of bioactive functionalized quinolinone molecules. | ||
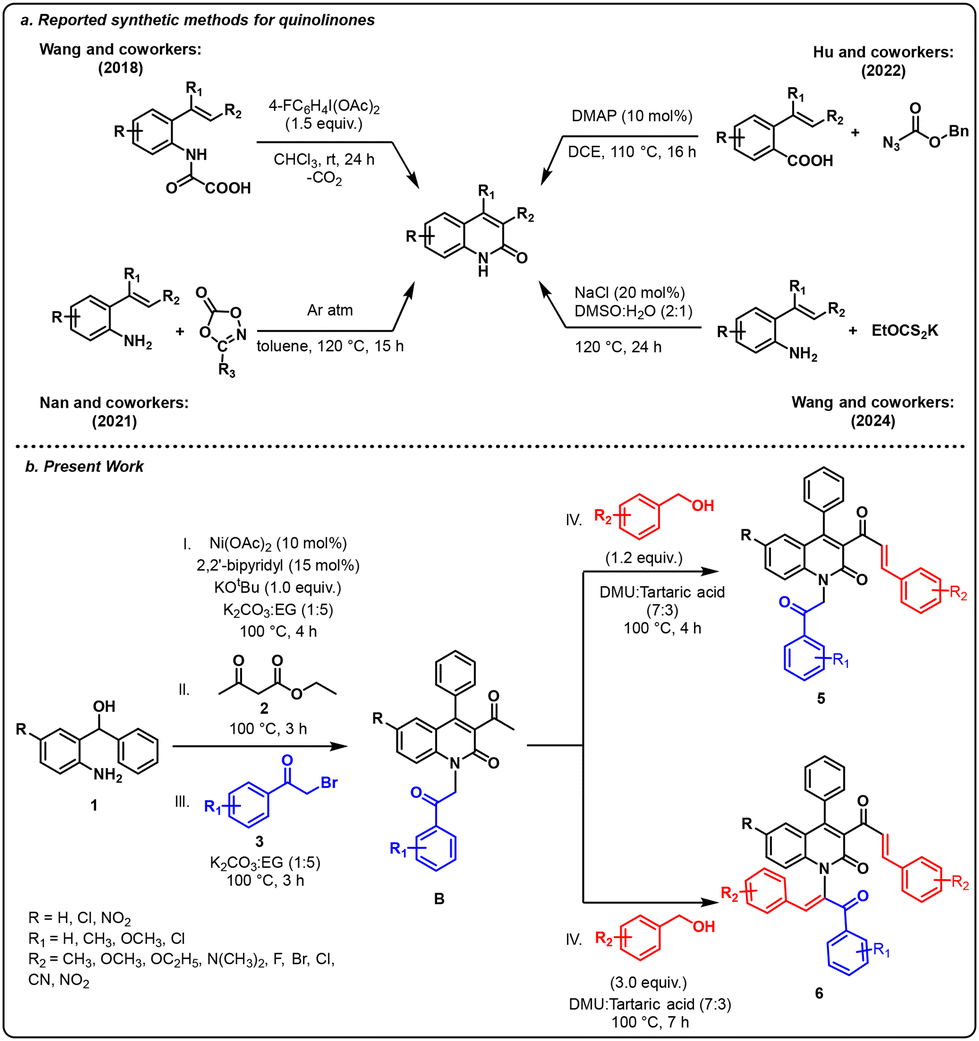 | ||
| Scheme 1 (a) Reported synthetic methods for quinolinones. (b) Present work. | ||
Results and discussion
To evaluate the practicality of the sequential reaction shown in Scheme 1, a preliminary examination was initiated using 2-amino-5-chlorobenzhydrol 1a (0.5 mmol, 1.0 equiv.), Ni(OAc)2 (10 mol%), 2,2′-bipyridyl (15 mol%), and KOtBu (1.0 equiv.) in dimethylsulfoxide (DMSO) at 100 °C for 15 h. Then ethyl acetoacetate 2a (0.5 mmol, 1.0 equiv.) was added, and the reaction was continued at 100 °C for 3 h. Subsequently, phenacyl bromide 3a (0.5 mmol, 1.0 equiv.) was added, and the reaction was allowed to proceed at 100 °C for 4 h. This was followed by the addition of benzyl alcohol 4a (0.6 mmol, 1.2 equiv.) and continued heating for an additional 5 h, affording only 22% of the 5a product (Table 1, entry 1). Based on the promising preliminary results, various organic solvents were tested. The evaluation revealed that organic solvents like DMF and toluene yielded less than 18% of the desired product (Table 1, entries 2 and 3). Then various ligands were successfully tested. When using ligands such as 1,10-phenanthroline (1,10-Phen), dimethylglyoxime (DMG) and Xantphos, we achieved less than 14% yield of the desired product (Table 1, entries 4–6). To improve the yield and general applicability of the optimized reaction conditions, several modifications were conducted. When the reaction was conducted in environmentally friendly solvents (DESs) such as K2CO3:glycerol (1:5), K2CO3:ethylene glycol (EG) (1:5), dimethylurea (DMU):PTSA (p-toluenesulfonic acid) (7:3), and ChCl (choline chloride):PTSA (1:1), 12–26% yields of the desired product 5a were observed (Table 1, entries 7–10). However, when using K2CO3:EG (1:5) as the DES, 81% yield of N-alkylated quinolinone B was achieved. To further increase the yield of 5a, DES-2 (ChCl:PTSA (1:1)) was introduced after the formation of N-alkylated quinolinone B and the yield of 5a was increased to 39% (Table 1, entry 11). Inspired by these results, we further optimized the reaction by employing TBAB (tetrabutyl ammonium bromide):PTSA (1:1) and DMU:PTSA (7:3) as DES-2, resulting in an increased product yield of 5a in the range of 41–46% (Table 1, entries 12 and 13). Based on these results, further investigations were conducted using DMU-based DES media such as DMU:citric acid (7:3), DMU:citraconic acid (7:3), DMU:succinic acid (7:3), DMU:malonic acid (7:3), DMU:oxalic acid (7:3) and DMU:tartaric acid (7:3) (Table 1, entries 14–19). From these trials, an 81% yield of 5a was achieved in the DMU:tartaric acid (7:3) DES medium (Table 1, entry 19). Inspired by this successful outcome, additional experiments were conducted using various bases and catalysts, but the results did not show any substantial improvement (Table 1, entries 20–27). A significant decrease in the yield of 5a was observed when the reaction temperature was either decreased or increased (Table 1, entries 28 and 29). Meanwhile, we examined the role of a catalyst, ligand and base. When the reaction was performed without a catalyst, ligand and base, the desired product 5a was not formed (Table 1, entries 30–32). Increasing the equivalents (2.0 equiv.) of the base under the reaction conditions did not impact product formation, whereas decreasing the loading of the catalyst or ligand affected the yield of the desired product (Table 1, entries 33–35).
a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Ligand (15 mol%) | Base (1.0 equiv.) | Solvent (1.5 ml)/DES-1 (500 mg w/w × 2)/DES-2 (300 mg w/w) | Temp (°C) | Time (h) |
5ae (%) |
|
a Standard reaction conditions: 5a:1a (0.5 mmol, 1.0 equiv.), Ni(OAc)2 (10 mol%), 2,2-bipyridyl (15 mol%), KOtBu (1.0 equiv.), and K2CO3:EG (1:5) (500 mg) were heated at 100 °C for 4 h. Then, 2a (0.5 mmol, 1.0 equiv.) was added and maintained at 100 °C for 3 h. Subsequently, 3a (0.5 mmol, 1.0 equiv.) and K2CO3:EG (1:5) (500 mg) were added and maintained at 100 °C for 3 h. Furthermore, alcohol 4a (0.6 mmol, 1.2 equiv.) and DMU:tartaric acid (7:3) (300 mg) were added and heated at 100 °C for 4 h.
b Base (2.0 equiv.).
c Ni(OAc)2 (5 mol%).
d 2,2-Bipyridyl (10 mol%).
e Isolated yields of 5a. NR = no reaction.
|
|||||||
| 1. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | DMSO | 100 | 27 | 22 |
| 2. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | DMF | 100 | 28 | 12 |
| 3. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | Toluene | 100 | 25 | 18 |
| 4. | Ni(OAc)2 | 1,10-Phen | KOtBu | DMSO | 100 | 25 | 14 |
| 5. | Ni(OAc)2 | DMG | KOtBu | DMSO | 100 | 25 | 11 |
| 6. | Ni(OAc)2 | Xantphos | KOtBu | DMSO | 100 | 25 | 8 |
| 7. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:Glycerol (1:5) |
100 | 18 | 21 |
| 8. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5) |
100 | 18 | 26 |
| 9. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | ChCl:PTSA (1:1) |
100 | 18 | 12 |
| 10. | Ni(OAc)2 | 2,2′-bipyridyl | KOtBu | DMU:PTSA (7:3) |
100 | 18 | 18 |
| 11. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/ChCl:PTSA (1:1) |
100 | 18 | 39 |
| 12. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/TBAB:PTSA (1:1) |
100 | 20 | 41 |
| 13. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:PTSA (7:3) |
100 | 18 | 46 |
| 14. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:citric acid (7:3) |
100 | 18 | 41 |
| 15. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:citraconic acid (7:3) |
100 | 18 | 69 |
| 16. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:succinic acid (7:3) |
100 | 18 | 46 |
| 17. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:malonic acid (7:3) |
100 | 16 | 61 |
| 18. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:oxalic acid (7:3) |
100 | 16 | 73 |
| 19. | Ni(OAc) 2 | 2,2′-Bipyridyl | KO t Bu |
K
2
CO
3
:EG (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5)/DMU:tartaric acid (7:3) :5)/DMU:tartaric acid (7:3) |
100 | 14 | 81 |
| 20. | Ni(OAc)2 | 2,2′-Bipyridyl | NaOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 77 |
| 21. | Ni(OAc)2 | 2,2′-Bipyridyl | Na2CO3 | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 56 |
| 22. | Ni(OAc)2 | 2,2′-Bipyridyl | NaOH | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 38 |
| 23. | Ni(OAc)2 | 2,2′-Bipyridyl | KOH | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 51 |
| 24. | NiCl2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 64 |
| 25. | NiSO4 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 12 |
| 26. | Ni(NO3)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 61 |
| 27. | NiBr2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 53 |
| 28. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
110 | 14 | 57 |
| 29. | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
90 | 14 | 63 |
| 30. | — | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | NR |
| 31. | Ni(OAc)2 | — | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | NR |
| 32. | Ni(OAc)2 | 2,2′-Bipyridyl | — | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | NR |
| 33.b | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
100 | 14 | 78 |
| 34.c | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
110 | 14 | 64 |
| 35.d | Ni(OAc)2 | 2,2′-Bipyridyl | KOtBu | K2CO3:EG (1:5)/DMU:tartaric acid (7:3) |
90 | 14 | 57 |

With the optimised reaction conditions established, our primary focus shifted to examining the reaction scope, as shown in Table 2. When the reaction was performed with various substituted benzhydrols 1, phenacyl bromides 3, and alcohols 4, moderate to good yields of the mono-alkenylated quinolinones (5a–5l) were obtained. Initially, we explored the scope of benzyl alcohol under the standard reaction conditions. When the reaction was conducted with electron-donating group (methyl and methoxy) substituted benzyl alcohol, we achieved 76–81% yields of the functionalized quinolinone products (5a–5c).
|
a Standard reaction conditions: 1 (0.5 mmol, 1.0 equiv.), Ni(OAc)2 (10 mol%), 2,2-bipyridyl (15 mol%), KOtBu (1.0 equiv.), and K2CO3:EG (1:5) (500 mg) were heated at 100 °C for 4 h. Then, 2 (0.5 mmol, 1.0 equiv.) was added and maintained at 100 °C for 3 h. Subsequently, 3 (0.5 mmol, 1.0 equiv.) and K2CO3:EG (1:5) (500 mg) were added and maintained at 100 °C for 3 h. Furthermore, alcohol 4 (0.6 mmol, 1.2 equiv.) and DMU:tartaric acid (7:3) (300 mg) were added and heated at 100 °C for 4 h.
b Isolated yields of 5.
|
|---|

|
In contrast, when we used electron-withdrawing (chloro) group substituted benzyl alcohols, the reaction yielded 73–77% of the corresponding products (5d and 5e). The ortho and para substituents (methyl and chloro) on the benzene ring of alcohols 4 significantly influenced the yields due to steric hindrance (5avs.5b and 5dvs.5e). Next, we investigated the scope of phenacyl bromides. Reactions of phenacyl bromides substituted with electron-donating (methoxy) and electron-withdrawing (chloro) groups when combined with benzyl alcohols featuring electron-withdrawing (chloro and fluoro) substitutions resulted in 74–78% yields of the corresponding final products (5f–5h). We then explored the reaction with other alkylating reagents. When the reaction was performed with benzyl chloride, it resulted in a 79% yield of the desired product (5i). Additionally, we also examined the reaction with strong electron-withdrawing group (nitro and cyano) substituted benzyl alcohols, which resulted in moderate yields of the desired products (5j and 5k). Finally, we also investigated the reaction with unsubstituted benzhydrol, which yielded 79% of the corresponding functionalized product (5l).
In order to synthesize compound 6a, a series of reactions were performed, as shown in Table 3. Under the standard conditions shown in Table 1, a sequential reaction was carried out with the addition of 2.0 equivalents of alcohol 4b, resulting in 61% yield of the desired product 6a (Table 3, entry 1). Extending the reaction time up to 17 h slightly improved the yield of the 6a product (Table 3, entries 2 and 3). Similarly, increasing the equivalents of alcohol 4b to 2.5–3.0 resulted in an increased quantitative yield of 6a (Table 3, entries 4 and 5). Finally, 81% yield of product 6a was achieved by adding 3.0 equivalents of alcohol 4b at 100 °C for 17 h (Table 3, entry 5). The effect of temperature was also examined, and lowering or raising the reaction temperature resulted in decreased yields of product 6a (Table 3, entries 6 and 7).
a
|
|
||||
|---|---|---|---|---|
| Entry | 4b (mmol) | Temp. (°C) | Time (h) |
6ab (%) |
|
a Standard reaction conditions: 6a:1a (0.5 mmol, 1.0 equiv.), Ni(OAc)2 (10 mol%), 2,2-bipyridyl (15 mol%), KOtBu (1.0 equiv.), and K2CO3:EG (1:5) (500 mg) were heated at 100 °C for 4 h. Then, 2a (0.5 mmol, 1.0 equiv.) was added and maintained at 100 °C for 3 h. Subsequently, 3a (0.5 mmol, 1.0 equiv.) and K2CO3:EG (1:5) (500 mg) were added and maintained at 100 °C for 3 h. Furthermore, alcohol 4b (1.5 mmol, 3.0 equiv.) and DMU:tartaric acid (7:3) (300 mg) were added and heated at 100 °C for 7 h.
b Isolated yields of 6a.
|
||||
| 1. | 2 | 100 | 14 | 61 |
| 2. | 2 | 100 | 15 | 63 |
| 3. | 2 | 100 | 17 | 68 |
| 4. | 2.5 | 100 | 17 | 76 |
| 5. | 3 | 100 | 17 | 81 |
| 6. | 3 | 80 | 17 | 49 |
| 7. | 3 | 120 | 17 | 53 |

Furthermore, the reaction scope was explored using the optimized conditions shown in Table 3 for the synthesis of bi-alkenylated quinolinones 6, as shown in Table 4 (6a–6p). When the reaction was performed with variously substituted phenacyl bromide (hydrogen and chloro) and various electron-donating group (methoxy, N,N-dimethyl, ethoxy and methyl) substituted alcohols, they afforded 75–84% yields of the desired bi-alkenylated quinolinones (6a–6j). The alcohols with electron-donating substituents (methoxy, methyl, and ethoxy) at the ortho, meta and para positions were well tolerated and yielded the corresponding desired products in moderate to good yields (6a–6i). Notably, alcohols with a methoxy substituent at the meta and para positions of the benzene ring were well suited for the reaction, with no notable influence on the yields of the corresponding products (6a–6d). Additionally, the di-substituted methoxy alcohols were also compatible with the reaction conditions, yielding the desired products in good yields (6e–6g). When the reaction was performed with substituted phenacyl bromide (H and Cl) and various electron-withdrawing group substituted alcohols (F, Cl, and Br), the desired bi-alkenylated quinolinones (6k–6n) were obtained in 67–75% yields. Alcohols 4 containing electron-donating substituents on the benzene ring resulted in higher yields of the corresponding products (6a–6j) compared to those with electron-withdrawing substituents (6k–6n). Given that the current reaction conditions are compatible with halogen functional groups, this feature can be advantageous for subsequent functionalization. Furthermore, when the reaction was carried out with phenacyl bromide containing an electron-donating group (methyl) and alcohols substituted with an electron-donating group (ethoxy), 79% yield of the corresponding product 6o was achieved. Additionally, the scope of benzhydrol was also examined. The reaction with benzhydrol containing a strong electron-withdrawing group (nitro) afforded 67% yield of the corresponding bi-alkenylated product 6p.
|
a Standard reaction conditions: 6:1 (0.5 mmol, 1.0 equiv.), Ni(OAc)2 (10 mol%), 2,2-bipyridyl (15 mol%), KOtBu (1.0 equiv.), and K2CO3:EG (1:5) (500 mg) were heated at 100 °C for 4 h. Then, 2 (0.5 mmol, 1.0 equiv.) was added and maintained at 100 °C for 3 h. Subsequently, 3 (0.5 mmol, 1.0 equiv.) and K2CO3:EG (1:5) (500 mg) were added and maintained at 100 °C for 3 h. Furthermore, alcohol 4 (1.5 mmol, 3.0 equiv.) and DMU:tartaric acid (7:3) (300 mg) were added and heated at 100 °C for 7 h.
b Isolated yields of 6.
|
|---|
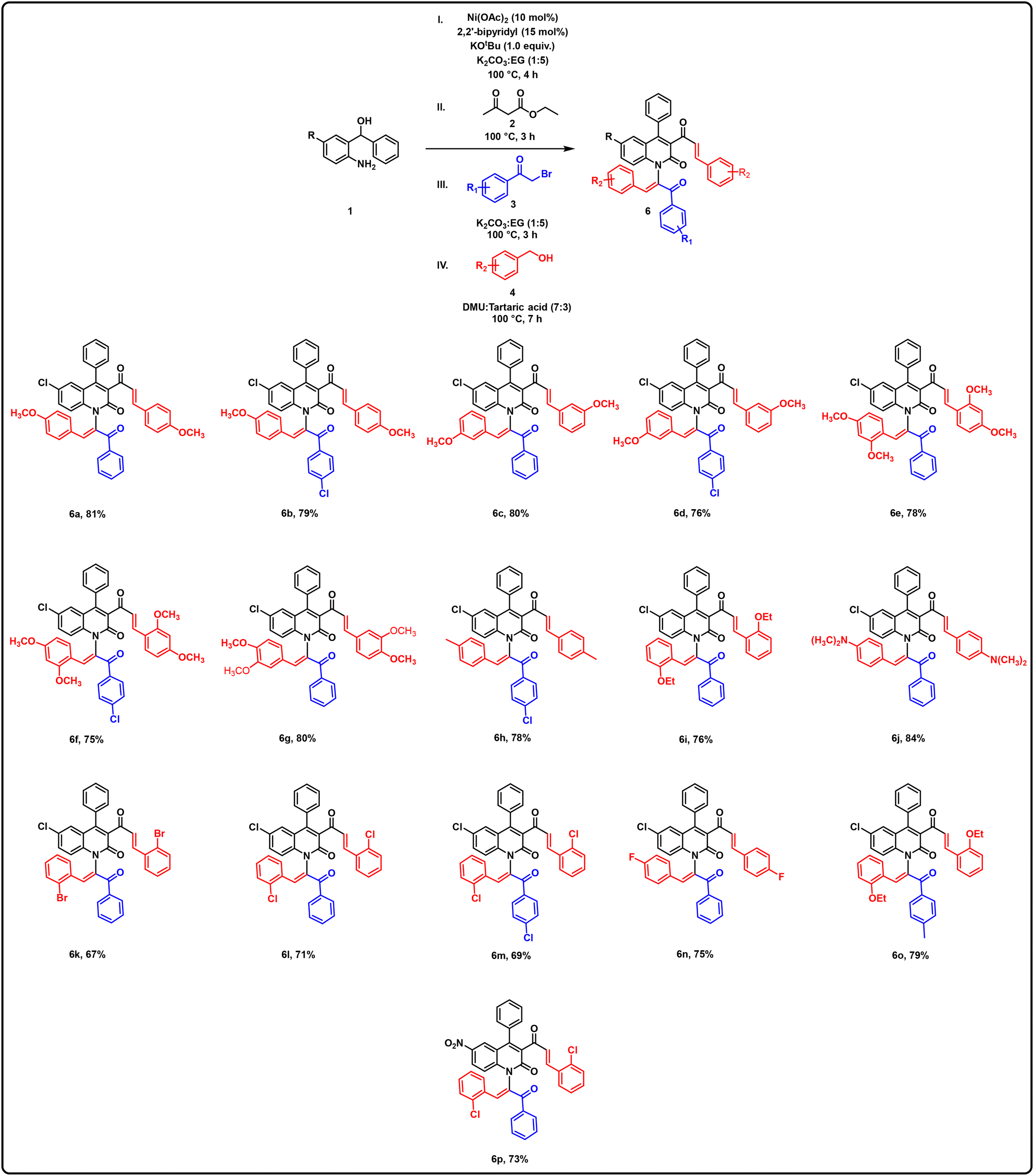
|
To gain a clearer insight into the reaction mechanism, control experiments were performed under the standard reaction conditions, as shown in Scheme 2. Initially, a model reaction was conducted using benzhydrol 1a as a starting material with the subsequent addition of Ni(OAc)2 (10 mol%), 2,2-bipyridyl (15 mol%), KOtBu and DES-1 (1:5), resulting in 94% yield of the benzophenone intermediate (1a′). Likewise, conducting the same reaction in DES-2 medium (7:3) led to the formation of only 12% of 1a′. From this observation, it is evident that DES-1 (a basic DES medium) plays a crucial role in facilitating the conversion of 1a to 1a′ (reaction condition 1). When 1a′ was reacted with ethyl acetoacetate 2a in the presence of DES-1 medium, 91% yield of intermediate A was obtained. In contrast, using DES-2 led to the formation of the 2-methyl quinoline ester product 7. This indicates that DES-1 (a basic DES medium) plays a crucial role in the formation of intermediate A. On the other hand, DES-2 facilitates the formation of the quinoline ester intermediate 7 (reaction condition 2). When the reaction was carried out using intermediate A and phenacyl bromide 3a in DES-1 medium (basic DES medium), intermediate B was obtained in 89% yield, whereas when the same reaction was performed in DES-2 medium (acidic DES medium), intermediate B was not observed. This signifies the pivotal role of DES-1 in the formation of intermediate B (reaction condition 3). Furthermore, when the reaction was performed using intermediate B and 2-chloro benzyl alcohol 4c by adding the catalyst, base, and ligand in DES-1 medium (basic DES medium), product 5e was not formed. However, when the same reaction was performed using DES-2 medium (acidic DES medium), we observed the formation of product 5e in 93% yield. From this observation, we identified the crucial role of DES-2 in facilitating the formation of 5e (reaction condition 4). Likewise, when the reaction was performed using intermediate B and aldehyde 4c′ in the presence of DES-2 medium, it yielded 84% of the 5e product (reaction condition 5). When the reaction was conducted using intermediate B and an excess of 4c (3.0 equiv.) in the presence of a catalyst, base, ligand, and DES-2 medium, it yielded 87% of the final product 6l (reaction condition 6). When the reaction was conducted using 5e and 4c (2.0 equiv.) in the presence of a catalyst, base, ligand, and DES-2, product 6l was formed in 89% yield (reaction condition 7). On the other hand, when the reaction was carried out using 5e and 4c′ (2.0 equiv.) in DES-2 medium, the yield of product 6l slightly decreased to 83% (reaction condition 7).
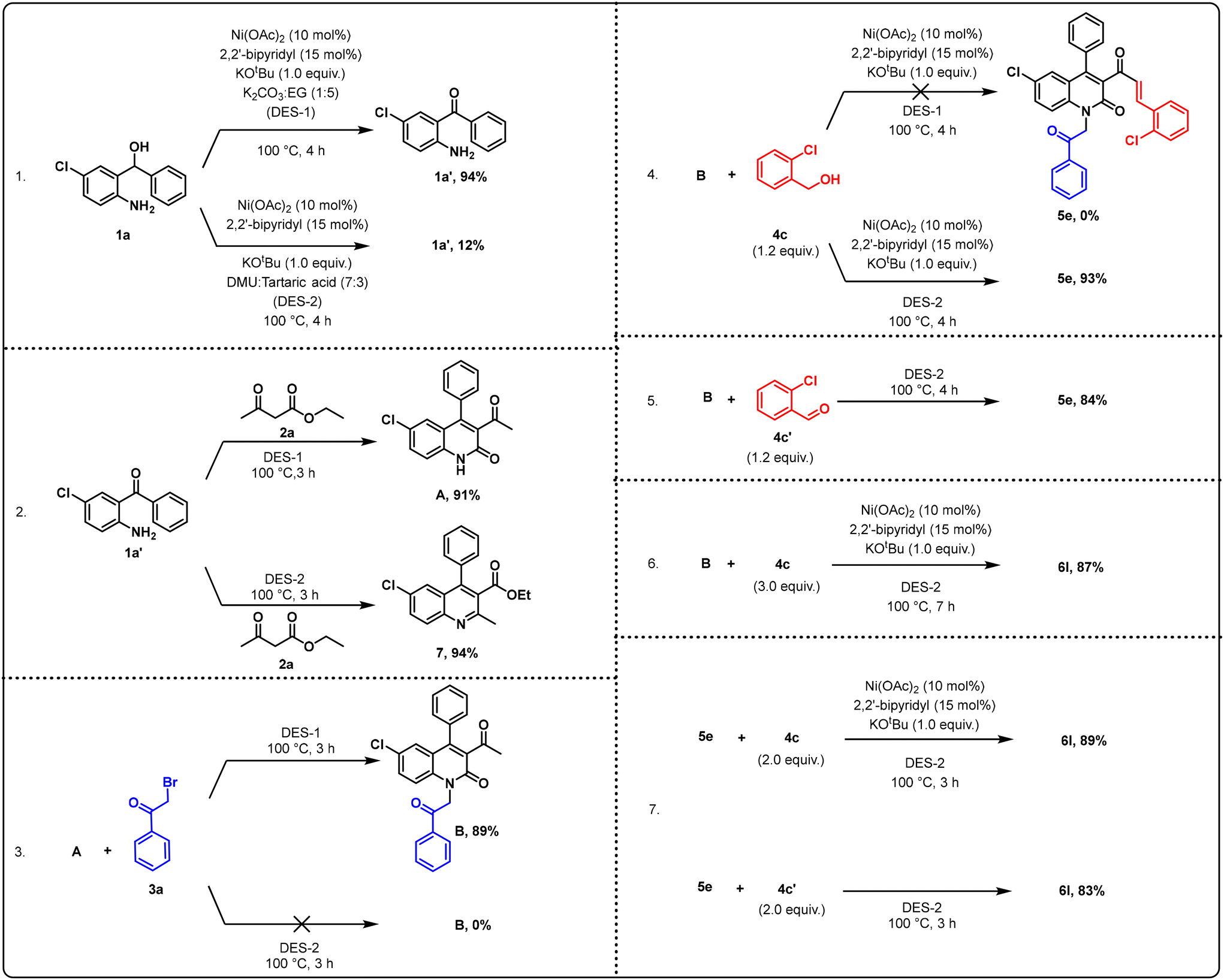 | ||
| Scheme 2 Control experiment study. | ||
Based on the control experiments and prior literature,49–54 a plausible reaction mechanism was proposed, as shown in Scheme 3. Initially, the nickel salt forms a complex (confirmed by HRMS, chromatogram provided in ESI 4.2†) with the ligands, aided by a base and the DES-1 medium. This nickel complex catalyzes dehydrogenation of benzhydrol 1a in DES-1 medium through the formation of an alkoxy nickel species (confirmed by HRMS, chromatogram provided in ESI 3.2†), followed by β-H elimination to yield benzophenone 1a′. Then, DES-1 facilitates the cyclisation of 1a′ with alkyl acetoacetate 2a, leading to the formation of 2-quinolinone intermediate A.55 Subsequently, intermediate A undergoes N-alkylation56 with phenacyl bromide 3, forming intermediate B. Then, DES-2 induces keto–enol tautomerism in the acetyl group of quinolinone intermediate Bvia H-bonding interactions with its oxygen atom, resulting in the formation of intermediate C. Meanwhile, DES-2 and Ni(II) catalysts assist in the dehydrogenation of alcohol 4, forming aldehyde 4′. Following this, intermediate C readily undergoes a Knoevenagel condensation57,58 with aldehyde 4′ to form the mono-alkenylated product 5. In addition, DES-2 facilitates a keto–enol tautomerism in the N-alkyl part of intermediate 5 to generate intermediate D. Intermediate D then reacts with another equivalent of aldehyde 4′ through a Knoevenagel condensation to form the bi-alkenylated quinolinone product 6.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
To highlight the scalability and practical applicability of the developed method, we conducted a bulk-scale reaction using 2-amino 5-chloro benzhydrol 1a (2.33 g, 10.0 mmol) as the starting material under the standard reaction conditions which afforded the desired product 6a in 78% yield (Scheme 4).
 | ||
| Scheme 4 Gram scale synthesis of 6a. | ||
Additionally, the reaction intermediate B and compound 6m were crystallized and analysed using the SC-XRD technique (see the ESI†), as shown in Fig. 2.
 | ||
| Fig. 2 (a) X-ray crystal structure of intermediate B CCDC (2374871, see the ESI†). (b) X-ray crystal structure of compound 6m CCDC (2388694, see the ESI†). | ||
Photophysical studies
The photophysical properties of the synthesized derivatives were studied, focusing on the electronic transitions of the molecules, as shown in Table 5. These properties arise from the conjugated delocalized electronic systems and the presence of various functional groups in the derivatives thus involving the π–π* and n–π* transitions.59,60 These transitions play a crucial role and offer valuable insights into the electronic structure of the molecules. The optical properties of the synthesized derivatives reveal that these compounds exhibit absorption in the range of 274–403 nm and emission in the range of 407–510 nm, as shown in Table 5. The calculated Stokes shift was in the range of 54–216 nm. Notably, in the 5 series, compound 5b showed the highest absorption at 352 nm, while in the 6 series, compound 6h exhibited the highest absorption at 403 nm. Similarly, in the 5 series, compound 5h showed the lowest absorption at 276 nm, and in the 6 series, compound 6i also displayed the lowest absorption at 274 nm.| Compound | λ abs (nm) | λ em (nm) | Stokes shift (nm) | ϕ F (%) |
|---|---|---|---|---|
| The absorption and emission spectra were recorded in DCM solvent, T = 293 K. The concentration of the compounds taken was 1.0 × 10−5 M. Finally, the fluorescence quantum yield was measured relative to a solution of quinine sulphate in 0.1 M H2SO4 (ϕF = 0.54). | ||||
| 5a | 308 | 436 | 128 | 0.23 |
| 5b | 352 | 473 | 121 | 0.30 |
| 5c | 282 | 439 | 157 | 4.76 |
| 5d | 310 | 439 | 129 | 0.39 |
| 5e | 304 | 439 | 135 | 11.13 |
| 5f | 288 | 491 | 203 | 1.59 |
| 5g | 288 | 441 | 153 | 1.19 |
| 5h | 276 | 441 | 165 | 2.58 |
| 5i | 315 | 407 | 92 | 3.54 |
| 5j | 299 | 410 | 111 | 7.78 |
| 5k | 302 | 415 | 113 | 7.93 |
| 5l | 307 | 413 | 106 | 2.85 |
| 6a | 332 | 461 | 129 | 0.91 |
| 6b | 332 | 460 | 128 | 0.75 |
| 6c | 332 | 494 | 162 | 1.37 |
| 6d | 289 | 430 | 141 | 0.98 |
| 6e | 353 | 442 | 89 | 0.65 |
| 6f | 353 | 440 | 87 | 4.16 |
| 6g | 295 | 410 | 115 | 7.08 |
| 6h | 403 | 457 | 54 | 0.55 |
| 6i | 274 | 445 | 171 | 1.80 |
| 6j | 311 | 439 | 128 | 7.06 |
| 6k | 302 | 507 | 205 | 1.47 |
| 6l | 278 | 439 | 161 | 15.50 |
| 6m | 279 | 439 | 160 | 3.58 |
| 6n | 294 | 510 | 216 | 0.57 |
| 6o | 302 | 408 | 106 | 0.36 |
| 6p | 315 | 417 | 102 | 3.41 |
In the emission studies, compounds 5b (473 nm) and 5f (491 nm) in the 5-series exhibited the highest emission, while compounds 6k (507 nm) and 6n (510 nm) in the 6-series showed the strongest emission. Additionally, compounds 5f and 6n displayed a larger Stokes shift (the difference between absorption and emission wavelengths), whereas compounds 5i and 6h had a smaller shift. Finally, we evaluated the quantum efficiency of the 5- and 6-series compounds to further refine the photophysical properties of the synthesized derivatives. Compounds 5e (11.13%) and 6l (15.50%) exhibited high quantum yield values, indicating enhanced fluorescence efficiency.61 The normalized absorption and emission spectra of the 5- and 6-series compounds are shown in Fig. 3 and 4.
 | ||
| Fig. 3 (a) Normalized absorption and (b) normalized emission spectra of compounds 5a–5l in DCM at 1.0 × 10−5 M concentration. | ||
 | ||
| Fig. 4 (a) Normalized absorption and (b) normalized emission spectra of compounds 6a–6p in DCM at 1.0 × 10−5 M concentration. | ||
Additionally, a solvatochromism study was also conducted to examine the influence of solvent polarity on the synthesized derivatives and the results are shown in Table 6. Their absorption and emission properties are altered based on their interaction with various solvent polarities. Based on this, we assessed the solvatochromic properties of compounds 5d and 6j using various solvents from non-polar to polar including pet-ether, ethyl acetate (EA), dioxane, methanol, DMF, and DMSO. The solvatochromic properties of compound 5d exhibited absorbance in the range of 296–303 nm and emission in the range of 404–506 nm. Compound 5d shows the highest emission (506 nm) in the dioxane solvent, which results in a redshift, signifying the formation of a more stable excited state.62,63 This occurrence is identified as positive solvatochromism. Conversely, a blue shift was observed in a non-polar solvent (petroleum ether), with a decreased emission value of 409 nm.
| Compd | Solvent | λ abs (nm) | λ em (nm) | ϕ F (%) | Stokes shift (nm) |
|---|---|---|---|---|---|
| The absorption and emission spectra were recorded in DCM solvent, T = 293 K. The concentration of the compounds taken was 1.0 × 10−5 M. Finally, the fluorescence quantum yield was measured relative to a solution of quinine sulphate in 0.1 M H2SO4 (ϕF = 0.54). | |||||
| 5d | Pet ether | 300 | 409 | 0.81 | 109 |
| EA | 296 | 404 | 0.11 | 108 | |
| Dioxane | 301 | 506 | 0.21 | 205 | |
| Methanol | 302 | 432 | 0.07 | 130 | |
| DMF | 301 | 416 | 0.64 | 115 | |
| DMSO | 303 | 412 | 1.67 | 109 | |
| 6j | Pet ether | 382 | 409 | 2.41 | 27 |
| EA | 390 | 475 | 0.18 | 85 | |
| Dioxane | 395 | 472 | 0.24 | 77 | |
| Methanol | 409 | 414 | 0.49 | 5 | |
| DMF | 401 | 492 | 0.05 | 91 | |
| DMSO | 402 | 469 | 0.11 | 67 | |
Likewise, compound 6i exhibited a red shift in a polar solvent (methanol), indicating positive solvatochromism, while a blue shift was observed in a non-polar solvent (petroleum ether). The maximum quantum efficiency for compounds 5d and 6j was found in DMSO and petroleum ether, respectively. The normalized absorption and emission spectra of compounds 5d and 6j are shown in Fig. 5 and 6.
 | ||
| Fig. 5 Solvatochromism properties of compound 5d. (a) Normalized absorption and (b) normalized emission spectra at 1.0 × 10−5 M concentration. | ||
 | ||
| Fig. 6 Solvatochromism properties of compound 6j. (a) Normalized absorption and (b) normalized emission spectra at 1.0 × 10−5 M concentration. | ||
Furthermore, the aggregation-induced emission behaviour of compound 6j was investigated through emission measurements in DMSO/water mixtures because of its excellent solubility in DMSO. The experiments involved varying the water fractions from 0 to 99% in DMSO/water mixtures. The results are shown in Fig. 7. The compound displayed a very weak emission around 470 nm in pure DMSO. The addition of water initially had no effect on the emission intensity, remaining unaltered up to a 50% water fraction. When fw went beyond 50%, a notable increase in the emission intensity was observed. When fw = 99%, an enhancement in the emission intensity was observed. It is believed that at a lower percentage of fw, the emission is driven by intra-molecular rotations,64 which become restricted in water-dominant dispersions (at higher fw percentages) due to the formation of nanoscopic aggregates (nanoparticles). This was also accompanied by a visible change in emission colour from yellowish orange to orange-red.
 | ||
| Fig. 7 (a) Changes in the fluorescence emission spectra of compound 6j (1 × 10−5 M, in DMSO) in the presence of different water fractions (fw 0 to 99%, v/v). (b) The corresponding fluorescence intensity ratio. | ||
In addition, the application of compound 6j was investigated, especially its use in security inks under acidic conditions,65,66 as shown in Fig. 8. Initially, the powder sample of compound 6j was dissolved in DCM solution and used to write the letters “FI” on Whatman filter paper. The written letters were displayed in a yellow colour. Upon fumigation with TFA vapour, the letters were transformed into a light-yellow colour, and after 2 minutes, the letters completely disappeared. Furthermore, the written letters reappeared after fumigation with NH3 vapour. Following TFA fumigation, the letters initially disappeared but reappeared in a 15 minute time interval without any additional treatment. From this study, it is observed that compound 6j can be used as rewritable optical recording media based on the reversibility of colour change.
 | ||
| Fig. 8 Photographic images of filter papers coated with compound 6j dissolved in DCM, viewed under normal light. | ||
Conclusion
In summary, a facile and eco-friendly nickel-catalyzed one-pot sequential synthesis of 28 novel N-substituted functionalized quinolinones has been developed. This method involves dehydrogenation, cyclization, N-alkylation, and α-alkenylation, utilizing substituted alcohols, alkyl acetoacetates, and α-bromo ketones, and achieves good yields. This approach is notable for several advantages, including the use of inexpensive metal catalysts, easily accessible feedstocks, mild reaction conditions, environmental sustainability, biodegradability, and operational simplicity. The mechanistic pathway for this novel approach has been thoroughly discussed, supported by control experiments and 1H NMR monitoring. Additionally, we demonstrated the generality and practical applicability of the reaction through bulk-scale experiments. Furthermore, we explored the photophysical properties of the synthesized compounds. Notably, compound 6j showed promising AIE behavior in a DMSO/water mixture and functions as a security ink under acidic conditions. Additionally, this approach not only enhances the versatile application of alkenylated quinolinones but also provides a new method for synthesizing heterocyclic compounds. We also believe that this method will be beneficial for the synthesis of a library of biologically significant quinolinone derivatives, which will assist in finding more potent bioactive molecules. Moreover, the synthesized products serve as valuable synthetic blocks for further functionalization, which is currently underway in our laboratory.Data availability
The data supporting the findings of this study are available within the article and/or its ESI.†Supporting data for this article are provided in the ESI,† which includes copies of the 1H NMR and 13C NMR spectra for all newly synthesized compounds, along with single crystal X-ray data for compound B (CCDC 2374871) and 6m (CCDC 2388694).†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank SIF-VIT for providing NMR, IR, HRMS and SC-XRD instrumentation facilities.References
- C. T. Alabaster, A. S. Bell, S. F. Campbell, P. Ellis, C. G. Henderson, D. S. Morris, D. A. Roberts, K. S. Ruddock, G. M. R. Samuels and M. H. Stefaniak, J. Med. Chem., 1989, 32, 575–583 CrossRef CAS PubMed.
- T. Fujioka, S. Teramoto, T. Mori, T. Hosokawa, T. Sumida, M. Tominaga and Y. Yabuuchi, J. Med. Chem., 1992, 35, 3607–3612 CrossRef CAS.
- S. Heeb, M. P. Fletcher, S. R. Chhabra, S. P. Diggle, P. Williams and M. Cámara, FEMS Microbiol. Rev., 2011, 35, 247–274 CrossRef CAS.
- R. Abonia, D. Insuasty, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, Eur. J. Med. Chem., 2012, 57, 29–40 CrossRef CAS PubMed.
- T. Shiro, T. Fukaya and M. Tobe, Eur. J. Med. Chem., 2015, 97, 397–408 CrossRef CAS PubMed.
- P. Dhiman, N. Arora, P. V. Thanikachalam and V. Monga, Bioorg. Chem., 2019, 92, 103291 CrossRef CAS PubMed.
- I. Phillips, A. King and K. Shannon, in The Quinolones, ed. V. T. Andriole, Academic Press, 3rd edn, 2000, vol. 3, pp. 99–137 Search PubMed.
- L. Deng, J. A. Deichert, S. Nguyen, I. S. Young and C. Han, Org. Lett., 2023, 25, 6710–6714 CrossRef CAS PubMed.
- S. Hu, J. Chen, J. X. Cao, S. S. Zhang, S. X. Gu and F. E. Chen, Bioorg. Chem., 2023, 19, 136 Search PubMed.
- V. Sharma, R. Das, D. K. Mehta, S. Gupta, K. N. Venugopala, R. Mailavaram, A. B. Nair, A. K. Shakya and P. K. Deb, Bioorg. Med. Chem., 2022, 59, 116674 CrossRef CAS PubMed.
- D. Jiang, J. Heterocycl. Chem., 2018, 55, 2003–2018 CrossRef CAS.
- E. Vessally, M. Babazadeh, K. Didehban, A. Hosseinian and L. Edjlali, Curr. Org. Chem., 2017, 21, 2561–2572 CrossRef CAS.
- W. P. Hong, I. Shin and H. N. Lim, Molecules, 2020, 25, 5450 CrossRef CAS PubMed.
- A. A. Aly, M. Ramadan, G. E. D. A. Abuo-Rahma, Y. A. M. M. Elshaier, M. A. I. Elbastawesy, A. B. Brown and S. Bräse, Adv. Heterocycl. Chem., 2021, 135, 147–196 CrossRef.
- P. S. Dube, L. J. Legoabe and R. M. Beteck, Mol. Divers., 2023, 27, 1501–1526 CrossRef CAS PubMed.
- L. B. Rao, C. Sreenivasulu, D. R. Kishore and G. Satyanarayana, Tetrahedron, 2022, 127, 133093 CrossRef CAS.
- A. Naeem, S. L. Badshah, M. Muska, N. Ahmad and K. Khan, Molecules, 2016, 21, 268 CrossRef PubMed.
- A.-G. A. El-Agamey and A. A. A. Attaia, Arch. Appl. Sci. Res., 2012, 4, 1339–1344 CAS.
- I. Kostopoulou, A. Diassakou, E. Kavetsou, E. Kritsi, P. Zoumpoulakis, E. Pontiki, D. Hadjipavlou-Litina and A. Detsi, Mol. Divers., 2021, 25, 723–740 CrossRef CAS.
- D. N. Toan, N. D. Thanh, M. X. Truong and D. T. Van, Arabian J. Chem., 2020, 13, 7860–7874 CrossRef CAS.
- W. P. Hong, I. Shin and H. N. Lim, Molecules, 2020, 25, 5450 CrossRef CAS PubMed.
- D. N. Toan, N. D. Thanh, M. X. Truong and D. T. Van, Med. Chem., 2020, 18, 36–50 CrossRef.
- H. Maruthesh, M. S. Katagi and B. P. Nandeshwarappa, Russ. J. Bioorg. Chem., 2023, 49, 1059–1067 CrossRef CAS.
- V. Kumar, M. Gohain, J. H. Van Tonder, S. Ponra, B. C. B. Bezuindenhoudt, O. M. Ntwaeaborwa and H. C. Swart, Opt. Mater., 2015, 50, 275–281 CrossRef CAS.
- W. M. Yip, Q. Yu, A. Tantipanjaporn, W. C. Chan, J. R. Deng, B. C. B. Ko and M. K. Wong, Org. Biomol. Chem., 2021, 19, 8507–8515 RSC.
- F. Ullah, S. Ullah, M. F. A. Khan, M. Mustaqeem, R. N. Paracha, M. F. ur Rehman, F. Kanwal, S. S. ul Hassan and S. Bungau, Molecules, 2022, 27, 6631 CrossRef CAS PubMed.
- H. Fan, P. Pan, Y. Zhang and W. Wang, Org. Lett., 2018, 20, 7929–7932 CrossRef CAS PubMed.
- J. Nan, P. Chen, X. Gong, Y. Hu, Q. Ma, B. Wang and Y. Ma, Org. Lett., 2021, 23, 3761–3766 CrossRef CAS PubMed.
- Y. Y. He, M. S. Zhu, Y. Gao and X. Q. Hu, Chem. Commun., 2022, 58, 11272–11275 RSC.
- Y. Zhang, Y. J. Chen, X. D. Yue, Y. L. Zhang, J. H. Jia, M. Li and X. C. Wang, Org. Lett., 2024, 26, 1985–1990 CrossRef CAS PubMed.
- A. Mandal and A. T. Khan, Org. Biomol. Chem., 2024, 22, 2339–2358 RSC.
- V. F. Batista, D. C. G. A. Pinto and A. M. S. Silva, ACS Sustainable Chem. Eng., 2016, 4, 4064–4078 CrossRef CAS.
- J. Song and B. Han, Natl. Sci. Rev., 2015, 2, 255–256 CrossRef.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Green Chem., 2016, 18, 2736–2744 RSC.
- C. Ruß and B. König, RSC Adv., 2012, 14, 2969 Search PubMed.
- C. Teja and F. R. Nawaz Khan, ACS Omega, 2019, 4, 8046–8055 CrossRef CAS PubMed.
- K. A. Pishro and M. H. Gonzalez, RSC Adv., 2024, 14, 14480–14504 RSC.
- B. B. Hansen, S. Spittle, B. Chen, D. Poe, Y. Zhang, J. M. Klein, A. Horton, L. Adhikari, T. Zelovich, B. W. Doherty, B. Gurkan, E. J. Maginn, A. Ragauskas, M. Dadmun, T. A. Zawodzinski, G. A. Baker, M. E. Tuckerman, R. F. Savinell and J. R. Sangoro, Chem. Rev., 2021, 121, 1232–1285 CrossRef CAS PubMed.
- C. Teja, A. Garg, G. K. Rohith, H. Roshini, S. Jena and F. R. Nawaz Khan, Polycyclic Aromat. Compd., 2022, 42, 4769–4779 CrossRef CAS.
- B. Tang, H. Zhang and K. H. Row, J. Sep. Sci., 2015, 38, 1053–1064 CrossRef CAS PubMed.
- A. E. Ünlü, A. Arlkaya and S. Takaç, Green Process. Synth., 2019, 8, 355–372 Search PubMed.
- D. Pavithra, K. R. Ethiraj and F. R. Nawaz Khan, Eur. J. Org. Chem., 2020, 7035–7050 CrossRef CAS.
- C. Teja and F. R. Nawaz Khan, ChemistrySelect, 2020, 5, 6470–6474 CrossRef CAS.
- K. Pari and F. R. Nawaz Khan, Org. Biomol. Chem., 2024, 22, 4163–4171 RSC.
- S. Suresh and F. R. Nawaz Khan, ACS Omega, 2024, 9, 36198–36219 CrossRef CAS PubMed.
- S. Suresh and F. R. Nawaz Khan, Tetrahedron Lett., 2024, 148, 155224 CrossRef CAS.
- S. Suresh and F. R. Nawaz Khan, ChemistrySelect, 2024, 9, e202402042 CrossRef CAS.
- S. Nagarajan and F. R. Nawaz Khan, ACS Omega, 2024, 9, 24464–24476 CrossRef CAS.
- I. A. Rather and R. Ali, ACS Omega, 2022, 7, 10649–10659 CrossRef CAS.
- I. A. Rather, S. H. Alotaibi, M. T. Alotaibi, M. Altaf and R. Ali, ACS Omega, 2022, 7, 35825–35833 CrossRef CAS PubMed.
- P. Keerthana and F. R. Nawaz Khan, Asian J. Org. Chem., 2024, 13, e202400192 CrossRef CAS.
- S. Parua, S. Das, R. Sikari, S. Sinha and N. D. Paul, J. Org. Chem., 2017, 82, 7165–7175 CrossRef CAS PubMed.
- D. Ye, Z. Liu, J. L. Sessler and C. Lei, Chem. Commun., 2020, 56, 11811–11814 RSC.
- S. Parua, R. Sikari, S. Sinha, S. Das, G. Chakraborty and N. D. Paul, Org. Biomol. Chem., 2018, 16, 274–284 RSC.
- X. Liu, X. Xin, D. Xiang, R. Zhang, S. Kumar, F. Zhou and D. Dong, Org. Biomol. Chem., 2012, 10, 5643–5646 RSC.
- D. Chen, Y. Bao, S. Yan, J. Wang, Y. Zhang and G. Li, Molecules, 2024, 29, 782 CrossRef CAS PubMed.
- S. S. Gawali, B. K. Pandia and C. Gunanathan, Org. Lett., 2019, 21, 3842–3847 CrossRef CAS.
- B. K. Pandia and C. Gunanathan, J. Org. Chem., 2021, 86, 9994–10005 CrossRef CAS.
- J. Cheng, K. Wei, X. Ma, X. Zhou and H. Xiang, J. Phys. Chem., 2013, 117, 16552–16563 CAS.
- H. Singla, S. Kumar, V. K. Maikhuri, K. Kavita and A. K. Prasad, ChemistrySelect, 2023, 8, e202203412 CrossRef CAS.
- D. Zając, D. Honisz, M. Łapkowski and J. Sołoducho, Molecules, 2019, 24, 2259 CrossRef PubMed.
- P. B. Kole, K. Sakthivel, S. J. Armaković, S. Armaković, M. Iqbal, F. V. Singh and S. P. Kollur, RSC Adv., 2024, 14, 16960–16970 RSC.
- S. Nigam and S. Rutan, Appl. Spectrosc., 2001, 55, 362A–370A CrossRef CAS.
- O. Anitha, M. Mathivanan, B. Tharmalingam, T. Thiruppathiraja, S. Ghorai, R. Natarajan, V. Thiagarajan, S. Lakshmipathi and B. Murugesapandian, Dyes Pigm., 2023, 212, 111091 CrossRef CAS.
- Y. Jiang, M. Liu, M. Wang, Y. Lei, Q. Ding, H. Wu and X. Huang, Org. Biomol. Chem., 2022, 20, 7770–7775 RSC.
- Megha, P. Kaur and K. Singh, Spectrochim. Acta, Part A, 2024, 307, 123649 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2374871 and 2388694. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01390e |
| This journal is © The Royal Society of Chemistry 2025 |