
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe behaviour of tricyclic fused host systems comprising seven-membered B-rings in mixed pyridines†
Benita
Barton
 *a,
Mino R.
Caira
*b,
Danica B.
Trollip
a and
Eric C.
Hosten
a
*a,
Mino R.
Caira
*b,
Danica B.
Trollip
a and
Eric C.
Hosten
a
aDepartment of Chemistry, Nelson Mandela University, PO Box 77000, Port Elizabeth, 6031, South Africa. E-mail: benita.barton@mandela.ac.za
bDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa. E-mail: mino.caira@uct.ac.za
First published on 20th October 2023
Abstract
In this work, the selectivity behaviour of two tricyclic fused host systems with seven-membered B-rings, namely N,N′-bis(5-phenyl-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H1) and N,N′-bis(5-phenyl-10,11-dihydro-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H2), was investigated in various mixtures of pyridine (PYR) and the three C-methylated pyridine isomers (2-, 3-, and 4-MP). It was first demonstrated that H1 possessed the ability to enclathrate all four pyridines in the single guest solvent experiments while H2 was only able to form complexes with PYR and 4MP. H2 showed significantly enhanced selectivities compared with H1, consistently preferring PYR, while 2MP was the favoured guest of H1. Selectivity profiles suggested that H1 has the ability to separate mixtures of 2MP/PYR when these contain 40% 2MP (K = 11.8). H2, on the other hand, was shown to have exceptional separatory potential for PYR/2MP and PYR/3MP mixtures even when the amount of PYR in these was as low as 20%. These host compounds, therefore, are able to separate some of these pyridyl mixtures with high efficiency. SCXRD analyses on five of the six complexes prepared here demonstrated that the reason for the preferential behaviours of H1 and H2 for 2MP and PYR, respectively, was the significantly shorter hydrogen bonding interactions present between the host and guest molecules in these complexes. Thermal analyses further showed that these two complexes were more thermally stable than those with the less preferred guest compounds.
1. Introduction
Each of the C-methylated pyridine analogues (2-, 3- and 4-methylpyridine, 2MP, 3MP and 4MP), also known as the picolines and pyridine bases, are chemicals with several important applications in the chemical industry. As examples, these nitrogen-containing liquids are used as building blocks towards the manufacture of dyes, textiles, agrochemicals, adhesives, pesticides and herbicides, and also serve as both solvents and bases in numerous chemical transformations.1–3While these methylated pyridines may be recovered from the coke oven after coking coal at elevated temperatures,4 they may also be synthesized on a commercial scale by means of a number of different strategies. One approach employs the gas phase methylation of pyridine (PYR) in the presence of various zeolites including mordenite, ZSM-22 and zeolite beta, and others.2,5 Additionally, the Chichibabin pyridine synthesis takes place in the presence of oxide catalysts, such as modified alumina or silica, with reagents acetaldehyde and ammonia or acrolein and ammonia.6 This dehydrocyclization reaction of acetaldehyde and ammonia may also be achieved over potassium salts of modified phosphoric acid catalysts.7
A major disadvantage of all of these methods for obtaining these pyridines is that, ultimately, a mixture of compounds is recovered, including unreacted PYR and 2-, 3- and 4-MP. The boiling points of PYR, 2-MP, 3-MP and 4-MP are 115, 129–130, 143–144 and 144.5–145.5 °C, respectively, and, clearly, subsequent separations in order to isolate these compounds in pure form are not trivial, with fractional distillations requiring high numbers of theoretical plates, not to mention the associated significant economic costs, more especially for the recovery of pure 3- and 4-MP.8–10 This challenge, therefore, has resulted in scientists exploring alternative methods to achieve these separations. Consequently, Zhao and co-workers considered dissociation extraction processes to effect some of these separations11 with some success. They investigated, amongst others, the concentration of the extractant and the temperature at which the extraction was carried out using p-toluenesulfonic acid in n-heptane. Other separation protocols included the employment of acetic or propionic acid in azeotropic experiments with MPs and 2,6-lutidine as well as electrophoresis.10,12
An alternative separation strategy for positional isomers is through supramolecular complexation involving crystalline host compounds that possess an extremely high selectivity for one particular guest isomer when crystallized from a mixture of isomers. The resultant inclusion compound of the host with the preferred guest species is stabilized by means of noncovalent interactions such as hydrogen bonding, π⋯π stacking and X–H⋯π interactions, amongst others, dependent upon the molecular structures of both host and guest species.13,14 Since such complexes are solids, a simple vacuum filtration effectively separates the host–guest isomer complex from the remaining isomers in solution. The included guest is readily separated from the complex using mild heat in distillation or, otherwise, chromatographic techniques. This method to effect such separations is an attractive one owing to its non-destructive nature, and the guest-free apohost compound may be recycled and reused continuously, ensuring a significantly greener separation strategy than many other current protocols. As an example, Nassimbeni et al. investigated the behaviour of the host compound 1,1,6,6-tetraphenylhexa-2,4-diyne-1,6-diol in mixed pyridines and noted that this compound possesses selectivity in such mixed solutions, alluding to the feasibility of efficient separations through host–guest chemistry.15
In our own laboratories, pursuits towards discovering optimal host compounds to address the challenge of separating and purifying the MP isomers most effectively by means of supramolecular chemistry have been ongoing. As such, we have investigated the behaviour of host compounds derived from tartaric acid,16,17 those synthesized from xanthone and thioxanthone,18–21 and those bearing a roof shape,22,23 as designed by Weber et al.,24 in such mixtures. The results of these explorations varied widely, dependent upon the design of the host compound, and different (and often complementary) selectivities were frequently noted in these experiments.
In the present work, the viability of employing the host compounds N,N′-bis(5-phenyl-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H1) and N,N′-bis(5-phenyl-10,11-dihydro-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H2) to effect the separations of the MPs (with added PYR), by means of supramolecular complexations, was investigated (Scheme 1) with the view to discovering the optimal host compounds for this process. The results of such experiments have not been reported on a prior occasion. All complexes, where crystal quality was suitable, were analysed by means of single crystal X-ray diffraction (SCXRD) experiments; thermoanalytical experiments were also employed as well as powder X-ray diffraction (PXRD), where applicable. The conclusions attained in this work are all provided herein.
 | ||
| Scheme 1 Structures of the two host compounds, N,N′-bis(5-phenyl-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H1) and N,N′-bis(5-phenyl-10,11-dihydro-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H2) and the pyridyl guest solvents pyridine (PYR), 2-methylpyridine (2MP), 3-methylpyridine (3MP) and 4-methylpyridine (4MP). | ||
2. Experimental
2.1 General
All starting and guest materials were purchased from Merck and were used without further purification.The 1H NMR experiments were carried out by means of a Bruker Ultrashield Plus 400 MHz spectrometer with CDCl3 as the deuterated solvent, and the data were analysed by means of MNOVA software.
All suitable single solvent complexes were subjected to SCXRD analyses. The applicable instrument was a Bruker Kappa Apex II diffractometer with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The data were collected using APEXII, whereas cell refinement and data-reduction were achieved by employing SAINT; numerical absorption corrections were carried out with SADABS.25 The structures were solved with SHELXT-2018/2 and refined by means of SHELXL-2018/3 (ref. 26) (using least-squares procedures) together with SHELXLE27 as the graphical interface. All non-hydrogen atoms were refined anisotropically, while the carbon- and oxygen-bound hydrogen atoms were inserted in idealized geometrical positions in a riding model; nitrogen-bound hydrogen atoms were found in the difference map and were allowed to refine freely. An alternative diffractometer was used for the complex of H1 with PYR. Intensity data were collected on a Bruker D8 VENTURE single crystal X-ray diffractometer using graphite-monochromated MoKα-radiation, with the crystal specimen cooled to 173(2) K with nitrogen vapour from a cryostream (Oxford Cryosystems). Data collection, performed with ω- and ϕ-scans of width 1.0°, was controlled using APEX3/v2019.1-0 (Bruker) software and refinement of the unit cell and data-reduction were performed with the program SAINT v8.40A (Bruker).28 Absorption corrections were applied using the multi-scan method with the program SADABS (2016/2).29 The structure was solved by direct methods and refined by full-matrix least-squares (programs in the SHELX suite).30 As a graphical user interface (GUI), version 4.0 of X-Seed (a program for supramolecular crystallography) was employed.31 In the final cycles of refinement, all non-hydrogen atoms were treated anisotropically, while H atoms were added in idealized positions in a riding model following their unequivocal location in successive difference Fourier maps. The crystal structures for H1·2(PYR), H1·2(2MP), H1·2(4MP), H2·2(PYR) and 2(H2)·3(4MP) were deposited at the Cambridge Crystallographic Data Centre (CCDC); CCDC numbers are 2218843, 2241126, 2241127, 2241128 and 2241129.
The PXRD experiment on 2(H2)·4MP was carried out by means of a Bruker D2 with a CuKα radiation source (λ = 1.5418 Å) and scans ranged from 5 to 50° 2θ at 0.02 deg per step and 0.5 s per step.
Two GC instruments, dependent upon their availability, were employed in order to quantify the guest compounds in the mixed complexes. Analyses were performed by means of either a Young Lin YL6500 GC equipped with an Agilent J&W Cyclosil-B column (30 m × 250 μm × 0.25 μm, calibrated) coupled to a flame ionization detector or an Agilent 7890A GC-Agilent 5975C VL mass spectrometer (GC-MS) equipped with the same column. In the former, the method involved an initial 2 min hold time followed by a heating rate of 30 °C min−1 to a temperature of 100 °C, which was then changed to a rate of 1.5 °C min−1 until 102 °C was attained. Finally, the temperature was increased to 103 °C at 0.5 °C min−1. The flow rate was 1.8 mL min−1 and the split ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80. In the latter, the method commenced with a temperature of 50 °C that was held there for 5 min and then ramped at 10 °C min−1 until 100 °C was reached. The flow rate was 1.5 mL min−1 and the split ratio was 1:80.
:80. In the latter, the method commenced with a temperature of 50 °C that was held there for 5 min and then ramped at 10 °C min−1 until 100 °C was reached. The flow rate was 1.5 mL min−1 and the split ratio was 1:80.
After recovery of the solids from the glass vials by means of vacuum filtration and washing with petroleum ether (40–60 °C), the crystals were patted dry in folded filter paper and thermal analyses were conducted on these without any further manipulation. The instrumentation used was either a TA SDT Q600 (with the data analysed using TA Universal Analysis 2000 software) or a Perkin Elmer STA6000 simultaneous thermal analyser (with the data analysed by means of Perkin Elmer Pyris 13 Thermal Analysis software). The samples were placed in open ceramic pans while an empty ceramic pan served as the reference. The purge gas was high purity nitrogen, and samples were heated from approximately 40 to 400 °C (for the TA SDT Q600 module system) and from 40 to 340 °C (for the Pyris system) with a heating rate of 10 °C min−1.
2.2 Synthesis of N,N′-bis(5-phenyl-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H1) and N,N′-bis(5-phenyl-10,11-dihydro-5-dibenzo[a,d]cycloheptenyl)ethylenediamine (H2)
The host compounds H1 and H2 were synthesized according to a previous report.322.3 Crystallization experiments employing single guest solvents
In order to determine whether host compounds H1 and H2 possessed enclathration ability for any of the pyridines, each one was crystallized from these potential guest solvents. Thus, H1 (0.05 g, 0.08 mmol) and H2 (0.04 g, 0.07 mmol) were independently dissolved in each of the pyridines (5 mmol) in glass vials. These were capped and stored at 4 °C which then allowed crystallization to occur. The crystals were isolated using vacuum filtration and crushed and washed with low boiling petroleum ether. Analysis was then performed by means of 1H NMR spectroscopy in order to observe whether complexation had occurred and, where successful, to calculate the host:guest (H:G) ratio of each complex by comparing the integrals of applicable host and guest resonance signals.
2.4 Crystallization experiments in equimolar mixed guest solutions
To establish whether H1 and H2 had selectivity for any of the pyridines, each one was crystallized from every possible combination of mixed guests where these were present in equimolar proportions. Therefore, H1 (0.05 g, 0.08 mmol) and H2 (0.04 g, 0.07 mmol) were dissolved in these equimolar mixtures (5 mmol combined amount) and the vials were capped and stored again at 4 °C. The crystals that formed in this way were isolated and treated in an identical manner to the single guest solvent experiments. Analysis was then performed by means of both 1H NMR spectroscopy (in order to determine the overall H:G ratios) and GC (so that the guest amounts in the crystals could be quantified).
2.5 Crystallization experiments in binary mixtures containing varying quantities of the two guest solvents
Since the pyridine mixtures as found in the chemical industry are not necessarily equimolar in nature, it was deemed prudent to investigate the selectivity behaviour of H1 and H2 in binary mixtures containing varying amounts of each of the two guest solvents present (GA and GB). As such, H1 (0.05 g, 0.08 mmol) and H2 (0.04 g, 0.07 mmol) were dissolved in solutions containing between 20:80 and 80:20 GA:GB (5 mmol combined amount). The vials were capped once more and stored at 4 °C which facilitated the crystallization process. Treatment of the crystals was the same as before, and analysis was performed by means of GC in order to quantify the amounts of GA and GB in the resultant complexes (ZA and ZB). A plot of ZA (or ZB) against XA (or XB), the amount of GA and GB in the original solution, allowed any host selectivity behaviour to be visualized under these changing conditions according to the equation by Pivovar et al.,33KGA:GB = ZGA/ZGB × XGB/XGA, where XGA + XGB = 1. In each of these plots, straight lines (KGA:GB = 1) were inserted which represent an unselective host compound.
2.6 Software
The program Mercury34 was used to prepare all unit cell, host–guest packing, host–guest interaction and void diagrams. In the latter case, the guest molecules were removed from the packing diagrams. The spaces that thus resulted were analysed by means of a probe with a 1.2 Å radius. This program further allowed for the analysis of all supramolecular contacts in each of the complexes.3. Results and discussion
3.1 Crystallization experiments employing single guest solvents
Table 1 contains the data obtained when host compounds H1 and H2 were crystallized from PYR and each of the MP isomers.| Guest |
H1:G |
H2:G |
|---|---|---|
|
a Host:guest (H:G) ratios were determined using 1H NMR spectroscopy.
|
||
| PYR | 1:2 |
1:2 |
| 2MP | 1:2 |
1:0 |
| 3MP | 1:1 |
1:0 |
| 4MP | 1:2 |
2:1 |
These single solvent experiments demonstrated that H1 possessed the ability to complex with all four of the guest solvents in this series (Table 1). H:G ratios were 1:2, 1:2, 1:1 and 1:2 for solvents PYR, 2MP, 3MP and 4MP, respectively. H2, on the other hand, did not include 2MP and 3MP, and only the apohost compound was recovered from these crystallization experiments. However, PYR and 4MP were enclathrated: the H:G ratio for the PYR-containing complex was 1:2, while 4MP was included with a 2:1 ratio. These 1H NMR spectra are provided in the ESI,† Fig. S1.
3.2 Crystallization experiments in equimolar mixed guest solutions
Tables 2 and 3 summarize the results obtained from competition experiments when host compounds H1 and H2 were crystallized from equimolar mixtures of PYR, 2MP, 3MP and 4MP. The preferred guest is indicated in bold text in each case, and the percentage estimated standard deviations (% e.s.d.s) are provided in parentheses since experiments were carried out in duplicate.| PYR | 2MP | 3MP | 4MP | Guest ratios (% e.s.d.s) | Overall H:G ratio |
|---|---|---|---|---|---|
|
a GC-MS and 1H NMR spectroscopy were used to obtain the G:G and overall H:G ratios, respectively.
b The competition experiments were conducted in duplicate and the % e.s.d. values are provided in parentheses.
|
|||||
| X | X | 8.8:91.2 (0.0) |
1:2 |
||
| X | X |
50.5:49.5 (3.2) |
3:1 |
||
| X | X | 43.1:56.9 (2.1) |
1:2 |
||
| X | X |
88.8:11.2 (0.8) |
1:3 |
||
| X | X |
63.5:36.5 (0.3) |
2:3 |
||
| X | X |
52.4:47.6 (0.2) |
4:1 |
||
| X | X | X | 7.3:84.4:8.3 (1.0:3.5:2.5) |
1:2 |
|
| X | X | X |
56.8:2.2:41.0 (2.2:0.4:1.9) |
1:2 |
|
| X | X | X |
35.3:32.4:32.3 (0.1:1.0:1.0) |
2:3 |
|
| X | X | X |
43.4:35.1:21.5 (1:1.9:0.9) |
1:2 |
|
| X | X | X | X | 27.0:32.1:17.7:23.2 (0.2:0.4:0.4:0.2) |
5:1 |
| PYR | 2MP | 3MP | 4MP | Guest ratios (% e.s.d.s) | Overall H:G ratio |
|---|---|---|---|---|---|
|
a These experiments were conducted in duplicate and % e.s.d. values are provided in parentheses.
b GC-MS and 1H NMR spectroscopy were used to obtain the guest and overall H:G ratios.
c No inclusion occurred and only the apohost was recovered from the experiment.
|
|||||
| X | X |
98.2:1.8 (1.9) |
2:3 |
||
| X | X |
97.9:2.1 (2.1) |
2:3 |
||
| X | X |
50.5:49.5 (2.3) |
3:4 |
||
| X | X | ||||
| X | X | 23.9:76.1 (0.7) |
3:4 |
||
| X | X | 28.9:71.1 (1.2) |
1:2 |
||
| X | X | X |
97.9:0.8:1.3 (2.2:0.8:1.4) |
1:2 |
|
| X | X | X |
53.5:1.0:45.5 (0:1:1) |
2:3 |
|
| X | X | X |
55.4:2.2:42.4 (0.7:2.3:1.6) |
1:2 |
|
| X | X | X |
44.7:28.0:27.3 (1.5:1.3:2.8) |
1:2 |
|
| X | X | X | X |
57.5:0.6:1.2:40.7(1.8:0.7:1.2:0.1) |
1:2 |
After GC analyses of the solids obtained from these equimolar guest mixtures, it was observed that 2MP was the guest most favoured by H1 (Table 2) when binary solutions were employed: the amount of 2MP present in these complexes was 91.2, 88.8 and 63.5% when the other guest present was PYR, 3MP and 4MP, respectively. In the absence of 2MP in binary solutions, the host compound displayed very modest to no tangible selectivity for either guest present: PYR/3MP, PYR/4MP and 3MP/4MP mixtures furnished crystals with only 50.5, 56.9 and 52.4% PYR, 4MP and 3MP, correspondingly. Furthermore, if 2MP was absent in the ternary experiment (PYR/3MP/4MP), then H1 also possessed essentially no selectivity (35.3/32.4/32.3%) for any of the guest solvents. From the remaining ternary experiments, it appeared as though the presence of both 2MP and 4MP resulted in poor H1 selectivities, in favour of PYR (56.8%, PYR/2MP/4MP) and 2MP (43.4%, 2MP/3MP/4MP). Only the PYR/2MP/3MP experiment produced exceptional results, and the mixed complex resulting from this equimolar solution already contained 84.4% 2MP. Finally, in the quaternary experiment, 2MP remained preferred but the affinity for this guest species was low (32.1%). The host selectivity was thus in the order 2MP (32.1%) > PYR (27.0%) > 4MP (23.2%) > 3MP (17.7%). Therefore, H1 may be a likely candidate for the separation of equimolar 2MP/PYR, 2MP/3MP and PYR/2MP/3MP mixtures, extracting significant amounts of 2MP from these solutions in host–guest chemistry experiments.
The overall H:G ratios in these experiments varied widely.
Contrastingly, the selectivity of H2 was always in favour of PYR in these equimolar experiments (Table 3) when this guest solvent was present. In fact, the selectivity for PYR was overwhelming when 4MP was absent in the binary and ternary solutions: PYR/2MP, PYR/3MP and PYR/2MP/3MP furnished mixed complexes with a near-complete selectivity for PYR (97.9–98.2%). The presence of PYR and 4MP in any of these mixtures, including the quaternary solution, resulted in a significant decline in the affinity of H2 for PYR, though the selectivity remained in favour of PYR: PYR/4MP, PYR/2MP/4MP, PYR/3MP/4MP and PYR/2MP/3MP/4MP solutions afforded mixed complexes with only between 50.5 and 57.5% PYR. In the absence of PYR, 4MP was usually moderately favoured, and crystals from 2MP/4MP and 3MP/4MP contained 76.1 and 71.1% 4MP. An exception was noted when the three MP isomers were mixed: here, 2MP was more selected, but the amount of 2MP in the complex was low (44.7%). Interestingly, only the apohost compound was recovered from the binary 2MP/3MP mixture. The host selectivity (from the quaternary experiment) was thus noted to be in the order PYR (57.5%) > 4MP (40.7%) > 3MP (1.2%) ≈ 2MP (0.6%).
The overall H:G ratios in these experiments also varied widely (as was observed in analogous experiments with H1 (Table 2)).
3.3 Crystallization experiments in binary mixtures containing varying quantities of the two guest solvents
The selectivity profiles that were obtained after GC analysis of the crystals that formed from binary mixtures in which the molar ratio of each guest was sequentially varied, and plotting Z against X, are provided in Fig. S2 (in the presence of PYR) and S3 (in the absence of PYR) for H1, and Fig. S4 (in the presence of PYR) and S5 (in the absence of PYR) for H2 in the ESI.† (Note that 2MP/3MP mixtures, in the case of H2, furnished only the apohost compound, and therefore the selectivity profile could not be constructed in this instance.)When H1 was presented with binary mixtures containing PYR (Fig. S2†), this guest remained largely disfavoured by the host compound. When 2MP/PYR mixtures contained 40, 60 and 80% 2MP, the recovered crystals were significantly enriched with 2MP, and 88.7, 92.8 and 95.8% of this guest were measured in the isolated solids, respectively (Fig. S2a†). The average selectivity coefficient (Kave) for the experiments in favour of 2MP was 8.7, while the highest K value (11.8) was calculated for the mixture that contained 40% 2MP. According to Nassimbeni and co-workers,35H1 may therefore serve as an excellent host compound for the separation of this particular mixture, since K at this point was greater than 10. Only when the solution contained small amounts of PYR (20%) was 2MP disfavoured, and the complex contained only 6.2% 2MP. The selectivity of H1 in 3MP/PYR and 4MP/PYR mixtures (Fig. S2b and c†), however, was not noteworthy, and all of the data points lie close to the line of no selectivity where K = 1. The averaged K values (in favour of 3MP and 4MP, respectively) were only 1.6 and 1.9, and host–guest chemistry strategies here would not be an effective separation method for such mixtures.
The selectivity profile that was obtained for H1 in 2MP/3MP mixtures (Fig. S3a†) demonstrates that this host compound was consistently selective for 2MP even at low concentrations of this guest solvent. Kave was modest (4.0), while the greatest K value was calculated in both the 40 and 60% 2MP binary mixtures (4.9). Since these values are significantly less than 10,35H1 cannot serve as an effective separatory tool for these mixtures. Fig. S3b† demonstrates that the preferential behaviour of H1 in 2MP/4MP mixtures was dependent upon the relative amounts of each guest present in the solution. When the mixture contained 60 and 80% 2MP, 80.7 and 94.8% 2MP were measured in the mixed complex. On the other hand, solutions enriched with 4MP (60 and 80%) afforded complexes with greater amounts of 4MP (79.1 and 84.3%). Experiments in favour of 2MP provided an average K value of 3.7 (the highest K value, 4.6, was calculated in the mixture containing 80% 2MP). In experiments that favoured 4MP, on the other hand, K was only 1.9 and 2.5 (the latter value was calculated for the mixture that contained 60% 4MP). These K values are too low for efficient separations of these solutions. As was the case in 2MP/4MP mixtures, when the solutions comprised 3MP and 4MP (Fig. S3c†), the host selectivity behaviour fluctuated, once more, according to the relative guest amounts present. 3MP was moderately preferred when the solution contained only 20% 4MP; the crystals then contained only 8.7% 4MP. However, at higher concentrations of 4MP (40, 60 and 80%), 4MP was then selected preferentially, and the mixed complexes contained 56.6, 76.6 and 87.7% 4MP, respectively. Kave, excluding the point in favour of 3MP, was only 2.0, and successful separations of these mixtures with H1 as the host compound are thus, once more, not viable.
In the case of H2 in solutions containing PYR and 2MP (Fig. S4a†) and PYR and 3MP (Fig. S4b†), remarkable selectivities were observed in favour of PYR, and this was unwavering across the concentration range. All of these experiments, extraordinarily, resulted in complexes with at least 95.0% PYR, even in solutions with low concentrations (20%) of this guest solvent. The averaged K values were 76.9 and 17.2 for PYR/2MP and PYR/3MP, respectively (note that in these calculations, K values could not be obtained when 100% PYR was found in the crystals, according to the mathematical expression for K). The greatest K value in PYR/2MP mixtures was an astounding 186.5 (the solution contained only 20% PYR), and that in PYR/3MP mixtures was 28.5 (when 40% PYR was present). Clearly, H2 is an excellent candidate to use to separate all mixtures of PYR/2MP and PYR/3MP when these contain 20% or more PYR. However, it is acknowledged that distillations would achieve similar results since PYR does boil at significantly lower temperatures than 2MP and 3MP. The selectivity profiles provided in both Fig. S4c (PYR/4MP) and S5b† (4MP/3MP) revealed H2 to be largely unselective for either guest species present since data points lie close to the K = 1 line of no selectivity. In these solutions, H2 would not be successful for any separations. In Fig. S5a† (2MP/4MP), the host behaviour changed depending on the concentrations of the two guest species present. At low concentrations of 2MP (20 and 40%), 4MP was favoured (the crystals contained only 18.6 and 23.5% 2MP), while 2MP was significantly preferred when 60 and 80% of this guest was present in the solution: the crystals that were isolated then contained 92.8 and 100.0% 2MP. The K value recorded for the mixture that contained 60% 2MP was 8.6 (this value could not be calculated in the solution containing 80% 2MP since the amount of 2MP in the crystals was 100.0%). Those experiments favouring 4MP had a Kave value of only 1.6, with the highest K value recorded being 2.2 when 60% of 4MP was present. H2 would thus only be successful as a separatory tool if mixtures contained 80% or more 2MP.
3.4 SCXRD analyses of formed complexes
All novel inclusion compounds with suitable quality crystals were analysed by means of SCXRD experiments. The only complex that could not be analysed by this technique was H1·3MP; the crystals in this case were too small despite numerous crystallization attempts at various temperatures and crystallization rates. ORTEP and similar figures for these crystal structures have been provided in the ESI,† Fig. S6. Note that in the case of the inclusion complex of H2 with 4MP, the H:G ratio of the crystal that was selected for the SCXRD experiment was 2:3, and thus differed from the single solvent recrystallization experiment (H:G 2:1, ascertained through 1H NMR spectroscopy, a bulk analytical method; this was confirmed (see later) by thermal analysis, also a bulk analytical technique). Clearly the selected crystal did not represent the bulk (in the ESI,† Fig. S7, is provided the experimental and calculated powder patterns for 2(H1)·4MP; the unit cell dimensions from both are similar but due to the drastic intensity differences, the composition of the material must be different (e.g., the same host compound but with different guest amounts with, e.g., different orientations, positions, etc.)). However, the aim of this work was to determine whether the two host compounds have the ability to facilitate the separation of these difficult-to-separate compounds, and thus this anomaly does not detract from the results that are provided here.
Complexes H1·2(PYR) and H2·2(PYR) displayed no disorder while the guest molecules in H1·2(2MP) were disordered over two orientations. In H1·2(4MP), both host and guest compounds displayed some disorder too, and guest molecules in 2(H2)·3(4MP) experienced disorder around an inversion centre.
The relevant crystallographic data for these SCXRD experiments are provided in Table 4. All complexes, except 2(H2)·3(4MP), which crystallized in the monoclinic crystal system and space group P21/c, were found to crystallize in the triclinic crystal system and space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) .
.
| H1·2(PYR) | H1·2(2MP) | H1·2(4MP) | H2·2(PYR) | 2(H2)·3(4MP) | |
|---|---|---|---|---|---|
| Chemical formula | C44H36N2·2(C5H5N) | C44H36N2·2(C6H7N) | C44H36N2·2(C6H7N) | C44H40N2·2(C5H5N) | 2(C44H40N2)·3(C6H7N) |
| Formula weight | 750.95 | 779.00 | 779.00 | 754.98 | 1472.94 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group |
P |
P |
P |
P |
P21/c |
| μ (Mo-Kα)/mm−1 | 0.074 | 0.071 | 0.069 | 0.072 | 0.071 |
| a/Å | 9.0392(4) | 8.7173(6) | 8.9471(10) | 8.9716(5) | 9.0013(3) |
| b/Å | 9.0518(3) | 11.2718(8) | 9.9740(11) | 9.2886(5) | 26.7346(9) |
| c/Å | 13.7313(6) | 12.6826(9) | 13.4375(14) | 13.8221(8) | 9.0416(3) |
| Alpha/° | 76.202(1) | 108.163(3) | 73.488(4) | 75.212(3) | 90 |
| Beta/° | 74.883(1) | 105.764(2) | 75.543(4) | 76.005(3) | 114.066(2) |
| Gamma/° | 67.225(1) | 104.337(2) | 74.301(4) | 66.684(3) | 90 |
| V/Å3 | 988.05(7) | 1061.65(13) | 1087.2(2) | 1009.80(10) | 1986.70(12) |
| Z | 1 | 1 | 1 | 1 | 1 |
| F(000) | 398 | 414 | 414 | 402 | 786 |
| Temp./K | 173 | 200 | 296 | 296 | 200 |
| Restraints | 0 | 105 | 178 | 0 | 105 |
| N ref | 6435 | 5285 | 5369 | 5002 | 4910 |
| N par | 266 | 342 | 346 | 268 | 318 |
| R | 0.0476 | 0.0392 | 0.0614 | 0.0406 | 0.0499 |
| wR2 | 0.1300 | 0.1095 | 0.1912 | 0.0974 | 0.1362 |
| S | 1.08 | 1.03 | 1.05 | 1.05 | 1.02 |
| θ min–max/° | 2.5, 31.7 | 1.8, 28.4 | 2.2, 28.3 | 2.4, 28.4 | 2.5, 28.3 |
| Tot. data | 57670 |
33628 |
39159 |
40261 |
47566 |
| Unique data | 6435 | 5285 | 5369 | 5002 | 4910 |
| Observed data [I > 2.0 sigma(I)] | 4672 | 4470 | 4428 | 4112 | 3894 |
| R int | 0.060 | 0.022 | 0.015 | 0.031 | 0.024 |
| Completeness | 0.994 | 0.998 | 0.999 | 0.995 | 1.000 |
| Min. resd. dens. (e Å−3) | −0.27 | −0.19 | −0.45 | −0.19 | −0.40 |
| Max. resd. dens. (e Å−3) | 0.33 | 0.30 | 0.40 | 0.23 | 0.38 |
H1·2(PYR). The asymmetric unit has half an H1 molecule lying about an inversion centre and a PYR molecule in a general position, linked to H1 by an N–H⋯N hydrogen bond (N16⋯N24, 3.293(2) Å).
H1·2(2MP). The asymmetric unit has half an H1 molecule lying about an inversion centre and a 2MP molecule in a general position, disordered over two interpenetrating sites, linked to H1 by N–H⋯N hydrogen bonds (N1⋯N4, 3.144(17) Å, and N1⋯N5, 3.16(3) Å).
H1·2(4MP). The asymmetric unit has half an H1 molecule lying about an inversion centre and a 4MP molecule in a general position, disordered over two interpenetrating sites, linked to H1 by N–H⋯N hydrogen bonds (N1⋯N4, 3.368(4) Å, and N1⋯N5, 3.211(5) Å).
H2·2(PYR). The asymmetric unit has half an H2 molecule lying about an inversion centre and a PYR molecule in a general position, linked to H2 by an N–H⋯N hydrogen bond (N1⋯N4, 3.256(2) Å).
2(H2)·3(4MP). The asymmetric unit has half an H2 molecule lying about an inversion centre and two 4MP components, one in a general position (with half occupancy) linked to H2 by an N–H⋯N hydrogen bond (N1⋯N4, 3.305(3) Å); the other 4MP molecule has 0.25 occupancy and is disordered about an inversion centre.
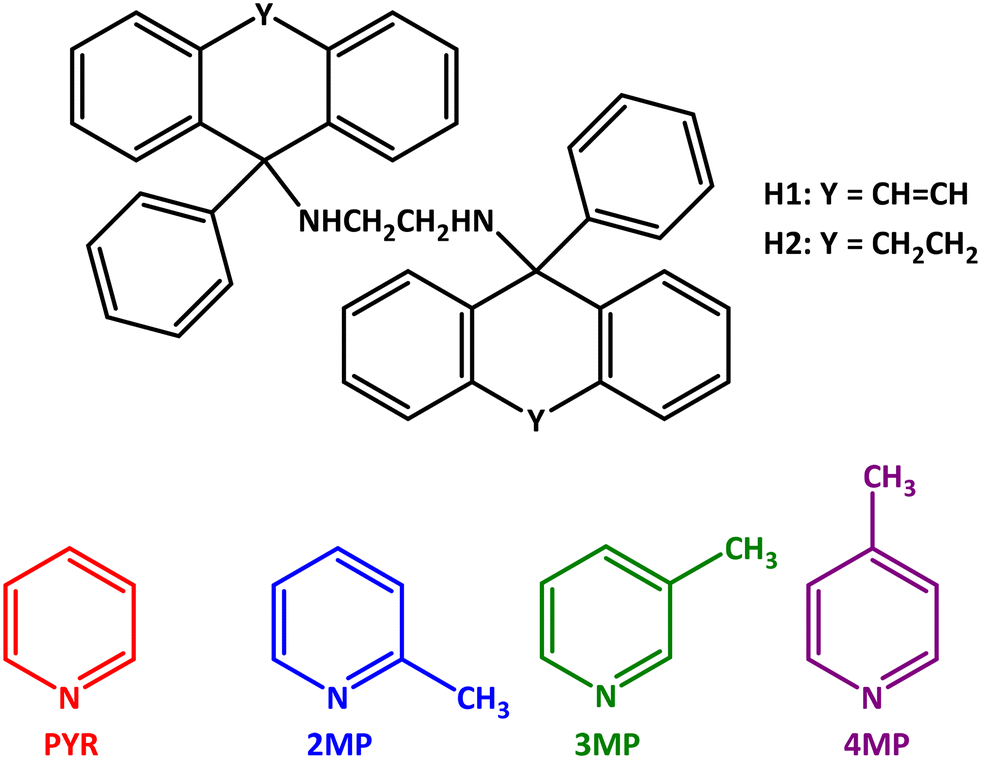
The unit cells of H1·2(PYR) (along [100]), H1·2(2MP) ([100]) and H1·2(4MP) ([100]), and H2·2(PYR) ([010]) and 2(H2)·3(4MP) ([100]) are depicted on the left-hand side in Fig. 1 (H1) and 2 (H2), respectively; the void (yellow) diagrams are also provided here (right-hand side), and two views of the voids are shown in each of Fig. 1a and c and 2a for clarity.
 | ||
| Fig. 1 Unit cells (left) and void diagrams (right) for a) H1·2(PYR), b) H1·2(2MP) and c) H1·2(4MP). | ||
 | ||
| Fig. 2 Unit cells (left) and void diagrams (right) for a) H2·2(PYR) and b) 2(H2)·3(4MP). | ||
Interestingly, both PYR-containing complexes (for H1 and H2) have very similar unit cell dimensions (Table 4) and the two host packing arrangements are therefore isostructural. However, this similarity in host packing is along two different axes as is witnessed in Fig. 1a and 2a (the structures are isostructural if you use a non-conventional unit cell by interchanging the a and b axes in one of these complexes). Also clear from these figures is that the guest molecules in H1·2(PYR) (Fig. 1a) and H1·2(4MP) (Fig. 1c) were accommodated in multidirectional channels, while 2MP in H1·2(2MP) (Fig. 1b) was found to reside in wide open channels that were parallel to the a-axis. In fact, the PYR molecules in H1·2(PYR) are arranged in close pairs and the distance between such pairs is relatively small, with the result that there is a continuity of guest molecules in three linear directions. In a similar fashion, PYR and 4MP were also housed in endless channels that assumed more than one direction in the crystals of the respective complexes with H2 (Fig. 2a and b).
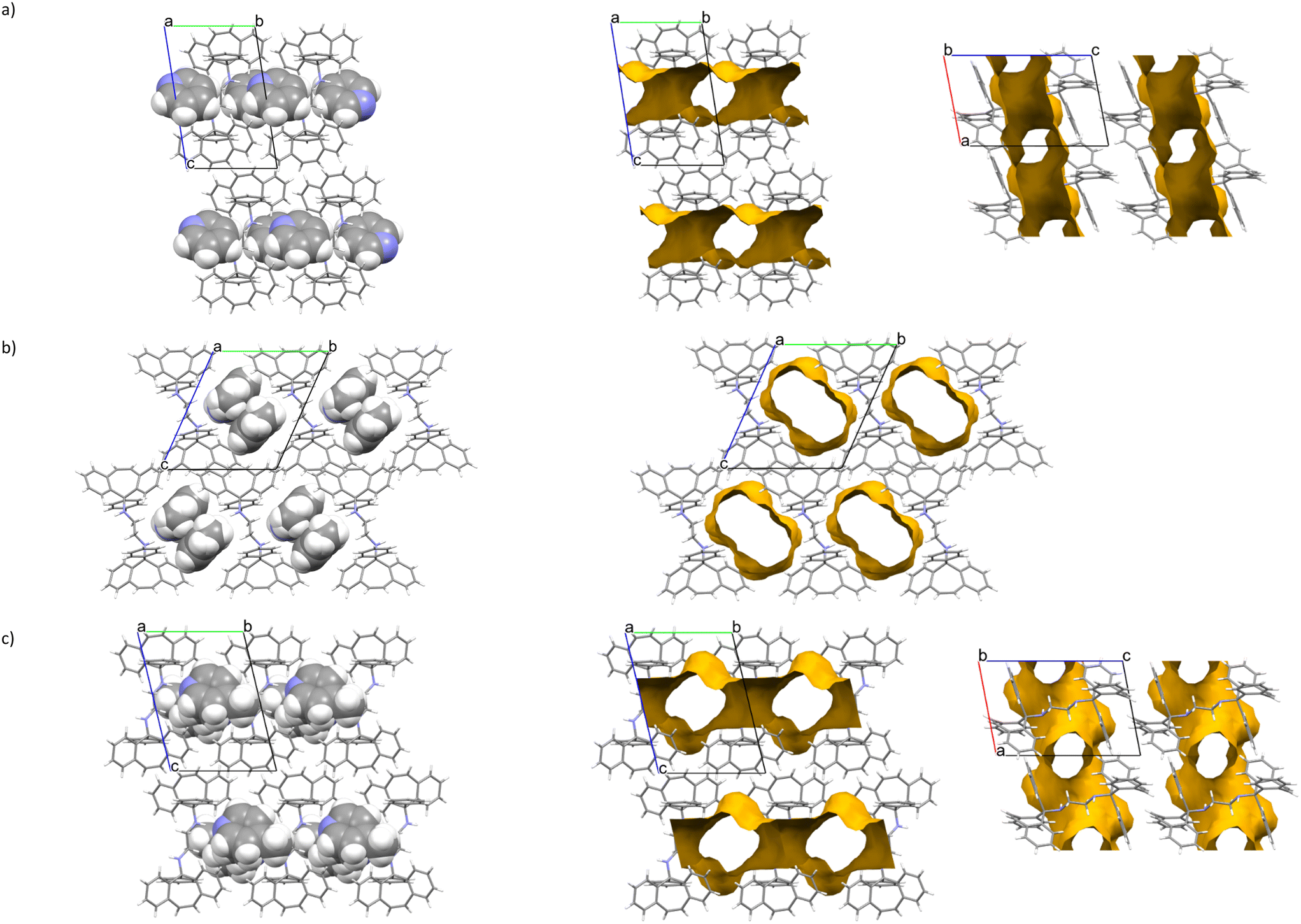
The noncovalent interactions present in the five complexes produced in this work were subsequently investigated. In H1·2(PYR) a classical (host)N–H⋯N–C(guest) hydrogen bond was observed. This is illustrated by means of two stereoviews in Fig. 3 (in both stick (left) and space-filling (right) representations) and the parameters were N⋯N 3.293(2) Å and H⋯N 2.39(2) Å, with the N–H⋯N angle being 173°. Despite all of the aromatic moieties present, no significant inter- or intramolecular (host)π⋯π(host) or (host)π⋯π(guest) contacts could be identified in this complex. However, two (host)C–H⋯π(guest) close contacts were observed, and these are illustrated, also by means of a stereoview, in Fig. 4 (in this figure the H-bonds that were mentioned earlier are also shown). The parameters for the two unique interactions (H⋯Cg, where Cg is the centroid of the pyridine molecule, and the C–H⋯Cg angle) are 2.95 Å, 145° and 2.70 Å, 163°, respectively. This complex also experienced two intramolecular non-classical C–H⋯N hydrogen bonds, and H⋯N distances were 2.40 and 2.39 Å and associated C–H⋯N angles 103 and 104°.
 | ||
| Fig. 3 Stereoview of the H1·2(PYR) complex unit showing host⋯guest hydrogen bonding, with atoms in stick representation (left) and space-filling mode (right). | ||
 | ||
| Fig. 4 Stereoview showing host⋯guest H-bonds (blue dotted lines) and the two unique C–H⋯π interactions (red dashed lines) between the host molecule and symmetry-generated pyridine molecules in the H1·2(PYR) complex. | ||
Fig. 5 shows two significant short stabilizing π⋯π interactions in the H1·2(2MP) complex involving two distinct host and guest molecules. The distances between the relevant ring centroids were 3.624(1) and 3.958(4) Å, respectively, with slippages of 0.854 and 1.789 Å. One of each of the guest⋯host and host⋯host C–H⋯π contacts was also identified (H⋯Cg 2.70 Å, 2.76 Å and C–H⋯Cg 151°, 150°) which are also illustrated in Fig. 5. Once more, classical (host)N–H⋯N–C(guest) hydrogen bonds were observed as well: both guest disorder components interacted with the host molecule in this way, and respective N⋯N and H⋯N distances measured 3.144(17), 3.16(3) Å and 2.28(2), 2.31(3) Å, correspondingly; the N–H⋯N angles were 158(1) and 156(1)°. Finally, as was the case in H1·2(PYR), two comparable intramolecular (host)C–H⋯N(host) interactions were identified and the H⋯N distances measured 2.40 and 2.37 Å; the respective C–H⋯N angles were 104 and 103°.
 | ||
| Fig. 5 The intermolecular (host)π⋯π(host) and (guest)π⋯π(guest) (green dashed lines) and the (guest)C–H⋯π(host) and (host)C–H⋯π(host) (magenta) intermolecular interactions in H1·2(2MP); distances are in Å. | ||
A subsequent analysis of the noncovalent interactions present in the H1·2(4MP) complex revealed that there were no significant guest⋯guest, guest⋯host or host⋯host π⋯π interactions present since all of these distance measurements were greater than 4.0 Å. Classical (host)N–H⋯N–C(guest) contacts were experienced by both disorder components of the guest molecule once more; N⋯N distances were 3.368(4) and 3.211(5) Å, and H⋯N 2.48(2) and 2.33(2) Å, while both the N–H⋯N angles measured 178(2)°. It is notable that the preferred guest compound of H1 (viz. 2MP), despite the N–H⋯N angles being somewhat smaller in that complex compared with that in H1·2(PYR) and H1·2(4MP), experienced statistically significantly shorter N⋯N distances (3.144(17) and 3.16(3) Å relative to 3.293(2) (H1·2(PYR), and 3.368(4) and 3.211(5) (H1·2(4MP)). Perhaps this observation plays some role in the preferential behaviour of H1 for 2MP. As was the case in the first two complexes, two intramolecular host contacts were also identified in H1·2(4MP), of the C–H⋯N type, and H⋯N distances for both were 2.40 Å and C–H⋯N angles were 104°.
For complexes involving H2, no significant π⋯π interactions were, once more, observed. In the H2·2(PYR) complex, however, two C–H⋯π contacts were noted, one between two host molecules and one between the host and guest species; H⋯Cg distances and C–H⋯Cg angles were 2.76 Å (140°) and 2.70 Å (167°), and Fig. 6 illustrates these. Once more, the host and guest molecules interacted by means of a classical (host)N–H⋯N–C(guest) hydrogen bonding interaction; the measurements for this interaction were 3.256(2) Å (N⋯N), 2.380(19) (H⋯N) and 171.8(15)° (N–H⋯N). Also present were the now ubiquitous two intramolecular (host)C–H⋯N(host) interactions (H⋯N 2.39, 2.33 Å and C–H⋯N 105, 106°).
 | ||
| Fig. 6 C–H⋯π interactions between a) C–H of a host methylene group and a phenyl ring in the same molecule, and (b) host and guest molecules in H2·2(PYR) (black dashed lines); distances are in Å. | ||
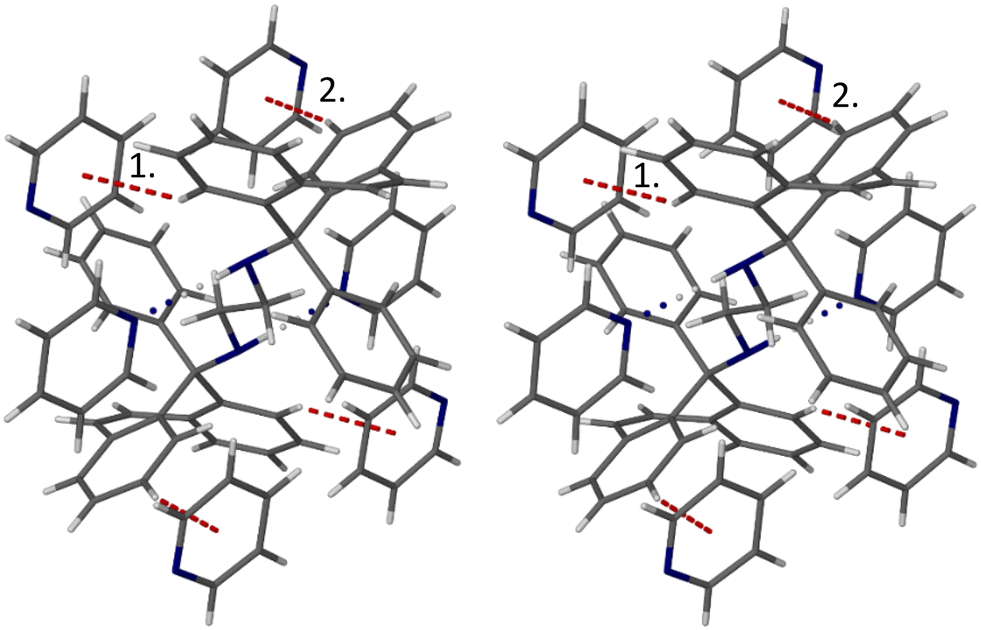
Four C–H⋯π interactions were observed in the 4MP-containing complex with H2. Fig. 7, a stereoview, is an illustration of three of these, one being a host intramolecular C–H⋯π interaction, another a (guest)C–H⋯π(host) contact, and finally, a (host)C–H⋯π(guest) interaction. The respective H⋯Cg distances measured 2.68, 2.89 and 2.69 Å, and associated C–H⋯Cg angles were 143, 140 and 152°, respectively.
 | ||
| Fig. 7 Stereoview depicting the intramolecular host C–H⋯π, intermolecular (guest)C–H⋯π(host) and intermolecular (host)C–H⋯π(guest) interactions (purple dashed lines) in 2(H2)·3(4MP); distances are in Å. | ||
Once more, the guest was bound in the crystal by means of a classical hydrogen bond ((host)N–H⋯N–C(guest)) that measured 3.305(3) Å (N⋯N) and 2.40(2) Å (H⋯N) with a corresponding angle of 169(2)° (N–H⋯N). The N⋯N hydrogen bond distance for the complex containing the preferred PYR guest species (H2·2(PYR)) is statistically significantly shorter (3.256(2) Å) than in the present instance (3.305(3) Å), and plausibly explains the affinity of H2 for PYR. Also present in the 2(H2)·3(4MP) complex are the intramolecular C–H⋯N non-classical hydrogen bonding interactions seen oftentimes before in these complexes, with H⋯N distances of 2.37 and 2.36 Å, and C–H⋯N angles of 104 and 106°.
Table S1 in the ESI† summarises the more important noncovalent interactions.
3.5 Thermal analysis
The thermogravimetric (TG), its derivative (DTG), and differential scanning calorimetric (DSC) traces after thermal analyses of the six pyridyl-containing complexes are provided (overlaid) in Fig. S8a–d (H1) and S9a and b (H2) in the ESI,† while the more important thermal data obtained from these are summarised in Table 5.| Complex | T on/°Ca | Calculated mass loss/% | Experimental mass loss/% |
|---|---|---|---|
| a T on is the onset temperature for the guest release process and serves as a measure of the thermal stability of the complex and was estimated from the DTG/TG. b The onset temperature of the guest release process commenced during sample preparation, and Ton and the experimental mass loss could thus not be measured. | |||
| H1·2(PYR) | 54.6 | 21.1 | 21.5 |
| H1·2(2MP) | 63.2 | 23.9 | 23.2 |
| H1·3MP | 13.6 | ||
| H1·2(4MP) | 23.9 | ||
| H2·2(PYR) | 76.5 | 21.0 | 19.8 |
| 2(H2)·4MP | 68.2 | 7.2 | 6.5 |
The mass losses experienced by the H1·2(PYR) (Fig. S8a†) and H1·2(2MP) (Fig. S8b†) complexes, where both H:G ratios were 1:2, were in close accordance with the expected mass losses (21.5 and 23.2% were measured while 21.1 and 23.9% were calculated, Table 5). However, in the case of both H1·3MP and H1·2(4MP) (Fig. S8c and d†), the mass losses that were expected (13.6 and 23.9%) were significantly higher than the measurements made in these experiments (10.1 and 7.0%). A plausible reason for this is that some guest may have escaped from the crystals during sample preparation, indicating that these two complexes were unstable at room temperature. While PYR was released in a multi-stepped manner from H1·2(PYR), the escape was in a simple single step for the 2MP-containing complex (in the latter case, the small inflection below 50 °C is attributed to the low boiling petroleum ether that was used to wash the crystals). Here, since the onset temperature (Ton) for the guest release process was the highest for H1·2(2MP) (63.2 compared with 54.6 °C for the PYR-containing complex, with the remaining two complexes being unstable under ambient conditions (3MP and 4MP)), this complex thus possessed the greatest thermal stability of the four, which agrees with the guest/guest competition experiments, where 2MP was demonstrated to be favoured by H1. This may be as a result of the shorter H-bond between host and guest molecules as observed from SCXRD data. Note that the guest release events for all four complexes were followed by the host melt and/or decomposition process as can be discerned in both the DSC and TG traces.
In the case of the H2·2(PYR) (Fig. S9a†) and 2(H2)·4MP (Fig. S9b†) complexes, expected (21.0 and 7.2%) mass losses were in close agreement with the experimentally-obtained measurements (19.8 and 6.5%). In the first of these, guest release occurred in two distinct steps while 4MP escaped in a singular event. Once more, and as was observed for the four complexes of H1, the preferred guest species of H2 (viz. PYR) was bound more tightly in the complex than 4MP, as was demonstrated by the greater Ton for H2·2(PYR) (76.5 °C) compared with that of 2(H2)·4MP (68.2 °C). Therefore, the complex containing the favoured PYR guest species formed the more stable complex, and this again was predicted by the guest/guest competition experiments (which favoured PYR) and was explained by SCXRD data (the host and PYR guest molecules in this case experienced statistically significantly shorter classical H-bonds than in 2(H2)·4MP). Finally, both TG traces demonstrated that host decomposition events followed that of the guest release processes.
4. Conclusion
Here, H2 was demonstrated to be a significantly more selective host compound than H1 in PYR and MP guest mixtures. In the single solvent experiments, H1 complexed with each of the four guest pyridines while H2 only formed complexes with PYR and 4MP. The equimolar binary guest experiments for H1 showed that 2MP remained the most favoured guest compound, while H2 preferred PYR. These results agreed with those from the equimolar ternary and quaternary guest competition experiments. Selectivity profiles constructed with H1 as the host compound demonstrated that it is able to separate mixtures of 2MP/PYR when these mixtures contain 40% 2MP (K = 11.8). Furthermore, H2 possessed an exceptional separation potential for PYR/2MP and PYR/3MP, in favour of PYR, even when the amount of PYR in these mixtures was low (20%). Numerous noncovalent interactions were identified by SCXRD analyses, and significantly shorter hydrogen bonding contacts in both H1·2(2MP) and H2·2(PYR) explained the affinity of these host compounds for 2MP and PYR, respectively. Additionally, thermal analyses showed that the complexes of H1 and H2 with their favoured guest compounds (2MP and PYR) had the greater thermal stabilities relative to the other complexes for each host compound. Therefore, both H1 and H2 do indeed have the ability, under certain conditions, to serve as candidates for the separation of some of these pyridine mixtures through host–guest chemistry strategies, as demonstrated by the results obtained in these investigations.Author contributions
Benita Barton: conceptualization; funding acquisition; methodology; project administration; resources; supervision; visualization; writing – original draft. Mino R. Caira: resources; visualization; formal analysis. Danica B. Trollip: investigation; methodology; validation. Eric C. Hosten: data curation; formal analysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support is acknowledged from the Nelson Mandela University and the National Research Foundation (NRF) of South Africa (NRF Grant number UID 144918). MRC thanks the University of Cape Town for access to research facilities.References
- E. Klingsberg, Heterocyclic compounds: Pyridine and its derivatives, Part 1, John Wiley & Sons, New York, 2019 Search PubMed.
- E. F. V. Scriven, J. E. Toomey and R. Murugan, Pyridine and pyridine derivatives, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York, 1996 Search PubMed.
- N. G. Grigor'eva, N. A. Filippova, M. I. Tselyutina and B. I. Kutepov, Synthesis of pyridine and methylpyridines over zeolite catalysts, Appl. Petrochem. Res., 2015, 5, 99–104 CrossRef.
- S. Shimizu, N. Watanabe, T. Kataoka, T. Shoji, N. Abe, S. Morishita and H. Ichimura Pyridine and pyridine derivative, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2000 Search PubMed.
- U. Kameswari, C. S. Swamy and C. N. Pillai, Methylation of pyridine over zeolites, Stud. Surf. Sci. Catal., 1994, 84, 1959–1964 CrossRef CAS.
- R. L. Frank and R. P. Seven, Pyridines. IV. A study of the Chichibabin synthesis, J. Am. Chem. Soc., 1949, 71, 2629–2635 CrossRef CAS.
- R. N. Senapati, D. D. Pathak, P. Dutta, H. Agarwalla and G. Sahu, Vapour phase synthesis of 2-methylpyridine and 4-methylpyridine over potassium salts of 12-tungstophosphoric acid, J. Sci. Ind. Res., 2022, 81, 814–820 CAS , https://nopr.niscpr.res.in/handle/123456789/60254, [Accessed 31 July 2023].
- V. Broughton, Reference Reviews, van Nostrand's Encyclopedia of Chemistry, 5th edn, 2006, vol. 20, pp. 45–46 Search PubMed.
- N. Cullinane, S. Chard and R. Meatyard, The preparation of methylpyridines by catalytic methods, Journal of the Society of Chemical Industry, 1948, 67, 142–143 CrossRef CAS.
- E. Coulson and J. Jones, Studies in coal tar bases. I. Separation of β- and γ-picolines and 2:6-lutidine, Journal of the Society of Chemical Industry, 1946, 65, 169–175 CrossRef CAS.
- H. Zhao, G. Xiao and J. Lu, Separation of picolines by dissociation extraction, J. Southeast Univ., Engl. Ed., 2005, 21, 68–72 CAS.
- S. Wren, Optimization of pH in the electrophoretic separation of 2-, 3-, and 4-methylpyridines, J. Microcolumn Sep., 1991, 3, 147–154 CrossRef CAS.
- J. L. Atwood and J. W. Steed, Encyclopedia of Supramolecular Chemistry, CRC Press, Marcel Dekker, Inc., New York, 2004, vol. 1 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd., USA, 2009 Search PubMed.
- J. Bacsa, M. R. Caira, A. Jacobs, L. R. Nassimbeni and F. Toda, Complexation with diol host compounds. Part 33. Inclusion and separation of pyridines by a diol host compound, Cryst. Eng., 2000, 3, 251–261 CrossRef CAS.
- B. Barton, M. R. Caira, E. C. Hosten and C. W. McCleland, A computational, X-ray crystallographic, and thermal stability analysis of TETROL and its pyridine and methylpyridine inclusion complexes, Tetrahedron, 2013, 69, 8713–8723 CrossRef CAS.
- B. Barton, E. C. Hosten and D. V. Jooste, Comparative investigation of the inclusion preferences of optically pure versus racemic TADDOL hosts for pyridine and isomeric methylpyridine guests, Tetrahedron, 2017, 73, 2662–2673 CrossRef CAS.
- B. Barton, L. de Jager and E. C. Hosten, Host proficiency of N,N'-bis(9-phenyl-9-thioxanthenyl)ethylenediamine for pyridine and the methylpyridine guests - a competition study, Supramol. Chem., 2018, 30, 61–71 CrossRef CAS.
- B. Barton, M. R. Caira, D. V. Jooste and E. C. Hosten, Investigation of the separation potential of xanthenyl- and thioxanthenyl-based host compounds for pyridine and isomeric picoline mixtures, J. Inclusion Phenom. Macrocyclic Chem., 2020, 98, 223–235 CrossRef CAS.
- B. Barton, M. R. Caira, D. V. Jooste and E. C. Hosten, Alternative purification protocols of mixed pyridines in the presence of trans-N,N′-bis(9-phenyl-9-xanthenyl)cyclohexane-1,4-diamine, J. Inclusion Phenom. Macrocyclic Chem., 2021, 99, 235–243 CrossRef CAS.
- B. Barton, L. de Jager, U. Senekal, E. Ferg and E. C. Hosten, Potential facile separation strategy for mixtures of 3- and 4-methylpyridine by employing N,N′-bis(9-phenyl-9-xanthenyl)ethylenediamine as an alternative host compound, J. Inclusion Phenom. Macrocyclic Chem., 2021, 100, 233–241 CrossRef CAS.
- B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, Selectivity considerations of host compound trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid when presented with pyridine and picoline mixtures: charge-assisted versus classical hydrogen bonding, CrystEngComm, 2022, 24, 4573–4583 RSC.
- B. Barton, M. R. Caira, U. Senekal and E. C. Hosten, Complementary host behaviour of three anthracenyl-derived roof-shaped compounds in mixed pyridines, CrystEngComm, 2023, 25, 1740–1754 RSC.
- E. Weber, T. Hens, O. Gallardo and I. Csöregh, Roof-shaped hydroxy hosts: synthesis, complex formation and X-ray crystal structures of inclusion compounds with EtOH, nitroethane and benzene, J. Chem. Soc., Perkin Trans. 2, 1996, 737–745 RSC.
- A. Bruker, APEX2, SADABS and SAINT, Bruker AXS Inc., Madison (WI), USA, 2010 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- U. Bruker, APEX3 v2019.1–0, SAINT V8.40A, Bruker AXS Inc., Madison (WI), USA, 2019 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, X-Seed−A software tool for supramolecular crystallography, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- B. Barton, R. Betz, M. R. Caira, E. C. Hosten, C. W. McCleland, P. L. Pohl and B. Taljaard, Clathrates of novel ethylenediamine derivatives: thermal, X-ray crystallographic and conformational analysis of inclusion complexes of N,N'-bis(5-phenyl-5-dibenzo[a,d]cycloheptenyl)ethylenediamine and its 10,11-dihydro analogue, Tetrahedron, 2016, 72, 7536–7551 CrossRef CAS.
- A. M. Pivovar, K. T. Holman and M. D. Ward, Shape-selective separation of molecular isomers with tunable hydrogen-bonded host frameworks, Chem. Mater., 2001, 13, 3018–3031 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: from visualization to analysis. Design and prediction, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- N. M. Sykes, H. Su, E. Weber, S. A. Bourne and L. R. Nassimbeni, Selective enclathration of methyl- and dimethylpiperidines by fluorenol hosts, Cryst. Growth Des., 2017, 17, 819–826 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The crystallographic data for H1·2(PYR), H1·2(2MP), H1·2(4MP), H2·2(PYR) and 2(H2)·3(4MP) were deposited at the Cambridge Crystallographic Data Centre (CCDC) and their CCDC numbers are 2218843, 2241126, 2241127, 2241128 and 2241129. The 1H NMR spectra for the six single solvent complexes, all selectivity profiles, relevant PXRD patterns, ORTEP and similar diagrams and thermograms have also been provided in the ESI. The noncovalent interactions present in these complexes have also been tabulated. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00811h |
| This journal is © The Royal Society of Chemistry 2023 |