
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the effect of substitution patterns on the symmetry of hydrogen-bonded supramolecular motifs in functionalized benzosiloxaboroles†
Krzysztof
Durka
 *a,
Adam
Zuba
a,
Paulina H.
Marek-Urban
ab,
Krzysztof
Nowicki
a,
Jakub
Drapała
a,
Krzysztof
Woźniak
b and
Sergiusz
Luliński
*a
*a,
Adam
Zuba
a,
Paulina H.
Marek-Urban
ab,
Krzysztof
Nowicki
a,
Jakub
Drapała
a,
Krzysztof
Woźniak
b and
Sergiusz
Luliński
*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: krzysztof.durka@pw.edu.pl; sergiusz.lulinski@pw.edu.pl
bDepartment of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
First published on 20th October 2023
Abstract
Crystal structures of a series of 26 functionalized 3-hydroxybenzo[c][1,2,3]siloxaboroles were compared taking into account electronic and steric effects of substituents at the aromatic ring on the hydrogen-bond (HB) motifs involving B–OH groups. The supramolecular assemblies of those compounds show strong variation depending on the number, position and type of substituents. Thus, HB dimers, trimers, tetramers and chains are formed. Most 7-substituted derivatives are isomorphous and crystallize in the I41/a tetragonal space group of symmetry featuring cyclic propeller-like tetramers as a characteristic structural motif. DFT calculations revealed that all observed HB motifs are characterized by similar stabilization energies ranging from −25 to −35 kJ mol−1 per molecule, which rationalizes the strong diversification of HB motifs in the studied structures.
Introduction
Arylboronic acids and their derivatives continue to attract enormous interest in various areas of chemistry and medicine. Benzoxaboroles are cyclic hemiesters of arylboronic acids which recently gained strong attention due to their promising and diverse biological activity (Scheme 1).1–4 They are characterized by improved thermodynamic stability and low toxicity. For instance, 5-Fluorobenzoxaborole (tavaborole) is currently used for the treatment of onychomycosis,5 while 5-(4′-cyanophenoxy)benzoxaborole (crisaborole) possesses strong anti-inflammatory activity and is used as a nonsteroidal topical agent for treatment of mild-to-moderate eczema.6,7 Another interesting example would be epetraborole, which is in a phase II/III clinical study now (NCT05327803) as an antibacterial agent against Mycobacterium avium complex lung disease.8 The knowledge of structural aspects of these compounds may be important, e.g., it may enable a deeper understanding of mechanistic aspects of their interactions with molecular targets. Thus, the crystal structures of various benzoxaboroles and related oxaboracyclic systems were determined followed by further studies concentrated on mechanistic aspects of their action. A Cambridge Structural Database (version 2022.3.0) search gives 50 hits of benzoxaboroles including structures functionalized with halogen atoms,9–11 alkoxy,12–16 aryloxy17 or formyl18–20 groups at various positions of aromatic rings. Derivatives bearing alkyl,13,21,22 aryl,23–27 heteroaryl28 or heterocyclic (morpholine, thiomorpholine, diazine etc.)19,29–33 groups at the C3 position were also structurally characterized. It is also worth noting that benzoxaboroles were used as co-crystal components with 4,4′-bipyridine and related HB acceptors.17,34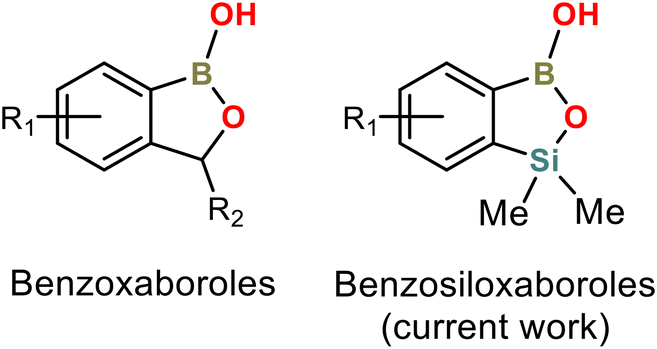 | ||
| Scheme 1 General molecular structures of benzoxa- and benzosiloxaboroles. | ||
Our group has been focused on the synthesis and physicochemical characterization of heteroelement analogues of benzoxaboroles, paying special attention to benzosiloxaboroles – a new class of benzo-fused heterocycles characterized by improved Lewis acidity, high stability and promising antimicrobial activity.35,36 They can be considered as silicon bioisosteres of benzoxaboroles, where the methylene group is replaced by the bulkier SiMe2 group (Scheme 1). Benzosiloxaboroles were identified as potent antifungal and antibacterial agents, especially against Gram-positive strains including S. aureus, S. epidermidis and E. faecalis.37 We have also found that they can be used as inhibitors of KPC/AmpC β-lactamases, which are enzymes responsible for antibiotic resistance developed in clinical bacterial strains. Benzosiloxaboroles also show high affinity towards biologically relevant diols such as dopamine, AMP and selected sugars, indicating their possible application in chemosensing devices.35,38 Synthetic routes are generally different between benzoxa- and benzosiloxaboroles, opening new possibilities for functionalization, especially at the C6 and C7 positions, e.g., in the vicinity of the silicon atom.39 Thus, over one hundred benzosiloxaboroles with diverse substitution patterns have already been obtained with the prospect of being able to synthesize hundreds more.40–42 We have already published several crystal structures of these compounds but we did not perform their detailed analysis. Herewith, we present a comprehensive study focused on the evaluation of substituent effect on crystal packing of these compounds. X-ray diffraction analysis was supplemented by relevant computational studies.
Results and discussion
Main structural motifs in benzoxaboroles
A CSD search revealed that benzoxaboroles tend to form R22(8)43 HB dimers due to mutual interactions of boronic groups resembling the motif characteristic for arylboronic acids (ArB(OH)2).44–49 However, in contrast to arylboronic acids which can act as double donors and double acceptors of HB, oxaboroles contain only one hydroxyl group, which suppresses the further propagation of HB interactions within crystal structures. The R22(8) dimeric motif was encountered in 20 out of 42 crystal structures of benzoxaboroles (erroneous structures and salts were excluded from the analysis). In the remaining cases, the B–OH group is arranged in the HB interaction with an R1 or R2 functional group (Scheme 2) or second crystal component, i.e., solvent molecule or co-crystal partner. In turn, the presence of a HB acceptor next to the boronic group favours an intramolecular HB.19,34 Taking into consideration only simple halogen, alkyl or alkoxy benzoxaborole derivatives, a dimeric motif featuring syn conformation of both B–OH groups is strongly preferred as it appears in 15 out of 17 crystal structures. In the remaining two structures (CSD refcodes: MIHLIB21 and QEXYUQ17), B–OH groups adopt an anti conformation, forming infinite molecular chains via repeatable HBs with endocyclic oxygen atoms. In the context of this study, the second structure deserves special attention as the oxaborole C3 carbon atom is substituted with two methyl groups which increase the steric hindrance in the vicinity of the endocyclic oxygen atom, weakening the tendency towards dimerization. Thus, the occurrence of other structural motifs can be expected in similar systems including benzosiloxaboroles. | ||
| Scheme 2 Main crystal motifs encountered in the crystal structures of benzoxaboroles according to a CSD search. | ||
Scope of investigated structures
In the current study we have investigated 27 crystal structures of benzosiloxaboroles. We have selected relatively simple compounds in order to understand the basic relation between molecular structure and the specificity of intermolecular contacts present in the respective crystal structures. Consequently, we measured 23 new crystal structures of benzosiloxaboroles substituted with a halogen atom (F, Cl, Br, I), CF3, CHO, CN, B(OH)2, and CO2Me groups at various positions of the aromatic ring (Table 1). They were abbreviated according to the position, quantity and type of substituent attached to the aromatic ring. For instance, 6,7-difluoro-1,1-dimethyl-3-hydroxybenzo[c][1,2,3]siloxaborole was abbreviated as 67-dF. Notably, compound 56-dF was found to crystallize in two polymorphic forms abbreviated as 56-dF-I and 56-dF-II. We have also included in this study 4 already published structures, i.e., 4567-tF,356-OMe-7-Cl,426-OTBDMS-7Cl39 and 4-CHO-67-dF.37 Although there are also several other known crystal structures of benzosiloxaboroles, they are characterized by more complex molecular and crystal structures based on multiple HB interactions with functional groups attached to the aromatic ring. Thus, they were excluded from the current analysis. Finally, we would like to point out that despite numerous attempts we have failed to grow single crystals of the unsubstituted benzosiloxaborole as, in contrast to functionalized derivatives, this compound is in the liquid state at ambient temperature.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Substitution pattern | Space group | Z′ | B–OH conform. | Ref. synth. | |||
| 1 | 2 | 3 | 4 | |||||
| a The structure contains one acetone molecule in the ASU. | ||||||||
| Mono-substituted | ||||||||
| 4-CF3 | CF3 | — | — | — | P21/c | 1 | Syn | New |
| 5-F | — | F | — | — | P21/n | 2 | Syn | New |
| 6-F | — | — | F | — | Pna21 | 2 | Anti | 38 |
| 6-Cl | — | — | Cl | — | P21/n | 2 | Syn | 38 |
| 6-CN | — | — | CN | — | P21/c | 2 | Syn | 43 |
| 6-CF3 | — | — | CF3 | — | P21/n | 2 | Syn | 38 |
| 7-F | — | — | — | F | I41/a | 1 | Syn | 40 |
| 7-Cl | — | — | — | Cl | I41/a | 1 | Syn | 38 |
| 7-Br | — | — | — | Br | I41/a | 1 | Syn | 38 |
| 7-CF3 | — | — | — | CF3 | I41/a | 1 | Syn | New |
| 7-CHO | — | — | — | CHO | I41/a | 1 | Syn | 40 |
| 7-TMS | — | — | — | SiMe3 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
2 | Syn | New |
| 7-I | — | — | — | I | Pbca | 1 | Syn | New |
| 7-BOH2 | — | — | — | B(OH)2 | P21/c | 1a | Anti | 38 |
| Di-substituted | ||||||||
| 4-CO2Me-7F | CO2Me | — | — | F | P21/n | 1 | Anti | 32 |
| 4-CN-7-F | CN | — | — | F | P21/c | 1 | Syn | 43 |
| 56-dF-I | — | F | F | — |
P |
3 | Syn | 36 |
| 56-dF-II | — | F | F | — | P21/c | 1 | Syn | 36 |
| 67-dF | — | — | F | F | P21/n | 3 | Syn + anti | 38 |
| 67-dCl | — | — | Cl | Cl | P21/n | 2 | Syn | 38 |
| 6-CHO-7F | — | — | CHO | F | P21/m | 1 | Syn | 38 |
| 6-OMe-7-Cl | — | — | OMe | Cl |
P |
4 | Syn | 41 |
| 6-OTBDMS-7Cl | — | — | OSiMe2tBu | Cl | P21/n | 1 | Syn | 41 |
| 56-dO | — |
|
— | P21/n | 1 | 38 | ||
| Tri-substituted | ||||||||
| 4-CHO-67-dF | CHO | — | F | F |
P |
1 | Anti | 43 |
| 57-dF-6-CHO | — | F | CHO | F |
P |
1 | Syn | 43 |
| Tetra-substituted | ||||||||
| 4567-tF | F | F | F | F | Pbcm | 1 | Syn | 36 |

Synthesis
The synthesis of most of the studied compounds was reported in our previous publications (proper references are given in Table 1). For the purposes of this study, we have obtained 5 new benzosiloxaboroles including 4-CF3, 5-F, 7-CF3, 7-TMS and 7-I. Compound 5-F was prepared from 2-bromo-4-fluoro-1-iodobenzene using a protocol involving I/Mg exchange,42 silylation with Me2Si(H)Cl and conversion of an intermediate arylsilane to the respective final product using a sequence of Br/Li exchange, boronation and hydrolysis (Scheme 3a). Both regioisomeric benzosiloxaboroles 4-CF3 and 7-CF3 were obtained from 2-bromobenzotrifluoride using a protocol similar to that elaborated for 5-F except that deprotonative lithiation with LTMP was performed prior to silylation (Scheme 3b).50 The key step in the synthesis of 7-CF3 was the basicity-gradient-driven thermally induced isomerization (“halogen dance” mechanism) of 2-bromo-3-(trifluoromethyl)phenyllithium to thermodynamically more stable 2-bromo-6-(trifluoromethyl)phenyllithium which gave rise to the desired arylsilane precursor pre-7-CF3. Compound 7-TMS was obtained from 1,3-dibromobenzene using a three-step sequence (Scheme 3c). First, 1,3-dibromo-2-(dimethylsilyl)benzene37 was converted to 1-bromo-2-(dimethylsilyl)-3-(trimethylsilyl)benzene pre-7-TMSvia Br/Li exchange and subsequent trapping of the intermediate aryllithium with Me3SiCl. Finally 7-TMS was obtained as described above for CF3-substituted derivatives. ipso-Iododesilylation of 7-TMS was cleanly accomplished using ICl in CH2Cl2 giving rise to the product 7-I. It should be noted that the reaction occurred chemoselectively at the C–SiMe3 bond whilst the adjacent C–SiMe2O remained intact.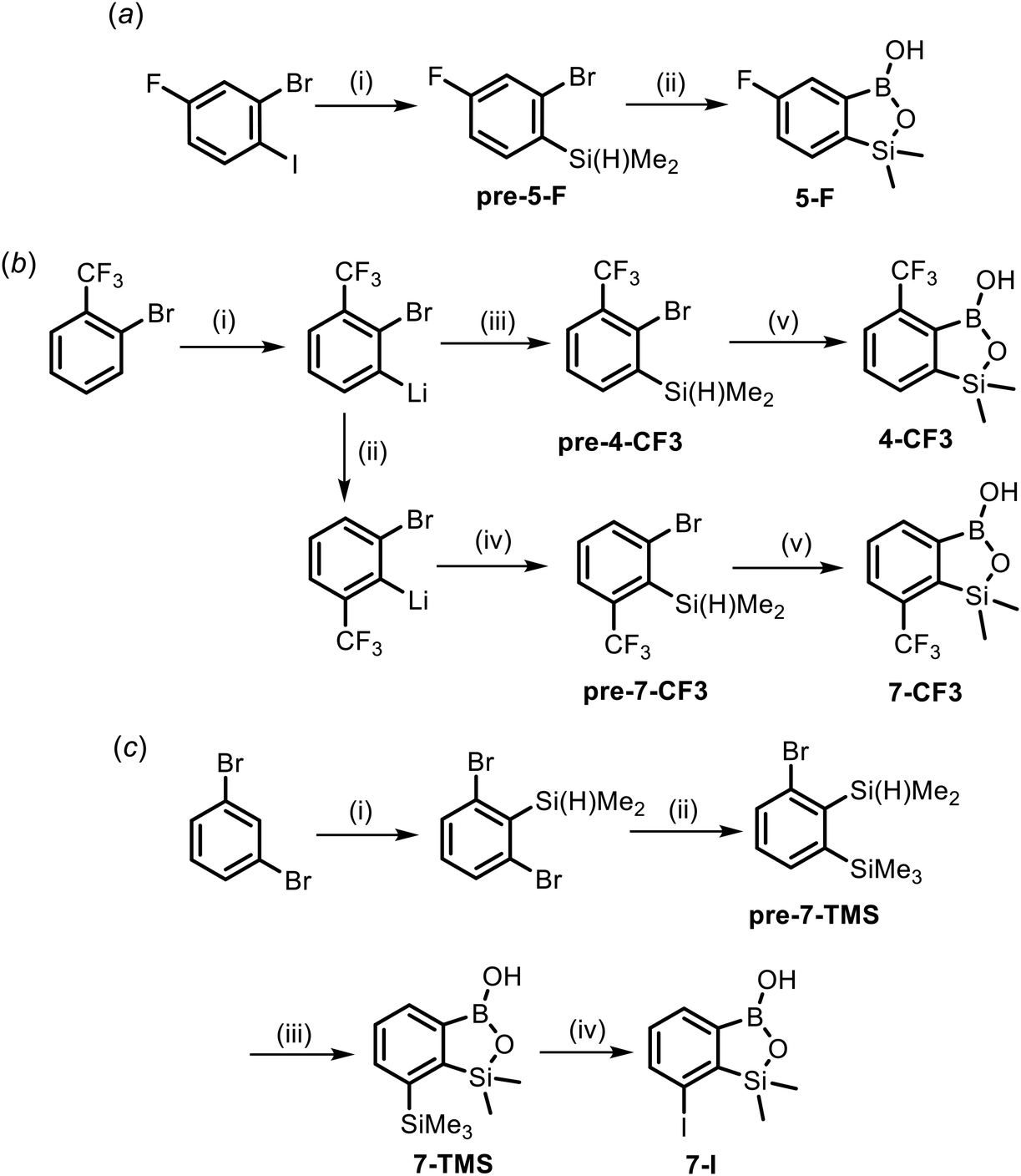 | ||
| Scheme 3 (a) Synthesis of benzosiloxaborole 5-F. Conditions: (i) (1) i-PrMgCl·LiCl, THF, −70 °C, (2) Me2Si(H)Cl; (ii) (1) t-BuLi, Et2O, −90 °C, (2) B(OMe)3, (3) 1 M aq. NaOH, (4) 1 M aq. HCl. (b) Synthesis of benzosiloxaboroles 4-CF3 and 7-CF3. Conditions: (i) LTMP, THF, −100 °C; (ii) ΔT (−75 °C); (iii) Me2Si(H)Cl, −100 °C; (iv) Me2Si(H)Cl, −75 °C; (v) (1) t-BuLi, Et2O, −90 °C, (2) B(OMe)3, (3) 1 M aq. NaOH, (4) 1 M aq. HCl. (c) Synthesis of benzosiloxaboroles 7-SiMe3 and 7-CF3. Conditions: (i) (1) LDA, THF, −75 °C, (2) Me2Si(H)Cl; (ii) (1) t-BuLi, Et2O, −90 °C, (2) Me3SiCl, THF, ΔT (25 °C); (iii) (1) t-BuLi, Et2O, −90 °C, (2) B(OMe)3, (3) 1 M aq. NaOH, (4) 1 M aq. HCl; (iv) (1) ICl, CH2Cl2, rt, (2) 1 M aq. Na2SO3. | ||
Molecular structures
Single crystals of benzosiloxaboroles were obtained by slow evaporation of the corresponding CHCl3 or acetone solutions under ambient air conditions. Benzosiloxaboroles form single crystals with well-defined faces and ordered crystal structures characterized by low R-factor values (usually below 5%). The molecular structures are presented in Fig. S1 and S2,† while crystal data and refinement parameters are summarized in Tables S1–S5.† Basic geometric parameters are presented in Tables S6 and S7.†The analysis of the molecular structures showed that the aromatic substituents only slightly affect the geometry of the siloxaborole ring. The C–B, Si–O and C–Si bond distances are in the ranges of 1.55–1.59 Å, 1.65–1.71 Å and 1.86–1.89 Å, respectively (Fig. 1). Endocyclic B–O bonds are slightly longer (1.36–1.41 Å) than exocyclic ones (1.33–1.37 Å) and the same relation is observed for benzoxaboroles. At this point it should be noted that B–O and B–C bond distances in arylboronic acids range from 1.32 to 1.40 Å and 1.54 to 1.62 Å, respectively (Fig. S6†). Thus, the basic geometric parameters of both benzoxa- and benzosiloxaboroles follow those observed for arylboronic acids. Regarding the oxaborole ring geometry, the presence of longer Si–O (benzosiloxaboroles) vs. C–O (benzoxaboroles) bonds is reflected by smaller B–O–Si (93°–96°) vs. B–O–C (109°–112°) bond angles. Conversely, endocyclic C–B–O bond angles are larger in benzosiloxaboroles (112°–114°) than in benzoxaboroles (106°–110°). These differences may also result in part from the presence of two methyl groups at the silicon atom compared to a simple methylene fragment in most benzoxaboroles. This so-called gem-dimethyl effect51,52 was also observed for benzoxaborole substituted with two methyl groups at the oxaborole C3 carbon atom.53 In accordance with our previous studies, the elongation of an endocyclic B–O bond in benzosiloxaboroles may be ascribed to some extent to a competitive Si–O bond hyperconjugation35 which reduces the basicity of the endocyclic oxygen atom despite the higher polarity of the Si–O vs. the C–O bond.54 Taking into consideration the lower basicity of the endocyclic oxygen atom and the steric effect of the SiMe2 group, it could be expected that at the expense of centrosymmetric dimers, other HB motifs would be relatively favored for benzosiloxaboroles.
 | ||
| Fig. 1 Histograms showing the distributions of (a) bond distances (Å) and (b) bond angles (°) within the siloxaborole ring. The analysis was performed for 27 crystal structures of benzosiloxaboroles (23 new and 4 contained in the CSD). Note that some structures consist of more than one molecule in the ASU (Table 1). | ||
Similar to benzoxaboroles, the BOH group in benzosiloxaboroles typically adopts the syn conformation. The anti conformation was naturally imposed by an intramolecular O–H⋯O HB interaction with an adjacent carbonyl group in 4-CO2Me-7F and 4-CHO-67-dF. Such a conformation was also observed for structures 6-F and 67-dF where a HB chain was the primary supramolecular motif.
Crystal structure analysis
The majority of the studied compounds crystallize in the centrosymmetric triclinic (P) and monoclinic (in most cases P21/c) crystal systems. In contrast, five out of eight 7-monosubstituted derivatives form isomorphous networks with I41/a crystal symmetry. Most of the studied systems crystallize in non-solvated forms and feature a compact molecular packing. Exceptionally, structure 7-BOH2 contains acetone molecules, while 6-F incorporates hexane molecules in its channel-type network. In most cases, the asymmetric unit (ASU) consists of one or two benzosiloxaborole molecules, although 56-dF-I and 67-dCl crystallize with 3 molecules in the ASU. In the case of 56-dF-I, the primary HB motif is a trimer, while the structure 67-dCl comprises two types of HB motifs, i.e., a dimer and a molecular chain. In contrast, the second polymorphic form 56-dF-II crystallizes with only one molecule in the ASU and forms a centrosymmetric R22(8) dimer. In the most peculiar case, 6-OMe-7-Cl, the ASU contains 4 molecules. Despite the fact that they possess the same conformation and are arranged in dimeric HB motifs, they have different crystal environments resulting from different involvement in secondary C–H⋯O, O⋯Cl and C(π)⋯B interactions.
The comparative analysis shows that benzosiloxaboroles offer a significantly larger variety of HB motifs with respect to benzoxaboroles. Undoubtedly, the most abundant motif is an R22(8) dimer involving two HBs formed by BOH groups (Fig. 2). However, it is observed in only 11 out of 26 considered structures (42%), i.e., less frequently compared to benzoxaboroles (48%). These differences become much more pronounced (50% vs. 88%) when only derivatives bearing simple substituents (halogen, CF3, SiMe3 and alkoxy) are to be considered. The dimers can involve either two molecules related by the center of symmetry or two symmetrically independent molecules. The latter case allows for a wider diversification of secondary intermolecular contacts. For example, in 6-Cl one molecule is involved in two sets of Oexo⋯B and C(Si)–H⋯C(π) interactions forming a discrete secondary dimeric motif, while its HB partner aggregates through C(Si)–H⋯C(π) interactions, forming an independent one-dimensional chain. Moreover, dimers comprising two symmetrically non-equivalent molecules usually deviate from a planar arrangement. This is especially visible for the dimer of 7-TMS, where the siloxaborole planes are twisted by 32(1)° (Fig. 2b).
 | ||
| Fig. 2 HB dimers in (a) 4-CF3 and (b) 7-TMS. Ellipsoids are drawn at 50% probability level. HBs are marked with red dashed lines. | ||
Unlike benzoxaboroles, benzosiloxaboroles show a strong tendency to form four-molecule assemblies. This is especially visible for the series of isomorphous derivatives substituted with fluorine, chlorine, bromine, trifluoromethyl and formyl groups at the 7-position, i.e., ortho with respect to the SiMe2 group. They crystallize in the I41/a tetragonal space group of symmetry and feature cyclic propeller-like R44(8) tetramers as a main structural motif. Tetramers adopt the chair conformation and are held by four sets of O–H⋯Oexo and C(Ar)–H⋯Oendo HBs (Fig. 3a). Although the aromatic substituents are outside the interaction region, we have found that they significantly affect the geometry of the HB inner structure. Specifically, the O⋯O distances decrease with the increase of the size of the substituent, making the entire tetramer more compact (Table 2). In order to validate the observed structural tendencies, we have obtained further two derivatives carrying bulkier substituents, i.e., iodine (7-I) and a trimethylsilyl group (7-TMS). However, both structures feature dimeric motifs, indicating that the formation of dimeric or tetrameric motifs can be controlled by the size of a substituent. Analysis of the crystal structures of 5-F and 67-dCl revealed a different tetrameric assembly (graph set notation D33(9)[R22(8)]). It consists of two molecules forming a central dimeric structure to which two other molecules are attached by lateral HBs (Fig. 3b). It is supported by the pairs of auxiliary C(Ar)–H⋯Oexo and C(Si)–H⋯Oendo interactions. A somewhat related ternary assembly motif (graph set notation D11(3)R22(8)) was found for 56-dF-I where one pendant siloxaborole molecule is attached to a central dimer (Fig. S3†).
 | ||
| Fig. 3 Two types of HB tetramers in (a) 7-F and (b) 5-F. | ||
| HB motive | d O⋯O/Å | E constint/kJ mol−1 | E optint/kJ mol−1 | |
|---|---|---|---|---|
| a The structure comprises two types of HB motifs – dimer (R22(8)) and chain (C22(6)). b Lateral HB with pendant benzosiloxaborole molecule from D33(9)[R22(8)] tetramer. c HB arranging exocyclic (exo) or endocyclic (endo) siloxaborole oxygen atoms. d HB with acetone molecule. e The intramolecular HB energy was estimated from the energy difference between two rotamers with CO2Me/CHO groups either coplanar or perpendicular to the siloxaborole ring plane. Thus only one value is provided. f Optimization led to a very different geometry from that observed in the crystal structure. | ||||
| 4-CF3 | Dimer R22(8) | 2.808(2) | −27.1 | −28.6 |
| 5-F | Tetramer D33(9)[R22(8)] | 2.733(1) | −33.9 | −35.8 |
| 2.761(1) (L)b | ||||
| 6-F | Chain C22(4) | 2.733(3) | −22.8/−33.2 | —f |
| 2.724(3) | ||||
| 6-Cl | Dimer R22(8) | 2.809(1) | −27.2 | −28.6 |
| 2.738(1) | ||||
| 6-CN | Dimer R22(8) | 2.753(2) | −27.0 | −28.1 |
| 2.781(2) | ||||
| 6-CF3 | Dimer R22(8) | 2.772(1) | −27.4 | −28.4 |
| 2.764(1) | ||||
| 7-F | Tetramer R44(8) | 2.750(2) | −31.2 | −31.5 |
| 7-Cl | Tetramer R44(8) | 2.710(2) | −31.4 | −31.7 |
| 7-Br | Tetramer R44(8) | 2.702(2) | −31.1 | −31.7 |
| 7-CF3 | Tetramer R44(8) | 2.685(1) | −31.8 | −31.8 |
| 7-CHO | Tetramer R44(8) | 2.717(2) | −32.8 | −32.8 |
| 7-TMS | Dimer R22(8) | 2.764(1)(endo)c | −27.3 | −29.3 |
| 2.815(1)(exo)c | ||||
| 7-I | Dimer R22(8) | 2.748(4) | −28.5 | −28.8 |
| 7-BOH2 | Chain C11(7)[R22(8)] | 2.696(1) (Ac)d | −31.9 | −33.0 |
| 2.814(1)(exo)c | ||||
| 2.748(1)(endo)c | ||||
| 4-CO2Me-7F | Intramolecular HB S11(7) | 2.638(4) | —e | −28.3f |
| 4-CN-7-F | Dimer R22(8) | 2.769(1) | −27.8 | −29.0 |
| 56-dF-I | Trimer D11(3)R22(8) | 2.841(3) (L)b | −30.9 | −33.4 |
| 2.781(3)(endo)c | ||||
| 2.708(3)(exo)c | ||||
| 56-dF-II | Dimer R22(8) | |||
| 67-dF | Dimer R22(8) + chain C22(6)a | 2.771(2) (R22(8)) | −26.0/−28.3 (R22(8)) | —f |
| 2.690(2) (C22(6)) | −25.5/−34.1 (C22(6)) | |||
| 2.733(2) (C22(6)) | ||||
| 67-dCl | Tetramer D33(9)[R22(8)] | 2.781(2) (L)b | −33.5 | −36.0 |
| 2.779(2) | ||||
| 6-CHO-7F | Chain C11(9) | 2.732(2) | −29.3 | −29.3 |
| 6-OMe-7-Cl | Dimer R22(8) | 2.767 | −27.2 | −28.6 |
| 2.782 | −26.8 | |||
| 2.753 | −28.5 | |||
| 2.787 | −26.9 | |||
| 6-OTBDMS-7Cl | Dimer R22(8) | 2.759 | −27.3 | −28.4 |
| 56-dO | Dimer R22(8) | 2.768(1) | −27.6 | −29.0 |
| 4-CHO-67-dF | Intramolecular HB S11(7) | 2.653 | —e | −27.7e |
| 6-CHO-57-dF | Chain C11(9) | 2.719(1) | −28.9 | −28.9 |
| 4567-tF | Dimer R22(8) | 2.744 | −28.5 | −29.9 |
Considering the chain HB motif in the crystal structure of 3,3-dimethylbenzoxaborole,17 it can be expected that benzosiloxaboroles should also arrange in linear assemblies. Indeed, the analysis of the crystal structure of 6-F revealed that the repeatable interactions of anti-oriented hydroxyl groups (dO⋯O = 2.718(3) Å) yielded an infinite zig-zag chain (C22(4), Fig. 4a). The adjacent chains are connected by weak C(Si)–H⋯F and C(Si)–H⋯C(π) interactions forming a 3-dimensional channel-type assembly filled with disordered hexane molecules (Fig. S3†). The introduction of another fluorine atom at the 7-position (67-dF) preserves the linearity of the main structural motif, but also reorganizes the molecular pattern through involvement of the endocyclic oxygen atom in the C22(6) HB chain. Consequently, it consists of molecules where exocyclic and endocyclic oxygen atoms alternately act as acceptors of the HB to the hydroxyl group (Fig. 4b). Interestingly, the 67-dF structure features also an independent R22(8) dimeric motif.
 | ||
| Fig. 4 HB chains in (a) 67-dF and (b) 6-F. | ||
Finally, the strong effect of functional groups possessing active HB sites is evident. For instance, in 7-BOH2 the heterodimeric interactions between the boronic group and the oxaborole ring lead to the formation of an infinite zig-zag C11(7)[R22(8)] chain (Fig. 5a). Both functional moieties adopt rare arrangements as the boronic group in the syn–syn configuration serves as a double HB donor to endo- and exocyclic oxaborole oxygen atoms while the anti-oriented oxaborole B–OH group acts as a HB donor to the acetone molecule. The HB distance with the endocyclic oxygen atom (dO⋯O = 2.748(1) Å) is much shorter with respect to the exocyclic one (dO⋯O = 2.814(1) Å). Furthermore, the HB interaction with acetone (dO⋯O = 2.696(1) Å) is one of the shortest in the studied series. In two other examples (6-CHO-7F, 6-CHO-57-dF), the supramolecular arrangement is governed by relatively strong HBs (6-CHO-7F: dO⋯O = 2.732(1) Å, 6-CHO-57-dF: dO⋯O = 2.719(1) Å) between boronic and formyl groups resembling the linear HB pattern (C11(9), Fig. 5b). Interestingly, in the case of 7-CHO, the formyl group is not involved in an intermolecular HB interaction with the BOH group. As already discussed, this structure retains an R44(8) tetrameric motif characteristic for other 7-monosubstituted derivatives. A quite different situation occurs when the carbonyl group is inserted at the ortho position relative to the B–OH group (4-position). For 4-CO2Me-7F and 4-CHO-67-dF structures, the boronic group adopts an anti-conformation and forms a strong intramolecular HB (S11(7)) with ester (dC(OMe)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯O = 2.636(3) Å) and formyl (dC(H)O⋯O = 2.653(5) Å) groups, respectively. Thus, both oxaborole oxygen atoms are accessible for interactions with HB donors. For 4-CO2Me-7F, they are bonded to aromatic C(Ar)–H protons (dC⋯O = 3.254(3) Å, 3.474(3) Å). The propagation of this motif along the [101] direction produces a molecular chain (Fig. 6). In the case of 4-CHO-67-dF weak HBs are formed with aromatic (dC⋯O = 3.440(5) Å) and formyl protons (dC⋯O = 3.199(5) Å), while the chain adopts a zig-zag pattern (Fig. S5†).
O⋯O = 2.636(3) Å) and formyl (dC(H)O⋯O = 2.653(5) Å) groups, respectively. Thus, both oxaborole oxygen atoms are accessible for interactions with HB donors. For 4-CO2Me-7F, they are bonded to aromatic C(Ar)–H protons (dC⋯O = 3.254(3) Å, 3.474(3) Å). The propagation of this motif along the [101] direction produces a molecular chain (Fig. 6). In the case of 4-CHO-67-dF weak HBs are formed with aromatic (dC⋯O = 3.440(5) Å) and formyl protons (dC⋯O = 3.199(5) Å), while the chain adopts a zig-zag pattern (Fig. S5†).
 | ||
| Fig. 5 HB molecular chains in (a) 6-CHO-7F and (b) 7-BOH2. | ||
 | ||
| Fig. 6 A fragment of the crystal structure of 4-CO2Me-7F showing the formation of intramolecular HBs and further molecular connections through C(Ar)-H⋯O interactions marked with grey dashed lines. | ||
Theoretical calculations
The comprehensive analysis of HB motifs in crystal structures of benzosiloxaboroles shows that the O⋯O distances are in the range of 2.684–2.861 Å, with a mean value of 2.750 Å and a standard deviation of 0.048 Å. This is comparable to benzoxaboroles where the O⋯O distances range from 2.704–2.867 Å with a mean value of 2.755 Å, and phenylboronic acids where the O⋯O distances range from ca. 2.67–2.90 Å with an average value of 2.75 Å. To further compare both classes of oxaboroles we have performed theoretical calculations using the M06-2X functional with the 6-311++G(d,p) basis set. The HB motifs were extracted from the corresponding crystal structures and then H atoms were optimized keeping the positions of other atoms fixed. In the second approach, the geometries of HB motifs were fully optimized (Table 2). It comes out that both procedures yielded very comparable interaction energy values, indicating that the geometries of HB motifs are only slightly affected by the crystal environment.We have also calculated the interaction energies for dimeric and tetrameric motifs for unsubstituted benzoxa- and benzosiloxaboroles (Table 3). The calculations revealed that HBs are slightly weaker in R22(8) dimeric and R44(8) tetrameric motifs of benzosiloxaborole, while the formation of a D33(9)[R22(8)] tetramer is energetically more advantageous, although the energy differences are rather small. The energy of R22(8) dimeric motifs in benzosiloxaboroles ranges from ca. −27 to −29 kJ mol−1 (per molecule) and is only slightly affected by the aromatic ring substitution. Comparing the interaction energies in R22(8) dimeric motifs in oxaboroles and phenylboronic acids (−22 to −25 kJ mol−1 per molecule),45,55,56 formation of dimers is more advantageous for the former systems due to the higher basicity of an endocyclic oxygen atom, thus serving as a better HB acceptor. Energy values estimated for intramolecular and intermolecular HBs with CHO, B(OH)2 and CO2Me functional groups are of a similar magnitude. Surprisingly, it appeared that the HB energy in the R44(8) tetramer is close to −31 kJ mol−1 per molecule, regardless of the substituent type. It is even higher when molecules are arranged in the D33(9)[R22(8)] tetrameric assembly (−34 kJ mol−1 per molecule). This implies that tetrameric motifs are energetically more favored compared to dimers. However, one should bear in mind that the occurrence of a particular crystal motif can be governed by the kinetic crystallization effects. For instance the fast crystallization of 56-dF from CHCl3 resulted in the formation of phase 56-dF-II comprising classical dimeric motifs. A slower crystallization from the same solvent yielded the polymorph 56-dF-I featuring a trimeric HB motif. Furthermore, aromatic substituents may promote specific intermolecular interactions including C–H⋯O, C–H⋯C(π), C(π)⋯B, O⋯B, C–H⋯F, C–H⋯Cl, halogen bonds, etc., thus changing the crystallization preferences.
| Benzoxaborole | Benzosiloxaborole | |
|---|---|---|
| Dimer R22(8) | −29.6 | −28.9 |
| Tetramer R44(8) | −33.6 | −31.3 |
| Tetramer D33(9)[R22(8)] | −34.8 | −36.1 |
In order to obtain deeper insight into the relative crystal stabilities, we have performed periodic calculations for selected isomeric benzosiloxaboroles bearing one fluorine (5-F, 6-F, 7-F), chlorine (6-Cl and 7-Cl) or trifluoromethyl (4-CF3, 6-CF3, 7-CF3) substituent. The obtained results are compared in Table 4. Considering the monofluorinated isomeric series, it is clear that the cohesive energies correspond to the interaction energies of the HB motif. Thus, 5-F based on the D33(9)[R22(8)] tetrameric HB structure is the most stable (Ecoh = −156 kJ mol−1), 7-F with the R44(8) tetrameric motif is characterized by slightly lower stability (Ecoh = −153 kJ mol−1) and 6-F, where molecules form a linear C22(4) chain motif, is less stable (Ecoh = −151 kJ mol−1). Similarly, the structure 7-Cl based on R44(8) tetramers is more advantageous (by 6 kJ mol−1) than 6-Cl with an R22(8) dimer. However, this trend is not preserved in CF3 benzosiloxaborole series, where the 7-CF3 structure (Ecoh = −179 kJ mol−1) with an energetically most stable R44(8) tetrameric motif is located in-between 4-CF3 (Ecoh = −182 kJ mol−1) and 6-CF3 (Ecoh = −174 kJ mol−1); both structures are based on R22(8) dimeric motifs. It should be noted that 4-CF3 is also characterized by the most compact structure within the CF3 series. This is reflected in a small lattice volume of 1120.6(6) Å3 (per molecule) compared to the other two systems [V(6-CF3) = 1152.9(1) Å3; V(6-CF3) = 1145.2(2) Å3). Such results indicate that the weak intermolecular interactions provide a similar contribution to the total stabilization energy, although some substituents and positions may significantly strengthen the lattice interactions.
| Structure/motif | 5-F/tetramer (D33(9)[R22(8)]) | 6-F/chain (C22(4)) | 7-F/tetramer (R44(8)) |
|---|---|---|---|
| E coh/kJ mol−1 | −156 | −151 | −153 |
| Structure/motif | 4-CF3/dimer (R22(8)) | 6-CF3/dimer (R22(8)) | 7-CF3/tetramer (R44(8)) |
| E coh/kJ mol−1 | −182 | −174 | −179 |
| Structure/motif | — | 6-Cl/dimer (R22(8)) | 7-Cl/tetramer (R44(8)) |
| E coh/kJ mol−1 | — | −155 | −161 |
Conclusions
A comparative study of 27 crystal structures of benzosiloxaboroles revealed that these compounds show higher structural diversity than benzoxaboroles. Besides R22(8) dimeric motifs, they also form trimers and tetramers as well as infinite molecular chains. This can be rationalized by similar stabilization energies of various types of aggregates. The appearance of a particular motif is affected by the substitution of the aromatic ring, although the lattice stabilization energies are comparable within the studied series and show correlation with energies of respective HB motifs. In this context, the crystallization kinetics can be of equal importance. Although the dimers are generally energetically less advantageous than tetramers, their appearance in crystal structures can be strongly promoted by the crystallization process. The monomer–dimer equilibrium can be affected by a substituent at the aromatic ring through its interaction with solvent molecules. Besides, benzosiloxaboroles, which are slightly less stabilized in their dimeric forms than benzoxaboroles, dissociate more easily, promoting the occurrence of other, energetically more favourable HB motifs.It should be stressed that the interactions with halogen substituents are generally weak and thus only marginally affect the primary HB motifs. Nevertheless, the analysis of crystal structures clearly showed that even small variations in the position and number of these substituents completely change the HB pattern. This was especially visible for fluorinated series of benzosiloxaboroles. The main crystal motifs range from R22(8) dimer (4567-tF, 67-dF, 56-dF-II), trimer (56-dF-I), R44(8) (7-F) and D33(9)[R22(8)] (5-F) tetramers to C22(4) (6-F) and C22(6) (67-dF) chains. Although it is difficult to rationalize the relationship between the molecular and the supramolecular structures for each of the discussed examples, we have found that 7-monosubstituted derivatives tend to form R44(8) cyclic tetrameric structures and the size of the substituent affects the geometry of a central HB motif.
Experimental section
General comments
Solvents used for reactions were dried by heating to reflux with sodium/benzophenone and distilled under argon. Selected aromatic starting materials and other reagents including alkyllithiums, 2,2,6,6-tetramethylpiperidine, trimethyl borate, chlorodimethylsilane, and chlorotrimethylsilane were used as received without further purification. In the 13C{1H} NMR spectrum of 7-I the resonance of boron-bound carbon atoms was not observed as a result of their broadening by a quadrupolar boron nucleus. 1H and 13C NMR chemical shifts are given relative to TMS using residual solvent resonances. 11B and 19F NMR chemical shifts are given relative to BF3·Et2O and CFCl3, respectively.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 40 mL) at −78 °C and the resulting suspension was stirred for 2 h. Chlorodimethylsilane (4.0 mL, 36.0 mmol) was added dropwise at −78 °C and the mixture was allowed to warm to rt and left overnight. Hexane (20 mL) was added and the mixture was quenched with water (20 mL). The organic phase was separated and the aqueous phase was extracted with Et2O (30 mL). The combined organic phase was dried with Na2SO4 and concentrated under reduced pressure. The oily residue was subjected to fractional distillation in vacuo to give crude 4-fluoro-2-bromo-1-(dimethylsilyl)benzene pre-5-F as a colorless liquid, b.p. 52–55 °C (5 × 10−3 mbar). Yield 6.2 g (89%). 1H NMR (300 MHz, CDCl3) δ 7.47 (dd, J = 8.3, 6.7 Hz, 1H), 7.33 (dd, J = 8.7, 2.4 Hz, 1H), 7.04 (td, J = 8.4, 2.4 Hz, 1H), 4.56 (hept, J = 3.7 Hz, 1H), 0.46 (d, J = 3.8 Hz, 6H) ppm. 13C{1H} NMR (75 MHz, CDCl3) δ 163.7 (d, J = 252.8 Hz), 137.9 (d, J = 8.0 Hz), 134.9 (d, J = 3.8 Hz), 130.8 (d, J = 8.9 Hz), 120.1 (d, J = 23.1 Hz), 114.1 (d, J = 19.4 Hz), −3.47 ppm. 19F NMR (376 MHz, CDCl3) δ −109.67 (td, J = 8.6, 6.8 Hz) ppm.
:1, 40 mL) at −78 °C and the resulting suspension was stirred for 2 h. Chlorodimethylsilane (4.0 mL, 36.0 mmol) was added dropwise at −78 °C and the mixture was allowed to warm to rt and left overnight. Hexane (20 mL) was added and the mixture was quenched with water (20 mL). The organic phase was separated and the aqueous phase was extracted with Et2O (30 mL). The combined organic phase was dried with Na2SO4 and concentrated under reduced pressure. The oily residue was subjected to fractional distillation in vacuo to give crude 4-fluoro-2-bromo-1-(dimethylsilyl)benzene pre-5-F as a colorless liquid, b.p. 52–55 °C (5 × 10−3 mbar). Yield 6.2 g (89%). 1H NMR (300 MHz, CDCl3) δ 7.47 (dd, J = 8.3, 6.7 Hz, 1H), 7.33 (dd, J = 8.7, 2.4 Hz, 1H), 7.04 (td, J = 8.4, 2.4 Hz, 1H), 4.56 (hept, J = 3.7 Hz, 1H), 0.46 (d, J = 3.8 Hz, 6H) ppm. 13C{1H} NMR (75 MHz, CDCl3) δ 163.7 (d, J = 252.8 Hz), 137.9 (d, J = 8.0 Hz), 134.9 (d, J = 3.8 Hz), 130.8 (d, J = 8.9 Hz), 120.1 (d, J = 23.1 Hz), 114.1 (d, J = 19.4 Hz), −3.47 ppm. 19F NMR (376 MHz, CDCl3) δ −109.67 (td, J = 8.6, 6.8 Hz) ppm.
A solution of pre-5-F (2.33 g, 10 mmol) in Et2O (10 mL) was added to a solution of t-BuLi (1.9 M, 6.9 mL, 13.0 mmol) in Et2O (30 mL) at −90 °C. The mixture was stirred for 30 min at −95 °C followed by dropwise addition of B(OMe)3 (1.65 mL; 15 mmol). The resulting mixture was allowed to warm to ca. −50 °C followed by hydrolysis with 1 M aq. NaOH (20 mL). The mixture was concentrated under reduced pressure and the remaining aqueous phase was washed with hexane followed by neutralization with aq. HCl (2 M, 20.0 mL). The mixture was extracted with hexane/Et2O (1:1, 30 mL). The organic phase was separated, dried with Na2SO4 and concentrated under reduced pressure to leave a viscous oily residue. It was crystallized in hexane at −60 °C. Filtration of the obtained precooled suspension afforded the product 5-F as a white crystalline solid (1.35 g, yield 69%). 1H NMR (400 MHz, CDCl3) δ 7.58 (dd, J = 8.0, 5.4 Hz, 1H), 7.49 (dd, J = 8.9, 2.4 Hz, 1H), 7.18 (ddd, J = 9.6, 8.0, 2.4 Hz, 1H), 6.13 (d, J = 6.4 Hz, 1H), 0.45 (s, 6H) ppm. 13C NMR (101 MHz, CDCl3) δ 164.6 (d, J = 249.7 Hz), 145.4 (d, J = 3.3 Hz), 143.3 (broad), 132.5 (d, J = 7.5 Hz), 118.5 (d, J = 21.3 Hz), 117.9 (d, J = 18.1 Hz), −0.4 ppm. 19F NMR (376 MHz, CDCl3) δ −111.40 (td, J = 9.2, 5.4 Hz) ppm. 11B NMR (96 MHz, CDCl3) δ 29.9 ppm.
A solution of pre-7-CF3 (2.83 g, 10 mmol) in Et2O (10 mL) was added to a solution of t-BuLi (1.9 M, 6.3 mL, 12.0 mmol) in Et2O (30 mL) at −90 °C. The mixture was stirred for 30 min at −95 °C followed by dropwise addition of B(OMe)3 (1.65 mL; 15 mmol). The resulting mixture was allowed to warm to ca. −50 °C followed by hydrolysis with 1 M aq. NaOH (15 mL) and by neutralization with aq. HCl (2 M, 20.0 mL). The organic phase was separated and the aqueous phase was extracted with Et2O (30 mL). The combined organic phase was concentrated under reduced pressure, the solid residue was mixed with water (20 mL) and the resulting white suspension was filtered. The solid was washed with water (3 × 10 mL) and hexane (10 mL) and dried under reduced pressure. The product 7-CF3 was obtained as a white powder. Yield 1.52 g (62%). 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 7.3 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.62–7.58 (m, 1H), 6.47 (s, 1H), 0.51 (q, J = 1.1 Hz, 6H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δ 147.4 (q, J = 3.4 Hz), 141.5 (broad), 134.8 (q, J = 1.4 Hz), 133.1 (q, J = 32.0 Hz), 130.2, 127.9 (q, J = 3.9 Hz), 125.1 (q J = 272.8 Hz), −0.92 (q, J = 1.8 Hz) ppm. 19F NMR (376 MHz, CDCl3) δ −60.86 ppm. 11B NMR (96 MHz, CDCl3) δ 30.4 ppm.
The subsequent conversion of pre-4-CF3 (2.83 g, 10.0 mmol) afforded the final product 4-CF3 as a white crystalline solid. Yield 2.11 g (86%). 1H NMR (400 MHz, CDCl3) δ 7.75–7.71 (m, 2H), 7.54 (t, J = 7.4 Hz, 1H), 5.54 (broad, 1H), 0.41 (s, 6H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δ 153.7, 137.9 (broad), 134.0, 133.2 (q, J = 31.5 Hz), 130.6, 126.7 (q, J = 5.5 Hz), 124.6 (q, J = 273.6 Hz), 0.8 ppm. 19F NMR (376 MHz, CDCl3) δ −60.14 ppm. 11B NMR (96 MHz, CDCl3) δ 29.2 ppm.
The combined filtrate was concentrated under reduced pressure and the oily residue was subjected to fractional distillation in vacuo to give crude 1-bromo-2-(dimethylsilyl)-3-(trimethylsilyl)benzene pre-7-TMS as a colorless liquid, b.p. 59–62 °C (10−3 mbar). Yield 5.05 g (88%). 1H NMR (400 MHz, CDCl3) δ 7.55 (dd, J = 7.4, 1.1 Hz, 1H), 7.53 (dd, J = 7.4, 1.1 Hz, 1H), 7.17 (dd, J = 8.0, 7.4 Hz, 1H), 4.87 (hept, J = 3.9 Hz, 1H), 0.54 (d, J = 3.9 Hz, 6H), 0.37 (s, 9H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δ 151.5, 144.9, 133.4, 133.3, 132.9, 130.1, 2.0, −2.8 ppm.
Compound pre-7-TMS (2.87 g, 10.0 mmol) was converted to 7-TMS using the protocol described for 4-CF3. The product was obtained as a white crystalline solid. Yield 1.80 g (72%). 1H NMR (300 MHz, CDCl3) δ 7.88 (dd, J = 7.3, 1.3 Hz, 1H), 7.76 (dd, J = 7.5, 1.3 Hz, 1H), 7.47 (t, J = 7.4 Hz, 1H), 6.40 (broad, 1H), 0.57 (s, 6H), 0.35 (s, 9H) ppm. 13C{1H} NMR (101 MHz, CDCl3) δ 155.8, 144.5, 140.2, 137.0, 132.0, 128.7, 1.1, 0.5 ppm. 11B NMR (96 MHz, CDCl3) δ 30.8 ppm.
Single-crystal X-ray diffraction
Single crystals were grown by a solvent-evaporation method under air from the CHCl3 or acetone solutions. A single crystal of 6-F was obtained using the diffusion method with the CHCl3/hexane solvent set. Single crystals of polymorphs 56-dF-I and 56-dF-II were obtained by slow/fast evaporation of a CHCl3 solution, respectively. However, the data quality of 56-dF-I was low. In all cases, a selected crystal was maintained at low temperature (100 K) with the use of an Oxford Cryosystems nitrogen gas-flow device. Exceptionally, structure 6-F was measured at 130 K. The crystal structures were established in a conventional way via X-ray data refinement employing the independent atom model (IAM). Data reduction and analysis were carried out with the CrysAlisPro suites of programs.57 All structures were solved by direct methods using SHELXS-9758 and refined using SHELXL-2016.59 All non-H atoms were refined anisotropically. All C–H H atoms were placed in calculated positions with C–H distances of 0.95 Å and Uiso(H) = 1.2Ueq(C). The positions of H atoms of hydroxy groups were located from a difference electron density maps. The O–H distances were fixed to 0.84 Å with a standard deviation of 0.01 Å and the directionality of O–H was refined freely. The Uiso parameter was set to 1.5Ueq with respect to oxygen atoms. The crystal structure of 6-F contains voids with disordered hexane molecules. The solvent contribution was subtracted from the data using SQUEEZE from the PLATON crystallographic package. In the case of 6-CHO-7F, the difference map revealed a slight disorder of the formyl group. However, the refinement showed that the contribution of a second component is very small (2.5%). Similarly, in the case of 7-Cl, the H2 atom from the O–H group was found to be disordered over two positions. The refinement showed that the occupancy factor for the alternate hydrogen position is only 20%. Thus, for both structures the disorder was not included in the final refinement procedure. All-important crystallographic data including measurement, reduction, structure solution and refinement details are placed in Tables S1–S5† or in the associated CIF files or can be retrieved from the Cambridge Crystallographic Data Centre as supplementary deposition no. 2259083 (4-CF3), 2259084 (4-CN-7-F), 2259085 (6-CF3), 2259086 (6-CHO-7F), 2259087 (6-Cl), 2259088 (6-CN), 2259089 (6-F), 2259090 (7-BOH2), 2259091 (7-Br), 2259092 (7-CF3), 2259093 (7-CHO), 2259094 (7-Cl), 2259095 (7-F), 2259096 (56-dO), 2259097 (56-dF-I), 2298596 (56-dF-II), 2259098 (57-dF-6-CHO), 2259099 (67-dCl), 2259100 (67-dF), 2166292 (4-CO2Me-7F), 2265514 (7-TMS), 2265515 (7-I), and 2265516 (5-F).Theoretical calculations
Theoretical calculations were performed at the M06-2X60/6-311++G(d,p)61 level of theory using Gaussian16 program.62 The HB motifs were extracted from the crystal structures. The positions of non-H atoms were fixed, while H atoms were fully optimised. In the second approach, the geometry of HB motifs was fully optimized. Following geometry optimization, the interaction energies were calculated using the supermolecular method including basis set superposition error (BSSE). The energy of an intramolecular HB was estimated from the difference between the electronic energies of two rotamers: one with a functional group coplanar with the oxaborole and aromatic rings, and the second with a functional group rotated by 90°. The energy of a HB chain was calculated taking two neighbouring molecules.The periodic calculations were performed within the CRYSTAL09 program package63 at the DFT(B3LYP) level of theory with the POB triple-zeta valence + polarization basis set (TZVP).64–67 Grimme dispersion correction was applied.68–70 Ghost atoms were selected up to 5 Å distance from the studied molecule in a crystal lattice and were used for the basis set superposition error estimation.71
Author contributions
K. D. and S. L.: conceptualization and supervision of the research, formal analysis, writing the original draft, writing – review & editing; A. Z.: DFT calculations; P. H. M.-U. and J. D.: X-ray crystallography; K. N.: synthesis; K. W.: analysis of crystal structures; S. L.: funding acquisition, project administration. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Centre (Poland) in the framework of the project UMO-2018/31/B/ST5/00210. This work was implemented as a part of the Operational Project Knowledge Education Development 2014–2020 co-financed by the European Social Fund (the TRIBIOCHEM interdisciplinary PhD programme (P. H. M.-U.)). Computational facilities were provided by the Wrocław Centre for Networking and Supercomputing (grant No. 285). We also acknowledge the support of the Warsaw University of Technology.References
- F. L. Rock, W. Mao, A. Yaremchuk, M. Tukalo, T. Crépin, H. Zhou, Y.-K. Zhang, V. Hernandez, T. Akama, S. J. Baker, J. J. Plattner, L. Shapiro, S. A. Martinis, S. J. Benkovic, S. Cusack and M. R. K. Alley, Science, 2007, 316, 1759–1761 CrossRef CAS PubMed.
- T. Akama, S. J. Baker, Y.-K. Zhang, V. Hernandez, H. Zhou, V. Sanders, Y. Freund, R. Kimura, K. R. Maples and J. J. Plattner, Bioorg. Med. Chem. Lett., 2009, 19, 2129–2132 CrossRef CAS PubMed.
- S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- A. Adamczyk-Woźniak, K. M. Borys and A. Sporzyński, Chem. Rev., 2015, 115, 5224–5247 CrossRef PubMed.
- N. Sharma and D. Sharma, J. Pharmacol. Pharmacother., 2015, 6, 236–239 CrossRef PubMed.
- L. McDowell and B. Olin, J. Pharm. Technol., 2019, 35, 172–178 CrossRef CAS PubMed.
- A. S. Paller, W. L. Tom, M. G. Lebwohl, R. L. Blumenthal, M. Boguniewicz, R. S. Call, L. F. Eichenfield, D. W. Forsha, W. C. Rees, E. L. Simpson, M. C. Spellman, L. F. Stein Gold, A. L. Zaenglein, M. H. Hughes, L. T. Zane and A. A. Hebert, J. Am. Acad. Dermatol., 2016, 75, 494–503 CrossRef CAS PubMed.
- J. R. Sullivan, A. Lupien, E. Kalthoff, C. Hamela, L. Taylor, K. A. Munro, T. M. Schmeing, L. Kremer and M. A. Behr, PLoS Pathog., 2021, 17, e1009965 CrossRef CAS PubMed.
- I. D. Madura, A. Adamczyk-Woźniak, M. Jakubczyk and A. Sporzyński, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67, o414–o415 CrossRef CAS PubMed.
- S. Sene, D. Berthomieu, B. Donnadieu, S. Richeter, J. Vezzani, D. Granier, S. Bégu, H. Mutin, C. Gervais and D. Laurencin, CrystEngComm, 2014, 16, 4999–5011 RSC.
- A. Adamczyk-Woźniak, M. K. Cabaj, P. M. Dominiak, P. Gajowiec, B. Gierczyk, J. Lipok, Ł. Popenda, G. Schroeder, E. Tomecka, P. Urbański, D. Wieczorek and A. Sporzyński, Bioorg. Chem., 2015, 60, 130–135 CrossRef PubMed.
- M. D
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) browski, P. Kurach, S. Luliński and J. Serwatowski, Appl. Organomet. Chem., 2007, 21, 234–238 CrossRef.
browski, P. Kurach, S. Luliński and J. Serwatowski, Appl. Organomet. Chem., 2007, 21, 234–238 CrossRef. - S. Kusano, S. Miyamoto, A. Matsuoka, Y. Yamada, R. Ishikawa and O. Hayashida, Eur. J. Org. Chem., 2020, 2020, 1598–1602 CrossRef.
- Y. Yamamoto, J. Ishii, H. Nishiyama and K. Itoh, J. Am. Chem. Soc., 2005, 127, 9625–9631 CrossRef CAS PubMed.
- Y.-L. Tan, A. J. P. White, D. A. Widdowson, R. Wilhelm and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 2001, 3269–3280 CAS.
- F. G. Vogt, G. R. Williams and R. C. B. Copley, J. Pharm. Sci., 2013, 102, 3705–3716 CrossRef CAS PubMed.
- G. Campillo-Alvarado, T. D. Didden, S. M. Oburn, D. C. Swenson and L. R. MacGillivray, Cryst. Growth Des., 2018, 18, 4416–4419 CrossRef CAS.
- S. Luliński and J. Serwatowski, J. Organomet. Chem., 2007, 692, 2924–2929 CrossRef.
- D. Wieczorek, E. Kaczorowska, M. Wiśniewska, I. D. Madura, M. Leśniak, J. Lipok and A. Adamczyk-Woźniak, Molecules, 2020, 25, 5999 CrossRef CAS PubMed.
- A. Adamczyk-Woźniak, K. Ejsmont, B. Gierczyk, E. Kaczorowska, A. Matuszewska, G. Schroeder, A. Sporzyński and B. Zarychta, J. Organomet. Chem., 2015, 788, 36–41 CrossRef.
- J. S. Kumar, C. M. Bashian, M. A. Corsello, S. C. Jonnalagadda and V. R. Mereddy, Tetrahedron Lett., 2010, 51, 4482–4485 CrossRef CAS PubMed.
- K. M. Wells, S. J. Mehrman, A. F. Abdel-Magid, C. Ferraro, L. Scott, H. M. Zhong, C. A. Teleha, S. Ballentine, X. Li, R. K. Russell, J. M. Spink, C. Diamond, S. Youells, Y. Zhang, F.-R. Tsay, S. Cesco-Cancia, S. M. Manzo and D. A. Beauchamp, Org. Process Res. Dev., 2015, 19, 1774–1783 CrossRef CAS.
- R. M. Al-Zoubi, M. S. Al-Zoubi, K. T. Jaradat and R. McDonald, Eur. J. Org. Chem., 2017, 2017, 5800–5808 CrossRef CAS.
- A. Adamczyk-Woźniak, M. K. Cyrański, M. Jakubczyk, P. Klimentowska, A. Koll, J. Kołodziejczak, G. Pojmaj, A. Żubrowska, G. Z. Żukowska and A. Sporzyński, J. Phys. Chem. A, 2010, 114, 2324–2330 CrossRef PubMed.
- J. Zhu, Y. Wei, D. Lin, C. Ou, L. Xie, Y. Zhao and W. Huang, Org. Biomol. Chem., 2015, 13, 11362–11368 RSC.
- G. Hazra, S. Maity, S. Bhowmick and P. Ghorai, Chem. Sci., 2017, 8, 3026–3030 RSC.
- V. L. Arcus, L. Main and B. K. Nicholson, J. Organomet. Chem., 1993, 460, 139–147 CrossRef CAS.
- A. Singh and R. Kumar, Chem. Commun., 2021, 57, 9708–9711 RSC.
- A. Sporzyński, M. Lewandowski, P. Rogowska and M. K. Cyrański, Appl. Organomet. Chem., 2005, 19, 1202–1203 CrossRef.
- A. Adamczyk-Woźniak, I. Madura, A. H. Velders and A. Sporzyński, Tetrahedron Lett., 2010, 51, 6181–6185 CrossRef.
- A. M. Dąbrowska, A. Adamczyk-Woźniak and I. D. Madura, CrystEngComm, 2022, 24, 3586–3596 RSC.
- A. Adamczyk-Woźniak, K. M. Borys, I. D. Madura, S. Michałek and A. Pawełko, Tetrahedron, 2013, 69, 8936–8942 CrossRef.
- K. M. Borys, A. Matuszewska, D. Wieczorek, K. Kopczyńska, J. Lipok, I. D. Madura and A. Adamczyk-Woźniak, J. Mol. Struct., 2019, 1181, 587–598 CrossRef CAS.
- G. Campillo-Alvarado, C. A. Staudt, M. J. Bak and L. R. MacGillivray, CrystEngComm, 2017, 19, 2983–2986 RSC.
- A. Brzozowska, P. Ćwik, K. Durka, T. Kliś, A. E. Laudy, S. Luliński, J. Serwatowski, S. Tyski, M. Urban and W. Wróblewski, Organometallics, 2015, 34, 2924–2932 CrossRef CAS.
- K. Nowicki, P. Pacholak and S. Luliński, Molecules, 2021, 26, 5464 CrossRef CAS PubMed.
- K. Durka, A. E. Laudy, Ł. Charzewski, M. Urban, K. Stępień, S. Tyski, K. A. Krzyśko and S. Luliński, Eur. J. Med. Chem., 2019, 171, 11–24 CrossRef CAS PubMed.
- P. Ćwik, P. Ciosek-Skibińska, M. Zabadaj, S. Luliński, K. Durka and W. Wróblewski, Sensors, 2020, 20, 3540 CrossRef PubMed.
- K. Durka, M. Urban, M. Czub, M. Dąbrowski, P. Tomaszewski and S. Luliński, Dalton Trans., 2018, 47, 3705–3716 RSC.
- P. Pacholak, J. Krajewska, P. Wińska, J. Dunikowska, U. Gogowska, J. Mierzejewska, K. Durka, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2021, 11, 25104–25121 RSC.
- J. Krajewska, K. Nowicki, K. Durka, P. H. Marek-Urban, P. Wińska, T. Stępniewski, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2022, 12, 23099–23117 RSC.
- M. Czub, K. Durka, S. Lulinski, J. Łosiewicz, J. Serwatowski, M. Urban and K. Woźniak, Eur. J. Org. Chem., 2017, 818–826 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- C. B. Aakeröy, J. Desper and B. Levin, CrystEngComm, 2005, 7, 102–107 RSC.
- I. D. Madura, K. Czerwińska and D. Sołdańska, Cryst. Growth Des., 2014, 14, 5912–5921 CrossRef CAS.
- K. Durka, K. N. Jarzembska, R. Kamiński, S. Luliński, J. Serwatowski and K. Woźniak, Cryst. Growth Des., 2012, 12, 3720–3734 CrossRef CAS.
- M. R. Shimpi, N. SeethaLekshmi and V. R. Pedireddi, Cryst. Growth Des., 2007, 7, 1958–1963 CrossRef CAS.
- J. J. Campos-Gaxiola, B. A. García-Grajeda, I. F. Hernández-Ahuactzi, J. A. Guerrero-Álvarez, H. Höpfl and A. Cruz-Enríquez, CrystEngComm, 2017, 19, 3760–3775 RSC.
- P. Rodríguez-Cuamatzi, O. I. Arillo-Flores, M. I. Bernal-Uruchurtu and H. Höpfl, Cryst. Growth Des., 2005, 5, 167–175 CrossRef.
- F. Mongin, O. Desponds and M. Schlosser, Tetrahedron Lett., 1996, 37, 2767–2770 CrossRef CAS.
- S. M. Bachrach, J. Org. Chem., 2008, 73, 2466–2468 CrossRef CAS PubMed.
- M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
- J. W. Tomsho, A. Pal, D. G. Hall and S. J. Benkovic, ACS Med. Chem. Lett., 2012, 3, 48–52 CrossRef CAS PubMed.
- F. Weinhold and R. West, J. Am. Chem. Soc., 2013, 135, 5762–5767 CrossRef CAS PubMed.
- M. K. Cyrański, A. Jezierska, P. Klimentowska, J. J. Panek and A. Sporzyński, J. Phys. Org. Chem., 2008, 21, 472–482 CrossRef.
- A. Adamczyk-Woźniak, M. K. Cyrański, K. Durka, J. T. Gozdalik, P. Klimentowska, R. Rusiecki, A. Sporzyński and D. Zarzeczańska, Crystals, 2019, 9, 109 CrossRef.
- O. D. Rigaku, CrysAlis PRO 1.171. 38.43, Rigaku Oxford Diffraction, Yarnton, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- R. Dovesi, R. Orlando, B. Civalleri, C. Roetti, V. R. Saunders and C. M. Zicovich-Wilson, Z. Kristallogr., 2005, 220, 571–573 CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
- B. Civalleri, C. M. Zicovich-Wilson, L. Valenzano and P. Ugliengo, CrystEngComm, 2008, 10, 405–410 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2004, 25, 1463–1473 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected single-crystal X-ray diffraction data, NMR spectra of new compounds. CCDC 2259083–2259100, 2265514–2265516, 2166292 and 2298596. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce00880k |
| This journal is © The Royal Society of Chemistry 2023 |