DOI:
10.1039/C5RA02241J
(Paper)
RSC Adv., 2015,
5, 25250-25257
Manganese oxide nanostructures: low-temperature selective synthesis and thermal conversion†
Received
5th February 2015
, Accepted 3rd March 2015
First published on 4th March 2015
Abstract
Nanostructured MnOx with crystalline phases of γ-MnOOH, δ-MnO2, α-Mn2O3, and Mn3O4 as well as rod, sheet, cube, and particle shapes were prepared via a facile, high-yield and low-temperature (60 °C) reduction route, without templates or surfactants. Selective preparation was achieved just by adjusting the amount of H2O2 or reactant concentration that controls simultaneously the growth thermodynamic and dynamic parameters of MnOx nanocrystals. Our results show that the bundle-like γ-MnOOH nanorods are formed through a lamellar growth, dissolution–recrystallization and oriented attachment process. Furthermore, the corresponding bundle-like β-MnO2, α-Mn2O3, and Mn3O4 nanorods were prepared by calcining these γ-MnOOH precursors under different atmospheres and temperatures. This synthetic procedure is simple, inexpensive and high-yield, and thus facilitates mass production of manganese oxide nanomaterials.
Introduction
Manganese oxides (MnOx) are of considerable importance in technological application, including catalysis,1 energy storage,2 microwave absorption,3 magnetism,4 biosensors,5 and ion exchange6 owing to their outstanding structural diversity combined with novel chemical and physical properties. A large number of manganese oxides are possible due to the availability of various oxidation states of manganese (II, III, and IV). Among the series of manganese oxides, MnO2 has proved to be a favorable material for electrochemical supercapacitors because of its satisfactory charge storage performance, natural abundance and low toxicity.7 As an active catalyst material, Mn2O3 can be used for organic pollutants and decomposition of nitrogen oxides from waste gases.8,9 On the other hand, the high disinfection capacity for bacteria and marked cytotoxic effect also makes Mn2O3 an alternative material for the development of new bactericides.10 Mn3O4 has been used as one of the raw materials in the manufacture of soft magnetic materials such as manganese zinc ferrite, which is useful for magnetic cores in transformers for power supply.4 Moreover, Mn3O4 is also known to be an active catalyst for the combustion of organic compounds in the temperature range of 373–773 K.11 For MnOOH, it shows outstanding absorption capacities for numerous trace elements from aqueous solution as well as a strong ability to oxidize natural and xenobiotic organic substances.12,13 Furthermore, MnOOH also is believed to be the most simple and practical precursor to prepare various manganese oxides, intercalation compounds and lithium manganese oxides for rechargeable lithium batteries.14,15
It is well-known that nanomaterials are more effective than bulk materials in applications for their extremely small feature size and large surface area.16,17 Selective synthesis of nanomaterials with controllable compositions and morphologies represents an increasingly important research direction in nanosciences and nanotechnologies because the intrinsic properties of nanostructures depend on their shape, phase, and size. Therefore, a range of preparation techniques have been exploited to synthesize high quality manganese oxide nanostructures. Up to now, manganese oxide nanostructures in the shapes of nanosheets,18 nanourchins,19 nanowires,20 nanoparticles,21 nanoplates22 and other nanostructures have been synthesized.23–25 For example, Kumar et al. have synthesized highly dispersed MnO2 nanoneedles by the application of ultrasound radiation.26 Mn2O3 nanoparticles were prepared by Thota et al. through a sol–gel method.27 In addition, Bai et al. have prepared branched mesoporous Mn3O4 nanorods through a two-step process including hydrothermal synthesis of branchy MnOOH precursors and followed by calcinations of the precursors at 300 °C under a N2 atmosphere.28 However, in most of the previously reported synthetic routes, few studies focused on the preparation of manganese oxide nanomaterials with various shapes and oxidation states by a controlled method. Facile and simultaneous control the oxidation states, crystal phases, and morphologies of nanomaterials remains challenging because of elusive interactive thermodynamic and kinetic parameters.29 Moreover, most of these synthetic methods show low yield, unavoidable disadvantages of complicated process and harsh synthesis conditions such as high temperature, high pressure and expensive equipments, so they are not suitable for mass production. Therefore, an easy, high-yield and low-temperature method needs to be developed for fabricating a large number of nanostructured manganese oxides.
In this paper, we report on a new, high-yield and low-temperature approach to the selective preparation of manganese oxide nanomaterials (γ-MnOOH, δ-MnO2, α-Mn2O3, and Mn3O4) with simultaneously controlled shape and crystallographic form. It was found that the oxidation states, crystal phases, and morphologies of the products were determined by the adding of H2O2, reactant concentration, reaction time, and reaction temperature. The possible growth mechanism of these bundle-like γ-MnOOH nanorods has been studied based on the experimental results. Furthermore, the bundle-like β-MnO2, α-Mn2O3, and Mn3O4 nanorods were also selectively produced by calcining the γ-MnOOH precursors under different atmosphere and temperature. This synthesis method of manganese oxide nanocrystals is simple and environmentally friendly, and it is easily scalable for industry.
Experimental section
All the reagents in this study were of analytical grade and used without further purification. In a typical synthesis of bundle-like γ-MnOOH nanorods, 10 mL H2O2 (30 wt%) was poured to 25 mL of a 1 M NaOH aqueous solution, then the NaOH–H2O2 solution was slowly added to 25 mL of the 0.5 M Mn(CH3COO)2·4H2O aqueous solution, and the dripping speed of NaOH–H2O2 solution was about 4 mL min−1. The resulting mixture was heated to 60 °C over 10–15 min and maintained for 24 h with vigorous stirring. After that, the solution was allowed to cool down to room temperature naturally. The resulting solid products were centrifuged and rinsed several times with absolute ethanol, then finally dried in a vacuum at 50 °C for 24 h. The yield of the products is about 1 g and can be easily enlarged by increasing the amount of the corresponding reactants. For the preparation of δ-MnO2 nanosheets, α-Mn2O3 nanocubes, and Mn3O4 nanoparticles, similar procedures were carried out except adjusting the amount of H2O2 or reactant concentration. In addition, the bundle-like γ-MnOOH nanorods were heated in tube furnace with different conditions could obtain various manganese oxide nanostructures. β-MnO2 and α-Mn2O3 nanorods could be prepared by treating the γ-MnOOH precursors at 350 °C and 500 °C for 4 h in air, respectively. Mn3O4 nanorods were obtained via treating the γ-MnOOH at 600 °C for 4 h under argon atmosphere. X-ray diffraction (XRD) patterns were collected on a Rigaku D/max-RA X-ray diffractometer using Cu Ka radiation (λ = 0.15406 nm). Transmission electron microscopy (TEM) images, high-resolution transmission electron microscopy (HRTEM) images, selected area electron diffraction (SAED) patterns and energy disperse spectroscopy (EDS) images were collected with a JEM-2200FS transmission electron microscope performed at 200 kV. The samples for TEM observation by dispersing the solid powder in ethanol ultrasonically and then dropping the solution onto carbon coated copper grids. The Raman measurements were carried out using a Raman spectrometer (Renishaw inVia) with a 633 nm He–Ne laser line as excitation. The pH values were measured by a pH meter.
Results and discussion
XRD patterns for the products synthesized at 24 h with different reaction temperatures are shown in Fig. 1. All diffraction peaks can be indexed to the monoclinic γ-MnOOH, which are consistent with the standard data from JCPDS card nO. 41-1379 (space group P21/c14; a = 0.5300 nm, b = 0.5278 nm, c = 0.5307 nm). In addition, there are no diffraction peaks corresponding to impurities, suggesting the high purity of the final products. In the case of product prepared at room temperature (RT) for 24 h, the peaks are broad and low. There is an increase in crystallinity with increasing the reaction temperature from RT to 60 °C as evidenced by appearance of sharper peaks. Further increasing the reaction temperature to 80 °C, the peaks have no obvious change. The samples prepared at different reaction temperatures were also analyzed by Raman spectra from 200 cm−1 to 800 cm−1, as shown in Fig. 2. Five main peaks at 360, 389, 530, 558, and 623 cm−1 are observed, which are good agreement with literature data for γ-MnOOH.30,31 These results demonstrate success in synthesizing the γ-MnOOH through a facile, efficient and low-temperature reduction route.
 |
| | Fig. 1 XRD patterns of the products prepared at 24 h. Reaction temperature: (a) RT, (b) 40 °C, (c) 60 °C, and (d) 80 °C. | |
 |
| | Fig. 2 Raman spectra of the products prepared at 24 h. Reaction temperature: (a) RT, (b) 40 °C, (c) 60 °C, and (d) 80 °C. | |
The morphologies, compositions and microstructures of γ-MnOOH synthesized at 60 °C were further investigated by TEM, EDS, and HRTEM. Fig. 3(a–c) show the TEM images of γ-MnOOH with different magnifications, we can see that the γ-MnOOH comprises a large number of nanorods with an average diameter of 10–30 nm. These uniform nanorods are parallel assembled into bundles with a diameter range of 0.3–5 μm and lengths of 2–15 μm. In the EDS pattern (Fig. 3(d)), Mn, O, C and Cu elements are detected. Among them, the signals for Cu and C elements are derive from the carbon coated copper grid. The atomic ration of manganese to oxygen is 0.51, which is very close to 0.5. It is very consistent with the stoichiometric composition of γ-MnOOH. Fig. 3(e) shows the γ-MnOOH nanorods and the corresponding SAED pattern. The SAED pattern demonstrates that the nanorods are single crystal with axis along the crystallographic [101]. We know that the γ-MnOOH is a tunnels-structured compound with interstitial (1 × 1) tunnels built by single chain of edge-sharing MnO6 octahedra along the [101] of the monoclinic unit-cell,32,33 the current γ-MnOOH nanorods are fast crystal grow along the tunnel direction, which is consistent with those observations for tunnels-structure manganese oxides.34,35 Fig. 3(f) shows the HRTEM image of Fig. 3(e), it displays the measured spacings of the lattice planes are 0.252 nm and 0.264 nm, which are consistent with the (111) and (020) planes of γ-MnOOH crystal, respectively. HRTEM analysis further indicates that high quality γ-MnOOH is synthesized successfully. Besides, the morphologies of γ-MnOOH synthesized at other temperatures (RT, 40 °C, and 80 °C) also have the bundle-like structures.
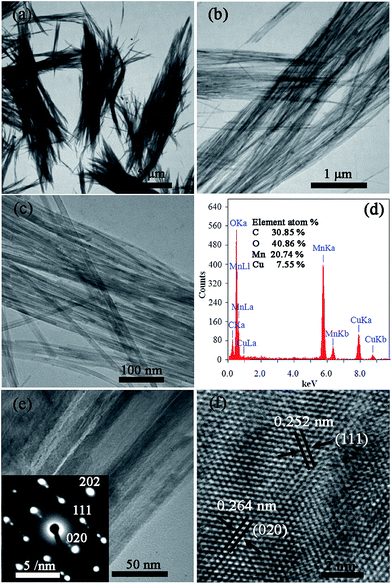 |
| | Fig. 3 (a–c) TEM images of the γ-MnOOH nanorods prepared at 60 °C, (d) the EDS pattern of γ-MnOOH nanorods, (e) TEM image of the γ-MnOOH nanorods and the corresponding SAED pattern (the inset), and (f) the HRTEM image of (e). | |
Our results show that the bundle-like γ-MnOOH nanorods are obtained under continuous stirring for 24 h. To study the effect of stirring on the structures and morphologies of the final products, the XRD patterns and TEM images of the products obtained after the mixing solution under stirring for 1 min and then aging for 24 h at 60 °C under static conditions are shown in Fig. 4. The XRD patterns also can be indexed to γ-MnOOH (Fig. 4(a)). However, the TEM observations (Fig. 4(b)) show that the bundle-like structures disappeared, the samples have a random nanorods morphologies with diameters of 10–30 nm and lengths ranging between 1 and 3 μm. This results indicate that stirring plays an important role in determining the morphologies of the γ-MnOOH, continuous stirring is needed to promote the interactions between the random nanorods and ultimately to form such bundle-like structures.
 |
| | Fig. 4 (a) XRD patterns and (b) TEM images of the products prepared after the mixing solution under stirring for 1 min and then aging for 24 h at 60 °C under static conditions. | |
To further investigate the formation process of the bundle-like γ-MnOOH nanorods, intermediates with various reaction times were collected and observed by TEM. At the initial stage, it can be clearly seen that there are many folds on the lamellae from the Fig. 5(a). As shown in Fig. 5(b), there is an interesting change in the morphologies of the samples synthesized at 10 min, which displays that the previously pleated lamellae grow to smooth lamellar structures and some small nanorods. The diameters and lengths of the nanorods are in the range of 10–30 nm and 100–300 nm, respectively. Prolonging the reaction time to 40 min, the amounts of smooth lamellar structures decreased and more oriented attachment of the nanorods are formed (Fig. 5(c)). These nanorods are parallel assembled together to form small bundle-like structures. After the reaction is further prolonged to 4 h (Fig. 5(d)), we can see that all the lamellar structures disappeared, the number of the small bundle-like structures increased, and these random small bundle-like structures with an average diameter of 100–300 nm and lengths of 2–3 μm spontaneously agglomerated together. When the reaction time increases up to 24 h (Fig. 5(e)), the larger and longer bundle-like structures are finally formed from the oriented attachment of these small bundle-like structures along their side surfaces. In addition, the pH changes with various reaction times are also examined in these experiments. In the current reaction, Mn2+ is oxidized by H2O2 to produce Mn3+ then combined with OH− that precipitates into γ-MnOOH. The pH value of the 0.5 M Mn(CH3COO)2·4H2O aqueous solution is 5.11 and that of NaOH–H2O2 solution is 10.63. The pH value of their initial mixture is 7.11, after stirring for 10 min, 40 min, 4 h and 24 h, the pH value of the solution is 6.35, 6.12, 6.07 and 6.02, respectively. The continuous acidification of the solution also indicates that generates γ-MnOOH precipitates. Based on the experimental results, there are mainly two possible formation mechanisms are used to explain the formation process: one is a dissolution-crystallization process which converts lamellae into primary nanorods at the beginning stage, the other is an oriented attachment process which aggregates nanorods along their side surfaces to form bundle-like products. In the synthesis system, hydroxyl ions hold the lamellae together through hydrogen bonding interactions and this weak and asymmetric hydrogen bonding gives these reactants perfect cleavage parallel to the lamellae.36–38 The lamellae structures are in a metastable state, and much evidence demonstrates that lamellar structures, regardless of whether they are artificial or natural, have a strong tendency to form wires/rods.39,40 Besides, the bundle formation might be related to the van der Waals interactions between the nanorods during the growth process. The schematic illustration of the formation mechanism for bundle-like γ-MnOOH nanorods is given in Fig. 5(f). Similarly, bundle-like γ-MnOOH nanorods are also obtained when comparative experiments are performed by substituting NaOH with the same mass of KOH and LiOH·H2O.
 |
| | Fig. 5 TEM images of the products synthesized at 60 °C collected at different times: (a) 1 min, (b) 10 min, (c) 40 min, (d) 4 h, and (e) 24 h. (f) Schematic illustration of the formation mechanism for bundle-like γ-MnOOH nanorods. | |
In our case, H2O2 is used to synthesize γ-MnOOH due to its oxidizing ability, addition of H2O2 causes complete oxidation of Mn2+ to Mn3+, favoring formation of γ-MnOOH. Fig. 6(a) presents the XRD patterns of as-synthesized products in the absence of H2O2. All of the peaks can be readily indexed to a pure phase of tetragonal Mn3O4, which are consistent with the standard data from JCPDS card no. 80-0382. In general, Mn3O4 contains mixed oxidation states of Mn2+ and Mn3+, which can be represented as MnO·Mn2O3. It is well known that the dissolved oxygen concentration in water is 0.267 mM under atmospheric conditions.41 Without adding H2O2, 0.5 M of Mn2+ is partially oxidized to Mn3+ by oxygen under atmospheric conditions, and Mn3O4 is obtained. Fig. 6(b) and (c) show the low and high magnification TEM images of the Mn3O4. It is observed that the Mn3O4 nanoparticles with uniform size distribution in the range of 20–30 nm. The HRTEM image (Fig. 6(d)) shows clear lattice fringes with a lattice spacing of 0.288 nm, which corresponded to the distance between the (200) crystal planes of Mn3O4. The inset of Fig. 6(d) is the corresponding the fast Fourier transform (FFT) pattern, indicating the single crystal Mn3O4 with a highly crystalline structure is synthesized. These results indicate that the H2O2 has certain influence on the structures and morphologies of the final products.
 |
| | Fig. 6 (a) XRD patterns of the as-synthesized products in the absence of H2O2, (b) low- and (c) high-magnification TEM images, (d) HRTEM image and the corresponding FFT pattern (the inset). | |
Fig. 7 shows the XRD patterns of the as-prepared products with different reactant concentrations. The amount of H2O2 (10 mL) and the concentration rate of Mn2+ to OH− (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) are keep constant. The XRD patterns of the products prepared at 0.5 M of Mn2+ are shown in Fig. 7(a), all diffraction peaks can be perfectly indexed to the γ-MnOOH. Decreasing the reaction concentration of Mn2+ to 0.1 M (Fig. 7(b)), it is observed that the products exhibit a mixed phase of both α-Mn2O3 (JCPDS card no. 71-0636, marked by *) and γ-MnOOH (JCPDS card no. 41-1379, marked by #). When the reaction concentration of Mn2+ is 0.04 M (Fig. 7(c)), all diffraction peaks can be exclusively indexed to a cubic phase α-Mn2O3. Fig. 8 shows the TEM and HRTEM images of the α-Mn2O3. In Fig. 8(a) and (b), we can see that the α-Mn2O3 nanoparticles have a cubic shape with edge lengths of 50–150 nm. The inset of Fig. 8(b) shows the HRTEM image of α-Mn2O3 nanocubes. The interplanar distance of the lattice fringes is 0.471 nm, corresponding to the (200) planes of α-Mn2O3. This indicates that the reactant concentration is an important factor in the preparation of MnOx nanostructures.
:2) are keep constant. The XRD patterns of the products prepared at 0.5 M of Mn2+ are shown in Fig. 7(a), all diffraction peaks can be perfectly indexed to the γ-MnOOH. Decreasing the reaction concentration of Mn2+ to 0.1 M (Fig. 7(b)), it is observed that the products exhibit a mixed phase of both α-Mn2O3 (JCPDS card no. 71-0636, marked by *) and γ-MnOOH (JCPDS card no. 41-1379, marked by #). When the reaction concentration of Mn2+ is 0.04 M (Fig. 7(c)), all diffraction peaks can be exclusively indexed to a cubic phase α-Mn2O3. Fig. 8 shows the TEM and HRTEM images of the α-Mn2O3. In Fig. 8(a) and (b), we can see that the α-Mn2O3 nanoparticles have a cubic shape with edge lengths of 50–150 nm. The inset of Fig. 8(b) shows the HRTEM image of α-Mn2O3 nanocubes. The interplanar distance of the lattice fringes is 0.471 nm, corresponding to the (200) planes of α-Mn2O3. This indicates that the reactant concentration is an important factor in the preparation of MnOx nanostructures.
 |
| | Fig. 7 XRD patterns of the as-prepared products with different reactant concentrations (CMn2+:COH− = 1:2). The reactant concentration of Mn2+: (a) 0.5 M, (b) 0.1 M, and (c) 0.04 M. | |
 |
| | Fig. 8 TEM and HRTEM images of the as-prepared α-Mn2O3, (a) low magnification TEM image, and (b) high magnification TEM image and the HRTEM image (the inset). | |
In order to future study the effect of the amount of NaOH on the final products. We conduct some experiments with different concentrations of OH− (the concentration of Mn2+ (0.5 M) is keep constant). When the concentration of OH− is 1 M, the obtained product is γ-MnOOH (Fig. 7(a)). Increasing the concentration of OH− to 1.5 M, the diffraction peaks of the product can be indexed to the monoclinic δ-MnO2 (Fig. 9(a)), which are consistent with the standard data from JCPDS card no. 43-1456. Fig. 9(b) and (c) show the low and high magnification TEM images of the δ-MnO2, which display the sheet shape of δ-MnO2. The inset of Fig. 9(c) represents the SAED pattern of these nanosheets. The SAED pattern consists of several diffused diffraction rings, which clearly indicates the formation of polycrystalline structure of δ-MnO2. Fig. 9(d) shows the HRTEM image of the δ-MnO2. The HRTEM image shows the interplanar spacing of the lattice fringes is 0.257 nm, corresponding to the (20![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) planes of the δ-MnO2. Our results clearly indicate that the amount of NaOH strongly affects the structures and morphologies of the MnOx nanostructures.
) planes of the δ-MnO2. Our results clearly indicate that the amount of NaOH strongly affects the structures and morphologies of the MnOx nanostructures.
 |
| | Fig. 9 (a) XRD patterns of the products prepared with 1.5 M OH−, (b) low magnification TEM image, (c) high magnification TEM image and the corresponding SAED pattern (the inset), and (d) HRTEM image. | |
It is well-known that metal hydroxides can convert to the corresponding metal oxides by dehydration under thermal annealing.28 The annealing parameters (i.e., atmosphere, time and temperature) are fundamental to control the oxidation states and morphologies of the final products. Therefore, we study the thermal-annealing behavior of the bundle-like γ-MnOOH nanorods under air and argon atmosphere, respectively. Fig. 10 shows the XRD patterns of the products prepared by calcining the γ-MnOOH precursors under different conditions. From Fig. 10(a) and (b), we could see that tetragonal β-MnO2 (JCPDS card no. 72-1984, marked by #) and cubic α-Mn2O3 (JCPDS card no. 71-0636, marked by *) were obtained by calcining the γ-MnOOH precursors in air at 350 °C and 500 °C for 4 h, respectively. In Fig. 10(c), all peaks of the XRD patterns of the product prepared by calcining the γ-MnOOH precursors at 600 °C for 4 h in argon are indexed to the tetragonal Mn3O4 (JCPDS card no. 80-0382, marked by +).
 |
| | Fig. 10 XRD patterns of the as-prepared products obtained by calcining the γ-MnOOH precursors under different conditions. (a) 350 °C for 4 h in air, (b) at 500 °C for 4 h in air, and (c) 600 °C for 4 h in argon. | |
Further detail characterization of the products obtained by calcining the bundle-like γ-MnOOH nanorods is carried out by TEM and HRTEM (Fig. 11). It can be clearly seen from these TEM images that the as-prepared β-MnO2, α-Mn2O3, and Mn3O4 samples preserve the rod-like morphologies of the γ-MnOOH precursors. The β-MnO2, α-Mn2O3 and Mn3O4 nanorods have an average diameter of 10–30 nm, 30–50 nm and 20–50 nm, respectively. These nanorods also are assembled into bundles, the bundle-like β-MnO2, α-Mn2O3 and Mn3O4 structures have a diameter range of 0.5–4 μm, 0.5–3 μm, 0.5–5 μm and lengths of 2–20 μm, 2–10 μm, 2–15 μm, respectively. To get more structural information about these bundle-like nanorods, the samples are observed with HRTEM and displayed in the Fig. 11(c), (f) and (i). From the Fig. 11(c), (f) and (i), we could see that the interplanar spacing of the lattice fringes is 0.310 nm, 0.272 nm, and 0.249 nm, corresponding to the (110) planes of the β-MnO2, (222) planes of the α-Mn2O3, and (211) planes of the Mn3O4, respectively. This indicates that high quality manganese oxide nanostructures also could be synthesized by calcining the bundle-like γ-MnOOH precursors under different conditions.
 |
| | Fig. 11 TEM and HRTEM images of the products prepared by heat-treatment of γ-MnOOH. (a–c) β-MnO2, (d–f) α-Mn2O3, and (g, h, i) Mn3O4. | |
In our experiments, selective preparation is achieved just by adjusting the amount of H2O2 or reactant concentration that control the oxidation states, crystal phases, and morphologies of MnOx nanocrystals by a facile and low-temperature synthetic route. Various manganese oxide nanostructures also could be obtained by the oxidation and dehydration of the γ-MnOOH precursors under different conditions of temperature and atmosphere. The possible chemical reactions can be formulated as follows:14
| |
 | (1) |
| |
 | (2) |
| |
 | (3) |
| |
 | (4) |
| |
 | (5) |
| |
 | (6) |
| |
 | (7) |
Conclusions
In summary, we developed a facile, high-yield and low-temperature synthetic route for the preparation of various nanostructured MnOx with crystalline phases of γ-MnOOH, δ-MnO2, α-Mn2O3, and Mn3O4 as well as shapes of the rods, sheets, cubes, and particles just by adjusting the amount of H2O2 or reactant concentration. In our synthesis system, the absence of H2O2 in the solution limits formation of Mn3+, while Mn2+ is partially oxidized to Mn3+ by oxygen under atmospheric conditions, favoring direct synthesis of Mn3O4 nanoparticles with uniform size distribution in the range of 20–30 nm. Addition of H2O2 causes complete oxidation of Mn2+ to Mn3+, favoring formation of bundle-like γ-MnOOH nanorods. These uniform γ-MnOOH nanorods with an average diameter of 10–30 nm are parallel assembled into bundles with a diameter range of 0.3–5 μm and lengths of 2–15 μm. Based on the TEM results, the formation mechanism of bundle-like γ-MnOOH nanorods through a lamellar growth, dissolution–recrystallization and oriented attachment process is proposed. Besides, at a low reactant concentration of Mn(CH3COO)2·4H2O and NaOH, the γ-MnOOH is converted to α-Mn2O3 nanocubes with edge lengths of 50–150 nm. And the δ-MnO2 nanosheets are also obtained by increasing the reaction concentration of NaOH. Furthermore, the bundle-like β-MnO2, α-Mn2O3, and Mn3O4 nanorods are synthesized by calcining the γ-MnOOH precursors under different heat treatment conditions. This synthetic route offers great opportunity for scale-up preparation of various shapes and structures of manganese oxides which may provide novel properties offering more applications in nanotechnology.
Acknowledgements
This work was supported financially by the National Basic Research Program of China (2011CB808200), the NSFC (11374120, 10979001, 51025206, 51032001, 51272224, and 21073071), and the Cheung Kong Scholars Programme of China.
Notes and references
- W. Sun, A. Hsu and R. R. Chen, J. Power Sources, 2011, 196, 627–635 CrossRef CAS PubMed.
- B. Ammundsen and J. Paulsen, Adv. Mater., 2001, 13, 943–956 CrossRef CAS.
- D. Yan, S. Cheng, R. F. Zhou, J. T. Chen, J. J. Feng, H. T. Feng, H. J. Li, Z. G. Wu, J. Wang and P. X. Yan, Nanotechnology, 2009, 20, 105706 CrossRef CAS PubMed.
- M. Drofenik, A. Znidarsic, M. Kristl, A. Kosak and D. Makovec, J. Mater. Sci., 2003, 38, 3063–3067 CrossRef CAS.
- X. L. Luo, A. Morrin, A. J. Killard and M. R. Smyth, Electroanalysis, 2006, 18, 319–326 CrossRef CAS.
- D. W. Kim, B. K. Jeon, N. S. Lee, C. S. Kim and H. I. Ryu, Talanta, 2002, 57, 701–705 CrossRef CAS.
- L. Wang, Y. L. Zheng, S. L. Chen, Y. H. Ye, F. G. Xu, H. L. Tan, Z. Li, H. Q. Hou and Y. H. Song, Electrochim. Acta, 2014, 135, 380–387 CrossRef CAS PubMed.
- M. Baldi, V. S. Escribano, J. M. G. Amores, F. Milella and G. Busca, Appl. Catal., B, 1998, 17, L175–L182 CrossRef CAS.
- T. Yamashita and A. Vannice, Appl. Catal., B, 1997, 13, 141–155 CrossRef CAS.
- M. S. Hassan, T. Amna, D. R. Pandeya, A. M. Hamza, Y. Y. Bing, H. Kim and M. Khil, Appl. Microbiol. Biotechnol., 2012, 95, 213–222 CrossRef CAS PubMed.
- M. Baldi, E. Finocchio, F. Milella and G. Busca, Appl. Catal., B, 1998, 16, 43–51 CrossRef CAS.
- X. S. Peng and I. Ichinose, Adv. Funct. Mater., 2011, 21, 2080–2087 CrossRef CAS.
- V. Q. Chiu and J. G. Hering, Environ. Sci. Technol., 2000, 34, 2029–2034 CrossRef CAS.
- Z. H. Yang, Y. C. Zhang, W. X. Zhang, X. Wang, Y. T. Qian, X. G. Wen and S. H. Yang, J. Solid State Chem., 2006, 179, 679–684 CrossRef CAS PubMed.
- T. Kanasaku, K. Amezawa and N. Yamamoto, Solid State Ionics, 2000, 133, 51–56 CrossRef CAS.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- M. S. Song, K. M. Lee, Y. R. Lee, I. Y. Kim, T. W. Kim, J. L. Gunjakar and S. Hwang, J. Phys. Chem. C, 2010, 114, 22134–22140 CAS.
- Z. Q. Li, Y. Ding, Y. J. Xiong and Y. Xie, Cryst. Growth Des., 2005, 5, 1953–1958 CAS.
- K. Song, J. Jung, Y. Heo, Y. C. Lee, K. Cho and Y. Kang, Phys. Chem. Chem. Phys., 2013, 15, 20075–20079 RSC.
- Y. F. Han, F. X. Chen, Z. Y. Zhong, K. Ramesh, L. W. Chen and E. Widjaja, J. Phys. Chem. B, 2006, 110, 24450–24456 CrossRef CAS PubMed.
- Y. J. Zhang, Y. Yan, X. Y. Wang, G. Li, D. R. Deng, L. Jiang, C. Y. Shu and C. R. Wang, Chem.–Eur. J., 2014, 20, 6126–6130 CrossRef CAS PubMed.
- J. Cao, Q. H. Mao, L. Shi and Y. T. Qian, J. Mater. Chem., 2011, 21, 16210–16215 RSC.
- J. H. Zhang, J. Du, H. L. Wang, J. L. Wang, Z. K. Qu and L. Jia, Mater. Lett., 2011, 65, 2565–2567 CrossRef CAS PubMed.
- G. Cheng, L. Yu, T. Lin, R. N. Yang, M. Sun, B. Lan, L. L. Yang and F. Z. Deng, J. Solid State Chem., 2014, 217, 57–63 CrossRef CAS PubMed.
- V. G. Kumar and K. B. Kim, Ultrason. Sonochem., 2006, 13, 549–556 CrossRef CAS PubMed.
- S. Thota, B. Prasad and J. Kumar, Mater. Sci. Eng., B, 2010, 167, 153–160 CrossRef CAS PubMed.
- Z. C. Bai, B. Sun, N. Fan, Z. C. Ju, M. H. Li, L. Q. Xu and Y. T. Qian, Chem.–Eur. J., 2012, 18, 5319–5324 CrossRef CAS PubMed.
- F. Y. Cheng, J. Shen, W. Q. Ji, Z. L. Tao and J. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 460–466 CAS.
- T. Gao, F. Krumeich, R. Nesper, H. Fjellvag and P. Norby, Inorg. Chem., 2009, 48, 6242–6250 CrossRef CAS PubMed.
- J. L. Zhou, L. Yu, M. Sun, B. Lan, F. Ye, J. He and Q. Yu, Mater. Lett., 2012, 79, 288–291 CrossRef CAS PubMed.
- T. Kohler, T. Armbruster and E. Libowitzky, J. Solid State Chem., 1997, 133, 486–500 CrossRef CAS.
- J. E. Post, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS.
- T. Gao, M. Glerup, F. Krumeich, R. Nesper, H. Fjellvag and P. Norby, J. Phys. Chem. C, 2008, 112, 13134–13140 CAS.
- X. F. Shen, Y. S. Ding, J. C. Hanson, M. Aindow and S. L. Suib, J. Am. Chem. Soc., 2006, 128, 4570–4571 CrossRef CAS PubMed.
- H. W. Hou, Y. Xie, Q. Yang, Q. X. Guo and C. R. Tan, Nanotechnology, 2005, 16, 741–745 CrossRef CAS.
- M. Z. B. Hussein, Z. Zainal, A. H. Yahaya and D. W. V. Foo, J. Controlled Release, 2002, 82, 417–427 CrossRef.
- C. H. Holm, C. R. Adams and J. A. Ibers, J. Phys. Chem., 1958, 62, 992–994 CrossRef CAS.
- X. Wang and Y. D. Li, Chem.–Eur. J., 2003, 9, 300–306 CrossRef CAS PubMed.
- J. Yang, J. H. Zeng, S. H. Yu, L. Yang, G. E. Zhou and Y. T. Qian, Chem. Mater., 2000, 12, 3259–3263 CrossRef CAS.
- E. Hosono, M. Ichihara and H. S. Zhou, Nanotechnology, 2008, 19, 395605 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra02241j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 

















