
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Di-magnesium and zinc catalysts for the copolymerization of phthalic anhydride and cyclohexene oxide†
Prabhjot K.
Saini
,
Charles
Romain
,
Yunqing
Zhu
and
Charlotte K.
Williams
*
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk
First published on 15th July 2014
Abstract
Two new homogeneous dinuclear catalysts for the ring-opening copolymerization of phthalic anhydride (PA)/cyclohexene oxide (CHO) and the terpolymerization of phthalic anhydride (PA)/cyclohexene oxide (CHO)/carbon dioxide (CO2) are reported. The catalysts are a di-magnesium (1) and a di-zinc complex (2), both are coordinated by the same macrocyclic ancillary ligand. Both catalysts show good polymerization control and activity (TOF = 97 (1) and 24 (2) h−1), with the di-magnesium complex (1) being approximately four times faster compared to the di-zinc (2) analogue. Their relative reactivity is closely related to that observed for well documented chromium salen/porphyrin catalysts. However, these results represent the first example of a well-defined magnesium catalyst which may be advantageous in terms of obviating use of co-catalysts, low cost, lack of colour and redox chemistry.
Introduction
Polyesters are a commodity produced on a 50 million tonne scale, annually.1 The most commonly applied route to prepare them is via condensation ‘AA + BB’ copolymerizations. However, these step growth syntheses are limited by a number of factors including: (1) the requirement for precise monomer stoichiometry in order to access high molecular weights; (2) the need for forcing conditions to drive the esterification reactions; and (3) the lack of polymerization control. Thus, the preparation of well-defined polyesters, as well as those with sophisticated molecular architectures and block copolymers, is complex, sometimes even impossible, using step-growth routes. The ring-opening polymerization (ROP) of cyclic esters offers a controlled polymerization route to aliphatic polyesters.2,3 However, there are only a limited range of polymerizable lactones, thereby narrowing the range of possible polymer structures.4 An attractive alternative is the ring-opening copolymerization (ROCOP) of epoxides and anhydrides (Scheme 1).5,6–12 This method is particularly desirable as it is highly controlled, and there is a wide variety of commodity epoxides/anhydrides which significantly broadens the range of polymers. Importantly, the ROCOP route enables the preparation of polyester backbones containing aromatic/semi-aromatic repeat units, which cannot be accessed using ROP but are useful to improve the polymers’ thermal–mechanical properties.6,9,10,12,13 Furthermore, the ROCOP route can be applied using a range of monomers derived from renewable resources,14 such as limonene oxide6,12 or maleic anhydride,6,7,9,10 which could be beneficial to improve the sustainability of the polymer manufacture.14,15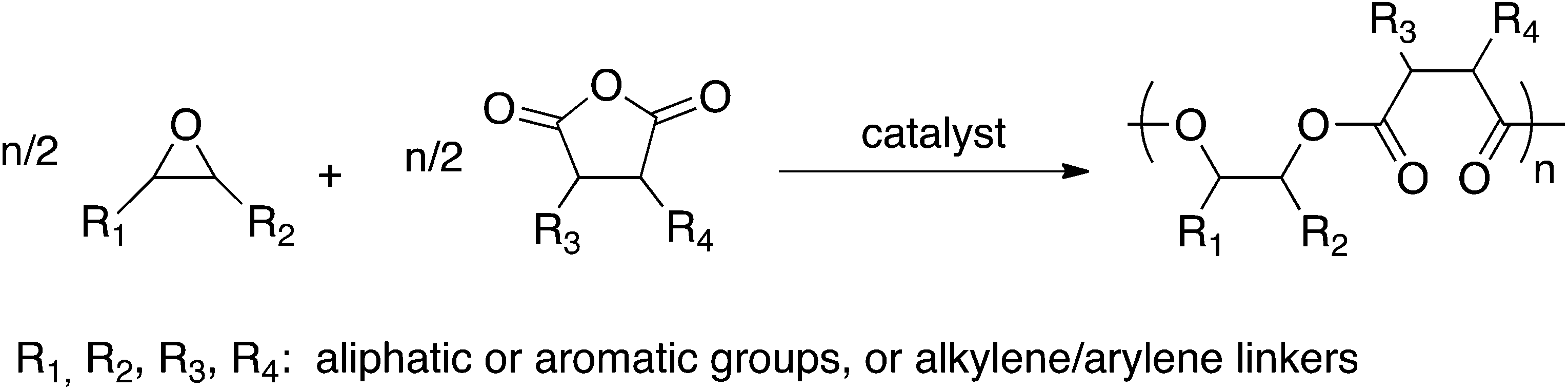 | ||
| Scheme 1 Illustrates the ROCOP (ring-opening copolymerization) of epoxides/anhydrides to afford polyesters. | ||
The ROCOP route is critically dependent on the selection of the metal catalyst which controls the polymerization rate, the degree of polymerization control and the monomer selectivity. While a plethora of catalysts are known for the ROP of cyclic esters,3 a far narrower range are known for epoxide/anhydride ROCOP. The homogeneous catalysts generally feature a Lewis acid metal centre(s), such as Zn(II), Cr(III), Co(III), Mn(III) or Al(III), either as homoleptic alkoxide/alkyl complexes16 or, more preferably, coordinated by ligands selected from salens9 and salans,17 β-diimines6,7 or porphyrins.8,10–12,18 Heterogeneous catalysts are also known and the most common type are double-metal cyanide (DMC) complexes.19 Generally, homogeneous heteroleptic metal alkoxides/carboxylate complexes are preferable in terms of polymerization control and selectivity. In such cases, the copolymerization is proposed to occur via a coordination–insertion mechanism whereby the metal alkoxide intermediate, formed by ring-opening of the epoxide, reacts with the anhydride, and the resulting metal carboxylate intermediate reacts with the epoxide to regenerate the metal alkoxide. Therefore, alternating copolymerization occurs by the continual cycling between metal alkoxide and carboxylate intermediates. Most of the active catalysts for epoxide/anhydride ROCOP are also effective for epoxide/CO2 ROCOP8,9,20 an attractive carbon dioxide consuming reaction, which also occurs via a related coordination–insertion pathway (with rapid interchange between metal alkoxide and carbonate intermediates). Combining the two ROCOP processes in a terpolymerization of epoxide/CO2/anhydride is of interest to generate new materials, however, there are only limited reports of homogenous catalysts for such terpolymerizations. These include β-diiminate zinc complexes and chromium porphyrin/salen/salophen complexes.8,9,20 The development of new terpolymerization ROCOP catalysts is of relevance in order to control the composition, and hence properties, of the copolymers.
Here, two examples of new zinc and magnesium homogeneous catalysts for the alternating copolymerization (ROCOP) of cyclohexene oxide (CHO) and phthalic anhydride (PA) are reported.
Results and discussion
ROCOP of cyclohexene oxide/phthalic anhydride
Recently, we reported catalysts 1 and 2 for the ROCOP of cyclohexene oxide with CO2 affording polycarbonates, at only one atmosphere of CO2, with very high selectivity for polymer formation.21,22–24 Given the similarities between the proposed pathways for the two ROCOP processes, 1 and 2 were tested for the copolymerization of cyclohexene oxide (CHO) and phthalic anhydride (PA) (Scheme 2 and Table 1).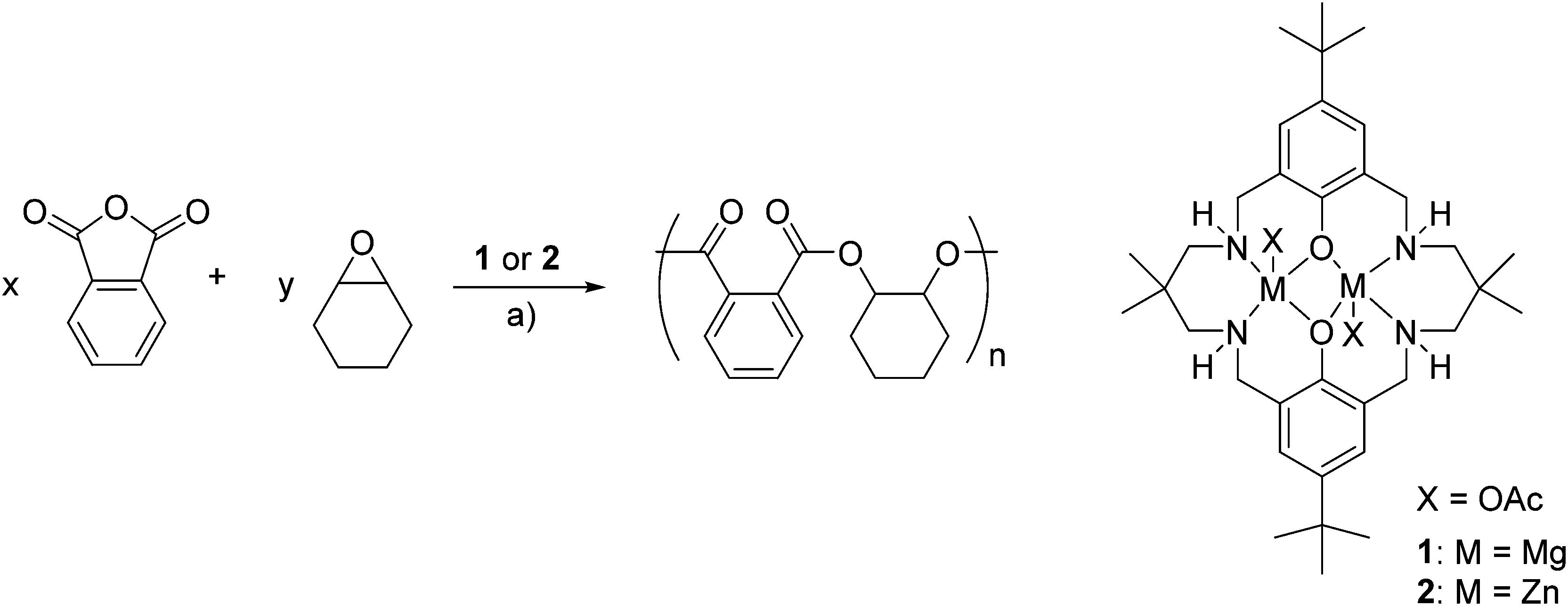 | ||
Scheme 2 Illustrates ROCOP of phthalic anhydride (PA) and cyclohexene oxide (CHO), initiated by complexes 1 or 2. Reagents and conditions (a): 100 °C, toluene, [PA] = 2.5 M, catalyst![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PA:CHO = 1:100:100 or neat CHO as the solvent, catalyst:PA:CHO = 1:100:800. :PA:CHO = 1:100:100 or neat CHO as the solvent, catalyst:PA:CHO = 1:100:800. | ||
| Run | Catalyst (Cat.) | Cat./PA/CHO | Solvent | t (h) | PA conv.a,b (%) | % ester linkagesc |
M
nd (g mol−1) |
M n Calc. (g mol−1) | PDId |
|---|---|---|---|---|---|---|---|---|---|
| Reactions were conducted at 100 °C.a Determined by 1H NMR spectroscopy (CDCl3) by integrating the normalized resonances for PA (7.97 ppm) and the phenylene signals in PE (7.30–7.83 ppm).b % error in PA conversion was <3% in all cases.c Determined by 1H NMR spectroscopy (CDCl3) by integrating the normalized resonances for ester linkages (4.80–5.26 ppm) and ether linkages (3.22–3.64 ppm).d Determined by SEC in THF, calibrated using polystyrene standards. | |||||||||
| 1 | 1 | 1/100/800 | Neat | 1 | 97 | >99 | 12670 |
11930 |
1.10 |
| 5470 | 1.06 | ||||||||
| 2 | 2 | 1/100/800 | Neat | 1 | 24 | >99 | 2570 | 5900 | 1.20 |
| 3 | 1 | 1/100/100 | Toluene | 22 | 19 | 83 | 3800 | 4670 | 1.11 |
| 4 | 2 | 1/100/100 | Toluene | 22 | 15 | 82 | 2250 | 3690 | 1.17 |
| 5 | 2 | 1/100/100 | Neat | 4 | 100 | >99 | 21170 |
12300 |
1.06 |
| 9100 | 1.08 | ||||||||
Polymerization in toluene solutions
In toluene solutions, both catalysts slowly afforded polyester, poly(1,2-cyclohexylene-1,2-phthalate) (PE), at temperatures of 100 °C ([PA]0 = 2.5 M). After 22 h, low conversions of PA were observed: 19 and 15% using 1 and 2, respectively (determined by comparison of the integrals of the aromatic protons in phthalic anhydride and the polyester, in the 1H NMR spectrum). Compared to neat conditions (see below section) the conversion values are much lower in toluene solutions, which is expected due to dilution factors.Considering the structure of the polymer, it is possible to form either perfectly alternating polyester structures by sequential epoxide/anhydride copolymerization or by sequential enchainment of epoxides, ether linkages may also form. The relative amounts of these different repeat units are usually analysed by comparing the integrals of signals in the 1H NMR spectra, however, it was discovered that when the sample dissolved was in CDCl3, the results were inconclusive as the ether signals overlapped (3.5–3.3 ppm) with the end group signals of the polyester (3.6–3.4 ppm). However, the 1H NMR spectra recorded in DMSO-d6 for a mixture of polyether and polyester showed no such overlap (Fig. S1:† polyester signals observed at 3.46 ppm and ether linkages at 3.59 ppm). Hence, for solutions of the polymer in DMSO-d6, the ether content can be determined by comparison of the relative integrals of the main chain and ether resonances. These spectra showed that in all cases there is a high content of ester linkages (>80%) with only moderate (<20%) contamination by ether linkages (a representative example of a polymer sample with ether linkage contamination is illustrated in Fig. S2.† The % ether linkages for all samples are reported in Table 2).
| Entry | Polymers | M n (PDI)a | PDI | % esterb | % carbonateb | % etherb | T g/°C | T d/°C |
|---|---|---|---|---|---|---|---|---|
| a Determined by SEC using polystyrene standards to calibrate the instrument. b Determined by 1H NMR spectroscopy by integrating the normalized resonances for ester (4.80–5.26 ppm), carbonate (4.40–4.80 ppm) and ether linkages (3.22–3.64 ppm). | ||||||||
| 1 | PCHC (Zn) | 4035 | 1.16 | 0 | >99 | <1 | 65 | 162 |
| 2 | PE (Zn) | 4200 | 1.14 | >99 | 0 | <1 | 57 | 316 |
| 3 | PE (Mg) | 12700 |
1.03 | >99 | 0 | <1 | 83 | 351 |
| 5500 | 1.08 | |||||||
| 4 | PE-PCHC (Zn) | 20000 |
1.01 | 30 | 56 | <1 | 104 | 199/317 |
| 9300 | 1.03 | |||||||
| 5 | PE-PCHC (Mg) | 19450 |
1.10 | 28 | 66 | <1 | 97 | 167/291 |
| 8400 | 1.06 | |||||||
The polyesters have low number averaged molecular weights, SEC analysis shows monomodal distributions with Mn < 5000 g mol−1 and narrow polydispersity indices (<1.2), due to the low conversion of PA (see Table 1). These values are in good agreement with the calculated values (without any calibration correction), assuming that, on average, one polymer chain is initiated per catalyst.24
Polymerizations in neat cyclohexene oxide
Polymerizations using cyclohexene oxide as both the monomer and the solvent showed substantially faster rates and higher conversions than in toluene solutions (Table 1, runs 1–2). Indeed, under these conditions it was possible to drive the polymerizations to complete consumption of anhydride (Table 1, runs 1 & 5). The magnesium containing catalyst 1 is approximately four times faster than the zinc analogue 2 (Table 1, entry 2). Catalyst 1 converts ∼97% of PA in 1 h, compared to 2 which converts 24% in 1 h, giving TOF = 97 h−1 and 24 h−1 for 1 and 2, respectively, based on PA consumption at 100 °C. This result is in line with the relative rates observed for ROCOP of CHO/CO2 where for the same catalysts 1 is six times faster than 2 (TOF = 152 h−1 and 25 h−1 for 1 and 2 at 100 °C, respectively).23In the case of the zinc catalyst 2, for CO2/CHO ROCOP the catalyst loading is 0.1 mol% and the TOF is 25 h−1, in contrast for PA/CHO ROCOP the catalyst loading is ten times higher (1 mol%) to achieve the same TOF (24 h−1): thus, CO2/CHO ROCOP is substantially faster than PA/CHO. In the case of the magnesium catalyst, the ROCOP of CHO/CO2 is around 1.5 times faster than CHO/PA at ten times lower catalyst loading. Considering the two different ROCOP catalytic cycles (Fig. 6), one explanation for this difference in rates may be a higher barrier to ring-opening of cyclohexene oxide by the zinc/magnesium carboxylate group (phthalate) (corresponding with a lower value for k2) compared to the zinc/magnesium carbonate group (corresponding to a higher value for k2′). Examining the results for other known catalysts reveals that there are rather few comparisons between the two ROCOP processes. In the case of [(BDI)ZnOAc], these catalysts show a lower activity for anhydride/epoxide compared to CO2/epoxide copolymerization. This reduction in rate was attributed to the faster insertion of the epoxide into the Zn–carbonate bond compared to the Zn–carboxylate bond.20
Generally, the activities of 1 and 2 (TOF = 152 h−1 and 25 h−1 in bulk, respectively) are similar to those reported for [(salen)MCl] and [(salophen)MCl] (M = Al, Cr, Co) homogeneous catalyst systems, species which additionally require ionic co-catalysts. These combined salen/salt system which show activity values in the range 125 < TON < 250 and 25 h−1 < TOF < 50 h−1, in solution, with complexes bearing Co and Cr being the most active.11 The highest activities are observed in bulk (at 130 °C) where values are observed for the TON = 250 and TOF = 100 h−1. Similarly, a metalloporphyrin catalyst [(TPP)CrCl], with DMAP as co-catalyst, shows comparable activities with TOF = 50 h−1 or 65 h−1 in solution or bulk, respectively.8 However, unlike these catalysts, 1 and 2 are effective without any additional co-catalyst, either in solution or in bulk. Catalysts 1 and 2 afford polymers with high ester-linkage contents; it is notable that metalloporphyrin or salen systems are known to form significant ether linkage contents, with very low activities, if applied without co-catalysts.8,11 In addition, such co-catalysts may be undesirable due to their ability to initiate side reactions and compromise the fidelity of the end groups.11 To the best of our knowledge, this is the first example of a well-defined magnesium complex for epoxide/anhydride ROCOP. Although one example of a homoleptic magnesium alkoxide catalyst (Mg(OEt)2) was reported previously, such species are known to aggregate25 and so the precise catalyst nuclearity and structure is not clear. Magnesium catalysts are attractive due to the low cost, low toxicity and abundance of the element. As an additional benefit most Mg complexes are colourless and inert to any redox chemistry.
Using neat CHO as the reaction medium, the polyesters formed using 1 and 2 show perfectly alternating structures, with no detectable ether linkage contamination (Table 1 and Fig. S1†). This high selectivity towards polyester formation suggests that these dinuclear catalysts have the correct balance of Lewis acidity (to aid epoxide and anhydride binding) and lability (to aid carboxylate or alkoxide attack of the epoxide or anhydride respectively). The polyesters have low molecular weights and bimodal molecular weight distributions, with the higher peak being approximately twice the Mn of the lower (Fig. S3†). Related bimodal molecular weight distributions were also observed for both 1 and 2 for CHO/CO2 ROCOP.22 Furthermore, the molecular weights obtained are somewhat lower than the calculated values, although the Mn values are calibrated using polystyrene standards. As the properties and behaviour of PE is likely quite different to that of polystyrene, the molecular weights are only indicative.8,9 However, it does appear that there is a general trend towards lower than expected molecular weights being observed for the products of epoxide/anhydride copolymerization. Other researchers have also observed that a range of different catalysts all produce polyesters of substantially lower molecular weights than would be expected;8 this reduction in Mn has been attributed to chain transfer reactions occurring with protic impurities, including water. Here, it is notable that increasing the quantity of CHO present (by up to 8 times versus catalyst), results in a substantial decrease in Mn, despite the polymerizations reaching higher overall conversions. This suggests that the epoxide is the source of some of the chain transfer agents; one possible species being cyclohexane diol (CHD) which could form by the reaction (catalysed) between CHO and any residual water. Every effort was made to exclude water from the reaction, including by drying and distilling the CHO, however, it should be appreciated that levels as low as 0.06 mol% (<10 ppm by mass) of residual water, versus the total amount of epoxide present, would be expected to result in the observed reductions of Mn.8 The bimodal weight distributions can be rationalised by the presence of mono-functional (acetate) and bifunctional (cyclohexane diol) initiating groups. Chains initiated from cyclohexane diol would be expect to propagate at the same rate as chains initiated from acetate groups, resulting in chain growth from both hydroxyl moieties and formation of a telechelic polymer.22 Thus, the higher Mn series is attributed to telechelic polyesters formed by initiation from cyclohexane diol, whilst the lower Mn series corresponded to chains initiated by acetate groups (from the catalyst).8 The MALDI-ToF spectrum of the polymer produced with complex 1 (Table 1, entry 1, Mn: 12670 (1.10) and 5470 (1.06) g mol−1) shows 2 series of peaks. These differ according to the end-groups: one series is α-acetyl-ω-hydroxyl and the other is α,ω-di-hydroxyl end-capped (Fig. 1). It should be noted that in the MALDI-ToF spectrum, the higher Mn series (12670 g mol−1 by SEC) is not fully observed (only the lower molecular weight tail, red circles), likely due to a lower propensity to volatilize commonly observed with this technique. The lower Mn series (5470 g mol−1 by SEC), end-capped with acetate groups, corresponds well with the MALDI-ToF series with Mn 3153 g mol−1.
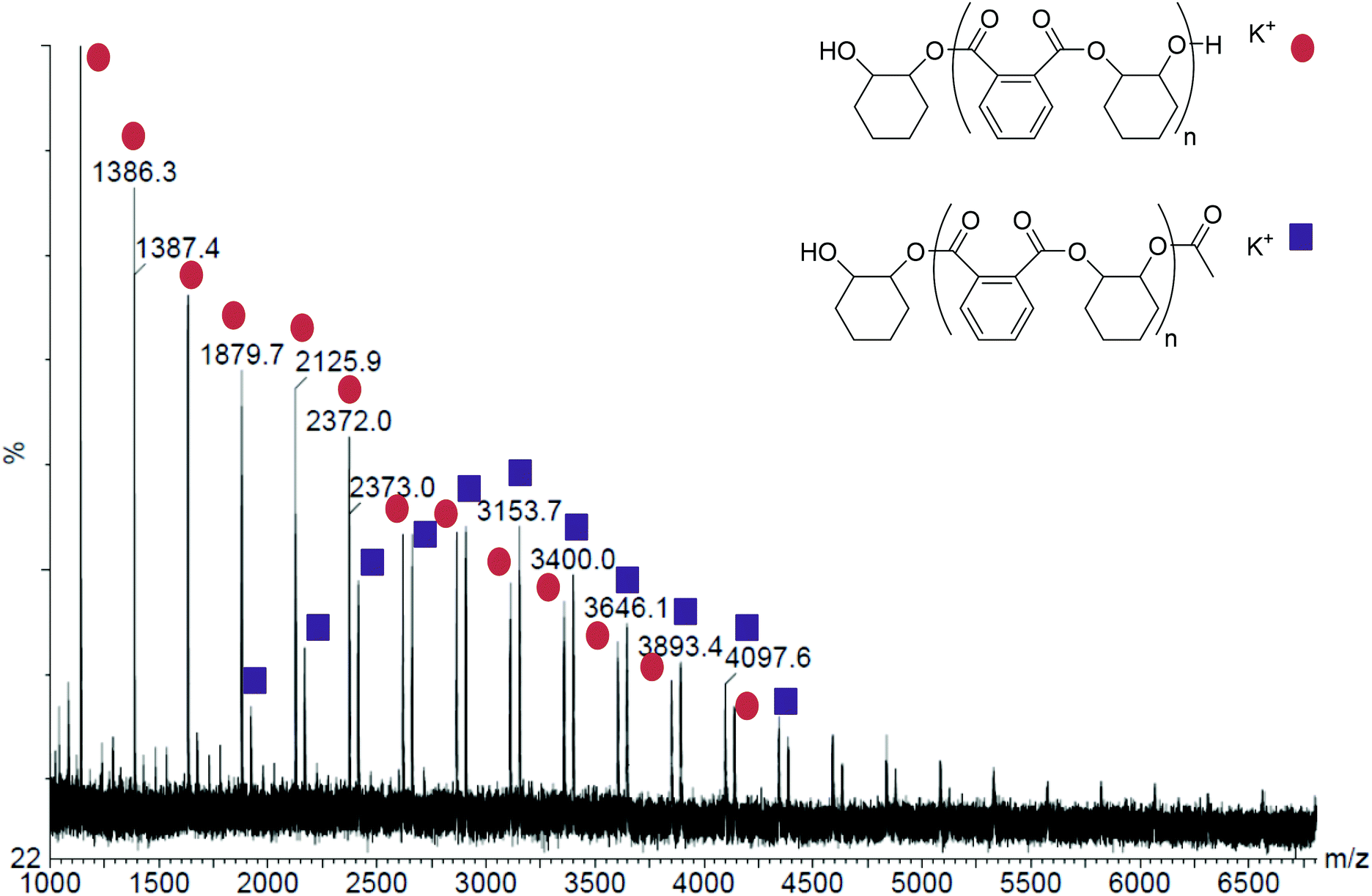 | ||
| Fig. 1 The MALDI-ToF spectrum of the polyester formed by complex 1 (Table 1, Run 1). | ||
Polymerization control and kinetic study
The polymerization control was monitored by taking aliquots and the evolution of molecular weights of the polyesters plotted against the PA conversion (Fig. 2 and S4†). This resulted in a linear correlation between the Mn and PA conversion for both catalysts, thereby signalling that both complexes were able to exert good polymerization control. Further support for well controlled polymerization comes from the narrow polydispersity indices of the polyesters produced.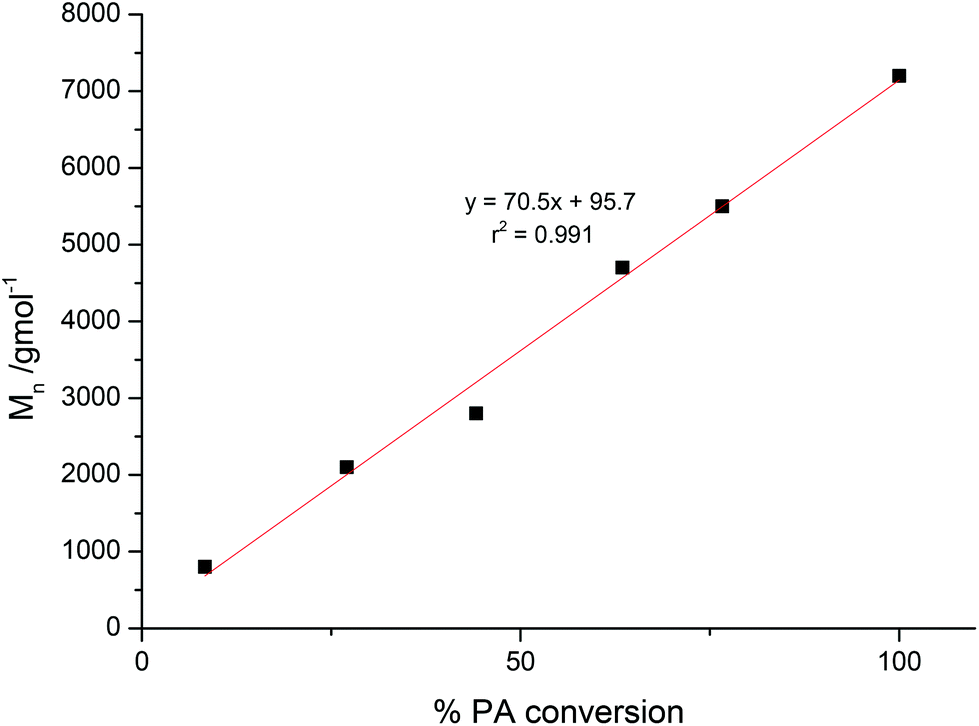 | ||
| Fig. 2 Evolution of Mn against PA conversion for ROCOP initiated by 1. Polymerization conditions: Cat.:PA:CHO = 1:100:800, 100 °C. At higher PA conversions SEC data becomes bimodal and, in these cases, the higher Mn of the two peaks is plotted. | ||
It was also of interest to investigate the polymerization kinetics and in particular the relationship between phthalic anhydride conversion and reaction time (Fig. 3a).
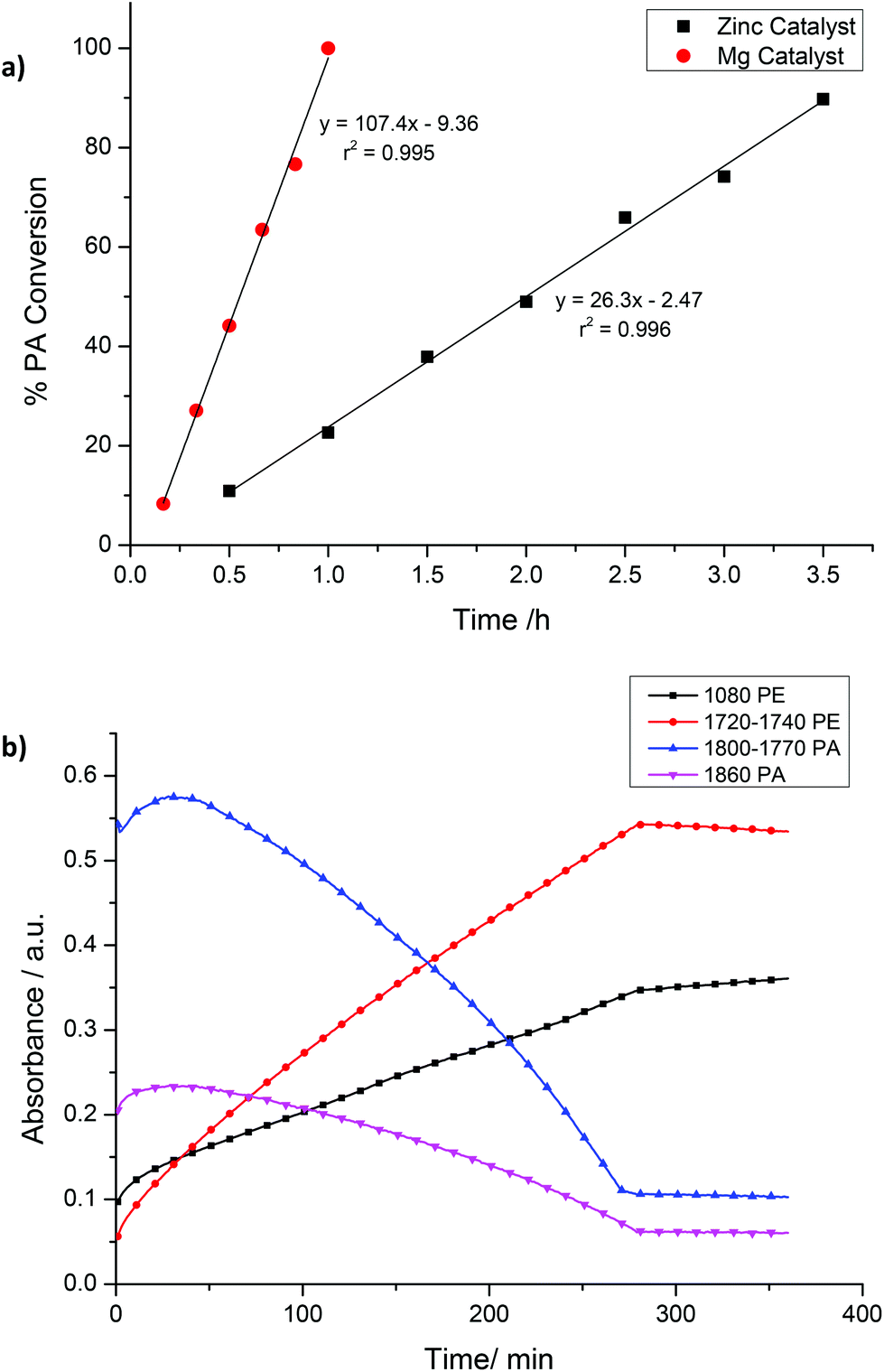 | ||
| Fig. 3 (a) Illustrates PA conversion (determined from the 1H NMR spectra) vs. time. (b) Illustrates the absorption intensity vs. time for various signals in the IR spectra for PA/CHO copolymerization using 2. Polymerization conditions: Cat. = 1 or 2, cat.:PA:CHO = 1:100:800, 100 °C. Where PA = phthalic anhydride and PE = polyester. Increase in PA concentration at start due to time required for PA to dissolve in the injected CHO. | ||
The 1H NMR data (Fig. 3a) show that the % conversion of PA increases linearly vs. time, a finding that is strongly indicative of a zero order dependence of the rate on PA concentration. Such a zero order rate dependence is also supported by monitoring of the polymerization using an in situ ATR-IR probe, which enables continual monitoring of the IR spectra as the polymerization progresses (Fig. 3b and S5†). Plotting the intensity of resonances associated with PA (1860 and 1800–1700 cm−1) also indicated there was a linear reduction in phthalic anhydride concentration. Thus, both NMR and IR spectroscopic data indicate that the rate of polymerization does not depend on the concentration of phthalic anhydride, suggesting that PA insertion occurs faster than epoxide ring-opening. In a previous polyester copolymerization study, the [(BDI)ZnOAc] catalyst also showed a zero order rate dependence on PA concentration.20
Terpolymerizations
The promising results for the ROCOP of CHO/PA prompted us to investigate the terpolymerization of CHO/PA/CO2, using CHO as the solvent, with catalysts 1 and 2 (Fig. 4).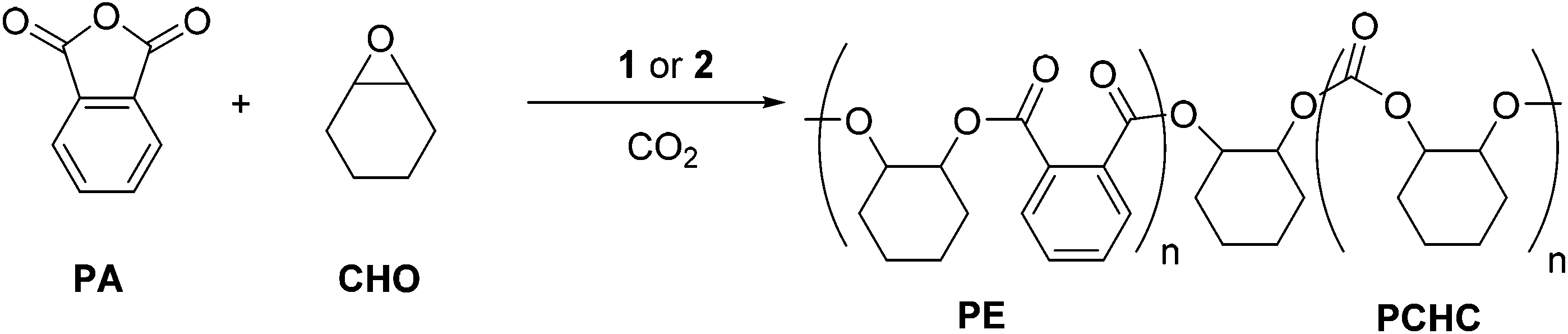 | ||
| Fig. 4 Illustrates the ROCOP terpolymerization of PA, CHO and CO2 to produce a copoly(ester-carbonate) (PE-PCHC), using catalyst 1 & 2 (catalyst structure illustrated in Scheme 1). | ||
A mixture of CHO/PA (800/100), under 1 bar of CO2, afforded well-defined block poly(ester-co-carbonates) with both catalysts 1 and 2. The polymerizations were monitored using the ATR-IR spectroscopic probe (Fig. 5 (2), S6 (1)†). In both cases, there are two clearly observable phases during which different monomers are enchained leading to the formation of the block copolymers. During the first phase, the concentration of anhydride decreases (1860 and 1800–1770 cm−1) and that of polyester (PE) increases (1720–1740 and 1080 cm−1). The concentration of polycarbonate (PCHC) is invariant (1014 and 1239–1176 cm−1), consistent with the first phase of the polymerization involving only PA/CHO copolymerization to give polyester. The slight increase in the intensity of the PCHC signal at 1014 cm−1 during this polyester forming phase is likely due to overlap of PCHC signals with polyester signals as they have similar stretch frequencies. After the PA has been fully consumed and the second phase of the polymerization begins. In this phase, the concentration of polycarbonate (PCHC) increases (1239–1176 and 1014 cm−1), but PA and polyester remain invariant (any apparent slight increase in signal intensity is due to the overlap of these frequencies with the PCHC frequencies). This is consistent with CHO/CO2 copolymerization occurring only after the PA is fully consumed and with the formation of a block copoly(ester-carbonate). In order to confirm this, aliquots were taken during the reaction. 1H NMR spectroscopy is used to determine the species present during different phases of the polymerization (Fig. S7 and S8†); during phase one only polyester is observed, and once PA consumption is complete (as evidenced by the loss of the signal at 7.9 ppm), the formation of PCHC occurs (as shown by the increase in intensity of the signal at 4.6 ppm). In the case of the magnesium catalyst 1, which is substantially faster than the zinc analogue, some carbonate repeat units do form once the conversion of phthalic anhydride exceeds 95% as shown in the 1H NMR spectra (Fig. S8†). In the case of the zinc catalyst 2, there is no evidence for any carbonate repeat units until the PA is completely consumed (Fig. S7†). Using both catalysts, there is <5% conversion to the cyclic carbonate by-product, demonstrating the high selectivity of the catalyst.
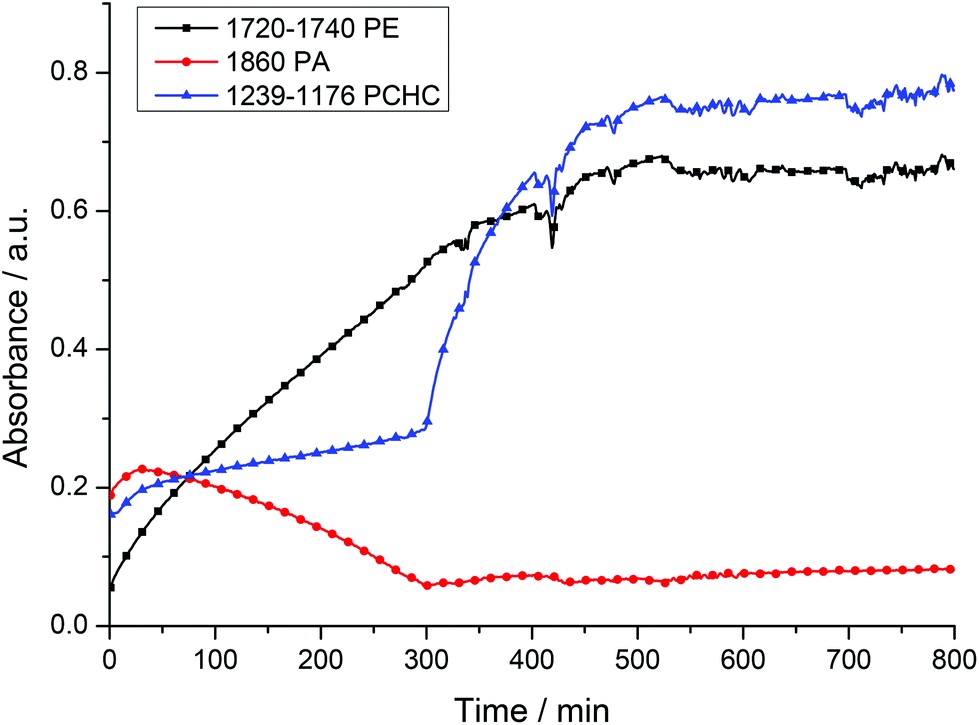 | ||
| Fig. 5 Shows the changes in the intensity of the ATR-FTIR resonances observed during ROCOP of PA, CHO and CO2 using complex 2. Polymerization conditions: Cat.:PA:CHO = 1:100:800 under 1 bar CO2 at 100 °C. The baseline ‘noise’ observed after 400 minutes results from an increase in sample viscosity due to polymerization reaching relatively higher conversions. Increase in PA concentration at start due to time required for PA to dissolve in the injected CHO. | ||
Similar monomer selectivity and block copolymer formation was previously observed for terpolymerizations using zinc β-diiminate,20 chromium porphyrin,8 chromium salen9 and chromium salophen8 catalysts. The observed selectivity is in accordance with the rate of insertion of anhydride being considerably faster than that of CO2 (k1 > k1′ in Fig. 6). Previous kinetic studies using catalyst 2 for CO2/CHO copolymerization have shown that there is a zero order dependence of the rate on CO2 pressure, over the range 1–40 bar.22 Thus, both the PA and CO2 insertion steps are pre-rate determining steps. Moreover, it is notable that the presence of the CO2 doesn't appear to significantly affect the polymerization kinetics of polyester formation; the complete consumption of PA occurs approximately as quickly as under a N2 atmosphere.
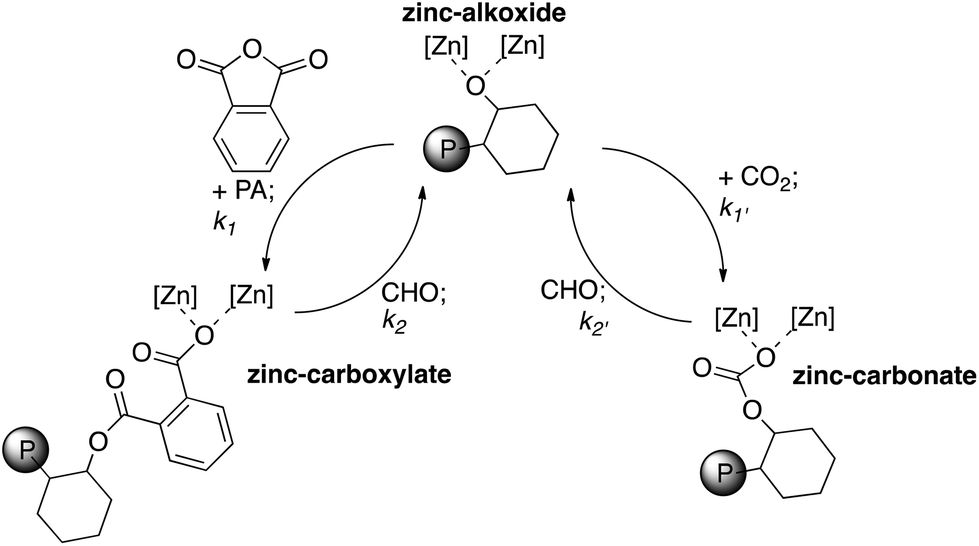 | ||
| Fig. 6 Illustrates the proposed pathways possible for metal alkoxide, carbonate and carboxylate intermediates during ROCOP. It is proposed that the relative order of rates is: k1 > k′1 ≫ k′2 > k2. Where [Zn] [Zn] = 2 (illustrated in Fig. 1) and P = growing polymer chain. | ||
Thus, the proposed elementary steps occurring during polymerization are illustrated in Fig. 6. The zinc alkoxide intermediate formed by ring-opening of the cyclohexene oxide can react either with phthalic anhydride or CO2. The rate of reaction with PA exceeds that of CO2, leading to rapid formation of the zinc carboxylate intermediate. The carboxylate reacts with CHO to re-generate the alkoxide. Only once all of the PA is consumed does the polymerization enter the second cycle (Fig. 6, RHS) whereby the alkoxide intermediate reacts with carbon dioxide to generate the polycarbonate block.
Polymer characterization
Thermal analyses of the polymers obtained using catalysts 1 and 2 revealed glass transition temperatures (Tg) of 57 and 83 °C for the polyester PE and 65 °C for the polycarbonate PCHC (produced using 2). The values for PE and PCHC are lower than the maximum values reported for these materials which are 107 °C and 115 °C, respectively.10,12 This is, likely, due to the lower molecular weights of the samples and/or unoptimised purification procedures.8,9 The block poly(ester-co-carbonates), PE-PCHC show only a single Tg at 97 and 104 °C, for polymers from 1 and 2, respectively. This indicates that the blocks are miscible, a related observation was made for block copoly(ester carbonates) by Duchateau et al.8 These block copolymers show a pronounced increase in Tg which has probably arisen due to the increases in molecular weights (Table 2).Conclusions
In conclusion, two new catalysts for the alternating copolymerization of cyclohexene oxide and phthalic anhydride are reported. These catalysts are di-magnesium and di-zinc macrocyclic complexes. The former is particularly significant because magnesium complexes are not yet well precedented for epoxide/anhydride ROCOP catalysis, despite their beneficial properties including low cost, lack of colour, lack of redox chemistry and abundance. The magnesium catalyst was four times faster than its zinc counterpart, which is in line with the relative rates observed with the same catalysts for the copolymerization of cyclohexene oxide/CO2. Both catalysts afford well controlled polymerizations, yielding polyesters with low molecular weights. Both complexes are also active for the terpolymerizations of cyclohexene oxide, phthalic anhydride and CO2: resulting in the formation of block copoly(ester-carbonates). The thermal properties of all the new polymers are reported, the terpolymers show a single glass transition above 100 °C, indicative of block miscibility. The differences between the catalysts, and the polymer products, for the two ROCOP processes illustrate the central importance of selecting the metal centre for this class of polymerizations. It also highlights the potential to control the rate, selectivity and polymer morphology by judicious choice of the metal catalyst. Future studies will exploit these findings to prepare a wide range of (co)polymers.Acknowledgements
Research funding was provided by the EPSRC (EP/K014070/1, EP/K035274/1 and EP/L017393/1) and by Grantham Institute for Climate Change and EIT Climate KIC (studentship to PKS).References
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- M. H. Chisholm and Z. P. Zhou, J. Mater. Chem., 2004, 14, 3081–3092 RSC; C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC; R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 RSC; J. M. Becker, R. J. Pounder and A. P. Dove, Macromol. Rapid Commun., 2010, 31, 1923–1937 CrossRef CAS PubMed; H. Seyednejad, A. H. Ghassemi, C. F. van Nostrum, T. Vermonden and W. E. Hennink, J. Controlled Release, 2011, 152, 168–176 CrossRef PubMed.
- T. Aida and S. Inoue, J. Am. Chem. Soc., 1985, 107, 1358–1364 CrossRef CAS; T. Aida, K. Sanuki and S. Inoue, Macromolecules, 1985, 18, 1049–1055 CrossRef; A. Bernard, C. Chatterjee and M. H. Chisholm, Polymer, 2013, 54, 2639–2646 CrossRef PubMed.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2011, 133, 10724–10727 CrossRef CAS PubMed.
- S. Huijser, E. HosseiniNejad, R. l. Sablong, C. D. Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning, R. Duchateau, E. Hosseininejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308–1313 RSC.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- C. E. Koning, R. J. Sablong, E. H. Nejad, R. Duchateau and P. Buijsen, Prog. Org. Coat., 2013, 76, 1704–1711 CrossRef CAS PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011 DOI:10.1038/ncomms1596.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- S. Inoue, K. Kitamura and T. Tsuruta, Makromol. Chem., 1969, 126, 250–265 CrossRef CAS; H. L. Hsieh, J. Macromol. Sci., Part A: Pure Appl. Chem., 1973, 7, 1525–1535 CrossRef; W. Kuran and A. Niestochowski, Polym. Bull., 1980, 2, 411–416 CrossRef; S. Takenouchi, A. Takasu, Y. Inai and T. Hirabayashi, Polym. J., 2002, 34, 36–42 CrossRef.
- J. Liu, Y.-Y. Bao, Y. Liu, W.-M. Ren and X.-B. Lu, Polym. Chem., 2013, 4, 1439–1444 RSC.
- T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39–48 CrossRef CAS; C. Chatterjee and M. H. Chisholm, Chem. Rec., 2013, 13, 549–560 CrossRef PubMed; N. D. Harrold, Y. Li and M. H. Chisholm, Macromolecules, 2013, 46, 692–698 CrossRef; C. Robert, T. Ohkawara and K. Nozaki, Chem. – Eur. J., 2014, 20, 4789–4795 CrossRef PubMed.
- Z. Hua, G. Qi and S. Chen, J. Appl. Polym. Sci., 2004, 93, 1788–1792 CrossRef CAS; X.-K. Sun, X.-H. Zhang, S. Chen, B.-Y. Du, Q. Wang, Z.-Q. Fan and G.-R. Qi, Polymer, 2010, 51, 5719–5725 CrossRef PubMed; H. S. Suh, J. Y. Ha, J. H. Yoon, C.-S. Ha, H. Suh and I. Kim, React. Funct. Polym., 2010, 70, 288–293 CrossRef PubMed.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 931–933 CrossRef CAS PubMed; M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef PubMed; M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef; A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC; C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef PubMed; P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 4164–4167 RSC.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredrickson and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- K. G. Caulton and L. G. Hubert-Pfalzgraf, Chem. Rev., 1990, 90, 969–995 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4py00748d |
| This journal is © The Royal Society of Chemistry 2014 |