
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modification of cellulose model surfaces by cationic polymer latexes prepared by RAFT-mediated surfactant-free emulsion polymerization†
Linn
Carlsson
ab,
Andreas
Fall
ab,
Isabelle
Chaduc
c,
Lars
Wågberg
ab,
Bernadette
Charleux
c,
Eva
Malmström
a,
Franck
D'Agosto
c,
Muriel
Lansalot
*c and
Anna
Carlmark
*a
aKTH Royal Institute of Technology, School of Chemical Science and Engineering, Fibre and Polymer Technology, Teknikringen 56–58, SE-100 44 Stockholm, Sweden. E-mail: annac@kth.se
bKTH Royal Institute of Technology, School of Chemical Science and Engineering, Wallenberg Wood Science Centre, Teknikringen 56–58, SE-100 44 Stockholm, Sweden
cUniversité de Lyon, Univ Lyon 1, CPE Lyon, CNRS, UMR 5265, C2P2 (Chemistry, Catalysis, Polymers & Processes), Team LCPP Bat 308F, 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: muriel.lansalot@univ-lyon1.fr
First published on 30th June 2014
Abstract
This paper presents the successful surface modification of a model cellulose substrate by the preparation and subsequent physical adsorption of cationic polymer latexes. The first part of the work introduces novel charged polymer nanoparticles constituted of amphiphilic block copolymers based on cationic poly(N,N-dimethylaminoethyl methacrylate-co-methacrylic acid) (P(DMAEMA-co-MAA)) as the hydrophilic segment, and poly(methyl methacrylate) (PMMA) as the hydrophobic segment. First, RAFT polymerization of N,N-dimethylaminoethyl methacrylate (DMAEMA) in water was performed at pH 7, below its pKa. The simultaneous hydrolysis of DMAEMA led to the formation of a statistical copolymer incorporating mainly protonated DMAEMA units and some deprotonated methacrylic acid units at pH 7. The following step was the RAFT-mediated surfactant-free emulsion polymerization of methyl methacrylate (MMA) using P(DMAEMA-co-MAA) as a hydrophilic macromolecular RAFT agent. During the synthesis, the formed amphiphilic block copolymers self-assembled into cationic latex nanoparticles by polymerization-induced self-assembly (PISA). The nanoparticles were found to increase in size with increasing molar mass of the hydrophobic block. The cationic latexes were subsequently adsorbed to cellulose model surfaces in a quartz crystal microbalance equipment with dissipation (QCM-D). The adsorbed amount, in mg m−2, increased with increasing size of the nanoparticles. This approach allows for physical surface modification of cellulose, utilizing a water suspension of particles for which both the surface chemistry and the surface structure can be altered in a well-defined way.
Introduction
During the last few decades, more environmentally friendly materials have attracted significant interest. Society is demanding high-tech products derived from renewable resources; for example high strength composites from reinforcing nanofibre-containing components are extremely interesting.1,2 In many of these applications polar, hydrophilic, naturally occurring materials have to be combined with non-polar carrier films or matrix polymers. To allow for a good compatibility in composites, and/or for tailored adhesion with the non-polar components, it is necessary to modify the surface of these materials.Cellulose is one of the most abundant renewable polymers in the world and has functional hydroxyl groups in the structure that can be used as chemical handles for further modification. By applying different pretreatments to cellulose it is possible to introduce either positive or negative charges into the cellulose fibres.2 In addition to the cellulose hydroxyl groups, these charges bring about a possibility to modify cellulose in several ways to allow for its application in, for example, biocomposites.1 By modifying the surface of cellulose, it can adapt different properties such as hydrophobicity3 or responsiveness to external stimuli.4 The surface modification can be performed by attaching small or larger molecules, such as polymers,3,5 employing either covalent linkages or physical attractions. The latter approach, for instance by electrostatic interactions, is a highly versatile method to tailor the properties of cellulose since the surface modification can be performed under mild conditions in water.6–8 Furthermore, this procedure may possibly be integrated and performed in conventional processes, for example in paper making. Alince et al.9–11 reported already in 1976 the favorable effect of addition of a poly(styrene-co-butadiene) latex for paper reinforcement. The same copolymer composition was employed to prepare nanocomposite films by casting latex dispersion with cellulose whiskers which considerably improved the mechanical properties.12 The adhesive behavior between latex particles of poly(styrene-co-butyl acrylate) and cellulose was also recently studied.13 Core–shell latexes of poly(butyl acrylate-co-styrene)/poly(2-ethylhexylacrylate-co-methyl methacrylate) have also been adsorbed to cellulose fibres, increasing their hydrophobicity and enabling the formation of polypropylene-based biocomposites with improved mechanical properties.14–16
In recent years, much work has been conducted in the field of reversible-deactivation radical polymerization (RDRP) techniques which provide possibilities to design well-defined homopolymers and block copolymers.17 Carlmark and Malmström pioneered the field of surface modification of cellulose by controlled covalent polymer grafting utilizing RDRP, in their case atom transfer radical polymerization (ATRP).3,5,18,19 Zhang et al. employed a poly(glycerol monomethacrylate) macromonomer obtained by ATRP as reactive steric stabilizer in emulsion polymerization to form latexes that were adsorbed to boronic-acid derivatized cellulose.20 In 2008, Perrier et al.21 were the first to exploit reversible addition–fragmentation chain transfer (RAFT) polymerization to surface modify cellulose.
Surface modification of cellulose by poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA)22,23 has recently been investigated, as this polymer is both thermo- and pH-responsive, which makes it interesting in a wide range of applications. PDMAEMA is antibacterial24 and could be used for gene-delivery,25 but also in other fields e.g. waste-water treatment26 and paints.27 Indeed, the tertiary amine group in PDMAEMA can either be protonated when below its pKa (close to 7.5, slightly chain length dependent),28 or permanently charged by quaternization (q-PDMAEMA). Several earlier investigations have employed grafting of PDMAEMA from cellulose or the potential interaction between q-PDMAEMA/cellulose after cationization of the PDMAEMA segment incorporated to obtain more complex architectures. As an example, surface modification has been performed by physical adsorption of q-PDMAEMA-b-PCL (poly(ε-caprolactone)) to cellulose model surfaces.22 In another study, cellulose nanofibrils have been modified with a cationic amphiphilic block copolymer consisting of q-PDMAEMA-b-polybutadiene.23 However, both studies employed organic solvents for the syntheses, which implies that a quaternization step is required as post-modification to obtain charges on the hydrophilic segment. Pei et al.29 recently reported block copolymers prepared by RAFT dispersion polymerization where different PDMAEMAs, also synthesized in organic solvent, were employed as macroRAFTs for polymerization of 2-phenylethyl methacrylate (PEMA). In addition, the use of a q-PDMAEMA macroRAFT (obtained from the aqueous RAFT polymerization of q-DMAEMA) was recently reported by Semsarilar et al.30 for the dispersion polymerization of 2-hydroxypropyl methacrylate in water, thus producing amphiphilic block copolymers.
The endeavor of producing new cellulose-based hybrid materials has resulted in large interest in amphiphilic block copolymers.22,31,32 These polymers are synthesized from a hydrophilic and a hydrophobic block, which can be tailored to be highly interesting as compatibilizers.31 However, the synthesis of amphiphilic block copolymers is demanding due to the difference in solubility between the blocks, and at least two (or more) steps with thorough purification between each step is often required. Recently, the development of RAFT,33,34 one of the most versatile RDRP techniques, in emulsion polymerization, has led to the facile water-based synthesis of amphiphilic block copolymers. The amphiphilic block copolymers self-assemble to form nanoparticles according to polymerization-induced self-assembly (PISA)35 as initially described by Hawkett et al.36 Originally, the first macromolecular RAFT agents (macroRAFT) were prepared in organic solvent. However, D'Agosto et al.37 recently performed PISA following a two-step, one-pot aqueous procedure. The synthesis of the hydrophilic macroRAFT is performed in water close to full monomer conversion and is directly followed by addition of the hydrophobic monomer in the same reactor. During the second step, polymer particles are formed, without the need for any additional surfactant. This simple, robust and eco-friendly strategy has been successfully applied to acrylic acid,38 methacrylic acid (MAA),39 and a mixture of MAA and poly(ethylene oxide) methyl ether methacrylate (PEOMA).40–42 Furthermore, different hydrophobic monomers such as styrenics and (meth)acrylics have been employed for the hydrophobic core, leading to many different compositions of amphiphilic block copolymers. In addition, different morphologies have been obtained,41–43 similar to those obtained by the post-polymerization self-assembly of amphiphilic block copolymers conducted at very low concentrations (<1 wt%).44 These syntheses, employing the PISA process, were performed under conventional aqueous emulsion polymerizations conditions, which are highly attractive from an industrial point of view due to the low environmental impact, the possibility of working at high solids contents at low viscosities, the fact that no work-up is required, as well as the ease of handling the final product.
In this work, we took advantage of the above-mentioned PISA strategy to design PDMAEMA-b-poly(methyl methacrylate) (PDMAEMA-b-PMMA) amphiphilic block copolymer nanoparticles that could interact with the negatively charged surface of cellulose fibres, enabled by the cationization of the PDMAEMA block. The RAFT polymerization technique has been utilized to polymerize DMAEMA in water at pH 7, below its pKa, in the presence of a RAFT agent, 4-cyano-4-thiothiopropylsulfanyl pentanoic acid (CTPPA), and the water soluble initiator 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA). The reaction conditions were such that DMAEMA was charged and sensitive to hydrolysis, which generated a small amount of deprotonated methacrylic acid. The resulting copolymer was thus mainly composed of positively charged DMAEMA units with some deprotonated methacrylic acid units and was denoted P(DMAEMA-co-MAA). Being overall positively charged, this macroRAFT was subsequently used to initiate the growth of a PMMA block and formed latex particles after emulsion polymerization of the hydrophobic monomer MMA. Furthermore, the synthesized latex nanoparticles were adsorbed on two different model surfaces, silica and cellulose, to demonstrate their potential for surface engineering.
Experimental section
Materials
N,N-Dimethylaminoethyl methacrylate (DMAEMA, Aldrich, 98%), methyl methacrylate (MMA, Acros, 99%), 2,2′-azobis(2-methylpropionamidine)dihydrochloride (AIBA, Aldrich, 97%), hydrochloric acid (HCl, VWR Prolabo, 35 wt%, technical grade), 1,3,5-trioxane (Aldrich, ≥99%), dimethyl sulfoxide (DMSO, 99.9%, Sigma), and N-methylmorpholine N-oxide (NMMO, Aldrich, 97%) were used as received. Water was deionized prior to use (Purelab Classic UV, Elga LabWater). The RAFT agent, 4-cyano-4-thiothiopropylsulfanyl pentanoic acid (CTPPA), was prepared according to literature procedures by reacting 4,4-azobis(4-cyanopentanoic acid) (ACPA) with bis(propylsulfanylthiocarbonyl)disulfide.45–47 Poly(vinyl amine) (PVAm, Lupamin 5095, Mw = 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was kindly supplied by BASF, Germany and dialyzed against deionized water and freeze-dried prior to its use in the preparation of the model cellulose surfaces.
000 g mol−1) was kindly supplied by BASF, Germany and dialyzed against deionized water and freeze-dried prior to its use in the preparation of the model cellulose surfaces.
The QCM crystals used were AT-cut crystals (5 MHz resonance frequency) with an active surface of sputtered silica (50 nm thickness) supplied by Q-sense AB. The cellulose fibres (Domsjö Dissolving Plus; Domsjö Aditya Birla AB, Domsjö, Sweden), used for the preparation of cellulose model surfaces, were pretreated by carboxymethylation to obtain anionic carboxylic charges (350 μeq. g−1) on the cellulose fibres prior to use according to a procedure developed and described by Wågberg et al.48
Instrumentation and methods
000 g mol−1) from PSS GmbH, using DMF (0.2 mL min−1) with 0.01 M LiBr as the mobile phase at 50 °C. A conventional calibration method was employed using narrow PMMA standards ranging from 700 to 2000000 g mol−1, and was used to determine the average molar masses (number-average molar mass, Mn, and weight-average molar mass, Mw) and the molar-mass dispersity (ĐM = Mw/Mn). Corrections for flow rate fluctuations were made using toluene as an internal standard. PSS WinGPC Unity software version 7.2 was used to process the data.
The number of particles per unit volume (Np) of the aqueous phase (mL−1 water) was calculated using the diameter obtained from TEM (Dn, nm) according to eqn (1), with τ (g mL−1 water) the solids content of the dispersed phase (τ = (mmacroRAFT + conversion × mMMA)/Vwater, with mmacroRAFT and mMMA the initial weight of P(DMAEMA-co-MAA) macroRAFT and MMA, respectively, Vwater the initial volume of water) and ρ the density of PMMA (1.18 g cm−3).
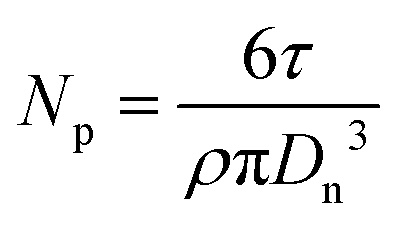 | (1) |
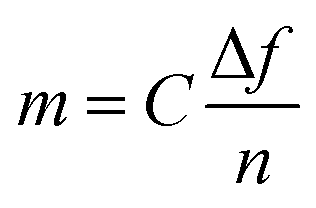 | (2) |
The dissipation is related to the viscoelastic properties of the adsorbed layer. A thin, rigid attached film is expected to yield a low change in dissipation. A more water-rich and mobile film is expected to yield a larger change in dissipation. The dissipation factor, D, is defined as:
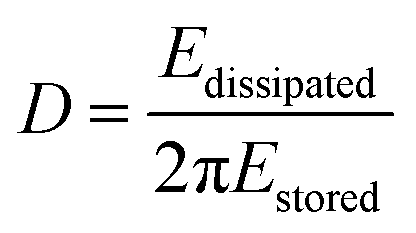 | (3) |
RAFT polymerization of DMAEMA in water
CTPPA (0.212 g, 0.766 mmol) and AIBA (21.2 mg, 76.6 μmol) were dissolved in 5.00 mL of water in a 25 mL round bottom flask equipped with a magnetic stirrer. DMAEMA (3.00 g, 18.1 mmol) and the internal reference for NMR analysis, 1,3,5-trioxane (0.272 g, 3.02 mmol), were added. The pH was adjusted to 7 by adding 35 wt% hydrochloric acid drop-wise and finally water to a total volume of 9.20 mL. The reaction mixture was degassed with argon in an ice bath for 30 minutes and thereafter immersed in a thermostated oil bath preheated to 70 °C. The monomer conversion was monitored by 1H NMR spectroscopy performed in D2O on samples withdrawn regularly during the polymerization by comparing the relative integrations of the protons of 1,3,5-trioxane and the vinylic protons of DMAEMA. The targeted number-average molar mass of PDMAEMA was 4000 g mol−1. The polymer was precipitated in cold acetone (0 °C) and dried under reduced pressure.Due to both the slight hydrolysis and the protonation of DMAEMA under the conditions employed, the resulting copolymer was mainly composed of charged DMAEMA units with some deprotonated methacrylic acid units (approximately 7%, see 1H-NMR, Fig. S1 and S2 in ESI†), and was denoted P(DMAEMA-co-MAA).
Theoretical number average molar masses were calculated using eqn (4):
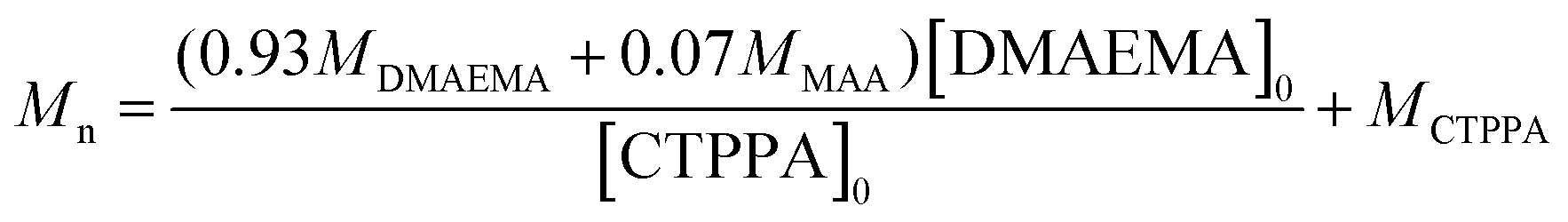 | (4) |
RAFT mediated emulsion polymerization of MMA in water
The synthesis is exemplified by entry 2 in Table 1. To a 25 mL round bottom flask equipped with a magnetic stirring bar, 0.393 g of P(DMAEMA-co-MAA) macroRAFT was added, followed by water. The added amount of water was adjusted so that the final concentrations of the macroRAFT and the monomer (MMA) were 6.00 mM and 2.12 M, respectively, corresponding to a solids content of 20 wt%. An aqueous solution of the initiator AIBA (3.4 g L−1) was prepared and 1 mL was added to the reaction mixture. The reaction mixture was degassed with argon on an ice bath for 30 minutes and thereafter immersed in a thermostated oil bath at 70 °C. The monomer conversion was followed by gravimetric analysis by withdrawing samples regularly during the polymerization. The molar mass and the molar-mass dispersity (ĐM) were determined by SEC in DMF.| Entrya | [macroRAFT]0 (mM H2O) | DPn,targetb | M n target (kg mol−1) | Reaction time (min) | Conv. (%) | τ (%) | SEC (DMF) | DLS | TEM | ζ Potentialh (mV) | Surface charge densityi (meq. g−1) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M n (kg mol−1) | Đ M | D h | PdIe | D n |
N
pg (L−1 water) |
|||||||||
| a All polymerizations were performed at 70 °C and pH 6. b Targeted DPn of the PMMA block. Calculations based on the targeted molar mass for the macroRAFT (3800 g mol−1). c Solids content. d Obtained by DMF-SEC using PMMA calibration. e Obtained by DLS. f Obtained by TEM. g Calculated from the number-average diameter obtained by TEM. h Measured in 10−3 M KCl at pH 6.5. i Determined by PET. | ||||||||||||||
| Latex 1 | 15 | 176 | 21.4 | 90 | 90 | 25 | 59 | 1.2 | 50 | 0.08 | 28 ± 5 | 8.70 × 1015 | 26.8 | 0.76 |
| Latex 2 | 7.3 | 353 | 39.1 | 80 | 83 | 20 | 116 | 1.4 | 68 | 0.05 | 44 ± 6 | 2.24 × 1015 | 36.5 | 0.43 |
| Latex 3 | 3.7 | 705 | 74.4 | 90 | 86 | 20 | 240 | 1.4 | 98 | 0.03 | 77 ± 6 | 4.18 × 1014 | 40.8 | 0.24 |
| Latex 4 | 1.9 | 1410 | 145 | 90 | 80 | 17 | 380 | 1.8 | 146 | 0.01 | 122 ± 11 | 1.05 × 1014 | 43.2 | 0.12 |
Preparation of the cellulose model surface
The procedure for preparation of model cellulose surfaces was developed and described by Wågberg et al.52 Prior to adsorption of the cellulose, the QCM silica coated crystals were rinsed in water and ethanol and cleaned in a plasma cleaner (PDC-32G, Harrick Scientific Corp., Ossining, New York, USA) with an effect of 100 W for 2 minutes under vacuum. Thereafter, the crystal was exposed to a PVAm solution (0.01 g L−1, pH 7.4) which was adsorbed as an anchoring layer. The carboxymethylated cellulose fibers (0.25 g) were dissolved in NMMO (12.5 g, 50 wt% solution in water) at 120 °C and then 37.5 g of DMSO was added to dilute the solution. Thereafter, the dissolved cellulose was spin-coated onto the PVAm coated QCM crystals and regenerated by exposure to Milli-Q water. The obtained cellulose model surface was expected to have a thickness of 30–40 nm according to the literature.52Preparation of latex dispersion for adsorption onto model surfaces
The latex dispersions were purified by dialysis in Milli-Q water and thereafter diluted to a concentration of 100 mg L−1 in Milli-Q water. All adsorptions were performed at pH 6 (pH was not adjusted).Results and discussion
Herein, we report a straightforward method for the synthesis of cationic latex nanoparticles designed for the surface modification of cellulose to achieve a hydrophobic surface with a specific topography determined by the size of the latex particles. The prepared charged latex particles were readily adsorbed onto negatively charged cellulose surfaces in aqueous solutions without any post modification of the latex.Synthesis of P(DMAEMA-co-MAA)-stabilized PMMA latexes
The successful synthesis of amphiphilic block copolymers, consisting of P(DMAEMA-co-MAA) as the hydrophilic and charged block and PMMA of varying molar masses as the hydrophobic block, is reported. Initially, CTPPA was employed as a chain transfer agent for the RAFT polymerization of DMAEMA in water (Scheme 1). It should be noted that CTPPA itself is not water-soluble; however, CTPPA is soluble in the hydrophilic monomer, DMAEMA, and can therefore be employed in an aqueous system.37 There are few earlier investigations available where DMAEMA has been polymerized in aqueous media by ATRP53,54 or by RAFT polymerization.55 However, all those studies lack structural analyses, i.e. NMR data. It is known that DMAEMA is sensitive towards hydrolysis,56 both under acidic and basic conditions, and that the hydrolysis is accelerated in the presence of charged polymers. However, the corresponding polymer, PDMAEMA, is not sensitive.28,57 Indeed, DMAEMA is advantageous compared to its acrylate counterpart, 2-(dimethylamino)ethyl acrylate (DMAEA), where both monomer and polymer poly(2-dimethylaminoethyl acrylate) (PDMAEA) are hydrolytically unstable.58 Self-catalyzed degradation of linear cationic PDMAEA in water, independently of molar mass and the pH of the solution, has been reported.59,60 Perrier et al.58 reported the hydrolysis of DMAEA upon polymerization in aqueous solution and speculated that the same behavior could occur for the methacrylate analogue but to a minor extent. We thus selected the more hydrolytically stable methacrylate, DMAEMA, for the aqueous RAFT polymerization to form the cationic hydrophilic block.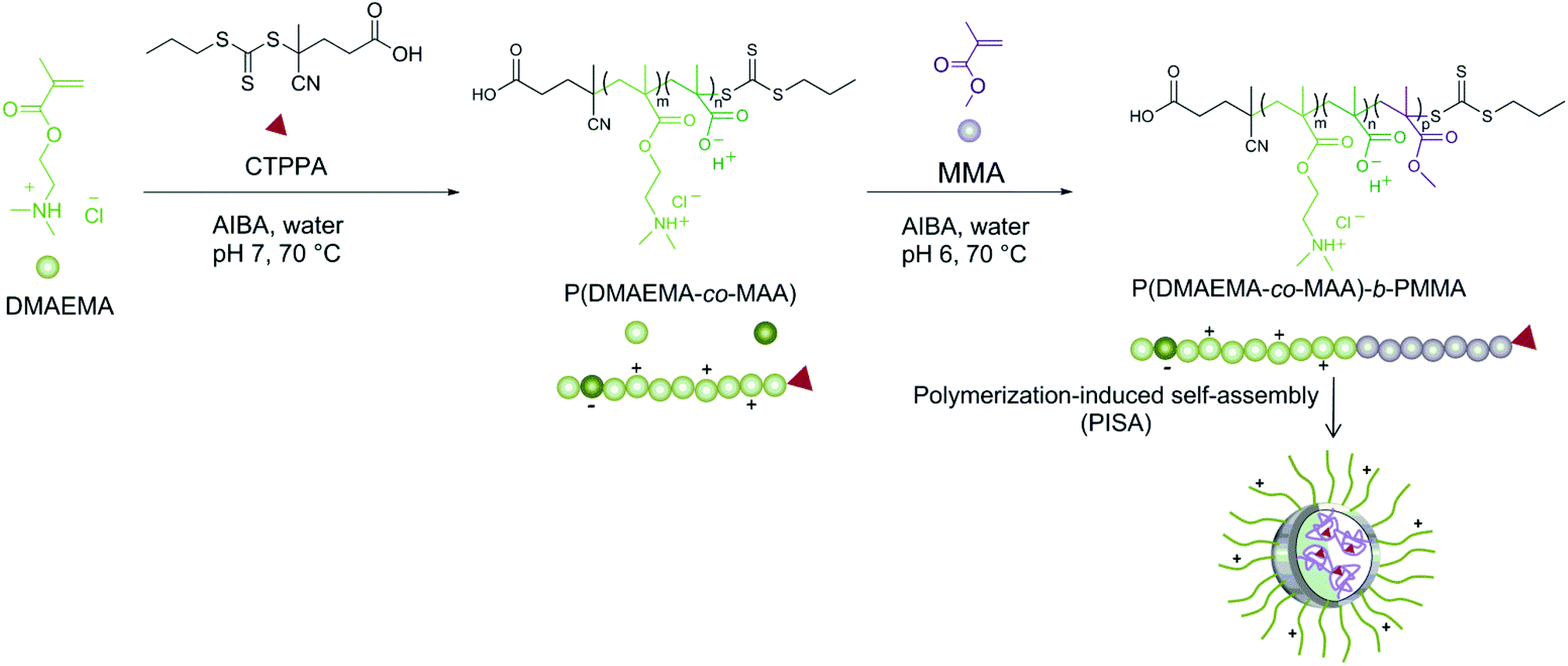 | ||
| Scheme 1 Schematic illustration of RAFT-mediated surfactant-free emulsion polymerization (PISA) of MMA to form P(DMAEMA-co-MAA)-decorated latex particles. | ||
Initial attempts to synthesize the macroRAFT species were performed without adjusting the pH (9.4). However, this resulted in substantial hydrolysis of DMAEMA which was verified by the formation of (deprotonated) methacrylic acid. By 1H-NMR, the two methylene (–CH2) protons in the (deprotonated) methacrylic acid (δ = 3.15 ppm and δ = 3.75 ppm) were observed. To circumvent this side-reaction, the pH was adjusted to 7 for the synthesis of the macroRAFT, a pH value for which the monomer is positively charged (pKa = 8.4).28 Consequently, the resulting latex particles would carry cationic charges originating from DMAEMA units that could interact with the anionic cellulose surface during the following adsorption step. Despite the decreased pH, MAA was still formed during the macroRAFT synthesis but to a lower extent. Approximately 7% of DMAEMA was hydrolyzed to MAA during the RAFT polymerization according to NMR (Fig. 2 in ESI†), resulting in the formation of a P(DMAEMA-co-MAA) copolymer carrying mainly cationized DMAEMA units and some deprotonated MAA units, thus providing an overall positively charged macroRAFT. Full conversion was reached in 2 h and no induction period was observed. However, the determination of molar mass and molar-mass dispersity could not be performed due to insolubility in DMF. Several attempts were made to analyze the macroRAFT by SEC using different eluents (DMF with LiBr, without and by adding base (NaOH), THF, and water with NH4OAc/NaNO3) but none of them were successful. This may be explained by the polyelectrolyte nature of the macroRAFT. However, the molar mass characteristics of the macroRAFT were successfully determined by MALDI-ToF MS (Fig. 3 in ESI†). The number average molar mass was determined to be 3900 g mol−1 in good agreement with the targeted molar mass, 3800 g mol−1 (re-calculated taking into account the presence of MAA units), and the molar-mass dispersity was reasonably narrow, ĐM = 1.22. Besides, the net charge density of the macroRAFT was determined by polyelectrolyte titration in Milli-Q water to be 4.2 meq. g−1.
The P(DMAEMA-co-MAA) macroRAFT was employed in the RAFT-mediated surfactant-free emulsion polymerization (PISA) of MMA to form P(DMAEMA-co-MAA)-decorated latex particles (Scheme 1). Table 1 presents the experimental conditions used for MMA polymerizations, targeting four different molar masses (DPn = 176, 353, 705 and 1410, for Latexes 1, 2, 3 and 4, respectively) by varying the initial amount of macroRAFT and keeping the amount of MMA constant. Thereby, the solids content varied from 25 to 17 wt% when the macroRAFT concentration decreased from 15 to 1.9 mM H2O, while the pH remained close to 6 in all cases. High monomer conversions were obtained after short reaction times for all performed polymerizations (Fig. 1a). Slightly lower conversion was obtained for Latex 4, DPn = 1410 (Table 1, entry 4). Unfortunately, as for the macroRAFT, it was difficult to monitor the evolution of molar mass with monomer conversion by SEC using common eluents (THF, water, chloroform or DMF) due to the insolubility of the block copolymers at low MMA conversions, owing to the dominating nature of the P(DMAEMA-co-MAA) hydrophilic segment. Other studies also report difficulties in the characterization of similar amphiphilic block copolymers that were soluble in DMF.30 The SEC traces for the final samples given in Fig. 1b showed that higher molar masses were obtained with increasing targeted DPn. The corresponding values of the molar masses can be found in Table 1, and were considerably higher than the targeted molar masses. There are several potential reasons for this; the SEC instrument was calibrated with linear PMMA standards, which possibly differ in hydrodynamic volume compared to the cationic P(DMAEMA-co-MAA)-b-PMMA polymer. Besides, the trithiocarbonate chain end of hydrophilic polymethacrylate type macroRAFT such as poly(methacrylic acid)38,61 has proven to be sensitive to hydrolysis, particularly at basic pH. P(DMAEMA-co-MAA) may follow this trend. The loss of active trithiocarbonate chain ends (during the macroRAFT synthesis or in the next emulsion polymerization step) may thus explain the higher molar mass observed for the resulting amphiphilic block copolymers. Furthermore, the charged nature of the macroRAFT, as already mentioned in the literature,30,38,62 may impede the first addition–fragmentation steps and thus disturb the self-assembly process.
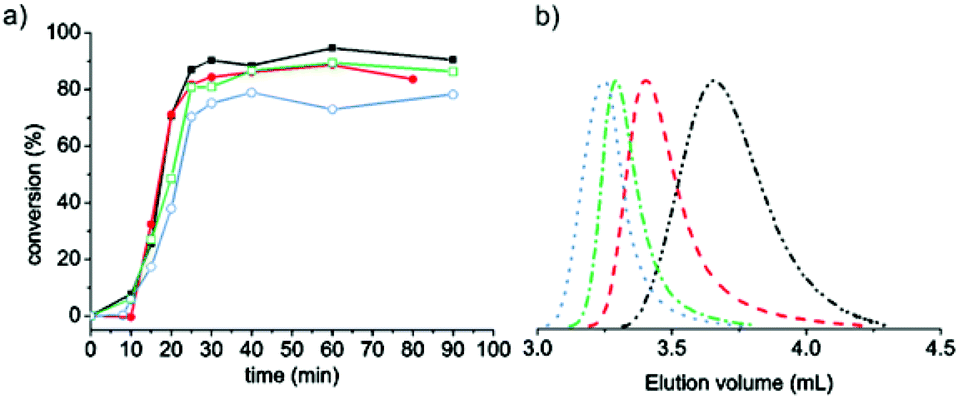 | ||
| Fig. 1 (a) Evolution of monomer conversion versus time for RAFT emulsion polymerizations of MMA mediated by P(DMAEMA-co-MAA) macroRAFT. Latex 1 (black filled squares), Latex 2 (red filled circles), Latex 3 (green hollow squares) and Latex 4 (blue hollow circles) and (b) SEC traces of final latex of Latexes 1–4, for different targeted polymerization degrees (DPn: 176 (black dash dot dotted line), 353 (red dashed line), 705 (green dashed-dotted line) and 1410 (blue dotted line). | ||
Table 1 presents the particle sizes and the polydispersity index, determined by DLS, and the ζ-potentials estimated from measurements of the electrophoretic mobility. The DLS measurements showed that the particle size increased with increasing molar mass of the PMMA block and that narrow size distributions were obtained. Further analyses by cryo-TEM (Fig. 2) indicated that spherical latex particles, uniform in size, were effectively formed. Moreover, by targeting a higher degree of polymerization for the hydrophobic segment, the size of the particles increased, which was in accordance with the results obtained by both DLS and SEC. The sizes of the particles measured by TEM were smaller compared to DLS. Most of the discrepancy between the particle sizes determined by the two different techniques could be attributed to the corona. The small particle sizes obtained, and all the abovementioned data, are consistent with the self-assembly of block copolymers and support a posteriori that the RAFT polymerization of DMAEMA performed in water at pH 7 was effectively controlled. The ζ-potentials show that all the latexes were positively charged despite the presence of deprotonated MAA units. The PET measurements (Table 1) indicated that the surface charges per gram material decreased, as expected, with an increased amount of non-charged PMMA, i.e. with the size of the particles.
 | ||
| Fig. 2 TEM images of the final P(DMAEMA-co-MAA)-b-PMMA latexes, obtained by RAFT-mediated surfactant-free emulsion polymerization (see Table 1 for details). | ||
Adsorption of cationic P(DMAEMA-co-MAA)-stabilized PMMA latexes onto cellulose and silicon wafers
The cationic latex nanoparticles were adsorbed onto negatively charged model surfaces of cellulose. When employing organic solvents for the synthesis of block copolymers incorporating a PDMAEMA segment, the quaternization of PDMAEMA is an additional step required for obtaining permanent charges. The only requirement with these pH-dependent P(DMAEMA-co-MAA)-b-PMMA latexes is to perform the adsorption at a pH leading to cationic particles, which is the case for a pH close to 6 (Table 1).Adsorption was investigated with the aid of QCM-D to evaluate the interaction between the cationic P(DMAEMA-co-MAA)-b-PMMA latex particles and the cellulose substrate.
Prior to measurements, latexes were diluted with Milli-Q water (100 mg L−1). The adsorption in the QCM-D was performed with a continuous flow of fresh particle dispersion until a stable baseline was obtained, indicating that saturation adsorption was reached. The adsorption was followed by a rinsing step to ensure that the adsorbed latexes were attached to the surface. As can be seen in Fig. 3, the significant decrease in frequency shows the adsorption of the cationic latexes to the negatively charged cellulose. The frequency is decreasing with increasing molar mass of the PMMA block in the latexes, and consequently, a decrease in the charge density of the latex with increasing the molar mass of the hydrophobic block. Thus, the adsorbed mass will increase with increasing size of the particles, prior to adsorption saturation,63 which is clearly demonstrated in Fig. 3. A change in the adsorption kinetics can also be detected for the latex with the longest hydrophobic block. The QCM-D permits a continuous flow of the solution through the equipment, allowing for a constant solution concentration of the polymer. However, the polymers have to diffuse over a stagnation layer to reach the model cellulose surface. The diffusion of the latex particles will be determined by the concentration gradient and the size of the latex particles. This size was determined by the size of the hydrophobic core of the particles and of the swollen polyelectrolyte layer on the particle surface. All the experiments were performed at a constant solids concentration. This leads to a rather complicated relationship for the samples investigated since the particle concentration will change with the size of the particles, the charge of the corona of the latex, and the size of the hydrophobic core of the latex. As is obvious in Fig. 3, Latex 4 contained the largest particles and was expected to have the slowest adsorption which was also detected. The initial adsorption kinetics for Latexes 2 and 3 were similar, whereas Latex 1 exhibited slower kinetics. Considering the change in particle concentration, as well as the size of the hydrophobic core, and that the charged corona was also changed, it is anticipated that the initial adsorption of Latex 2 and Latex 3 should be similar. However, as the adsorbed amount increased, the interaction within the adsorbed layer will also affect the adsorption kinetics, as the saturation adsorption was reached. The higher charge of Latex 1 can also explain the adsorption behavior of this species. In Fig. 3 it is shown that there was a small initial increase in frequency and also a decrease in the dissipation, indicating that deswelling of the cellulose occurred, which has also been reported earlier in the literature.64 The adsorbed amount is dependent both on the charges and on the size of the particles as discussed earlier. The driving force for the adsorption is the release of counterions, as the cationic latex adsorbs to the charges on cellulose, which results in a large gain in entropy. As the surface charge of the latex decreases the adsorbed amount, for a complete ion-exchange, will increase. As demonstrated in Table 2, an inverse relationship between the adsorbed amount and charge density of the latex particles is indeed found. Furthermore, since the polymer chains in the particles will not be able to change their conformation, as flexible polyelectrolytes do when they are adsorbed at the solid–liquid interface, there will be an increase in the adsorbed amount due to a much thicker adsorbed layer for the large latex particles. Consequently, the larger size and lower charge of the latex will have a twofold effect on the adsorbed amount of latex.
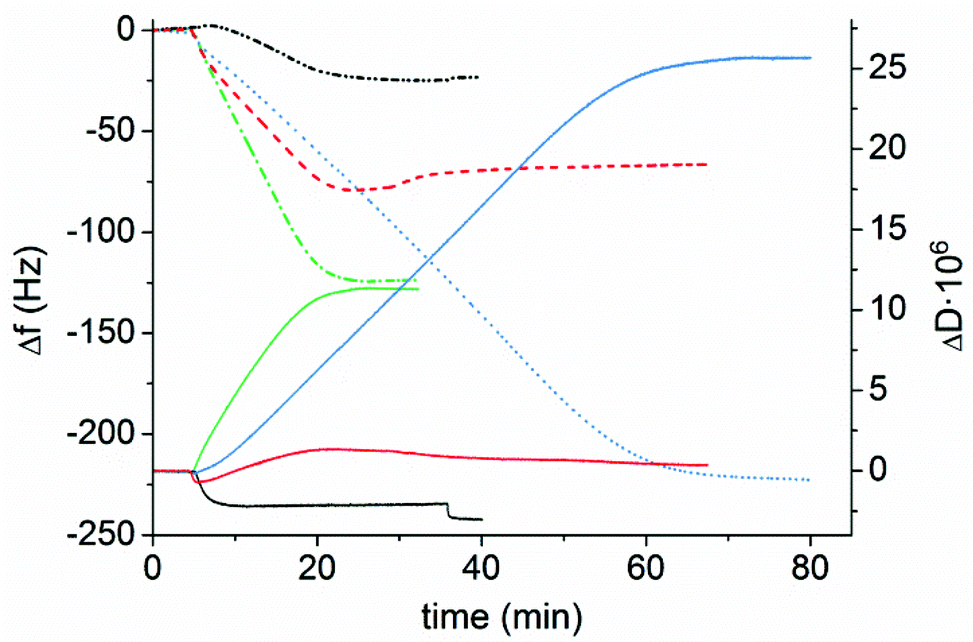 | ||
| Fig. 3 Adsorption of P(DMAEMA-co-MAA)-b-PMMA latex particles on a cellulose model surface in the QCM-D. Latex 1 (black dash dot dotted line), Latex 2 (red dashed line), Latex 3 (green dashed-dotted line) and Latex 4 (blue dotted line) correspond to the frequency for the four samples, and the solid lines Latex 1 (black), Latex 2 (red), Latex 3 (green) and Latex 4 (blue) correspond to the dissipation for the four different samples. The concentration of the latex dispersion was kept at 100 mg L−1. | ||
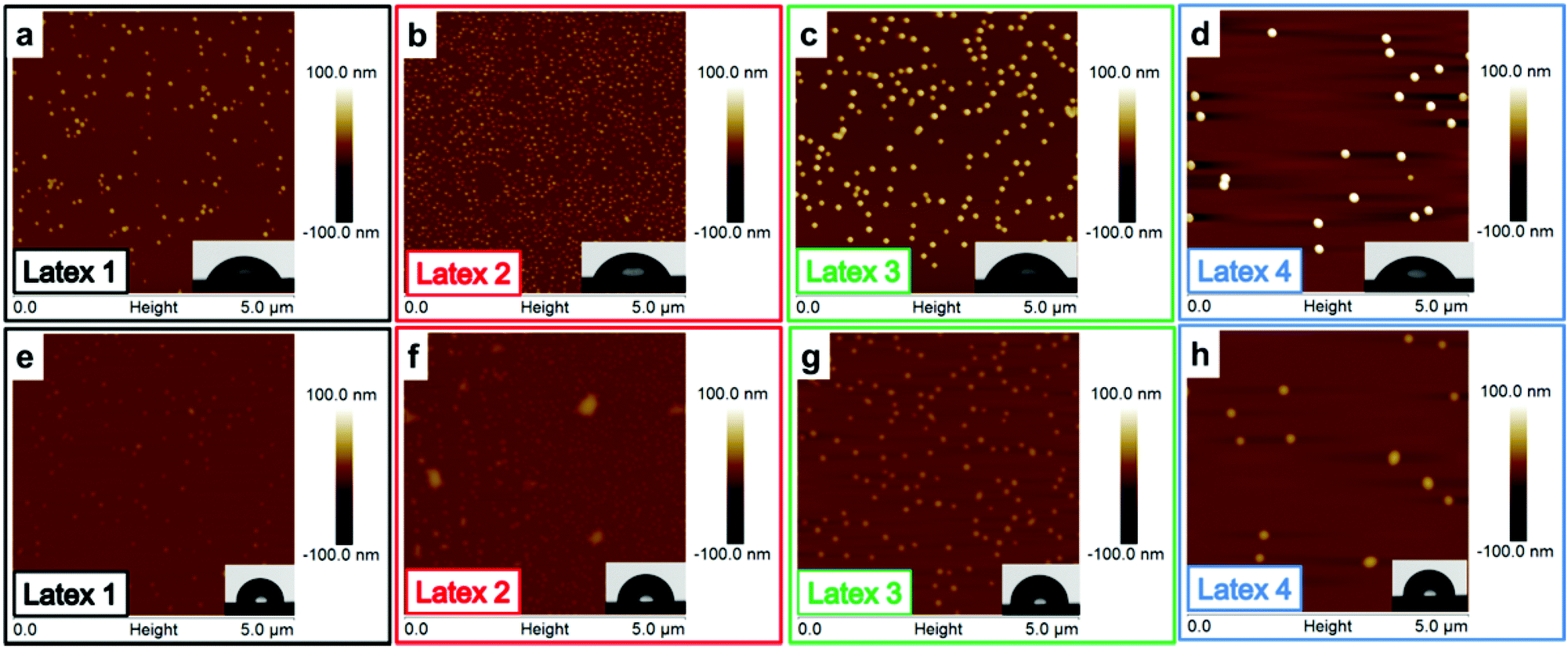
The dissipation is providing information about the viscoelastic properties of the adsorbed layer including the surface-bound solvent. By increasing the length of the PMMA block, the dissipation increased, except for the shortest PMMA block where a decrease in dissipation was shown. This is most probably due to the mild deswelling of the cellulose surface, caused by the latex with highest charge density (Latex 1) that was discussed earlier. By studying the AFM images (Fig. 4a–d), it can clearly be seen that a substantial amount of latex was adsorbed to the cellulose model surfaces. The cellulose was nearly completely covered with latex particles. This high coverage is in accordance with the large decrease in frequency observed in the QCM-D measurements. In order to examine the wetting properties of the surfaces, contact angle (CA) measurements on latex saturated cellulose model surfaces were performed. The contact angles varied between 64 and 71° which are significantly higher than 25° which is the CA for a neat cellulose model surface (Table 3). The latex modified surfaces were annealed at 160 °C for 2 h which caused the contact angle to increase to 93–98° (Table 3). The heat treatment was used to increase the mobility of the polymer chains of the particles which enabled the charged hydrophilic polymer to move to the negatively charged cellulose model surface. Hence, the PMMA hydrophobic core was exposed to the air–solid interface which increased the contact angle (Table 3). The AFM images (Fig. 4e–h) show that the height of the particles decreased after annealing, but the particle shape was maintained. This was expected since the mobility of the polymer chains increases above the glass transition temperature of PMMA. The glass transition temperatures for the various latexes were determined by DSC and varied from 116 to 121 °C (Fig. S4 and Table S1 in ESI†). The average roughness values of the entire surface can be found in Table 3. The roughness data show that the annealed surfaces have a lower roughness than the non-heated surfaces. This indicates that the particles collapsed to a certain extent during the heat treatment.
 | ||
| Fig. 4 AFM images of adsorbed cationic P(DMAEMA-co-MAA)-b-PMMA latex particles onto cellulose model surfaces formed on QCM crystals. Insert pictures show the contact angles. The four upper images (a–d) show the surfaces after adsorption of latex (100 mg L−1) and the lower four images (e–h) were analysed after annealing of the corresponding surfaces at 160 °C for 2 h. | ||
| Entry | CA (°) | CA (°), Δ | R q | R q, Δ |
|---|---|---|---|---|
| Neat cellulose model surface | 25 ± 1 | — | — | — |
| Latex 1 | 66 ± 4 | 93 ± 2 | 14 | 4.6 |
| Latex 2 | 71 ± 2 | 93 ± 3 | 12 | 4.9 |
| Latex 3 | 67 ± 2 | 95 ± 2 | 14 | 4.2 |
| Latex 4 | 64 ± 6 | 98 ± 1 | 32 | 11 |
Due to the large amount of adsorbed latex on the cellulose model surfaces, it was not possible to observe individual latex nanoparticles. Therefore, as a control experiment, the latexes were deposited on silicon wafers, using a short adsorption time to obtain lower particle coverage. Hence, the individual particles were easier to visualize. Apart from the shorter adsorption time, the surface roughness was an important difference between the silica surfaces and the cellulose model surfaces. The silica surface is considerably smoother, providing a smaller surface area which will affect the adsorption and improve the visualization of the individual particles. On the silica surfaces (Fig. 5), the particles were clearly, individually separated and all particles were spherically shaped. As expected, the particle sizes determined by AFM were similar to those previously obtained with DLS and cryo-TEM (Table 1). The full coverage of the cellulose model surface is probably due to the high affinity between the latex particles and the cellulose and the more extended adsorption times for the cellulose surfaces. The particles are expected to repel each other due to electrosteric repulsion. However, the latex–cellulose attraction, i.e. the gain in entropy upon release of counterions, is dominating, which gives rise to a high particle coverage for the cellulose model surface which has a considerably higher charge in comparison to the silicon wafer. Contact angle measurements were also performed for the silicon wafers, showing contact angles between 48 and 60° for the different samples. Those values are in accordance with the earlier published data where Yu et al. reported CA of 50° on PDMAEMA-grafted silica surfaces.65 In the same work, block copolymers of PDMAEMA-b-PMMA have also been grafted yielding CAs of 65°, and the contact angle for the homopolymer PMMA grafted surfaces was 70°. Upon annealing, a similar behavior was observed for the silica surfaces as that for the cellulose model surfaces: the height of the particles decreased upon heating due to the annealing of the particles (Table 4). As expected, the contact angles increased due to the exposure of the PMMA and the measured CAs were within the interval 85–93° for all samples (Table 4). Those values were higher than earlier reported values in the literature, but could possibly be explained by a larger surface roughness compared with the grafted surfaces, which could increase these contact angles.
 | ||
| Fig. 5 AFM images of adsorbed cationic P(DMAEMA-co-MAA)-b-PMMA latex particles on silica surfaces. The four upper images (a–d) depict the surfaces after adsorption of latex (100 mg L−1) for 10 s and the four lower images (e–h) after annealing at 160 °C for 2 h. | ||
| Entry | CA (°) | CA (°), Δ | h p (nm) | h p (nm), Δ | R q | R q, Δ |
|---|---|---|---|---|---|---|
| Latex 1 | 54 ± 8 | 87 ± 3 | 35 ± 7 | 12 ± 3 | 5.8 | 1.7 |
| Latex 2 | 60 ± 2 | 85 ± 3 | 24 ± 10 | 7 ± 4 | 8.2 | 3.4 |
| Latex 3 | 48 ± 5 | 85 ± 1 | 58 ± 22 | 23 ± 3 | 13 | 4.7 |
| Latex 4 | 51 ± 6 | 93 ± 2 | 102 ± 11 | 40 ± 15 | 13 | 4.2 |
Conclusions
In this work, the successful synthesis of amphiphilic block copolymers, P(DMAEMA-co-MAA)-b-PMMA, has been performed by surfactant-free RAFT-mediated emulsion polymerization based on the PISA process. The properties and characteristics of four cationic latexes with different PMMA molar masses have been investigated. The stable latex nanoparticles were all spherical, showing a narrow size-distribution with different sizes of the hydrophobic cores. Adsorption was performed at a pH at which the particles were charged and large amounts were successfully adsorbed to cellulose model surfaces in a QCM-D equipment, which considerably changed the character of the modified surface. The mass adsorbed to the cellulose model surfaces was found to increase with increasing molar mass of the hydrophobic PMMA block, i.e. the size of the latex nanoparticles, and with decreasing surface charge density of the latex with increasing length of the PMMA block. Annealing of the surfaces resulted in an increased hydrophobic character both for silica substrates and cellulose model surfaces. Furthermore, the pronounced affinity between the latex particles and the cellulose is highly promising for the development of biocomposites on a larger scale where no solvent exchange, or post-functionalization, is necessary. Besides, this kind of latex is highly interesting for papermaking since its characteristics can be tailored and it is produced without any low molar mass additives that may migrate from the product.Acknowledgements
Pierre-Yves Dugas (C2P2, LCPP Team) is greatly acknowledged for his assistance with TEM. L.C. would like to thank Svensk Franska Stiftelsen, the Swedish Research Council FORMAS and Wallenberg Wood Science Centre (WWSC) for financial support. B.C. thanks the Institut Universitaire de France for nominating her as a senior member. L.W. is grateful for financial support from WWSC.Notes and references
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1 CrossRef CAS
.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358 CrossRef CAS PubMed
- A. Carlmark, Macromol. Chem. Phys., 2013, 214, 1539 CrossRef CAS
- E. Larsson, C. C. Sanchez, C. Porsch, E. Karabulut, L. Wågberg and A. Carlmark, Eur. Polym. J., 2013, 49, 2689 CrossRef CAS PubMed
- E. Malmström and A. Carlmark, Polym. Chem., 2012, 3, 1702 RSC
- G. Decher, Science, 1997, 277, 1232 CrossRef CAS
- G. Decher and J. Hong, Chem. Phys., 1991, 95 Search PubMed
-
G. Fleer, Polymers at Interfaces, Chapman & Hall, London, 1st edn, 1993 Search PubMed
- B. Alince, J. Appl. Polym. Sci., 1979, 23, 549 CrossRef CAS
- B. Alince, M. Inoue and A. A. Robertson, J. Appl. Polym. Sci., 1979, 23, 539 CrossRef CAS
- B. Alince, M. Inoue and A. A. Robertson, J. Appl. Polym. Sci., 1976, 20, 2209 CrossRef CAS
- V. Favier, G. R. Canova, J. Y. Cavaillé, H. Chanzy, A. Dufresne and C. Gauthier, Polym. Adv. Technol., 1995, 6, 351 CrossRef CAS
- C. Lidenmark, T. Pettersson, O. J. Karlsson, S. M. Notley, M. Norgren and H. Edlund, Int. J. Adhes. Adhes., 2013, 44, 250 CrossRef CAS PubMed
- Y. Pan, M. Z. Wang and H. Xiao, Compos. Sci. Technol., 2013, 77, 81 CrossRef CAS PubMed
- Y. Pan, H. Xiao and Z. Song, Cellulose, 2013, 20, 485 CrossRef CAS
- Y. Pan, H. Xiao, Y. Zhao and Z. Wang, Carbohydr. Polym., 2013, 95, 428 CrossRef CAS PubMed
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS PubMed
- A. Carlmark, E. Larsson and E. Malmström, Eur. Polym. J., 2012, 48, 1646 CrossRef CAS PubMed
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900 CrossRef CAS PubMed
- D. Zhang, K. L. Thompson, R. Pelton and S. P. Armes, Langmuir, 2010, 26, 17237 CrossRef CAS PubMed
- D. Roy, J. T. Guthrie and S. Perrier, Soft Matter, 2008, 4, 145 RSC
- S. Utsel, C. Bruce, T. Pettersson, L. Fogelström, A. Carlmark, E. Malmström and L. Wågberg, ACS Appl. Mater. Interfaces, 2012, 4, 6796 CAS
- M. Wang, A. Olszewska, A. Walther, J.-M. Malho, F. H. Schacher, J. Ruokolainen, M. Ankerfors, J. Laine, L. A. Berglund, M. Österberg and O. Ikkala, Biomacromolecules, 2011, 12, 2074 CrossRef CAS PubMed
- L.-A. B. Rawlinson, S. a. M. Ryan, G. Mantovani, J. A. Syrett, D. M. Haddleton and D. J. Brayden, Biomacromolecules, 2009, 11, 443 CrossRef PubMed
- A. P. Majewski, U. Stahlschmidt, V. Jérôme, R. Freitag, A. H. E. Müller and H. Schmalz, Biomacromolecules, 2013, 14, 3081 CrossRef CAS PubMed
- S. Zhu, N. Yang and D. Zhang, Mater. Chem. Phys., 2009, 113, 784 CrossRef CAS PubMed
-
A. Doroszkowski and V. J. Pavey, Imperial Chemical Industries PLC, UK, EP0061833 A1, 1982, p. 21 Search PubMed
- P. van de Wetering, N. J. Zuidam, M. J. van Steenbergen, O. A. G. J. van der Houwen, W. J. M. Underberg and W. E. Hennink, Macromolecules, 1998, 31, 8063 CrossRef CAS
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342 RSC
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416 CrossRef CAS PubMed
- S. Utsel, A. Carlmark, T. Pettersson, M. Bergström, E. E. Malmström and L. Wågberg, Eur. Polym. J., 2012, 48, 1195 CrossRef CAS PubMed
- K. Littunen, U. Hippi, T. Saarinen and J. Seppälä, Carbohydr. Polym., 2013, 91, 183 CrossRef CAS PubMed
-
C. Barner-Kowollic, Handbook of RAFT-polymerization, Wiley-VCH, Weinheim, 2008 Search PubMed
- M. Semsarilar and S. Perrier, Nat. Chem., 2010, 2, 811 CrossRef CAS PubMed
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753 CrossRef CAS
- C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such and B. S. Hawkett, Macromolecules, 2005, 38, 2191 CrossRef CAS
- I. Chaduc, W. Zhang, J. Rieger, M. Lansalot, F. D'Agosto and B. Charleux, Macromolecular Rapid Communications, 2011, 32, 1270 CrossRef CAS PubMed
- I. Chaduc, A. Crepet, O. Boyron, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2013, 46, 6013 CrossRef CAS
- I. Chaduc, M. Girod, R. Antoine, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2012, 45, 5881 CrossRef CAS
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 7584 CrossRef CAS
- W. Zhang, F. D'Agosto, O. Boyron, J. Rieger and B. Charleux, Macromolecules, 2012, 45, 4075 CrossRef CAS
- W. Zhang, F. D'Agosto, P.-Y. Dugas, J. Rieger and B. Charleux, Polymer, 2013, 54, 2011 CrossRef CAS PubMed
- E. Groison, S. Brusseau, F. D'Agosto, S. Magnet, R. Inoubli, L. Couvreur and B. Charleux, ACS Macro Letters, 2012, 1, 47 CrossRef CAS
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC
- G. Bouhadir, N. Legrand, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1999, 40, 277 CrossRef CAS
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435 CrossRef CAS
- T. Boursier, I. Chaduc, J. Rieger, F. D'Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355 RSC
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784 CrossRef PubMed
- D. Horn, Prog. Colloid Polym. Sci., 1978, 65, 251 CAS
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS
- C. Aulin, I. Varga, P. M. Claesson, L. Wågberg and T. Lindström, Langmuir, 2008, 24, 2509 CrossRef CAS PubMed
- S. Gunnars, L. Wågberg and M. A. Cohen Stuart, Cellulose, 2002, 9, 239 CrossRef CAS
- F. Zeng, Y. Shen, S. Zhu and R. Pelton, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3821 CrossRef CAS
- T. Xing, J. Liu and S. Li, Text. Res. J., 2013, 83, 363 CrossRef PubMed
- Q. Xiong, P. Ni, F. Zhang and Z. Yu, Polym. Bull., 2004, 53, 1 CrossRef CAS
- N. A. Kuznetsova, O. A. Kazantsev, K. V. Shirshin, T. A. Khokhlova and A. P. Malyshev, Russ. J. Appl. Chem., 2003, 76, 1117 CrossRef CAS
- X. Jiang, M. C. Lok and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2077 CrossRef CAS PubMed
- W. Zhao, P. Fonsny, P. FitzGerald, G. G. Warr and S. Perrier, Polym. Chem., 2013, 4, 2140 RSC
- N. P. Truong, Z. Jia, M. Burges, N. A. J. McMillan and M. J. Monteiro, Biomacromolecules, 2011, 12, 1876 CrossRef CAS PubMed
- P. Cotanda, D. B. Wright, M. Tyler and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3333 CrossRef CAS
- I. Chaduc, M. Lansalot, F. D'Agosto and B. Charleux, Macromolecules, 2012, 45, 1241 CrossRef CAS
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2012, 28, 914 CrossRef CAS PubMed
- L. Winter, L. Wågberg, L. Ödberg and T. Lindström, J. Colloid Interface Sci., 1986, 111, 537 CrossRef CAS
- L.-E. Enarsson and L. Wågberg, Biomacromolecules, 2009, 10, 134 CrossRef CAS PubMed
- W. H. Yu, E. T. Kang, K. G. Neoh and S. Zhu, J. Phys. Chem. B, 2003, 107, 10198 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Determination of the degree of hydrolysis of DMAEMA during its RAFT polymerization in water by 1H NMR and the molar mass of P(DMAEMA-co-MAA) by MALDI-ToF, determination of the glass transition temperatures of P(DMAEMA-co-MAA) and Latexes 1–4 by DSC and details of the adsorption measurements in the QCM-D. See DOI: 10.1039/c4py00675e |
| This journal is © The Royal Society of Chemistry 2014 |