
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVersatile NHC-based zinc and magnesium complexes for the synthesis and chemical recycling of aliphatic polyesters and polycarbonates†
Federica
Tufano
a,
Federica
Santulli
a,
Concetta
Liguori
a,
Giuseppe
Santoriello
a,
Ida
Ritacco
a,
Lucia
Caporaso
ab,
Fabia
Grisi
 ab,
Mina
Mazzeo
ab and
Marina
Lamberti
*ab
ab,
Mina
Mazzeo
ab and
Marina
Lamberti
*ab
aDepartment of Chemistry and Biology “Adolfo Zambelli” University of Salerno, via Giovanni Paolo II, 132, 84084 Fisciano, SA, Italy. E-mail: mlamberti@unisa.it
bCIRCC, Interuniversity Consortium Chemical Reactivity and Catalysis, Bari 70126, Italy
First published on 10th December 2024
Abstract
Backbone substituted NHC derivatives of zinc and magnesium, in the presence of alcohol initiators, have been shown to be effective catalysts for the ring-opening polymerization of different cyclic monomers, efficiently furnishing sustainable aliphatic polyesters and polycarbonates. Comparing the behaviour of complexes with the same ligands but different metals, the order of the activity was found to be dependent on the monomer, with magnesium complexes showing a higher activity in the ROP of ε-caprolactone (ε-CL), trimethylene carbonate (TMC) and 1-methyl-trimethylene carbonate (Me-TMC), while the zinc complexes polymerized L-lactide (L-LA) and 2,2-dimethyl-trimethylene carbonate (DTC) more efficiently than their magnesium counterparts. Also the symmetry of the backbone substituents on the NHC ligand influenced the activity in the ROP with the syn substituents inducing a higher activity for both metals and for all the monomers with respect to the ligand with the anti symmetry. Kinetic studies demonstrated the polymerization reactions of L-LA, ε-CL and TMC to proceed via a mechanism first order in monomer concentration, with the four zinc and magnesium complexes. The obtained polymers possessed controlled molecular masses and dispersities dependent on the metal (generally higher for polymers obtained by magnesium complexes). The results of MALDI-ToF and NMR analysis confirmed the controlled nature of the present catalytic systems, where side reactions, such as inter- and intramolecular transesterifications, were minimized during the polymerization. Zinc and magnesium complexes were also tested in preliminary alcoholysis experiments of PLLA samples, and found able to promote these reactions of polylactide upcycling.
Introduction
Leading researchers in the field of synthetic polymers recently described the invention of plastic as a blessing and a curse, at the same time.1 Just as the innumerable advantages of plastic materials are undeniable, due to their versatility, lightness and low cost, equally the environmental crisis of which plastic itself is one of the main culprits is there for all to see. Since it is beyond doubt that also a society without plastic would be unsustainable,2 the polymer community is working both on the production of polymers that are more sustainable for the environment3 and on the implementation of recycling techniques for plastic materials which are already on the market.4In this context, aliphatic polyesters and aliphatic polycarbonates occupy a relevant position, as they are obtained from renewable resources and are biodegradable, thus fully satisfying the criteria to be included in the category of biopolymers.5
The most common and effective route to obtain well-defined aliphatic polyesters6 and aliphatic polycarbonates7 is the ring-opening polymerization (ROP) of the corresponding cyclic monomers catalysed by well-defined metal complexes. More interesting are sustainable metal catalysts, that is, catalysts based upon inexpensive biocompatible and environmentally friendly metals, essential aspects when these polymers are applied in the fields of biomedicine and pharmaceuticals.8,9
Magnesium and zinc are essential elements for humans, playing a central role in a large number of enzymatic reactions. Their common oxidation state is +2 with closed shell configurations (Zn2+, [Ar]3d104s0; Mg2+, [Ne]3s0), showing very similar ionic radii over a range of coordination numbers. Despite their strong similarities, magnesium and zinc compounds often show different reactivities in catalysis.10
Thus, a number of magnesium and zinc complexes containing various ancillary ligands, such as β-diketiminate, phenolate, and N-heterocyclic carbenes, have been synthesized, some of them demonstrating high activities and good control in the production of aliphatic polyesters11–22 and/or aliphatic polycarbonates.23–29
Both classes of polymers have all the prerequisites to become fully compatible with a model of circular economy for plastics, however the strategies for their end-of-life management are still not well planned.4,30 In this context, chemical recycling could offer great opportunities to convert these materials into the starting monomers,31,32 or, alternatively, into high added value products.33 Several examples have been recently reported describing the chemical recycling of PLA by green processes based on efficient and non-toxic catalysts.34 Particularly, homogeneous zinc complexes are emerging as the most efficient catalysts for the chemical alcoholysis of PLA to obtain alkyl lactates.18,20,35–41
Among the different classes of ancillary ligands explored as a coordinative environment for the synthesis of homogeneous organometallic catalysts, NHC ligands have gained considerable attention as versatile and powerful ligands because of their unique stereoelectronic properties associated with a high degree of modularity.42–45 Examples of zinc complexes, supported by NHC ligands having different architectures, were described by different researchers as active catalysts in the ROP of cyclic esters and/or cyclic carbonates.27,28,46–49 In addition, Dagorne and co-workers reported that zinc complexes, stabilized by NHC ligands bearing sulfur or oxygen atoms as additional donors, promoted also the alcoholysis of PLA.48
Less developed are NHC–magnesium complexes,50,51 as the increased electropositivity of magnesium makes the NHC–metal bond of these species weaker and more labile, strongly limiting their applications in catalysis.52–56 Indeed, to the best of our knowledge, only a few examples of magnesium complexes coordinated to NHC ligands able to promote the ROP of cyclic esters have been reported to date.47,49,57
We recently reported backbone substituted NHC-based zinc complexes as active catalysts in the ring-opening polymerization of both lactide58 and six-membered cyclic carbonates.59 NHC ligands that combine syn- or anti-phenyl groups on the backbone with o-tolyl N-substituents have been selected for the possibility they offer of creating differently shaped reactive pockets around the metal, which may affect the activity and selectivity of the resulting catalysts. In fact, the precise stereochemical arrangement of the phenyl groups on the backbone can, in turn, induce a preferred relative orientation of the N-tolyl groups, thus providing stable NHC conformers that define the available space near the metal in a different way.60–62 Furthermore, the configuration of the backbone also seems to affect the electronic properties of the NHC ligands, and, consequently, the catalytic behaviour of the corresponding catalysts.58,63,64 In this contribution, we extend our interest in the synthesis of aliphatic polyesters and polycarbonates to magnesium complexes and describe polymerization experiments carried out to compare the activity of zinc and magnesium based catalysts (Fig. 1).
 | ||
| Fig. 1 Structures of complexes, monomers and initiators used in this work. | ||
Furthermore, we investigate the performances of the zinc and magnesium complexes in the methanolysis reaction of PLLA samples.
Results and discussion
Synthesis and characterization of complexes
NHC proligands61 and zinc complexes 1a and 2a58 were synthesized according to reported procedures (Scheme 1). | ||
| Scheme 1 Schematic pathway for the synthesis of NHC–zinc and –magnesium complexes. | ||
Magnesium complexes 1b and 2b were synthesized in benzene by direct reaction (Scheme 1) between the corresponding carbene salt and 1 equiv. of Mg[N(TMS)2]2.
After stirring the reaction mixture for one hour at room temperature, complexes 1b and 2b were recovered, by evaporation of the solvent under vacuum, as whitish powders in good yields (1b: 85%; 2b: 80%). Both complexes were fully characterized by NMR spectroscopy (Fig. S3–S12†). The 1H NMR spectrum of complex 1b (Fig. 2), recorded in C6D6 at room temperature, reveals a CS symmetry in solution with eight signals for the hydrogen atoms of the ligand (two signals are superimposed) and one singlet for the proton resonance of the methyl groups bound to silicon. The disappearance of the resonance due to the proton of the precarbenic position of the imidazolium salt suggested the formation of the complex in which the NHC ligand is coordinated to magnesium. COSY and NOESY NMR analysis allowed the full assignment of the proton signals (Fig. S3–S7†). Unlike what was previously observed with analogous zinc complexes 1a and 2a,58 the signal of the methyl groups bound to silicon shows an intensity that is double that expected, while the signal of the amine formed during the reaction has a negligible intensity compared to that expected from the liberation of an equivalent of free amine. Even after removing the solvent and drying the solid product for a few hours under vacuum, the integral value of this signal does not decrease. From this observation, we hypothesized that the amine produced during the reaction remains coordinated to magnesium and forms a hydrogen bond with the amide group which makes them interchangeable, according to the structure represented in the inset of Fig. 2. Subsequently, the spectrum of the magnesium complex was recorded in THF-d8, wondering whether this coordinating molecule was able to replace the amine. Indeed, in this case, the 1H NMR spectrum shows two different signals at low chemical shift, although the partial overlap of the signals does not allow a reliable integration.
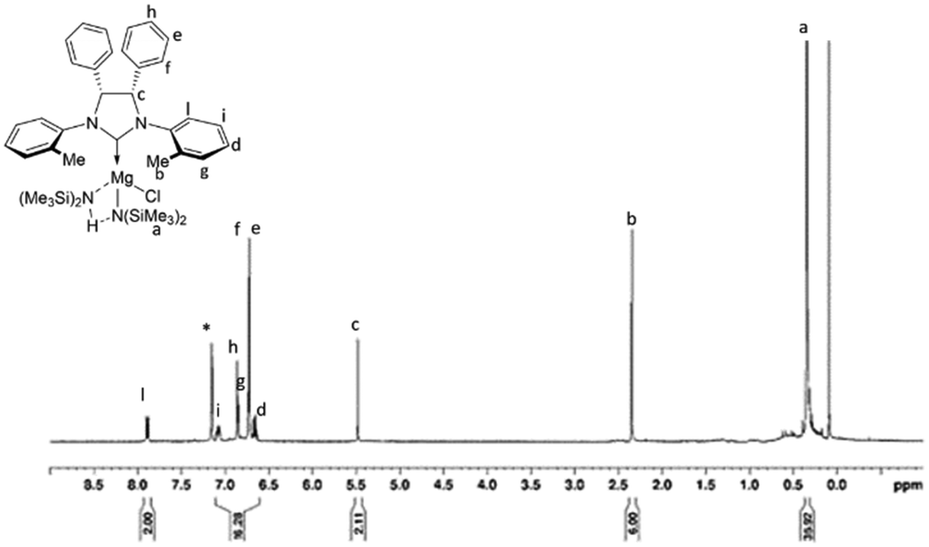 | ||
| Fig. 2 1H NMR spectrum of magnesium complex 1b (20 °C, 600 MHz, C6D6). | ||
To get more structural information on magnesium complex 1b, the diffusion coefficients were determined by the 2D version of the PGSE experiment (DOSY, Diffusion Ordered SpectroscopY) in both C6D6 (Fig. 3) and THF-d8 solution (Fig. S13†), using TMSS as an internal standard. The results are summarized in Table S1.† The obtained diffusion coefficients were then converted into molecular masses and the values were compared with the molecular masses of: the monomeric complex, the monomeric complex with an additional molecule (amine or THF) and the dimeric species. For complex 1b the best agreement of the molecular mass estimated by DOSY carried out in C6D6 (886 Da) was found with the theoretical mass of the monomeric complex with the coordinated amine (786 Da), in agreement with what we hypothesized using 1H NMR data. On the other hand, the molecular mass estimated by DOSY in THF-d8 (605 Da) is very close to that of the monomeric complex (624 Da), although more reasonably the species present in the solution of this coordinating solvent will have a magnesium coordinated also to a THF molecule (corresponding to a molecular mass of 696 Da) which displaced the amine. Confirming the displacement, the diffusion coefficient associated with the signal of the amine was different from that of the main species (Fig. S13†), and corresponded to an estimated molecular mass (190 Da) in good agreement with the effective molecular mass of the amine (162 Da). Finally, as can be seen from Table S1,† for zinc complex 1a the value of the mass estimated via DOSY NMR (Fig. S14†) in C6D6 (553 Da) is closer to that of the mass of the monomeric complex (665 Da), as expected.
 | ||
| Fig. 3 DOSY NMR spectrum of complex 1b with TMSS as an internal standard (400 MHz, 25 °C, C6D6). | ||
Thus, we may conclude that in solution of non-coordinating solvents, the amine (HN(SiMe3)2) formed during the synthesis of the organometallic species remains coordinated to the metal for both magnesium complexes while moving away from the metal for zinc complexes. To rationalize this different behavior of the syn and anti magnesium (1b and 2b) and zinc (1a and 2a) complexes, DFT calculations were performed.
Therefore, the formation energy of the monomeric Zn and Mg complexes and those of corresponding complexes with the amine coordinated were computed and compared (see Scheme 2 and Fig. 4).‡
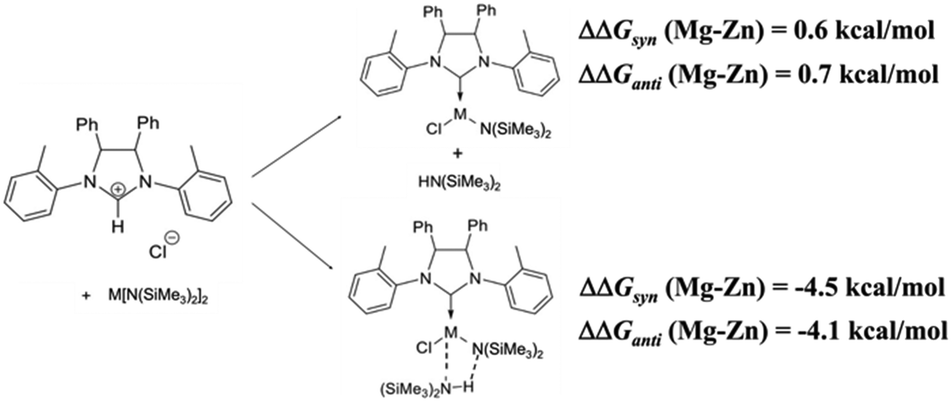 | ||
| Scheme 2 Free energy difference in benzene as a solvent, calculated for the formation of monomeric complexes of M = Zn and Mg (ΔΔGsyn (Mg–Zn) and ΔΔGanti (Mg–Zn), respectively) with the reactants and reaction products at an infinite distance. | ||
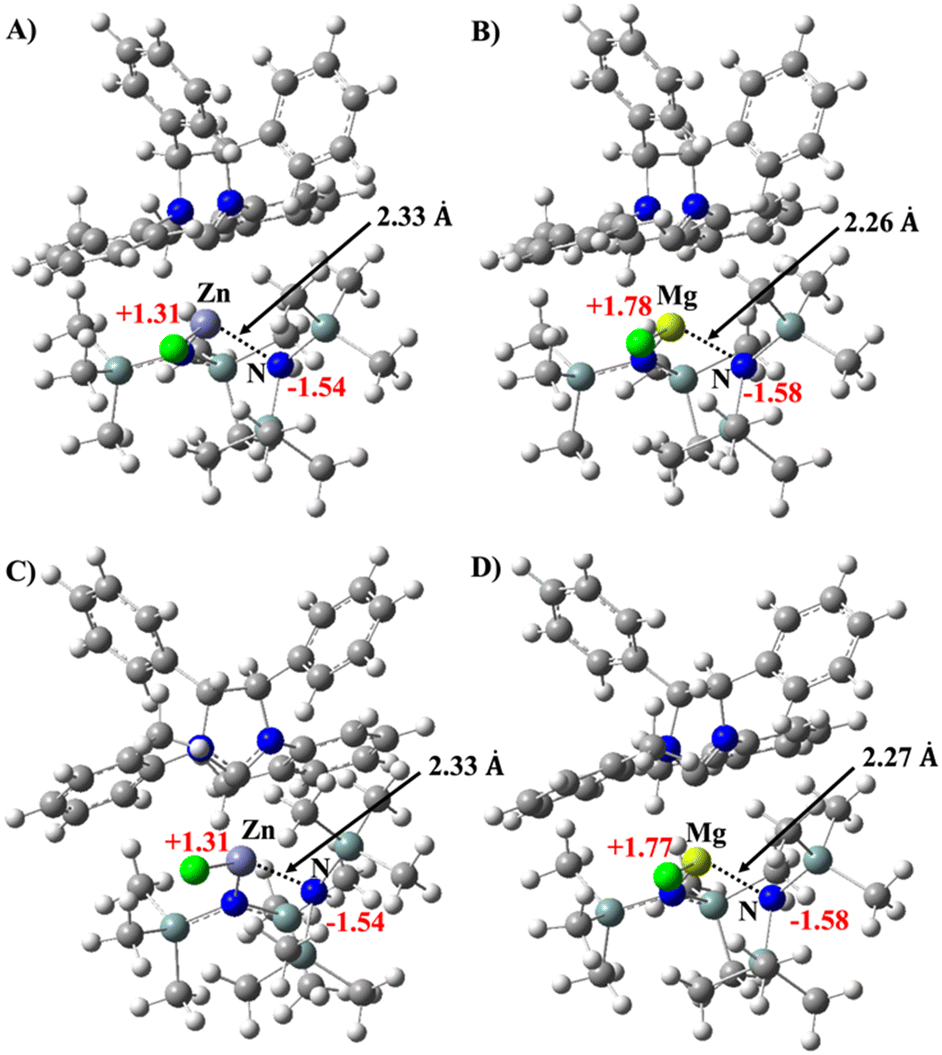 | ||
| Fig. 4 Optimized structure of the syn (panel A and B) and anti (panel C and D) monomeric complexes of Zn and Mg with the coordinated amine. The structures are represented in balls and sticks. C, H, N, Si, Cl, Zn and Mg are depicted in grey, white, blue, light green, green, light purple and yellow, respectively. The bond distances are reported in angstrom (Å) and shown in black, while the NBO charge values of the metals and of the amine nitrogen are in red. | ||
The calculations, in agreement with the experimental results, show that the formation of the syn and anti monomeric complexes was favored for Zn by about 1 kcal mol−1, while the formation of the amine coordinated complexes is favored for Mg by about 5 kcal mol−1.
In line with this result, the analysis of the most stable geometries of the syn and anti complexes of Zn and Mg shows that the interaction of the metal with the nitrogen of the coordinated amine is stronger for the magnesium complexes with a shorter M–N distance (compare 2.33 Å for Zn, Fig. 4A and C, with 2.26/2.27 Å for the corresponding complexes of Mg, Fig. 4B and D). This is a consequence of the difference in charge of the two metal cations. As in Fig. 4, the positive partial charge of Mg is about 0.5 higher than that of Zn, which increases the interaction of Mg with the amine, favoring coordination at the metal center. Furthermore, calculations carried out on magnesium complex 1b show that the replacement of the amine coordinated to the metal by the THF solvent molecule is favored by approximately 13 kcal mol−1 (Table S2 and Scheme S1†), in agreement with the experimental evidence obtained by NMR analyses.
Ring-opening polymerization of cyclic monomers
Having observed a distinguishable behavior of the magnesium complex in different deuterated solvents, we decided to begin this study on the activity of our complexes in the ROP experiments, by verifying the activity of magnesium complex 1b as the polymerization solvent varies. Polymerization tests of TMC (100 equivalents of monomer) with magnesium complex 1b and one equivalent of benzyl alcohol as the initiator§ were carried out at room temperature and selecting toluene, dichloromethane and THF, as classical solvents, in addition to methyl–THF and 1,3-dioxolane as green solvents (Table S4†). We observed the activity of the catalytic system constituted by complex 1b/BnOH to decrease in the order: toluene > THF > 1,3-dioxolane > DCM > Me–THF (see Table S4† for more details). Delighted by the fact that our catalytic system showed good activity in all cases, even in green solvents, we decided to use THF as the solvent to compare the behavior of the four complexes in the ring-opening polymerization of different cyclic esters and cyclic carbonates (Scheme 3 and Table 1) as the activity was higher in this solvent. During the polymerization, the product mixtures were analysed by 1H NMR spectroscopy, with the quantities of polymers and monomers determined by integrating the appropriate resonances (whose chemical shift values are specified in the ESI† part). | ||
| Scheme 3 ROP of the cyclic esters and cyclic carbonates promoted by NHC-based zinc and magnesium complexes 1a–2a and 1b–2b. | ||
| Entry | Complex | Monomer (equiv.) | Initiator | Temp. (°C) | Time | Conv.b (%) | M thn (kDa) | M expn (kDa) | Đ | k obs (h−1) |
|---|---|---|---|---|---|---|---|---|---|---|
a General conditions: polymerization reactions were carried out in THF except when differently specified. Complex = 8 μmol for cyclic carbonates, 15 μmol for cyclic esters; one equivalent of alcohol.
b Determined by 1H NMR spectral data.
c
M
thn (kDa) = MMmonomer × ([monomer]0/[Cat]0) × monomer conversion × 10−3.
d Experimental Mn (in kDa) and Mw/Mn (Đ) values were determined by SEC in THF using polystyrene standards and corrected using a factor of 0.58 for PLA,65 0.56 for PCL66 and 0.73 for PTMC (as indicated in the literature67 for PTMC with theoretical mass in the range 5000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). Experimental Mn (in g mol−1) values indicated in italic have been determined by MALDI analysis.
e Polymerization experiments carried out under bulk conditions. 000). Experimental Mn (in g mol−1) values indicated in italic have been determined by MALDI analysis.
e Polymerization experiments carried out under bulk conditions.
|
||||||||||
| 1 | 1a | L-LA (100) | iPrOH | 20 | 2.5 h | 84 | 12.1 | 8.5 | 1.27 | 0.80 |
| 2 | 1b | L-LA (100) | iPrOH | 20 | 6 h | 55 | 7.9 | 6.3 | 1.25 | 0.14 |
| 3 | 2a | L-LA (100) | iPrOH | 20 | 6 h | 61 | 8.8 | 8.2 | 1.21 | 0.23 |
| 4 | 2b | L-LA (100) | iPrOH | 20 | 6 h | 49 | 7.2 | 6.3 | 1.18 | 0.11 |
| 5 | 1a | ε-CL (100) | iPrOH | 20 | 7 h | 80 | 9.1 | 10.1 | 1.19 | 0.17 |
| 6 | 1b | ε-CL (100) | iPrOH | 20 | 15 min | 99 | 11.3 | 10.3 | 1.90 | 13.6 |
| 7 | 2a | ε-CL (100) | iPrOH | 20 | 9 h | 7 | 0.8 | 9.5 × 10−3 | ||
| 8 | 2b | ε-CL (100) | iPrOH | 20 | 30 min | 99 | 11.3 | 6.7 | 1.6 | 8.8 |
| 9 | 1a | TMC (100) | BnOH | 20 | 1 h | 92 | 9.4 | 10.5 | 1.45 | 2.7 |
| 10 | 1b | TMC (100) | BnOH | 20 | 10 min | 95 | 9.7 | 9.9 | 1.76 | 42.3 |
| 11 | 2a | TMC (100) | BnOH | 20 | 5.5 h | 96 | 9.8 | 8.8 | 1.30 | 0.57 |
| 12 | 2b | TMC (100) | BnOH | 20 | 15 min | 96 | 9.8 | 12.4 | 1.63 | 10.1 |
| 13e | 1a | DTC (50) | BnOH | 160 | 1 h | 79 | 5.1 | 4.6 | ||
| 14e | 1b | DTC (50) | BnOH | 160 | 1 h | 94 | 6.1 | 5.7 | ||
| 15e | 1a | Me-TMC (50) | BnOH | 70 | 1 h | 75 | 3.8 | 5.9 | ||
| 16e | 1b | Me-TMC (50) | BnOH | 70 | 3.5 h | 75 | 4.5 | 4.3 | ||
| 17e | 1a | Me-TMC (50) | BnOH | 160 | 30 min | 91 | 5.3 | 6.2 | ||
| 18e | 1b | Me-TMC (50) | BnOH | 160 | 30 min | 84 | 4.9 | 5.7 | ||
Initially, we examined the effectiveness of the zinc and magnesium complexes for the ring-opening polymerization of two cyclic esters, such as L-lactide (L-LA) (entries 1–4) and ε-caprolactone (ε-CL) (entries 5–8) by adding one equivalent of isopropanol (iPrOH) as an initiator, in THF as a solvent and at 20 °C.
The four complexes were found active in the synthesis of both polylactide and polycaprolactone, however while in L-LA polymerization zinc complexes were more active than the analogue magnesium complexes, and the opposite order of activity was observed in the ROP of ε-caprolactone. In all cases, comparing complexes with the same metal but different NHC ligands, it may be inferred that better performance is induced to the catalysts by NHC ligand L1 with respect to L2.
Subsequently, the performance of the four complexes was checked in the ROP of TMC (100 equivalents) in the presence of one equivalent of benzyl alcohol as the initiator, working in THF at 20 °C. Also in this case, the complexes were all able to produce poly(trimethylene carbonate), with magnesium complexes showing activities higher than those of the corresponding zinc complexes, for both NHC ligands. Meanwhile, the influence of the two different ligands was the same as that observed in the ROP of the cyclic esters and already observed in our previous paper.58
Finally, the activities of complexes 1a and 1b were compared in the ROP of two substituted cyclic carbonates, which were synthesized by a green procedure starting from biorenewable starting materials, such as CO2 and the opportune diol.68,69 For these monomers the polymerizations were carried out under bulk conditions at 70 and 160 °C, for 1-methyl-trimethylene carbonate (Me-TMC) and 2,2-dimethyl-trimethylene carbonate (DTC), respectively. Zinc complex 1a showed a higher activity than magnesium with Me-TMC (entries 15 and 16), while magnesium 1b was more active with DTC (entries 13 and 14). Wondering if the temperature could have an effect on the detachment of the amine from the magnesium and hence on its activity, we carried out the ROP of Me-TMC also at 160 °C with both complexes (entries 17 and 18). In this case, almost complete conversions were registered after 30 minutes. Moreover, although at this higher temperature the difference in activity between the two complexes is smaller, zinc complex 1a retained a slightly higher efficiency than magnesium complex 2a.
To determine the catalytic performance of all the complexes, kinetic studies were carried out. The apparent rate constants (kobs) (where kobs = kp[Cat]x, and kp is the propagation rate constant) were obtained from the slope of the semilogarithmic plots of the monomer conversion (ln([Monomer]0/[Monomer]t)) versus time, shown in Fig. 5 and summarized in Table 1. A linear relationship was observed in all cases, indicating first-order kinetics in monomer concentration. Comparing the behaviour of zinc and magnesium complexes in the ROP of the same monomer, the catalytic activity of the tested complexes decreased in the order: 1a > 2a > 1b > 2b for L-LA and 1b > 2b > 1a > 2a for both ε-CL and TMC.
 | ||
| Fig. 5 First-order kinetic plots for the consumption of L-LA, ε-CL and TMC by complex 1a (orange full squares, entries 1, 5 and 9 in Table 1), complex 1b (green empty squares, entries 2, 5 and 10 in Table 1), complex 2a (purple full circles, entries 3, 6 and 11 in Table 1) and complex 2b (blue empty circles, entries 4, 7 and 12 in Table 1) in combination with an equivalent of BnOH or iPrOH as initiators, in tetrahydrofuran at room temperature. | ||
When comparing the most active catalytic systems reported in the literature, such as, for example, for the ROP of lactide, the hyperactive zinc complexes supported by macrocyclic ligands reported by Williams,70 the zinc guanidinate complexes reported by Herres-Pawlis71 and the phenoxy–imino–pyridine zinc complexes reported by some of us,18 or even the more recent (thio)urea anion organocatalysts by Waymouth72 which were found to be highly efficient catalytic systems for fast and ultra-selective ROP of various cyclic monomers, it is clear that the activity of our catalytic systems is somewhat lower. However, when these results are compared with similar zinc and magnesium complexes bearing NHC-type ligands,28,46–49 the performance of our systems is either comparable or even superior (as shown in Table S6†).
Moreover, beyond the activity values themselves, we found the dependence of activity on both the metal and the monomer particularly interesting. In fact, in most literature reports comparing zinc and magnesium complexes with the same ancillary ligand as catalysts in ROP processes, magnesium species typically show higher activity, regardless of the monomer used, whether cyclic esters73–77 or cyclic carbonates.25,78
The obtained polymers were fully characterized by NMR, MALDI-ToF (Fig. S17–S22 and S25–S29†) and SEC analysis. 1H NMR spectra of polylactide and polycaprolactone samples, in addition to the signals expected for the proton of the repeating monomeric units, clearly showed the existence of –OCH(CH3)2 and HO(CH2)5CO–/HOCH(CH3)CO– (for PCL and for PLLA, respectively) as exclusive chain end groups. Coherently, the 1H NMR spectra of polycarbonate polymers showed signals which can be associated with benzyloxy α-chain ends and with the methylene protons of ω-chain ends (for Me-TMC also a signal associated with methine protons of ω-chain ends was observed, see Fig. S25†). Thus, for all polymerization reactions, the NMR characterization suggests the initiation reaction from the alcohol initiator and the termination reaction by hydrolysis.
The MALDI spectra of the polymers obtained from the magnesium complexes show a prevalent distribution with intervals in Da equal to the mass of the monomer unit and corresponding to the polymer chains with the end groups expected from the initiation with the alcohol used as an initiator (isopropanol for the cyclic esters and benzyl alcohol for cyclic carbonates) and termination by hydrolysis, in agreement with that observed via NMR analysis. However, smaller distributions attributable to secondary events (i.e. transesterifications and/or hydrolysis) are observable in the same MALDI spectra. This result indicates that the polymerizations promoted by the magnesium complexes are less controlled than those promoted by the zinc complexes (whose MALDIs did not show significant secondary distributions). The same conclusion can be drawn from the observation of the dispersity values obtained through SEC analysis: in fact, these values are, in most cases, higher for polymerizations promoted by magnesium. Finally, as indicated in Table 1, the molecular weights determined experimentally by SEC and/or MALDI are in fair agreement with theoretical molecular weights.
Mechanistic studies via NMR analysis
In our previous studies, we observed that NHC–zinc complex 1a by reaction with one equivalent of an alcohol in C6D6 forms the corresponding carbene alcoholic adduct and a zinc species.58,59 Here, we carried out the same stoichiometric reactions with magnesium complex 1b with one equivalent of benzyl alcohol in three different deuterated solvents, such as C6D6, THF-d8 and CD2Cl2 and, for comparison, the same reactions, in the same solvents, were performed also with zinc complex 1a (Fig. 6 and S30 and S31†). In all cases, the quantitative conversion of the metal complex into the benzyloxy adduct was observed in the time necessary to acquire the NMR spectra, as indicated by the pattern of this species (already reported in C6D6) and by the presence of the same signals in the spectra of the experiments carried out with zinc and magnesium complexes, in each solvent. To confirm that the alcohol adduct was involved in the polymerization reaction, 15 equivalents of TMC were added to the reaction mixture obtained by mixing complex 1b with benzyl alcohol in THF-d8. The NMR spectrum obtained after a few minutes shows the disappearance of the signals of both the alcohol adduct and the added monomer and the appearance of new signals attributable to a new form of the alcohol adduct with the PTMC chain bonded in place of the benzoxide group (Fig. S32†).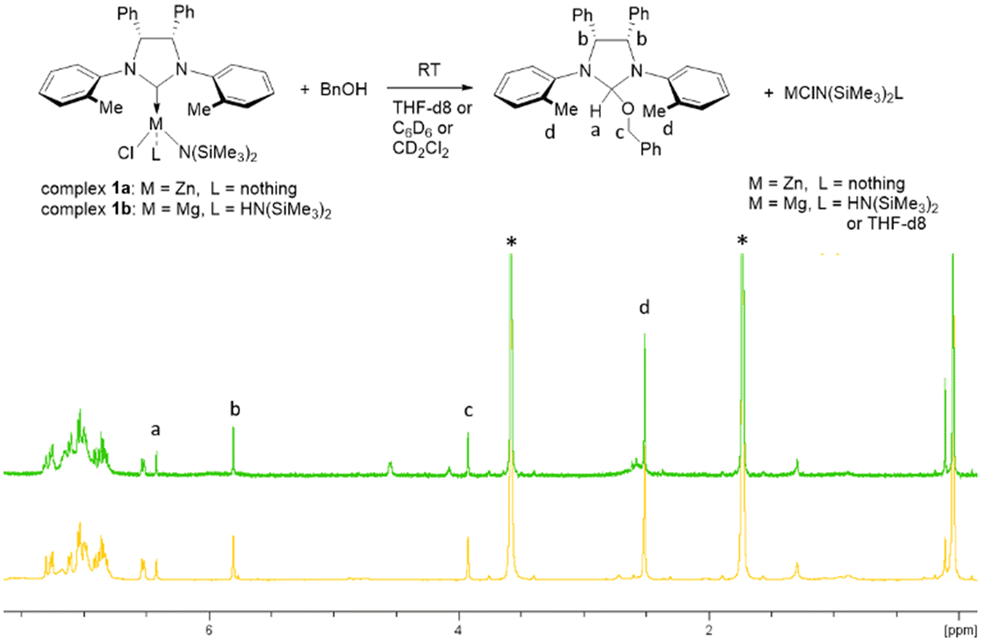 | ||
| Fig. 6 Up: Schematic reaction of complexes 1a and 1b with BnOH in different deuterated solvents. Down: 1H NMR spectra of the products obtained from the reaction of complex 1a (green spectrum) and complex 1b (orange spectrum) with one equivalent of BnOH in THF-d8 (peaks denoted with *) (400 MHz, 20 °C). | ||
With the aim to have more information on the active mechanism with these catalytic systems and also a clearer comparison between the activities of the zinc and magnesium complexes (which differ not only for the metal but also for the presence of a neutral amine coordinated to magnesium), we conducted a series of new polymerization experiments whose results are summarized in Table S7 of the ESI.†
We chose L-lactide and trimethylene carbonate as reference monomers, thus we first investigated whether the isopropoxide adduct L1HOiPr, synthesized ad hoc, was active in the ROP of these two monomers.79 By carrying out the reactions under the same conditions used with the metal complexes, no conversion of the monomers into the corresponding polymers was found in both cases, suggesting that the alcohol adduct requires the cooperation of the metal species to promote the reactions. Then, we conducted the ROP of trimethylene carbonate with three different catalytic systems: i.e., complex 1a with 1 equivalent of iPrOH, the isopropoxy adduct with 0.5 equivalents of ZnCl2 and 0.5 equivalents of Zn[N(TMS)2]2 and finally the isopropoxy adduct with one equivalent of Zn[N(TMS)2]2. It is worth mentioning that the second experiment was conducted by premixing equal amounts of the two metal precursors in order to induce the formation of the mixed chloride/amide zinc species which is believed to generate together with the alcohol adduct (see the reaction in Fig. 6). The same polymerizations were carried out with magnesium-based systems. And, finally, the same six catalytic systems were used in the ROP of L-LA, under the same reaction conditions. First of all, these control experiments highlight the cooperative action of the alcohol adduct with the metal species, also indicating the importance of the nature of the metallic species. In fact, the systems consisting of the alcohol adduct and the equimolar mixture of the two metal precursors show activity and polymerization control similar to those found with the metal complex/alcohol combination, while the catalytic systems consisting of the isopropoxy adduct and an equivalent of the metal amide are the most active catalysts but do not show efficient control over the polymerization processes. In line with this, magnesium amide alone shows a high activity in the ROP of both monomers but at the expense of control over the molecular masses of the obtained polymers. Another interesting result is that magnesium-based systems are more active than zinc-based ones for the TMC polymerizations, while the reverse order of activity is observed in the polymerization of L-LA. This aspect, highlighted in the kinetic graphs shown in Fig. S33,† allows us to state that this order of activity depends on the metal and not on the additional equivalent of amine coordinated to magnesium.
These results lead us to hypothesize that also these magnesium-based catalytic systems promote the polymerizations of cyclic esters and cyclic carbonates via a dual mechanism (Scheme 4), already reported for other NHC-based systems in the literature57,80,81 and by us for complex 1a. The principal role of the Lewis acidic metal species is the activation of the monomer by coordination of the carbonylic oxygen, while the free carbene, in equilibrium with the alcoholic adduct (bearing the isopropoxy group or the growing chain), catalyzes the polymerization by a monomer-activated mechanism. The free carbene can behave both as a base and as a nucleophile according, respectively, to pathways a) and b) shown in Scheme 4. In both cases, a polymer chain with the same end groups is obtained. DFT studies will be conducted in this direction to discriminate between the two different possibilities.
 | ||
| Scheme 4 Proposed mechanism for the ROP of cyclic monomers promoted by zinc and magnesium complexes in combination with an alcohol. In “pathway a”, the free NHC acts as a base while in “pathway b” the NHC acts as a nucleophile. In both cases the metal species operates as the Lewis acid which activates the monomer. | ||
Since magnesium complexes compared to zinc complexes showed higher activity in the polymerization of ε-caprolactone, trimethylene carbonate, and 2,2-dimethyl-trimethylene carbonate, while a reverse order of activity was recorded with L-lactide and 1-methyl-trimethylene carbonate, we tentatively hypothesize the intervention of steric effects, which slow down the polymerization promoted by the magnesium complexes with the monomers having a substituent close to their carbonyl moiety.
Chemical recycling of PLLA
The zinc complexes and magnesium complex 2b were also tested as catalysts in the chemical recycling, via methanolysis, of PLLA. Generally, in solution, the alcoholysis proceeds by a random scission of the polymer chains18 to produce oligomers that are progressively converted into the final product, methyl lactate (Me-LA), a compound that found large application as a green solvent (Scheme 5). The species formed during the reaction can be easily quantified by analysis of the methine region (ca. δ = 4.2–5.2 ppm) of the 1H NMR spectra. The efficiency of the process can be estimated by parameters such as conversion, selectivity, and yield of methyl lactate (XInt, SMe-LA, YMe-LA, respectively) that are evaluated by sample checks of the reaction mixtures (see the ESI† for more details). Initially, the experiments were carried out under the same reaction conditions described in the literature for the NHC zinc complexes, i.e. with a zinc complex:ester units of the PLLA:methanol molar ratio of 1:200:100. For all tests, a PLLA sample (Mn = 30 kDa, Đ = 1.2), opportunely synthesized and purified to remove catalytic residuals, was used. The PLLA samples were dissolved in DCM/MeOH solutions, and the reactions were carried out in the glove box, at room temperature. The results are reported in Table 2. Under these conditions, complex 1a was found to be scarcely active: after 24 hours only 6% of the polymer was converted to oligomeric species, while no evidence of the formation of methyl lactate emerged from the 1H NMR spectrum of the reaction mixture (entry 1, Table 2). Differently, in the presence of 2a, after the same time, about 56% of the polymer was oligomerized with a yield of 20% in Me-LA (entry 2, Table 2). This result is similar to that obtained by Dagorne with dinuclear zinc–N-heterocyclic carbene complexes for which a yield of about 30% of methyl lactate was obtained after 24 hours starting from a PLA sample of Mn = 18 kDa.48
 | ||
| Scheme 5 Methanolysis reactions promoted by complexes 1a, 2a and 2b. | ||
| Entry | Cat | DCM (mL) | Time (h) | X Int (%) | S Me-LA (%) | Y Me-LA (%) |
|---|---|---|---|---|---|---|
| a All reactions were carried out by using 14 μmol of catalyst (0.5 mol% relative to ester linkages), 2.8 mmol of PLLA in 1.4 mL of DCM, with 1.4 mmol of MeOH at room temperature. b Determined by 1H NMR spectroscopy. c 10 μmol of catalyst (1 mol% relative to ester linkages), 1.0 mmol of PLLA and 1.0 mL of methanol were used. d PLLA from a commercial plastic cup. | ||||||
| 1 | 1a | 1.4 | 24 | 6 | <1 | <1 |
| 2 | 2a | 1.4 | 24 | 56 | 36 | 19 |
| 3 | 2b | 1.4 | 24 | 15 | 10 | 2 |
| 4c | 1a | — | 24 | 57 | 100 | 57 |
| 5c | 2a | — | 19 | 92 | 100 | 92 |
| 6c,d | 2a | — | 19 | 70 | 100 | 70 |
| 7c,d | 2b | — | 19 | 100 | 100 | 100 |
| 8c | L1HOiPr | — | 2 | 5 | 100 | 5 |
| 9c | Zn[N(SiMe3)2]2 | — | 2 | 40 | 100 | 40 |
| 10c | L1HOiPr/Zn[N(SiMe3)2]2 | — | 2 | 95 | 100 | 95 |
A lower activity was achieved with magnesium complex 2b (entry 3, Table 2), reflecting the same reactivity scale observed in the ROP of L-LA (see entries 3 and 4, Table 1). In all cases, the alcoholysis performed in solution gave mixtures of oligomers and Me-LA, coherently with a mechanism of random scission of the polymeric chains by the catalytic system (Scheme 5). On the other hand, when the alcoholysis reactions were performed in neat methanol (entries 3 and 4, Table 2), Me-LA was produced selectively (Scheme 5), even at low conversions, as a consequence of the unzipping mechanism of the polymer starting from the chain ends, as discussed in previous work.18 Interestingly, in the absence of solvent, the performance of both catalysts improved significantly as clearly evident by comparing entries 3 and 4 with entries 1 and 2. Zinc complex 2a was able to convert quantitatively the whole amount of PLLA in methyl lactate after 19 hours (compare entries 2 and 5, Table 2), and preserved its activity (entry 6, Table 2) in the degradation of a post-consumer PLLA sample (Mn = 53 kDa).
Analogous results were obtained with magnesium complex 2b. The examples of well-defined Mg(II) catalysts active in the solvolysis of polyesters are quite rare; recently Jones reported catalen Mg(II)-complexes that allowed the methanolysis of 76% of a PLA sample (Mn = 45 kDa) after 8 hours at 80 °C.82
As previously described, mechanistic studies performed to unravel the nature of the active species in the ROP reactions revealed that the reaction between 1a and one equivalent of the benzyl alcohol led to the formation of an equimolar mixture of the corresponding alcoholic adduct (L1HOBn) and ZnClN(SiMe3)2, whereas both species are involved in the catalysis. Reasonably, the formation of the same species can be invoked also during the alcoholysis reactions of PLLA. To verify the involvement of both species in this catalysis, three alcoholysis experiments were performed: with the purposely synthesized L1HOiPr (see ESI†), with Zn[N(SiMe3)2]2 and with their equimolar mixture. The activity of the alcoholic adduct alone was very scarce, after 2 hours only 5% of the PLLA sample was converted (entry 8 in Table 2). On the other hand, zinc amide was much more active (entry 9 in Table 2), as previously reported.35,83,84 Nicely, the best activity was achieved when the combination of these species was used as the catalytic system, confirming the beneficial effect of their cooperation also in the alcoholysis reactions (entry 10 in Table 2).
Interestingly, the activity scale of the two zinc complexes in the alcoholysis reaction was the opposite with respect to that observed in the ROP of lactide. Both electronic and steric aspects could be accountable for the observed behavior. As already discussed in previous studies,58 the different stereochemistry of phenyls on the backbone influences the donating capacity of carbene ligands toward the zinc atom, as well as that of the carbon atom between the two nitrogen atoms in the corresponding alcoholic adduct. At the same time, steric factors could be involved, related to the binding of the complexes with different symmetry of the backbone substituents on the NHC ligand, with the different substrates (i.e., the monomer and the polymer chain). The syn and anti symmetric alcoholic adducts, deriving from complexes 1a and 2a respectively, would interact differently with the s-cis lactones and s-trans esters of interchain esters, favouring the interaction with the monomer or the polymer chain.85
Experimental
General methods
All manipulations of air- and/or water-sensitive compounds were performed under a dry nitrogen atmosphere by using a Braun Labmaster glovebox or standard Schlenk techniques. The glassware used in the synthesis of metal complexes and in the polymerization reactions was dried in an oven at 120 °C overnight. All chemicals were purchased from Merck while trimethylene carbonate was purchased from TCI. Benzene, toluene, THF and hexane were distilled over sodium benzophenone.Dichloromethane was distilled over calcium hydride. CD2Cl2, THF-d8 and C6D6 were dried over activated 3 Å molecular sieves. Benzyl alcohol and isopropyl alcohol were dried by refluxing over sodium. L-Lactide was crystallized in toluene and then dried on P2O5. ε-Caprolactone was dried over CaH2 and distilled under nitrogen. TMC was purified twice by recrystallization from dry THF and stored in the refrigerator. All other solvents and chemicals were commercially available and used as received unless otherwise stated. The synthesis and characterization of proligands, zinc complexes and substituted cyclic carbonates (Me-TMC and DTC) are described in the ESI.† NMR spectra were measured with Bruker AVANCE spectrometers operating at 300, 400 and 600 MHz, at 25 °C. Chemical shifts δ are given in ppm relative to the residual solvent peak of the used deuterated solvent. Molecular masses (Mn and Mw) and their dispersities (Mw/Mn) were measured by gel permeation chromatography (SEC), using THF as the eluent (1.0 mL min−1) and narrow polystyrene standards were used as the reference. MALDI-ToF mass spectra were recorded using a Bruker solariX XR Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7 T refrigerated actively shielded superconducting magnet (Bruker Biospin, Wissembourg, France). The samples were prepared at a concentration of 1.0 mg mL−1 in THF, while the matrix (DCTB, trans-2-[3-(4-tertbutylphenyl)-2-methyl-2-propenylidene] malononitrile) was mixed at a concentration of 10.0 mg mL−1.
Synthesis of complex 1b
The reaction was carried out in a glove box: in a vial, 0.100 g (2.273 × 10−4 mol) of NHC proligand was weighed, dissolved in 4 mL of anhydrous benzene and transferred into a 20 mL vial, equipped with a magnetic stirrer. In another vial, 0.0784 g (2.273 × 10−4 mol) of Mg[N(TMS)2]2 was weighed and dissolved in 4 mL of anhydrous benzene. The solution of the metal precursor was transferred to the solution of the salt and the mixture was left stirring for one hour at room temperature. The solvent was subsequently removed under reduced pressure and the complex obtained was a white powdery solid (yield = 85%). 1H NMR (400 MHz, C6D6, 298 K): δ 0.34 (s, 18H, Si(CH3)3), 2.35 (s, 6H, CH3), 5.48 (s, 2H, CH2), 6.66 (t, 2H, Ar–H), 6.73 (d, 2H, Ar–H), 6.85 (tr, 2H, Ar–H), 6.84 (d, 2H, Ar–H), 6.86 (d, 2H, Ar–H), 7.07 (t, 2H, Ar–H), 7.88 (d, 2H, Ar–H). 13C NMR (100.6 MHz, C6D6, 298 K): δ 6.37 (Si(CH3)3), 19.55 (2 CH3), 73.22 (2 CH), 127.24 (2 CH) 127.97 (2 CH), 128.23 (2 CH) 128.76 (2 CH), 128.81 (2 CH), 129.89 (2 CH), 132.05 (2 CH), 132.19 (2 Cq), 134.12 (2 Cq), 137.61 (2 Cq-N), 209.49 (C carbenic).Synthesis of complex 2b
The same procedure used for the synthesis of complex 1b was followed. In this case 0.100 g (2.273 × 10−4 mol) of the NHC proligand and 0.0784 g (2.273 × 10−4 mol) of Mg[N(TMS)2]2 were used. The complex was obtained as a yellow powdery solid (yield = 80%). 1H NMR (400 MHz, C6D6, 298 K): δ 0.35 (s, 18H, Si(CH3)3), 2.21 (s, 6H, CH3), 5.41 (s, 2H, CH2), 6.76 (d, 2H, Ar–H), 6.80 (d, 2H, Ar–H), 6.88 (tr, 2H, Ar–H), 7.01 (tr, 2H, Ar–H), 7.10 (d, 2H, Ar–H), 7.19 (d, 2H, Ar–H), 8.06 (d, 2H, Ar–H). 13C NMR (100.6 MHz, C6D6, 298 K): δ 6.86 (Si(CH3)3), 20.26 (2 CH3), 76.92 (2 CH), 127.61 (2 CH) 129.15 (2 CH), 129.66 (2 CH), 129.80 (2 CH), 129.85 (2 CH), 130.43 (2 CH), 132.67 (2 CH), 133.37 (2 Cq), 138.15 (2 Cq), 138.54 (2 Cq-N), 208.45 (C carbenic).Synthesis of the isopropoxide adduct L1HOiPr
The reaction was carried out in the glove-box in a nitrogen atmosphere. A THF suspension (1.25 mL) of KH (0.0238 g, 5.93 × 10−4 mol) was added to a THF suspension (4.4 mL) of the L1HCl salt (0.200 g, 4.55 × 10−4 mol). Once the addition was complete, the reaction mixture was left to stir for 5 minutes at room temperature. Subsequently, a solution of iPrOH (0.0251 g, 4.2 × 10−4 mol) in THF (1.0 mL) was added and the reaction mixture was left under stirring for 1 hour. Then, the reaction mixture was filtered to remove residual salts and the solvent was removed under vacuum. The product was obtained as a white powdery solid (yield = 58%). 1H NMR (400 MHz, C6D6, 298 K): δ 0.53 (d, 6H, CH3), 2.61 (s, 6H, CH3), 3.00 (m, H, CH), 5.73 (s, 2H, CH), 6.22 (s, H, CH), 6.72 (t, 2H, Ar–H), 6.83 (t, 6H, Ar–H), 6.97 (t, 2H, Ar–H), 7.02 (t, 2H, Ar–H), 7.13 (t, 2H, Ar–H), 7.35 (d, 2H, Ar–H). 13C NMR (150 MHz, C6D6, 298 K): δ 19.64 (2 CH3), 23.13 (2 CH3), 67.24 (2 CH), 71.05 (CH), 100.53 (CH), 124.75 (2 CH) 125.58 (2 CH), 126.88 (2 CH) 127.32 (2 CH), 128.02 (2 CH), 129.21 (2 CH), 131.45 (2 CH), 134.26 (2 Cq), 139.69 (2 Cq), 142.93 (2 CqN).Computational details
All the DFT geometry optimizations were performed by using the Gaussian16 package86 at the B3LYP-D3(BJ)87,88 level of theory, using the quasi relativistic LANL2DZ ECP effective core potential89 for Zn and Mg and the 6-311g (d,p) basis set for C, H, N, Cl, and Si atoms. The reported free energies were obtained by adding the thermal correction in the gas-phase to the electronic energy in solvent (SMD model) calculated via single point energy calculations in benzene (ΔGBENZ) at the M06 (ref. 90) level. The electronic configuration of the systems was described by using the standard triple-ζ TZVP91 basis set with a polarization function of Ahlrichs and co-worker for the atoms of the main groups and LANL2DZ ECP pseudopotential for Zn and Mg. The ΔΔG values in benzene as a solvent were computed as the energy difference between the ΔGs of the Mg complexes and the ΔGs of the Zn complexes. The charge distribution in the complexes was calculated using the natural bond orbital (NBO) analysis.General polymerization procedure
Procedure for methanolysis performed in DCM solution
The zinc complex (14 μmol) was transferred into a 4 mL vial, equipped with a magnetic stirrer, and was dissolved in 0.4 ml of DCM. The polymer (200 mg, 2.8 mmol) was weighed into another 4 mL vial and then dissolved in 1 ml of DCM. The solution of the complex was added to the polymer solution and left stirring for 5 minutes: after this time, methanol (56.3 μl, 1.4 mmol) was added and the reaction mixture was stirred at room temperature for the predicted time. An aliquot of the reaction mixture was transferred in a vial containing wet CD2Cl2 and was analyzed by 1H NMR spectroscopy.Procedure for methanolysis performed under solvent free conditions
The zinc complex (10 μmol) was weighed into a 4 mL vial then the polymer (72 mg, 1 mmol) and 1 mL of methanol were added. The solution was left stirring for the desired time before stopping and carrying out the analysis via1H NMR spectroscopy.Conclusions
In this work, complexes based on zinc and magnesium, supported by N-heterocyclic carbene ligands, were synthesized and characterized by NMR spectroscopy. NMR spectra show that the amine formed during the synthesis remains in the coordination sphere of magnesium complexes, but not the zinc complexes. This different behavior was rationalized by DFT calculations. The theoretical results highlight that there is a greater positive charge on magnesium compared to zinc, which stabilizes the interaction of magnesium complexes with the amine.Subsequently, the complexes were tested in the ring-opening polymerization of different cyclic esters and cyclic carbonates derived from renewable sources. The versatility of the complexes was demonstrated by the fact that in all cases the expected polymers were produced. These biopolymers have been characterized using different techniques, suggesting that all the complexes exert a fair control in the polymerization processes, with the zinc complexes performing slightly better than the magnesium species. Kinetic studies showed a first-order dependence on monomer concentration (L-LA, ε-CL and TMC), for all the complexes. NMR studies to identify the nature of the species formed during the reactions and polymerization experiments with different catalytic systems that mimic the species formed in situ have led us to hypothesize a dual activation mechanism, in which the cooperation between the alcohol adduct and the mixed metal species is essential for controlling the polymerization process. Interestingly, an order of activity of magnesium vs. zinc complexes depending on the monomer nature was found. Tentatively, we hypothesized that steric effects occur, however, this unexpected result prompts us to examine the reaction mechanism through in-depth computational studies that will be conducted and published in due time. Finally, studies on methanolysis reactions of PLLA samples underlined the versatility of zinc and magnesium complexes, capable of promoting also these polyester upcycling reactions, which are strategically important from the point of view of the environmental sustainability, as they close the life cycle of the polymer itself.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: M. L.; methodology: F. T., F. S., C. L., G. S., I. R.; investigation: M. L., L. C., M. M. and F. G.; visualization: M. L., L. C., M. M. and F. G.; data curation: M. L. and L. C.; writing – review and editing: M. L., L. C., F. G. and M. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Patrizia Iannece for elemental analysis, Dr Patrizia Oliva for NMR technical assistance, Dr Patrizia Iannece for MALDI analysis and Dr Mariagrazia Napoli for SEC analysis. Alessandra Monzo is recognized for the preliminary studies carried out during her Bachelor internship. The authors are grateful for funding from the Università degli Studi di Salerno (FARB grant: ORSA231338). I. R., G. S., and L. C. would like to thank the Supercomputing Centre CINECA, Bologna, Italy, for providing computing time.Notes and references
- L. T. J. Korley, A. J. McNeil and G. W. Coates, Acc. Chem. Res., 2022, 55, 2543–2544 CrossRef CAS PubMed.
- A. Buchard and T. Junkers, Polym. Chem., 2022, 13, 1785–1786 RSC.
- Y. D. Y. L. Getzler and R. T. Mathers, Acc. Chem. Res., 2022, 55, 1869–1878 CrossRef CAS PubMed.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- W. Yu, E. Maynard, V. Chiaradia, M. C. Arno and A. P. Dove, Chem. Rev., 2021, 121, 10865–10907 CrossRef CAS PubMed.
- M. S. Holzwarth and B. Plietker, ChemCatChem, 2013, 5, 1650–1679 CrossRef CAS.
- D. Pappalardo, T. Mathisen and A. Finne-Wistrand, Biomacromolecules, 2019, 20, 1465–1477 CrossRef CAS PubMed.
- A. Friedrich, J. Eyselein, J. Langer and S. Harder, Organometallics, 2021, 40, 448–457 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845–11854 CrossRef CAS.
- P. D. Knight, A. J. P. White and C. K. Williams, Inorg. Chem., 2008, 47, 11711–11719 CrossRef CAS PubMed.
- M. D. Jones, M. G. Davidson, C. G. Keir, L. M. Hughes, M. F. Mahon and D. C. Apperley, Eur. J. Inorg. Chem., 2009, 635–642 CrossRef CAS.
- Y. Yang, H. Wang and H. Ma, Inorg. Chem., 2015, 54, 5839–5854 CrossRef CAS PubMed.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, Jr., M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, Dalton Trans., 2013, 42, 13036–13047 RSC.
- F. Santulli, M. Lamberti and M. Mazzeo, ChemSusChem, 2021, 14, 5470–5475 CrossRef CAS PubMed.
- F. Santulli, F. Bruno, M. Mazzeo and M. Lamberti, ChemCatChem, 2023, 15, e202300498 CrossRef CAS.
- F. Santulli, G. Gravina, M. Lamberti, C. Tedesco and M. Mazzeo, Mol. Catal., 2022, 528, 112480 CrossRef CAS.
- M. Fuchs, S. Schmitz, P. M. Schaefer, T. Secker, A. Metz, A. N. Ksiazkiewicz, A. Pich, P. Koegerler, K. Y. Monakhov and S. Herres-Pawlis, Eur. Polym. J., 2020, 122, 109302 CrossRef CAS.
- S. Ghosh, P. M. Schaefer, D. Dittrich, C. Scheiper, P. Steiniger, G. Fink, A. N. Ksiazkiewicz, A. Tjaberings, C. Woelper, A. H. Groeschel, A. Pich, S. Herres-Pawlis and S. Schulz, ChemistryOpen, 2019, 8, 951–960 CrossRef CAS PubMed.
- M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Chem. – Eur. J., 2008, 14, 8772–8775 CrossRef CAS PubMed.
- M. Helou, O. Miserque, J.-M. Brusson, J.-F. Carpentier and S. M. Guillaume, Adv. Synth. Catal., 2009, 351, 1312–1324 CrossRef CAS.
- D. J. Darensbourg, W. Choi, P. Ganguly and C. P. Richers, Macromolecules, 2006, 39, 4374–4379 CrossRef CAS.
- C. Fliedel, V. Rosa, F. M. Alves, A. M. Martins, T. Aviles and S. Dagorne, Dalton Trans., 2015, 44, 12376–12387 RSC.
- G. Schnee, C. Fliedel, T. Aviles and S. Dagorne, Eur. J. Inorg. Chem., 2013, 2013, 3699–3709 CrossRef CAS.
- C. Fliedel, S. Mameri, S. Dagorne and T. Aviles, Appl. Organomet. Chem., 2014, 28, 504–511 CrossRef CAS.
- G. L. Gregory, G. S. Sulley, J. Kimpel, M. Lagodzinska, L. Hafele, L. P. Carrodeguas and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202210748 CrossRef CAS PubMed.
- N. K. Kalita and M. Hakkarainen, Curr. Opin. Green Sustainable Chem., 2023, 40, 100751 CrossRef CAS.
- C. F. Gallin, W.-W. Lee and J. A. Byers, Angew. Chem., Int. Ed., 2023, 62, e202303762 CrossRef CAS PubMed.
- L. Cederholm, J. Wohlert, P. Olsen, M. Hakkarainen and K. Odelius, Angew. Chem., Int. Ed., 2022, 61, e202204531 CrossRef CAS PubMed.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124, 2617–2650 CrossRef CAS PubMed.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- F. Santulli, M. Lamberti, A. Annunziata, R. C. Lastra and M. Mazzeo, Catalysts, 2022, 12, 1193 CrossRef CAS.
- S. D'Aniello, S. Lavieville, F. Santulli, M. Simon, M. Sellitto, C. Tedesco, C. M. Thomas and M. Mazzeo, Catal. Sci. Technol., 2022, 12, 6142–6154 RSC.
- M. Fuchs, M. Walbeck, E. Jagla, A. Hoffmann and S. Herres-Pawlis, ChemPlusChem, 2022, 87, e202200029 CrossRef CAS PubMed.
- T. Becker, A. Hermann, N. Saritas, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2024, e202400933 CrossRef CAS PubMed.
- M. Fuchs, P. M. Schafer, W. Wagner, I. Krumm, M. Walbeck, R. Dietrich, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2023, 16, e202300192 CrossRef CAS PubMed.
- J. Stewart, M. Fuchs, J. Payne, O. Driscoll, G. Kociok-Kohn, B. D. Ward, S. Herres-Pawlis and M. D. Jones, RSC Adv., 2022, 12, 1416–1424 RSC.
- A. Hermann, T. Becker, M. A. Schafer, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2022, 15, e202201075 CrossRef CAS PubMed.
- I. Jain and P. Malik, Eur. Polym. J., 2021, 150, 110412 CrossRef CAS.
- M. Jalal, B. Hammouti, R. Touzani, A. Aouniti and I. Ozdemir, Mater. Today: Proc., 2020, 31, S122 CAS.
- S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley VCH, Mannheim, Germany, 1st edn, 2014 Search PubMed.
- S. Diez-Gonzalez, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, London, UK, 1st edn, 2010 Search PubMed.
- L. R. Collins, L. A. Moffat, M. F. Mahon, M. D. Jones and M. K. Whittlesey, Polyhedron, 2016, 103, 121–125 CrossRef CAS.
- P. L. Arnold, I. J. Casely, Z. R. Turner, R. Bellabarba and R. B. Tooze, Dalton Trans., 2009, 7236–7247, 10.1039/b907034f.
- C. Fliedel, D. Vila-Vicosa, M. J. Calhorda, S. Dagorne and T. Aviles, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS.
- N. Ferrentino, F. Franco, F. Grisi, S. Pragliola, M. Mazzeo and C. Costabile, Mol. Catal., 2022, 533, 112799 CrossRef CAS.
- S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114, 8747–8774 CrossRef CAS PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- B. Bantu, G. Manohar Pawar, K. Wurst, U. Decker, A. M. Schmidt and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2009, 1970–1976 CrossRef CAS.
- S. Okamoto, H. Ishikawa, Y. Shibata and Y.-i. Suhara, Tetrahedron Lett., 2010, 51, 5704–5707 CrossRef CAS.
- A. Baishya, M. K. Barman, T. Peddarao and S. Nembenna, J. Organomet. Chem., 2014, 769, 112–118 CrossRef CAS.
- A. Baishya, T. Peddarao and S. Nembenna, Dalton Trans., 2017, 46, 5880–5887 RSC.
- S. Mondal, S. Sarkar, D. Mallick and D. Mukherjee, Polyhedron, 2024, 251, 116849 CrossRef CAS.
- S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS PubMed.
- F. Tufano, F. Santulli, F. Grisi and M. Lamberti, ChemCatChem, 2022, 14, e202200962 CrossRef CAS.
- F. Tufano, C. Napolitano, M. Mazzeo, F. Grisi and M. Lamberti, Biomacromolecules, 2024, 25, 4523–4534 CrossRef CAS PubMed.
- T. J. Seiders, D. W. Ward and R. H. Grubbs, Org. Lett., 2001, 3, 3225–3228 CrossRef CAS PubMed.
- A. Perfetto, C. Costabile, P. Longo, V. Bertolasi and F. Grisi, Chem. – Eur. J., 2013, 19, 10492–10496 CrossRef CAS PubMed.
- A. Perfetto, C. Costabile, P. Longo and F. Grisi, Organometallics, 2014, 33, 2747–2759 CrossRef CAS.
- V. Paradiso, V. Bertolasi and F. Grisi, Organometallics, 2014, 33, 5932–5935 CrossRef CAS.
- C. Ambrosio, V. Paradiso, C. Costabile, V. Bertolasi, T. Caruso and F. Grisi, Dalton Trans., 2018, 47, 6615–6627 RSC.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- M. Save, M. Schappacher and A. Soum, Macromol. Chem. Phys., 2002, 203, 889–899 CrossRef CAS.
- I. Palard, M. Schappacher, B. Belloncle, A. Soum and S. M. Guillaume, Chem. – Eur. J., 2007, 13, 1511–1521 CrossRef CAS PubMed.
- G. L. Gregory, M. Ulmann and A. Buchard, RSC Adv., 2015, 5, 39404–39408 RSC.
- T. M. McGuire, E. M. Lopez-Vidal, G. L. Gregory and A. Buchard, J. CO2 Util., 2018, 27, 283–288 CrossRef CAS.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- A. Hermann, S. Hill, A. Metz, J. Heck, A. Hoffmann, L. Hartmann and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2020, 59, 21778–21784 CrossRef CAS PubMed.
- J. Zhang, K. H. Lui, R. Zunino, Y. Jia, R. Morodo, N. Warlin, J. L. Hedrick, G. Talarico and R. M. Waymouth, J. Am. Chem. Soc., 2024, 146, 22295–22305 CrossRef CAS PubMed.
- R. M. Slattery, A. E. Stahl, K. R. Brereton, A. L. Rheingold, D. B. Green and J. M. Fritsch, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 48–59 CrossRef CAS.
- R. Duan, C. Hu, Z. Sun, X. Pang and X. Chen, ACS Omega, 2018, 3, 11703–11709 CrossRef CAS PubMed.
- I. D'Auria, C. Tedesco, M. Mazzeo and C. Pellecchia, Dalton Trans., 2017, 46, 12217–12225 RSC.
- V. Balasanthiran, M. H. Chisholm, K. Choojun and C. B. Durr, Dalton Trans., 2014, 43, 2781–2788 RSC.
- M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS PubMed.
- M. Huang, C. Pan and H. Ma, Dalton Trans., 2015, 44, 12420–12431 RSC.
- A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881–2883, 10.1039/b601393g.
- A. Balint and S. Naumann, Polym. Chem., 2021, 12, 5320–5327 RSC.
- M. L. McGraw and E. Y. X. Chen, Macromolecules, 2020, 53, 6102–6122 CrossRef CAS.
- J. Payne, P. McKeown, O. Driscoll, G. Kociok-Kohn, E. A. C. Emanuelsson and M. D. Jones, Polym. Chem., 2021, 12, 1086–1096 RSC.
- R. Yang, G. Xu, C. Lv, B. Dong, L. Zhou and Q. Wang, ACS Sustainable Chem. Eng., 2020, 8, 18347–18353 CrossRef CAS.
- Q. Liu, R. Yang, B. Dong, H. Sun, G. Xu and Q. Wang, Polym. Degrad. Stab., 2024, 222, 110706 CrossRef CAS.
- X. Zhang, G. O. Jones, J. L. Hedrick and R. M. Waymouth, Nat. Chem., 2016, 8, 1047–1053 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, Wallingford, E.U.A., 2016 Search PubMed.
- A.-R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of analytical methods, experimental data for polymerization and depolymerization, MALDI-TOF-MS results, and DFT details. See DOI: https://doi.org/10.1039/d4cy01353k |
| ‡ To overcome the entropy error that is made when comparing reactions with different molecularities, we report the relative energies of formation of the anti and syn zinc and magnesium complexes for each type of reaction. |
| § Our previous observations with zinc complex 1a highlighted the critical role of the initiator in achieving controlled polymerization. The same result has been obtained when using magnesium complex 1b (for more details, see Table S3†); thus, in this work, we carried out all the polymerization in the presence of alcohol. |
| This journal is © The Royal Society of Chemistry 2025 |