
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient prediction of the local electronic structure of ionic liquids from low-cost calculations†
Frances K.
Towers Tompkins
 a,
Lewis G.
Parker
a,
Richard M.
Fogarty
b,
Jake M.
Seymour
a,
Rebecca
Rowe
b,
Robert G.
Palgrave
c,
Richard P.
Matthews
d,
Roger A.
Bennett
a,
Patricia A.
Hunt
e and
Kevin R. J.
Lovelock
*a
a,
Lewis G.
Parker
a,
Richard M.
Fogarty
b,
Jake M.
Seymour
a,
Rebecca
Rowe
b,
Robert G.
Palgrave
c,
Richard P.
Matthews
d,
Roger A.
Bennett
a,
Patricia A.
Hunt
e and
Kevin R. J.
Lovelock
*a
aDepartment of Chemistry, University of Reading, Reading, UK. E-mail: k.r.j.lovelock@reading.ac.uk
bDepartment of Chemistry, Imperial College London, UK
cDepartment of Chemistry, University College London, UK
dSchool of Health, Sport and Bioscience, University of East London, UK
eSchool of Chemical and Physical Sciences, Victoria University of Wellington, New Zealand
First published on 18th March 2025
Abstract
Understanding and predicting ionic liquid (IL) electronic structure is crucial for their development, as local, atomic-scale electrostatic interactions control both the ion–ion and ion–dipole interactions that underpin all applications of ILs. Core-level binding energies, EB(core), from X-ray photoelectron spectroscopy (XPS) experiments capture the electrostatic potentials at nuclei, thus offering significant insight into IL local electronic structure. However, our ability to measure XPS for the many thousands of possible ILs is limited. Here we use an extensive experimental XPS dataset comprised of 44 ILs to comprehensively validate the ability of a very low-cost and technically accessible calculation method, lone-ion-SMD (solvation model based on density) density functional theory (DFT), to produce high quality EB(core) for 14 cations and 30 anions. Our method removes the need for expensive and technically challenging calculation methods to obtain EB(core), thus giving the possibility to efficiently predict local electronic structure and understand electrostatic interactions at the atomic scale. We demonstrate the ability of the lone-ion SMD method to predict the speciation of halometallate anions in ILs.
1. Introduction
Knowledge of local electronic structure within ionic liquids (ILs) is vital to understand and predict electrostatic interactions, which control both the ion–ion and ion–dipole interactions that underpin all applications of ILs.1–7 Electrostatic interactions are of vital importance in ILs for e.g. anion–metal cation interactions in potential battery electrolytes,8 and cation–substrate interactions in cellulose processing.9As it is not possible to experimentally test the very large number of possible ILs due to cost, computational screening offers an attractive option for identifying anions and cations with the optimum local electronic structure. However, validation against experimental data is required to demonstrate that the computational screening method captures IL local electronic structure properties and give confidence in predictions.
Element-specific core-level binding energy of an electron, EB(core), from core-level X-ray photoelectron spectroscopy (XPS)10–12 is a local, atomic-level electronic structure descriptor that quantifies non-covalent electrostatic interaction strengths in ILs, e.g. the overall anion basicity/electron donor ability.10–12 Furthermore, differences in EB(core) correlate very well with the electrostatic potential at a nucleus (Vn);12–14Vn is used in the organic chemistry community as a descriptor for non-covalent electrostatic interaction strengths.15–17 Experimental EB(core,exp.) values are available for many elements for ILs from a diverse selection of anions (e.g., [NTf2]− = bis[(trifluoromethane)sulfonyl]imide and halometallate anions) and cations (mainly organic imidazolium and onium cations),10–12,18–25 and give consistent EB(core,exp.) values across different experimental apparatus and research groups.10,11,18–23,25
E B(core,exp.) can be used for validation of computational screening methods for IL local electronic structure properties because differences in EB(core,exp.) are driven mainly by local bonding/interactions. Contributions to EB(core,exp.) can potentially come from: (i) ground-state effects (in XPS called initial-state effects) which are determined by the local bonding/interactions, and (ii) the influence of the core-hole created during the photoemission event (in XPS called final-state effects) which is controlled by the relaxation of other core and valence electrons. It is often assumed that final-state contributions are vital,26 but there is increasing evidence that ground-state contributions dominate differences in EB(core,exp.).12,14,27 For 17 small organic molecules, a good correlation found between gas-phase EB(N 1s,exp.) and ground-state calculated EB(N 1s, calc.) (i.e., no core-hole included in the calculations) from density functional theory (DFT).27 We recently demonstrated that EB(core,exp.) and EB(core,calc.) (with no core-hole included in the calculations) from ab initio molecular dynamics (AIMD) correlate very well for three [C8C1Im][A] ILs; structures of ILs from AIMD are expected to reflect very well the IL solvation structure.12,14 These findings show that EB(core,exp.) can be used to validate differences in EB(core,calc.) (when no core-hole is included in the calculations), as differences in EB(core,exp.) are driven mainly by differences in local bonding/interactions, with final-state effect contributions being far less important. Any significant difference between EB(core,exp.) and EB(core,calc.) would be due to an inability of the calculation to capture the IL solvation structure and therefore the local electronic structure; any final-state contributions to EB(core,exp.) would be expected to be relatively small.
Screening ILs using EB(core) as a descriptor requires a method to calculate EB(core,calc.) with a good level of accuracy while still being relatively low-cost and technically undemanding. Using AIMD will give high quality IL solvation structures as noted above, but is very expensive and technically demanding, meaning that AIMD is not suitable to obtain EB(core) for screening purposes. Classical molecular dynamics (MD) simulations are lower cost than AIMD to obtain structures, and links between calculated atomic charges from structures derived from MD simulations and XPS EB(core) have been found,28 but MD simulations are not ideal for screening ILs as they are still technically demanding and can be expensive, especially if polarisable force-fields are used to improve inter-ion interactions. Ion-pairs computed using (gas phase) density functional theory (DFT) show a good (qualitative) visual match to experimental EB(core), however, the number of reported comparisons is very small,20,29 and ion-pair calculations require knowledge of the IL anion–cation structure.30–32
Using a lone-ion, an ion with no other explicit ions solvated to it, computed within a generalised solvation environment, such as SMD (solvation model based on density)33 offers a potential solution to the problem of balancing cost and accuracy. Lone-ion-SMD calculations are very small-scale, relatively inexpensive and technically undemanding calculations. We recently found using lone-ion-SMD calculations that 39 ILs gave excellent linear correlations between calculated and experimental EB(valence); but no data was given for EB(core).34 Moreover, preliminary results for onium cations and halozincate anions in ILs gave a good (qualitative) visual match between experimental and calculated XP spectra, but no quantitative comparisons of EB(core) were made.25,35
In this article we validate EB(core,calc.) using both quantitative and qualitative (visual) methods for lone-ion-SMD DFT calculations against benchmark XPS. We collate and publish, mostly for the first time here, a significant quantity of largely our own EB(core) data for 44 ILs. Seven elements (C, N, O, F, P, S, Cl) across 14 cations and 27 anions are examined, leading to XPS for 44 ILs being validated. This allows us to demonstrate that small-scale calculations using SMD capture the solvation environment of ILs and therefore give good quality EB(core,calc.), which are descriptors of local electronic structure.
We present a case study to demonstrate the ability of lone-ion-SMD DFT calculations to identify speciation in a chlorobismuthate IL by comparison to experimental XPS data, a challenging undertaking given that Bi is a heavy metal that can expand its coordination sphere and a lone pair that could be either stereochemically active or inactive. Speciation is not easy to determine for closed shell, spectroscopically quiet metals, e.g. Bi3+; standard spectroscopic methods (e.g. UV-vis, NMR) cannot supply sufficient insight. Furthermore, the femtosecond timescale for a single X-ray photoemission event means XPS provides a snapshot of the chlorobismuthate anions present in the liquid-phase without having to consider dynamics of the nuclei during the timescale of the X-ray photoemission event, which is a significant positive for speciation studies. We have previously demonstrated that this approach works well for halozincate-based ILs where halide atoms bridge between two zinc atoms but no lone pairs are present.25
2. Experimental
2.1. Experimental methods
XPS for 39 ILs (Table S1, ESI†) have previously been published, where the focus was mainly on valence XPS, as well as one aqueous solution (Table S2, ESI†).12,13,36,37 Core-level XP spectra were published mainly to demonstrate IL purity so most EB(core) values are published here for the first time for these 39 ILs.
2.2. Computational methods: small-scale DFT
DFT calculations were carried out for 14 cations (Table S3, ESI†) and 30 anions (Table S4, ESI†) at the B3LYP-D3(BJ) level, using Becke's three-parameter exchange functional in combination with the Lee, Yang and Parr correlation functional (B3LYP) as implemented in the Gaussian 09 and Gaussian 16 suite of programs.38–41 Grimme's D3 dispersion correction with Becke–Johnson damping was used to account for dispersion.42–45 The 6-311+G(d,p) basis set was employed for lighter atoms (H, C, N, O, F, P, S, Cl, Fe, Co, Ni, Zn), except for calculations undertaken to test the effects of varying the basis set which are discussed further below.46–49 LANL2DZ and LANL2DZdp pseudo potentials and the associated basis sets were employed for the heavier atoms In and Sn respectively.50 Data for the chlorobismuthate anions was taken from calculations reported in ref. 51; cc-pVDZ-pp (scalar relativistic) pseudopotentials and aug-cc-pVDZ associated basis sets were employed for the heavy Bi atoms,52,53 and the aug-cc-pVDZ basis set was employed for Cl in these calculations.Optimisations were carried out without symmetry constraints. The self-consistent field (SCF) convergence criteria were 10−9 on the density matrix and 10−7 on the energy matrix (scf = conver = 9). The numerical integration grid was a pruned grid with 99 radial shells and 590 angular points per shell (int = ultrafine). Frequency analysis was carried out for all optimised structures, which are confirmed as minima by the absence of imaginary modes.
[C4C1Im][PF6] parameters were used for calculations for all ions. The SMD parameters employed are shown in Table S5 (ESI†).
Structures were optimised as individual, isolated ions (referred to as lone-ion). Structures optimised in the gas phase are identified as GP structures while those optimised using SMD are referred to as SMD structures.
2.3. Computational methods: producing calculated XP spectra
To produce calculated XP spectra for core-levels that have spin–orbit coupling (Cl 2p, S 2p, P 2p), each calculated EB(core) was adjusted using EB separation and area factors given in Table S6 (ESI†). Once the adjusted calculated EB(core) were obtained, a Gaussian–Lorentzian product (GLP) function was applied to each calculated EB(core) data point using eqn (1) and then summed to produce calculated XPS data. The mixing parameter, m, and function width, F, were set to the values given in Table S6 (ESI†).To produce calculated XP spectra for core-levels that do not have spin–orbit coupling (F 1s, O 1s, N 1s, C 1s), a GLP function was applied to each calculated EB data point for each core-level using eqn (1) and then summed to produce calculated XPS data. E is the calculated EB for each core-level and x is EB at which the intensity is calculated. The mixing parameter, m, and function width, F, were set to the values given in Table S6 (ESI†).
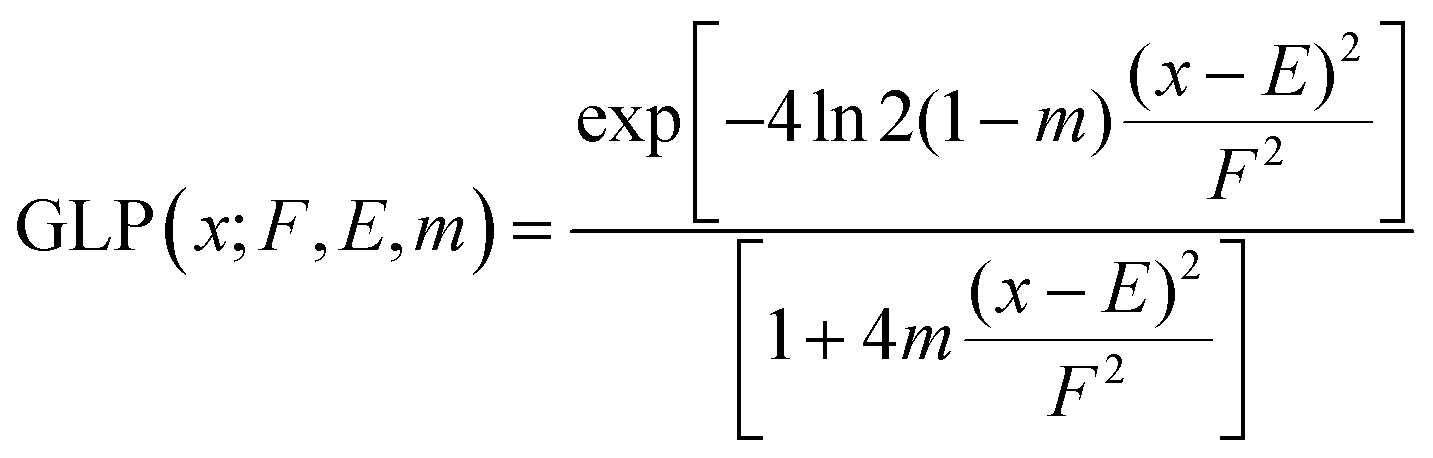 | (1) |
2.4. Data analysis
All experimental XP spectra were fitted using CasaXPS™ software.54 Fitting was carried out using a Shirley background and GL30 line shapes (70% Gaussian, 30% Lorentzian). Peak constraints used are outlined in the ESI,† Section S4.In ESI,† Table S8, the core-level used for charge referencing is given. The experimental uncertainty in EB(core,exp.) is ±0.10 eV.19
To align calculated XP spectra to experimental XP spectra, for each core-level the average difference between the EB(core,exp.) and EB(core,calc.) were used (average EB values are given in ESI,† Tables S10–S16 and S18). The average difference for each core-level represents the average final-state contribution for that core-level (along with other systematic errors from DFT). The calculated uncertainty in lone-ion-SMD EB(core,calc.) is ±0.20 eV (based on a combination of the experimental uncertainty in EB(core,exp.) and the standard error of the sample mean for different lone-ion-SMD EB(core,calc.), ESI,† Table S18).
3. Results and discussion
3.1. Validation of lone-ion-SMD for calculating IL core-level XPS
The excellent ability of lone-ion-SMD core-level calculations to capture core-level electronic structure is demonstrated here for both cations and anions (Fig. 1–3). | ||
| Fig. 1 Calculated binding energy, EB(calc.) from lone-ion-SMD against experimental XPS binding energy, EB(exp.) for: (a) C 1s, (b) N 1s, (c) S 2p3/2, (d) O 1s, (e) F 1s, (f) Cl 2p3/2, (g) P 2p3/2. m is the gradient of each line-of-best fit. The experimental uncertainty is ±0.10 eV; the calculated uncertainty is ±0.20 eV. | ||
 | ||
| Fig. 2 Experimental and calculated Cl 2p XPS for [C8C1Im][A] where A = Cl−, [ZnCl4]2−, [SnCl3]− and [InCl4]−: (top) experimental Cl 2p XPS (vertically offset for clarity); (middle) calculated Cl 2p XPS for lone-ion-SMD of the anions (EB(correction) = +5.18 eV, vertically offset for clarity); (bottom) calculated Cl 2p XPS for lone-ion-gas-phase (GP) of the anions (EB(correction) = +10.20 eV, vertically offset for clarity). Experimental XP spectra are area normalised and charge referenced using methods given in ESI,† Section S5. | ||
 | ||
| Fig. 3 Experimental and calculated C 1s XPS: (a) (top) experimental C 1s XPS for [N4,1,1,1][NTf2]; (middle) calculated C 1s XPS for lone-ion-SMD of [N4,1,1,1]+ and [NTf2]− (EB(correction) = +8.37 eV); (bottom) calculated C 1s XPS for lone-ion-gas-phase (GP) of [N4,1,1,1]+ and [NTf2]− (EB(correction) = +4.10 eV), with the cation contribution vertically offset for clarity; (b) (top) experimental C 1s XPS for four [CnC1Im][NTf2] ILs (where n = 2, 4, 6, 8); (middle) calculated C 1s XPS for lone-ion-SMD of four [CnC1Im]+ cations (where n = 2, 4, 6, 8) and the [NTf2]− anion (EB(correction) = +8.37 eV); (bottom) calculated C 1s XPS for lone-ion-gas-phase (GP) of [CnC1Im]+ (where n = 2, 4, 8) and [NTf2]− (EB(correction) = +4.10 eV), with the cation contributions vertically offset for clarity. Experimental XP spectra are area normalised and charge referenced using methods given in ESI,† Section S5. | ||
 | ||
| Fig. 4 Experimental and calculated P 2p XPS for four P-containing ILs and four lone P-containing lone ions: (top) experimental P 2p XPS for and [C4C1Im][PF6], [C2C1Im][FAP], [C4C1Im][Me2PO4] and [P6,6,6,14][NTf2] (vertically offset for clarity); (bottom) calculated P 2p XPS for lone-ion-SMD of the ions [PF6]−, [FAP]−, [Me2PO4]− and [P6,6,6,14]+ (EB(correction) = +3.23 eV, vertically offset for clarity). Experimental XP spectra are area normalised and charge referenced using methods given in ESI,† Section S5. | ||
Calculated XP spectra from lone-ion-SMD for anions give very good visual matches to experimental XP spectra for anions, including when 1- and 2-anions are considered, with the same single EB(correction) = +5.18 eV. A lone-ion-SMD Cl 2p calculation for the 2-anion [ZnCl4]2− matches almost perfectly to the experimental XP spectra both visually relative to other anions (Fig. 2, top and middle).
Calculated XP spectra from lone-ion-SMD for cations versus anions give very good matches to experimental XP spectra, showing that lone-ion-SMD calculations capture the electronic structure of the cation versus the anion very well in most cases. Lone-ion-SMD C 1s core-level calculations for [N4,1,1,1][NTf2] and [CnC1Im][NTf2] match visually, with the same single EB(correction) = +8.37 eV, almost perfectly to the experimental XP spectra for [N4,1,1,1][NTf2] and [CnC1Im][NTf2] respectively (Fig. 3 top and middle). Furthermore, lone-ion-SMD P 2p core-level calculations for [PF6]−, [FAP]−, [Me2PO4]− and [P6,6,6,14]+ match visually almost perfectly to the appropriate experimental XP spectra, all with the same single EB(correction) = +3.23 eV (Fig. 4).
We now compare EB(C 1s) in different cationic environments in more detail. A lone-ion-SMD C 1s calculation for the cation [N4,1,1,1]+ matches visually almost perfectly to the shape of the experimental XP spectrum for [N4,1,1,1]+ (with the anion [NTf2]−, Fig. 3a top and middle respectively), with EB(CC–N 1s) larger than EB(Calkyl 1s) for both experimental and calculated data. For [CnC1Im]+, the visual match is excellent for the EB(C 1s) order: EB(C2 1s) > EB(Chetero 1s) > EB(Calkyl 1s) (Fig. S6 and S7, ESI†), which matches to all C 1s fitting models used for IL XPS.18,37,55 However, for [CnC1Im]+ the EB(C 1s) differences are slightly larger for EB(C 1s,calc.) than EB(C 1s,exp.) (Fig. S6 and S7, ESI†). Similar results were obtained from AIMD-DFT calculations for three [C8C1Im][A] ILs (see Fig. S7 for a visual comparison of lone-ion-SMD and AIMD C 1s data, ESI†),12 demonstrating that these differences are likely due to a combination of the SMD method not quite capturing the effect of the solvation environment on the cations and final-state effect contributions not accounted for in the ground-state calculations of EB(Ccation 1s).
Lone-ion-GP calculations perform very poorly compared to lone-ion-SMD calculations, demonstrating the importance of using an SMD in lone-ion calculations. The lone-ion-GP C 1s calculations for [N4,1,1,1][NTf2] and [CnC1Im][NTf2] provide very poor matches to the corresponding experimental C 1s XP spectra (Fig. 3a top and bottom). ΔEB for EB(CCF 1s) from the anion relative to EB(Chetero 1s) from the cation was ∼+6 eV from experiments and ∼−2 eV from lone-ion-GP calculations, a huge mismatch (Fig. 3a, top and bottom). Furthermore, lone-ion-GP C 1s calculations for the cationic alkyls give very poor visual matches to the experimental C 1s XP spectra for both [N4,1,1,1]+ and [CnC1Im]+ (Fig. 3a and b, top and bottom). Lone-ion-GP calculations perform terribly at capturing the overall ion electronic structure. The lone-ion-GP calculations gave EB(Cl 2p) differences that are clearly far too large compared to the experimental EB(Cl 2p) differences, and also get the EB order completely wrong compared to the experimental EB data, e.g. EB(Cl 2p3/2,calc.) for [ZnCl4]2− smaller than Cl−, (Fig. 2, top and bottom).
We have found excellent matches for experimental and calculated XPS using a relatively modest basis set – 6-311+G(d,p) for the light atoms; the basis set choice had an impact on absolute EB(core,calc.) but little impact on differences in EB(core,calc.). Comparisons of tetraalkylammonium cations ([N4,1,1,1]+, [C4C1Pyrr]+, [C8C1Pyrr]+) for the 6-311+G(d,p) data published here and def2-QZVPP employed in ref. 35 show the differences in EB(N 1s,calc.) are very similar irrespective of the basis set identity, and very similar to the experimental differences in EB(N 1s,exp.) (Fig. S17, ESI†). Note that for EB(N 1s) the EB(correction) = +9.76 eV for the 6-311+G(d,p) basis set and EB(correction) = +6.37 eV for the def2-QZVPP basis set (data taken from ref. 35), demonstrating that the impact on absolute EB(core,calc.) is relatively strong but the impact on relative EB(core,calc.) is very little. Furthermore, comparisons of larger basis sets, def2-QZVPP and def2-TZVPP, against each other for the calculated Cl 2p XPS of chlorozincate anions showed minimal impact, with small absolute EB differences of less than 1 eV and much smaller relative EB differences than 1 eV.25
Our calculation method allows easy and efficient prediction of EB(core,exp.) for the seven core-levels for which EB(correction) has been determined. A constant EB(correction) = EB(core,exp.) − EB(core,calc.) for each core-level is required to align the experimental and calculated XP spectra (Fig. 2–4 and Fig. S6–S15, ESI†). For each core-level the EB(correction) needed is calculated from the average of individual EB(correction); the average EB(correction) values are given in ESI,† Table S18, which are calculated using the individual EB(correction) values given ESI,† Tables S10–S16. For example, a single EB(correction) = +15.84 eV is needed for the calculated XPS to give excellent visual matches to experimental XPS (Fig. S14, ESI†). The effectiveness of a single EB(correction) for each core-level across different chemical systems, including both cations and anions, indicates a high transferability for the EB(correction) parameter to a wide range of ions. Our results for calculations with different basis sets demonstrate that an EB(correction) value is needed for each core-level for each different basis set/functional combination (Fig. S17, ESI†). This situation is analogous to the calculations performed to aid interpretation of NMR spectroscopy data, where standard corrections in ppm are available for different basis set/functional combinations.56 The order of the average EB(correction), which is F 1s > O 1s > N 1s > C 1s > Cl 2p > S 2p > P 2p, matches the order of average EB(core,exp.) for each element (Table S18, ESI†). This increasing average EB(correction) with increasing average EB(core,exp.) is likely due to a deficiency in the DFT calculations, but could also be due to the increasing importance of final-state effects for core-levels with larger average EB(core,exp.).
3.2. Using calculated EB(core) to determine halometallate anion speciation
One use of the combination of calculated and experimental XPS is to investigate halometallate anion speciation; here we demonstrate this approach for the very challenging case of an equimolar 1 × BiCl3 + 1 × [C8C1Im]Cl mixture (Fig. 5), i.e., a mole fraction of x = 0.50 for BiCl3. In this case, plausible anion structures include [BiCl4]− (tetrahedral or sawhorse), [Bi2Cl8]2− and [Bi3Cl12]3−.51 | ||
| Fig. 5 Experimental Cl 2p XPS for x = 0.5 BiCl3 dissolved in [C8C1Im]Cl (top, middle and bottom) and calculated Cl 2p XPS for lone-ion-SMD of the anions (EB(correction) = +4.74 eV): (top) [Bi2Cl8]2−; (middle) [Bi3Cl12]3−; (bottom) [BiCl4]−. The experimental XP spectrum is charge referenced using methods given in ESI,† Section S5. The structures are given on the right-hand side. | ||
Our experimental XPS results conclusively rule out the presence of free Cl− anion. A Cl 2p3/2 peak due to Cl− would occur at EB(Cl 2p3/2,exp.) ≈ 197 eV,19 and no such contribution is evident (Fig. 5). Therefore, our experimental XPS results conclusively rule out [Bi2Cl7]− as the chlorobismuthate species, as for 1 × BiCl3 + 1 × [C8C1Im]Cl the presence of [Bi2Cl7]− would also give Cl−, and there is clearly no Cl− present in XPS of 1 × BiCl3 + 1 × [C8C1Im]Cl (Fig. 5). Furthermore, the calculated XP spectrum for 1 × [Bi2Cl7]− + 1 × Cl− gives a poor visual match to the experimental XPS for 1 × BiCl3 + 1 × [C8C1Im]Cl (Fig. S16, ESI†).
Contrary to a “first glance” visual interpretation of the experimental XPS (Fig. 5), it has been determined that more than one Cl electronic environment is present. The experimental Cl 2p spectrum for 1 × BiCl3 + 1 × [C8C1Im]Cl appears to give a single Cl electronic environment. A single Cl electronic environment on standard lab XPS apparatus gives a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 peak area ratio for Cl 2p with a EB peak separation of 1.60 ± 0.02 eV and a full width at half maximum (FWHM) of ∼0.9 eV, as found for Cl−, [ZnCl4]2−, [SnCl3]−etc. (ref. 25). For 1 × BiCl3 + 1 × [C8C1Im]Cl, the FWHM when fitted with two components with the appropriate spin–orbit coupling constraints (2:1 peak area ratio and EB peak separation = 1.60 eV, Table S6, ESI†), i.e. potentially a single Cl electronic environment, gave FWHM for the two components of 1.1 eV, significantly larger than the 0.9 eV FWHM that we have observed for one Cl environment.25 We were unable to produce a peak fitting model with two Cl electronic environments (i.e. four components); this observation does not rule out the presence of two different Cl electronic environments, it means that the two different Cl electronic environments have relatively similar EB(Cl 2p).
:1 peak area ratio for Cl 2p with a EB peak separation of 1.60 ± 0.02 eV and a full width at half maximum (FWHM) of ∼0.9 eV, as found for Cl−, [ZnCl4]2−, [SnCl3]−etc. (ref. 25). For 1 × BiCl3 + 1 × [C8C1Im]Cl, the FWHM when fitted with two components with the appropriate spin–orbit coupling constraints (2:1 peak area ratio and EB peak separation = 1.60 eV, Table S6, ESI†), i.e. potentially a single Cl electronic environment, gave FWHM for the two components of 1.1 eV, significantly larger than the 0.9 eV FWHM that we have observed for one Cl environment.25 We were unable to produce a peak fitting model with two Cl electronic environments (i.e. four components); this observation does not rule out the presence of two different Cl electronic environments, it means that the two different Cl electronic environments have relatively similar EB(Cl 2p).
Our experimental XPS results conclusively rule out [BiCl4]−. Firstly, the most stable structure for [BiCl4]− (determined using differences in calculated total energies51), a seesaw/sawhorse structure, gives a poor visual match to the experimental Cl 2p XPS (Fig. 5 bottom) as seesaw/sawhorse [BiCl4]− gives two very different Cl electronic environments, with a substantial EB difference of 0.9 eV (Table S17, ESI†). Secondly, tetrahedral [BiCl4]− with a stereochemically inactive lone pair (found to be unfavourable by calculated differences in total energies51) is not present, as only one Cl electronic environment would be observed for such a highly symmetric structure.
We assign the speciation for 1 × BiCl3 + 1 × [C8C1Im]Cl as [Bi2Cl8]2−, as the best visual match of experimental and calculated Cl 2p XP spectra is for [Bi2Cl8]2− (Fig. 5 top). There is a good visual match between experimental and calculated Cl 2p XPS for [Bi3Cl12]3− (Fig. 5 middle), suggesting that [Bi2Cl8]2− is the primary anion present but a small quantity of [Bi3Cl12]3− may also be present. [Bi2Cl8]2− gives two relatively similar Cl electronic environments, with a small EB difference of 0.4 eV (Table S15, ESI†), which matches to our unsuccessful attempts to fit two Cl electronic environments in the Cl 2p XP spectrum. The EB(correction) needed to obtain a very good visual match for both [Bi2Cl8]2− and [Bi3Cl12]3−, +4.74 eV (Fig. 5 top and middle), is very similar to the EB(correction) needed for a good visual match for all Cl-containing anions, +5.18 eV (Fig. 2 middle).
The total energies for the lone-ion-SMD calculations of [Bi2Cl8]2− and [Bi3Cl12]3− were both 8 kJ mol−1 more stable than of [BiCl4]−,51 matching well to our XPS finding of no [BiCl4]− present. However, such relatively small total energy differences, especially for [Bi2Cl8]2−versus [Bi3Cl12]3−, highlight the challenges of using total energies from small-scale calculations to determine halometallate speciation.
4. Summary and conclusions
We have presented small-scale calculations that can efficiently predict IL local electronic structure to a good level of accuracy without the need for experimental data. EB(core) calculated using inexpensive and technically accessible DFT calculations employing an SMD on lone-ions to capture solvation for 14 cations and 27 anions are successfully validated against an extensive experimental XPS dataset of 44 ILs using quantitative and qualitative comparisons. EB(core) values represent a descriptor for the ability of an atom to form electrostatic interactions. EB(core) has great potential as a molecular-level local descriptor for non-covalent electrostatic interaction strengths in ILs, especially given the dearth of molecular-level descriptors available for ILs. This demonstration opens significant opportunities.The experimental dataset published here is ideal for validation of IL calculations of EB(core). The experimental dataset given here includes seven elements, and EB(core,exp) data is available for other elements, e.g., Zn 2p and Br 3d for halometallate anions in ILs,25 giving multiple opportunities to validate calculations. If calculations are validated, then the user can have high confidence that the local electronic structure is captured in their calculations, and it is highly likely that the calculations capture the liquid-phase solvation environment of ILs.
Our findings give the potential for screening ILs for specific desired electronic properties. For example, calculating EB(core) can allow screening for atoms in ions that will give strong electrostatic interactions.
Our combination of experimental XPS data and lone-ion-SMD calculations shows great promise for determining the speciation of relatively complex metal-containing anions in ionic liquids. Through a combination of calculated and experimental spectra we find strong evidence that [Bi2Cl8]2− is the dominant species; a finding which would be difficult to determine without both the experimental and computational results.
Our dataset of EB(core) values can be used as input data for quantitative structure–property relationships (QSPR)57 of ILs, a field that is expected to grow significantly in the coming years; the strong links of EB(core) and the electrostatic potential at a nucleus (Vn)14 make EB(core) values particularly attractive in this area. Both (or just one or the other) experimental and calculated EB(core) can be used. Using just experiments or just calculations is simple; no EB(correction) is needed for the calculations. If obtaining EB(core) from a mixture of experiments and calculations, option one is use our calculation method (same basis set, functional and SMD parameters) for the ion or ions in question and use the appropriate EB(correction) value for the core-level in question, option two is to use a different calculation method which would require a range of reference ions to be calculated to allow EB(correction) to be obtained for the core-level in question.
Data availability
Data for this article, including X-ray photoelectron spectroscopy experimental data and log files for all calculated structures, are available at University of Reading Research Data Archive, DOI: 10.17864/1947.001411. Analysed data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
KRJL acknowledges support from a Royal Society University Research Fellowship (URF\R\150353, URF\R\211005, RGF\EA\180089, RGF\R1\180053, RF\ERE\210061 and RF\ERE\231015). RPM acknowledges support from the Royal Society of Chemistry through the RSC Research Fund grant (R21-4762139998).References
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 RSC.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. J. Zheng, S. G. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- T. Zhou, C. M. Gui, L. G. Sun, Y. X. Hu, H. Lyu, Z. H. Wang, Z. Song and G. Q. Yu, Chem. Rev., 2023, 123, 12170–12253 CrossRef CAS PubMed.
- Z. Y. Zhao, H. Li and X. Gao, Chem. Rev., 2023, 124, 2651–2698 CrossRef PubMed.
- J. L. Shamshina and R. D. Rogers, Chem. Rev., 2023, 123, 11894–11953 CrossRef CAS PubMed.
- X. Q. Li, K. Chen, R. L. Guo and Z. Wei, Chem. Rev., 2023, 123, 10432–10467 CrossRef CAS PubMed.
- G. X. Li, K. Chen, Z. G. Lei and Z. Wei, Chem. Rev., 2023, 123, 10258–10301 CrossRef CAS PubMed.
- N. Takenaka, S. Ko, A. Kitada and A. Yamada, Nat. Commun., 2024, 15, 1319 CrossRef CAS PubMed.
- B. L. Lu, A. R. Xu and J. J. Wang, Green Chem., 2014, 16, 1326–1335 RSC.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wölfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H. P. Steinrück, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed.
- B. B. Hurisso, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 17737–17748 RSC.
- E. Gousseva, F. K. Towers Tompkins, J. M. Seymour, L. G. Parker, C. J. Clarke, R. G. Palgrave, R. A. Bennett, R. Grau-Crespo and K. R. J. Lovelock, J. Phys. Chem. B, 2024, 128, 5030–5043 CrossRef CAS PubMed.
- E. Gousseva, S. D. Midgley, J. M. Seymour, R. Seidel, R. Grau-Crespo and K. R. J. Lovelock, J. Phys. Chem. B, 2022, 126, 10500–10509 CrossRef CAS PubMed.
- F. K. Towers Tompkins, E. Gousseva, R. A. Bennett, R. Grau-Crespo and K. R. J. Lovelock, 2025, in preparation.
- C. H. Suresh, G. S. Remya and P. K. Anjalikrishna, Wiley Interdiscip. Rev.-Comput. Mol. Sci., 2022, 12, e1601 CrossRef CAS.
- C. H. Suresh and S. Anila, Acc. Chem. Res., 2023, 56, 1884–1895 CrossRef CAS PubMed.
- B. Galabov, S. Ilieva, G. Koleva, W. D. Allen, H. F. Schaefer and P. V. Schleyer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 37–55 CAS.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H. P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158–5190 CrossRef CAS PubMed.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. L. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- M. Reinmöller, A. Ulbrich, T. Ikari, J. Preiss, O. Höfft, F. Endres, S. Krischok and W. J. D. Beenken, Phys. Chem. Chem. Phys., 2011, 13, 19526–19533 RSC.
- A. W. Taylor, S. Men, C. J. Clarke and P. Licence, RSC Adv., 2013, 3, 9436–9445 Search PubMed.
- R. M. Fogarty, R. P. Matthews, C. R. Ashworth, A. Brandt-Talbot, R. G. Palgrave, R. A. Bourne, T. V. Hoogerstraete, P. A. Hunt and K. R. J. Lovelock, J. Chem. Phys., 2018, 148, 193817 CrossRef PubMed.
- R. M. Fogarty, R. Rowe, R. P. Matthews, M. T. Clough, C. R. Ashworth, A. Brandt, P. J. Corbett, R. G. Palgrave, E. F. Smith, R. A. Bourne, T. W. Chamberlain, P. B. J. Thompson, P. A. Hunt and K. R. J. Lovelock, Faraday Discuss., 2018, 206, 183–201 RSC.
- F. K. T. Tompkins, L. G. Parker, R. M. Fogarty, J. M. Seymour, E. Gousseva, D. C. Grinter, R. G. Palgrave, C. D. Smith, R. A. Bennett, R. P. Matthews and K. R. J. Lovelock, Chem. Commun., 2024, 60, 10756–10759 RSC.
- J. M. Seymour, E. Gousseva, F. K. T. Tompkins, L. G. Parker, N. O. Alblewi, C. J. Clarke, S. Hayama, R. G. Palgrave, R. A. Bennett, R. P. Matthews and K. R. J. Lovelock, Faraday Discuss., 2024, 253, 251–272 RSC.
- W. F. Egelhoff, Surf. Sci. Rep., 1987, 6, 253–415 CrossRef.
- N. P. Bellafont, F. Illas and P. S. Bagus, Phys. Chem. Chem. Phys., 2015, 17, 4015–4019 RSC.
- N. V. S. Avula, A. Mondal and S. Balasubramanian, J. Phys. Chem. Lett., 2018, 9, 3511–3516 CrossRef CAS PubMed.
- M. Lembinen, E. Nommiste, H. Ers, B. Docampo-Alvarez, J. Kruusma, E. Lust and V. B. Ivanistsev, Int. J. Quantum Chem., 2020, 120, e26247 CrossRef CAS.
- P. A. Hunt and I. R. Gould, J. Phys. Chem. A, 2006, 110, 2269–2282 CrossRef CAS PubMed.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2014, 16, 3238–3253 RSC.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437–14453 RSC.
- V. S. Bernales, A. V. Marenich, R. Contreras, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2012, 116, 9122–9129 CrossRef CAS PubMed.
- R. M. Fogarty, R. P. Matthews, P. A. Hunt and K. R. J. Lovelock, 2025 10.1039/D5CP00214A.
- F. K. Towers Tompkins, L. G. Parker, R. M. Fogarty, J. M. Seymour, E. Gousseva, D. C. Grinter, R. G. Palgrave, C. D. Smith, R. A. Bennett, R. P. Matthews and K. R. J. Lovelock, Chem. Commun., 2024, 60, 10756–10759 RSC.
- R. M. Fogarty, R. G. Palgrave, R. A. Bourne, K. Handrup, I. J. Villar-Garcia, D. J. Payne, P. A. Hunt and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2019, 21, 18893–18910 RSC.
- J. M. Seymour, E. Gousseva, A. I. Large, C. J. Clarke, P. Licence, R. M. Fogarty, D. A. Duncan, P. Ferrer, F. Venturini, R. A. Bennett, R. G. Palgrave and K. R. J. Lovelock, Phys. Chem. Chem. Phys., 2021, 23, 20957–20973 RSC.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2006, 124, 014104 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- R. Rowe, K. R. J. Lovelock and P. A. Hunt, J. Chem. Phys., 2021, 155, 014501 CrossRef CAS PubMed.
- K. A. Peterson, J. Chem. Phys., 2003, 119, 11099–11112 CrossRef CAS.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- C. J. Clarke, S. Maxwell-Hogg, E. F. Smith, R. R. Hawker, J. B. Harper and P. Licence, Phys. Chem. Chem. Phys., 2019, 21, 114–123 RSC.
- M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2012, 112, 1839–1862 CrossRef CAS PubMed.
- S. Koutsoukos, F. Philippi, F. Malaret and T. Welton, Chem. Sci., 2021, 12, 6820–6843 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cp00892a |
| This journal is © the Owner Societies 2025 |