
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom-temperature phosphorescence in crystalline bifurcating halogen⋯halogen synthon containing chromophore†
Suvarna
Sujilkumar
 ,
Agaja
Kalyani
and
Mahesh
Hariharan
*
,
Agaja
Kalyani
and
Mahesh
Hariharan
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Maruthamala P.O., Vithura, Thiruvananthapuram, Kerala 695551, India. E-mail: mahesh@iisertvm.ac.in
First published on 11th March 2025
Abstract
Herein, we report room-temperature phosphorescence in a brominated dihydropentacene derivative having bifurcating bromine synthon in the crystalline state. Delayed emission experiments and insights from theoretical investigations have corroborated the feasibility of long-lived triplet excitons, offering promising implications for synthon-stabilized, metal-free, organic phosphors.
Room-temperature phosphorescence (RTP) in purely organic chromophores offers a wide range of applications over their organometallic counterparts owing to its biocompatibility, environmental safety, and cost-effective synthesis.1,2 Efficient RTP can be achieved by enhancing the rate of intersystem crossing (ISC) and suppressing the molecular motions in the molecule.3 The incorporation of halogen atoms is primarily known to facilitate phosphorescence in organic chromophores as a consequence of the heavy atom effect (HAE) and the resulting enhancement of spin–orbit coupling (SOC) between the singlet and triplet excited states.4 Additionally, the presence of various intermolecular noncovalent halogen interactions within the three-dimensional (3D) crystalline framework enables the restriction of molecular motions and suppresses triplet exciton quenching, thereby promoting efficient phosphorescence.5,6 The crucial role played by exciton interactions on photoluminous efficiency has driven significant research to uncover the correlation between intermolecular packing arrangements and the resultant RTP efficiency in the crystalline state.5,7
Supramolecular synthons are repeating structural motifs arising from noncovalent intermolecular interactions and are crucial in governing the crystal packing and concomitant photophysical properties.8 Synthon interactions are the outcome of various noncovalent forces, including halogen bonding, hydrogen bonding, π⋯π interactions, halogen⋯π interactions, cation/anion⋯π interactions, which are pivotal in determining the stability and assembly of the crystal structure.9,10 The halogen⋯halogen interactions originate from the anisotropic distribution of electron density on the halogen atom, which creates an attractive interaction between its σ-hole and a nucleophilic site, either on another halogen atom in a different molecule or within the same molecule.11 The existence of these inter- and intramolecular furcating halogen⋯halogen (X⋯X) interactions within the crystal packing helps to mitigate nonradiative deactivation by providing structural rigidity to the crystal structure and leads to efficient phosphorescence.12,13 The supramolecular arrangement of monomers in the crystal, governed by electronic coupling, critically influences luminescence properties, with weaker coupling leading to enhanced radiative emission rates.14 As a result, understanding the origin and nature of noncovalent interactions and excitonic coupling in crystalline assembly is essential for the precise design and efficient harvesting of RTP from supramolecular synthons.
Our group has been in constant pursuit of realizing the packing, luminescence properties, and charge transfer exciton dynamics of unique crystalline architectures, which motivated us to exploit synthon architectures to harvest RTP.9,15 Herein, we report the observation of RTP in a bifurcating synthon of crystalline organic derivative. The origin of long-lived red-shifted delayed emission was conclusively determined to be phosphorescence in nature by temperature-dependent gated emission and quantum chemical calculations. Further, through computational analysis, we quantified the electronic communication between the monomers, which was found to be dominantly Coulombic in nature with null short-range charge transfer coupling. Current work aims to design organic phosphor architectures with enhanced lifetime for emergent applications in imaging and optoelectronics.
The 6,13-bis(dibromomethylene)-6,13-dihydropentacene, namely, P–Br4, was synthesized and characterized following the modified procedure (Fig. 1a and Scheme S1 and Fig. S1–S5, ESI†).16 The slow evaporation of the solvent in a supersaturated solution of P–Br4 in dichloromethane yielded crystals of P–Br4, which upon single crystal X-ray diffraction, revealed a monoclinic unit cell with the Pc space group (Fig. 1b and Table S1, ESI†). The needle-like crystals of the P–Br4 viewed along the a-axis resembled a butterfly-type architecture (Fig. 2a and Fig. S6, ESI†). P–Br4 crystal images captured under UV illumination and after UV lamp removal showed no significant afterglow (Fig. S7, ESI†). The crystal structure analysis revealed the existence of alternatively packed bifurcating synthons formed through two Br⋯Br interactions emanating from the same center with distance (dBr⋯Br) as 3.64 Å and 3.76 Å (Fig. 1c and Fig. S8, ESI†). Three different types of dimers with centroid to centroid distance of 7.15 Å (dimer1), 7.90 Å (dimer2), and 12.32 Å (dimer3) were identified through detailed analysis of the crystal structure (Fig. S9, ESI†). To quantitatively understand the prevalent noncovalent interactions within the identified dimers, symmetry-adapted perturbation theory (SAPT(0)) was carried out at the aug-cc-pVDZ level of theory.17 The SAPT(0) results revealed the prominent role of dispersion forces in stabilizing all the identified dimers with negative SAPT(0) values (Table S2, ESI†). The noncovalent interaction (NCI) analysis was performed to visualize the noncovalent interactions in P–Br4 crystals by creating an index using electron density.18 The interaction region marked by a green disc between the hydrogen and the bromine atoms in the isosurface plot signifies a stabilizing van der Waals interaction characterized by low electron density, and the red-filled areas at the center of the rings indicate repulsive and destabilizing interactions (Fig. 2b). Furthermore, the NCI analysis of the synthon dimer of P–Br4 elucidated the stabilizing role of intermolecular Br⋯Br interactions in directing the synthon formation and regulating crystal packing (Fig. S10, ESI†). The relative strength of intramolecular interactions within the crystalline architecture was evaluated with the aid of Hirshfeld surface analysis.19 The percentage of contributions to the Hirshfeld surface areas of various intramolecular interactions is shown as a histogram chart in Fig. 2c. The H⋯Br interaction was found to be the dominant contributor to the total interaction, with a contribution of 35.2% (Fig. S11, ESI†).
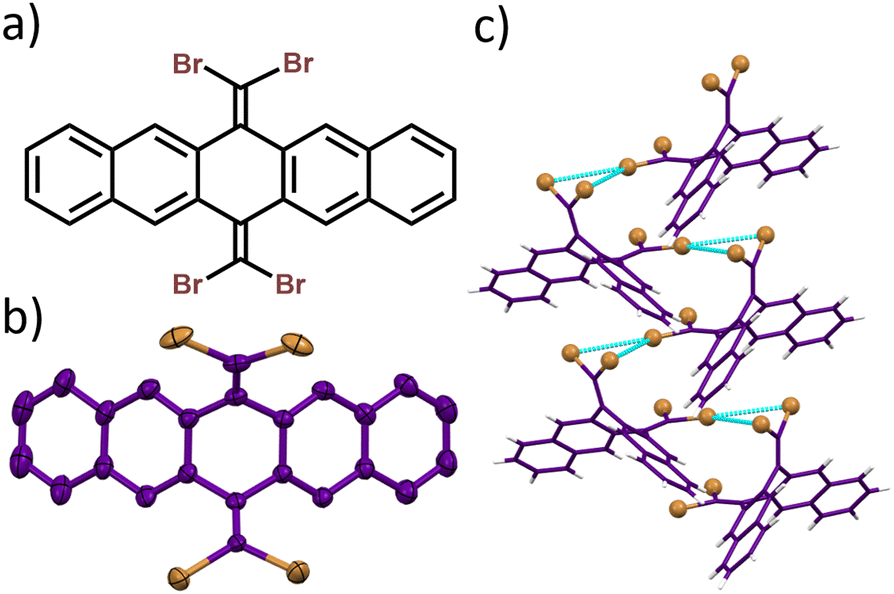 | ||
| Fig. 1 (a) Molecular structure and (b) crystal structure of P–Br4. (c) Bifurcating bromine synthons formed in P–Br4 crystals. | ||
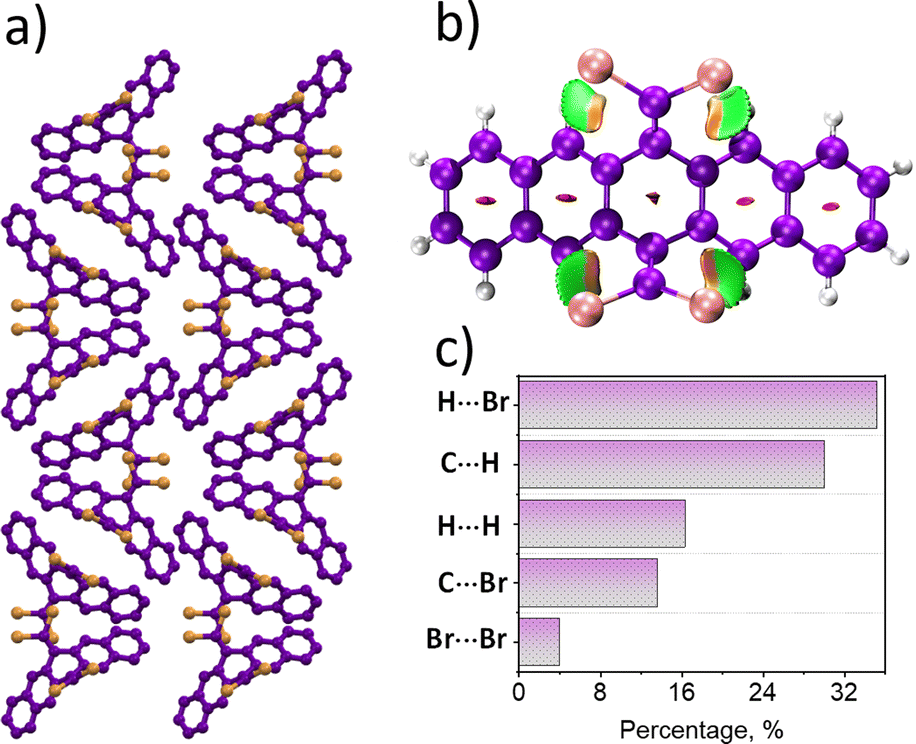 | ||
| Fig. 2 (a) Crystal packing viewed along a axis (b) NCI analysis and (c) Hirshfeld surface analysis chart of P–Br4 crystal. | ||
The energy framework analysis was carried out to visualize and quantify the type of interaction energies involved in the supramolecular crystalline assembly. The key interaction energies, including electrostatic, polarization, repulsion, and dispersion energies, were calculated using the benchmarked CE-B3LYP/6-31G(d,p) model (Table S4, ESI†).20 The thicker cylinder depicted in the energy framework further reinforced the supreme role of dispersion energy in intermolecular stabilization (Fig. S12, ESI†). Bader's quantum theory of atoms in molecules (QTAIM) analysis was employed to quantitatively validate the description of the noncovalent interaction based on the calculated topological properties.21 The interatomic interactions are characterized by representative points known as bond critical points (BCPs). The QTAIM analysis of the P–Br4 dimer revealed BCPs corresponding to the intermolecular Br···Br interactions that constitute the synthon and are defined as (3, −1) BCPs (Fig. S13, ESI†). The electron density values (ρ(r)) at the BCP determine the bond strength, with higher values signifying stronger bonds. A positive value of ∇2ρ(r) indicates noncovalent interactions, while a negative value suggests a polar covalent bond. The (ρ(r)) values and its Laplacian (∇2ρ(r)) were calculated for the bond and ring critical points involved in the synthon interaction (Table S5 and Fig. S14, ESI†). For Br⋯Br interactions, ρ(r) values at the BCPs range from 0.003 to 0.008 a.u. while ∇2ρ(r) ranges from 0.008 to 0.02 a.u. The low ρ(r) values and small positive Laplacian values observed indicate a reduced electronic charge density between the nuclei, suggesting weak to moderately closed shell noncovalent halogen interactions within the dimer. Furthermore, the analysis identified a (3, +1) ring critical point (RCP), indicating the formation of a bifurcating bromine synthon resulting from the noncovalent interaction between three Br atoms (Fig. S14, ESI†). Similarly, the QTAIM topological plots of dimer2 and dimer3 revealed bromine interacting with the dihydropentacene carbon atoms and the hydrogen atoms (Fig. S15, ESI†). The electrostatic surface potential (ESP) maps provided a qualitative representation of the electron density distribution within the crystal structure.22 The red regions within the dihydropentacene core and near the bromine atoms indicate areas of high electron density or negative electrostatic potential, while the blue portions represent σ-holes with low electron density (Fig. S16, ESI†). These electron-rich bromine atoms participate in noncovalent interactions such as halogen bonding by interacting with σ-holes of other molecules, thereby stabilizing the crystal packing.9
To probe the excitonic interaction prevalent in the crystalline assembly, the long-range Coulombic coupling (JCoul) and short-range charge transfer coupling (JCT) between the dimers were computed using CATNIP 1.9.23 The synthon-assisted P–Br4 exhibited a dominant JCoul of 602.10 cm−1 (Table S6, ESI†). The electron transfer integral (te) and hole transfer integrals (th) of the identified dimers, stemming from LUMO–LUMO and HOMO–HOMO orbital overlap of the monomers, respectively, were computed using the CATNIP software package (see ESI,† Section S1.8). Based on the computed charge transfer integrals, the short-range charge transfer coupling value was calculated and found to be JCT = −4.88 cm−1. The negligible JCT is possibly due to the larger interchromophoric distance and the highly sensitive nature of CT-mediated excitonic coupling (Tables S6, ESI†). The null JCT character effectively ruled out the possibility of having a significant charge transfer character, thereby mitigating the nonradiative processes in the crystal framework.24 Additionally, the Theodore analysis was performed, strengthening the negligible CT character attributing to the singlet excited states (Fig. S17 and Table S7, ESI†). The analysis of anisotropic charge carrier mobility provided insights into the charge transport properties of the P–Br4 crystal system. The hole and electron mobility were observed to be 0.0687 and 0.163 cm2 V−1 s−1, respectively (Fig. S18, ESI†). Consequently, our quantum chemical investigations collectively suggest that the noncovalent halogen bonding interactions effectively stabilize the crystalline molecular structure of P–Br4.
The spectroscopic investigation of P–Br4 was carried out at the monomeric solution state and crystalline state. The electronic absorption and emission spectra of P–Br4 were recorded in tetrahydrofuran (THF) at room temperature. The absorption spectrum spanned from ∼230–330 nm with a maximum of 281 nm (Fig. S19a, ESI†). The emission spectrum of P–Br4 in THF was obtained by exciting the sample at 310 nm and displayed a distinct emission band with a maximum intensity centered around 315 nm (Fig. S19b, ESI†). The Kubelka–Munk transformed diffuse reflectance spectrum of the P–Br4 crystal spanned from ∼190–390 nm, and the steady-state emission spectrum spread over ∼334–514 nm with an emission maximum of ∼376 nm (Fig. 3 and Fig. S20, ESI†). The fluorescence quantum yield of P–Br4 in the THF solution was determined using the relative method with quinine sulphate and in the crystalline state using the absolute method with an integrating sphere. As a consequence of the incorporation of four Br atoms in the pentacene-6,13-dione core, the fluorescence quantum yield was found to be <1% in both the solution and crystalline state. The negligible fluorescence quantum yield observed for P–Br4 suggests the possibility of alternative decay pathways upon photoexcitation, such as ISC. In a bid to understand the delayed emission characteristics of the P–Br4 crystal, the phosphorescence spectrum was acquired at room temperature by exciting at 310 nm with a delay time of 50 μs. The P–Br4 crystals demonstrated a long-lived red-shifted delayed emission band at ∼500 nm compared to the prompt emission at 376 nm (Fig. 3). Remarkably, a longer decay lifetime of 143.4 μs was observed for P–Br4 crystals at room temperature suggesting the luminescence emanating from the triplet manifold (Fig. S21, ESI†). Additionally, the gated emission measurements were performed at 77 K temperature. The phosphorescence intensity observed at 77 K is enhanced compared to room temperature, confirming the phosphorescent nature of P–Br4, as the suppression of non-radiative losses at lower temperatures enhances radiative emission (Fig. S22a, ESI†). Consequently, the phosphorescence lifetime has enhanced ∼4.6 times at 77 K compared to room temperature, attaining a value of 0.66 ms, indicating significantly higher phosphorescence efficiency of P–Br4 at cryogenic temperature (Fig. S22b, ESI†).
 | ||
| Fig. 3 Steady-state emission and phosphorescence emission spectra of P–Br4 crystal. | ||
The feasibility of ISC leading to RTP in P–Br4 was further investigated by theoretical models. The ground state geometry optimization of P–Br4 has been performed using density functional theory (DFT) at the CAM-B3LYP/def2svp level of theory. The efficiency of the ISC rate in between the excited singlet (S) and triplet states (T) is determined by a smaller energy gap (ES–T) and a higher magnitude of spin–orbit coupling (SOC) constants. Vertical excitation energies for the singlet and triplet excited states were computed using time-dependent density-functional theory (TD-DFT) and Tamm–Dancoff approximation (TDA), respectively. The computed energies for S1 (3.759 eV) and T6 (3.734 eV) states were observed to have the minimum energy gap with ES1–T6 = 0.024 eV (Table S8, ESI†). The SOC constant between the singlet and triplet states in proximity was computed using the PySOC package at the CAM-B3LYP/def2svp level of theory.25 A significantly high value of the SOC matrix element 〈S1|Hsoc|T6〉 = 35.94 cm−1 and near degeneracy between the S1 and T6 excited states suggests strong facilitation for ISC (Table S9, ESI†). In addition, the natural transition orbital (NTO) visualization enabled us to validate the character of radiation-less transition between S1 and T6 excited states in P–Br4 (Fig. 4a). The S1 state transition of P–Br4 is characterized by a π–π* excitation. In contrast, the T6 state exhibits a redistribution of electron density, with the electronic cloud centered on the Br atom displaying a moderate rotational displacement relative to the core ring of P–Br4. This change in orbital character can facilitate effective SOC and enhance the ISC transition in the P–Br4 crystal system.26
 | ||
| Fig. 4 (a) Natural transition orbitals (NTOs) for the S1 and T6 excited states for P–Br4. (b) Showing the proposed Jablonski diagram of P–Br4. | ||
In conclusion, we report the observation of room-temperature phosphorescence in a bifurcating halogen synthon-stabilized chromophore, P–Br4. Theoretical calculations, including QTAIM, Hirshfeld, NCI, and SAPT, substantiated the stabilizing interaction present in the crystalline architecture. Steady-state and gated emission experiments unambiguously confirmed the presence of RTP in the crystalline state. The computational analysis highlighted the crucial role of negligible charge transfer coupling and noncovalent halogen bonding in minimizing nonradiative relaxation routes while simultaneously accelerating the possibility of ISC. This work sheds light on the utilization of organic RTP materials for various potential applications in imaging and optoelectronics.
M. H. acknowledges MoE-STARS/STARS-2/2023-0770 for financial support. S. S. acknowledges PMRF for financial assistance. We kindly thank Mr Alex P. Andrews for the X-ray diffraction analyses. We thank the Padmanabha HPC cluster at IISER TVM for the computing time.
Data availability
All the experimental data were provided in the ESI.†Conflicts of interest
There are no conflicts to declare.References
- Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- X. Meng, Q. Hu, X. Wang, T. Ma, W. Liu, X. Zhu and C. Ye, J. Mater. Chem. C, 2022, 10, 17620–17627 RSC.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- J. Zhou, L. Stojanović, A. A. Berezin, T. Battisti, A. Gill, B. M. Kariuki, D. Bonifazi, R. Crespo-Otero, M. R. Wasielewski and Y.-L. Wu, Chem. Sci., 2021, 12, 767–773 RSC.
- L. Xiao, Y. Wu, Z. Yu, Z. Xu, J. Li, Y. Liu, J. Yao and H. Fu, Chem. – Eur. J., 2018, 24, 1801–1805 CrossRef CAS PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- M. A. Niyas, R. Ramakrishnan, V. Vijay, E. Sebastian and M. Hariharan, J. Am. Chem. Soc., 2019, 141, 4536–4540 CrossRef CAS PubMed.
- N. G. Nair, K. Prasad, H. C. Babu and M. Hariharan, Cryst. Growth Des., 2022, 22, 2318–2327 CrossRef CAS.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao, J. Shi, B. Tong, Z. Cai and Y. Dong, Angew. Chem., Int. Ed., 2022, 61, e202200236 CrossRef CAS PubMed.
- L. Xiao and H. Fu, Chem. – Eur. J., 2019, 25, 714–723 CrossRef CAS PubMed.
- A. Zhao, X. Wu, X. Jiang, J. Gao, J. Wang and W. Shen, Phys. Chem. Chem. Phys., 2021, 23, 26813–26821 RSC.
- R. Ramakrishnan, M. A. Niyas, M. P. Lijina and M. Hariharan, Acc. Chem. Res., 2019, 52, 3075–3086 CrossRef CAS PubMed.
- A. A. Gorodetsky, C.-Y. Chiu, T. Schiros, M. Palma, M. Cox, Z. Jia, W. Sattler, I. Kymissis, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7909–7912 CrossRef CAS PubMed.
- K. Szalewicz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 254–272 CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- R. F. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- H. Shi, W. Yao, W. Ye, H. Ma, W. Huang and Z. An, Acc. Chem. Res., 2022, 55, 3445–3459 CrossRef CAS PubMed.
- C. Lin, T. Kim, J. D. Schultz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2022, 14, 786–793 CrossRef CAS PubMed.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2414288. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00496a |
| This journal is © The Royal Society of Chemistry 2025 |