
Selenium-doped carbon materials: synthesis and applications for sustainable technologies†
Sławomir
Dyjak
 *a,
Bartłomiej J.
Jankiewicz
b,
Stanisław
Kaniecki
a and
Wojciech
Kiciński
*a
*a,
Bartłomiej J.
Jankiewicz
b,
Stanisław
Kaniecki
a and
Wojciech
Kiciński
*a
aInstitute of Chemistry, Military University of Technology, gen. Sylwestra Kaliskiego 2, 00-908 Warsaw, Poland. E-mail: slawomir.dyjak@wat.edu.pl; wojciech.kicinski@wat.edu.pl
bInstitute of Optoelectronics, Military University of Technology, gen. Sylwestra Kaliskiego 2, 00-908 Warsaw, Poland
First published on 30th January 2024
Abstract
Heteroatom-doping of carbonaceous materials and (nano)structures permits alteration of their fundamental physicochemical properties and thus amplifies the range of their potential applications. While nitrogen-doping dominated this research area, doping and co-doping with other non-metal and metal elements proved to be equally efficient strategies to tune carbonaceous materials’ characteristics. Heavier chalcogens are considered atypical carbon dopants due to their large atomic radii, high polarizability, and electronegativity comparable to that of carbon. Se-doped carbons are much less common than their S-doped counterparts, yet recently, they appeared as attractive functional materials for a range of sustainable technologies. This review depicts progress in Se-doped and co-doped carbonaceous materials from 2012 to the present. The report is divided into three major parts: first, the versatile chemistry of selenium and the significance of Se-doping is presented, followed by a variety of preparation methods, and finally, prominent examples of advanced applications in the fields of heterogeneous (electro)catalysis, energy conversion/storage, and pollutant neutralization.
1. Introduction
Carbonaceous materials of graphenic structure,1i.e., made of hexagonally arranged, sp2-bonded carbon atoms, are exceptional in at least two ways. Firstly, the versatile chemistry of a carbon element permits construction of endless chains, sheets, and three-dimensional (3D) structures, delivering a virtually unlimited number of (nano)structures and morphologies. Secondly, the fact that materials built of sp2-bonded carbon atoms exhibit unique physicochemical stability is equally important. As the sp2 framework of graphenic carbons possesses exceptional chemical stability, the substitution of carbon atoms by a wide range of heteroatoms allows fine-tuning of their fundamental properties. Nitrogen-doped and co-doped carbonaceous materials are by far the most commonly investigated doped carbons. This is not only because N can easily substitute the carbon atom in the hexagonal lattice without much distortion of its geometry, but the N-dopant is also very stable thermally and withstands high heat treatment temperatures (HTTs).2 The similarity of nitrogen and carbon atoms’ covalent radii (0.71 Å for N and 0.73 Å for C-sp2)3 enables easy substitution, but a significant difference in electronegativity (3.04 for N vs. 2.55 for C; the Pauling scale) leads to N-doping-induced charge transfers from carbon atoms to the adjacent N atoms, and this causes increased reactivity of N-doped carbon materials.4,5 Consequently, N-doped and co-doped carbons became a class of new promising functional materials especially attractive for heterogeneous (electro)catalysis.6–8Utilizing the benefits of N-doping, a new type of metal-free heteroatom-doped carbonaceous material has been developed and studied as catalysts for various chemical processes with particular emphasis on oxygen and hydrogen electrocatalysis. Metal-free carbon-based catalysis, also known as carbocatalysis, has emerged as a fascinating chapter in the field of carbon science and technology.9 It was quickly realized that besides single N-doping, co-doping and/or doping with other elements such as 3p (e.g., P or S) and 4p (e.g., Ge or Se) elements can further boost the (electro)chemical activity of heteroatom-doped graphenic carbons. Sulfur-doped carbonaceous materials gained particular attention, and a few review papers have already been published on this topic.10–12 Unlike nitrogen, chalcogens such as S and Se exhibit much bigger covalent atomic radii (1.05 and 1.20 Å for S and Se, respectively), yet their electron affinities are not that far off carbon (2.58 and 2.55 for S and Se, respectively). In consequence, doping heavier chalcogens into the graphene layer causes severe geometrical distortion, but the carbon-chalcogen bonds are not expected to be as polarized as carbon–nitrogen bonds. While nitrogen forms energetically very stable graphitic (tertiary N) structures fully infused in the hexagonally arranged, sp2-bonded carbon structure, chalcogens preferentially occupy edge and defect sites. A fundamental question therefore arises: what does it really mean to dope graphenic carbon with a heteroatom?
2. Carbon doping, functionalization, or decoration – is there a difference?
Heteroatoms can be adsorbed on the surface of a graphene layer by physical intermolecular forces or chemisorbed by creating covalent bonds with carbon atoms. Recently, P. A. Denis asked a very relevant question: “When can an atom be considered a dopant?”.13 When one describes carbon-doped materials, explicit differentiation between functionalization, adsorption (physisorption), or substitution must be discussed. Substitutional doping occurs when a foreign element is embedded in the graphene framework by replacing a carbon atom. In other words, it is chemical adsorption (chemisorption) in a single vacancy by creating covalent bonds with three sp2-hybridized carbon atoms. Only B and N are regarded as the true graphene dopants, as they can replace carbon atoms in the hexagonal plane in such a manner without creating much deformation. In contrast, chalcogens (e.g., O, S, Se) are preferentially located at the edges and larger vacancies. Halogens (e.g., F, Cl, Br), which form a single bond with carbon, are considered covalent functional groups. Oxygen is a very special case as a carbon dopant. Carbon-based materials such as nanoporous carbons and carbon nanostructures usually occur as fine powders and possess chemically heterogeneous surfaces because they easily chemisorb oxygen from the atmosphere. In fact, besides hydrogen, oxygen functional groups are predominant on carbon surfaces and, in many instances, may determine their chemical surface properties.14 While oxygen forms a profusion of various functional groups on the surface of carbon materials, a structure where an oxygen atom would replace a carbon atom in a graphene layer and would be fully infused into a hexagonally arranged, sp2-bonded system is unknown. Long-lasting scrutiny of the chemical structure of graphene oxide (GO) indeed proved that chemisorbed oxygen exists in graphenic carbons only as covalently bonded functional groups and edge sites. It should be noted here that most of the so-called graphene materials reported in the literature were prepared via the reduction of GO, and hence by default, all carbon materials based on GO and reduced graphene oxide (rGO) are functionalized with oxygen. In the case of graphene research, the term doping is also used in relation to adatoms, i.e., physically adsorbed atoms. This is because the adsorption of atoms at the graphene surface is considered a surface transfer doping (charge transfer from the adsorbed dopant to graphene). In contrast to substitutional doping, surface transfer doping of graphene is fully reversible. This terminology is sometimes confusing when it comes to heteroatom-doped carbon materials. As carbon materials are often porous structures or occur as nanostructures with void space (e.g., carbon nanotubes), they can also be impregnated with non-carbon media, yet impregnation is different from chemically bonded dopants. Moreover, the term decoration is commonly used in the field of carbon materials and (nano)structures. Decoration usually refers to nanoparticles (NPs) and clusters deposited on carbon surfaces and can involve charge transfer and even covalent bonding. The term decoration may be used when particles of specific functions are supported on carbonaceous substrates.15,16 In such composites the carbon support enhances the desired functions of the deposited particles. However, heteroatom-doped carbonaceous materials are not composites. Herein, within the scope of this review, the term doping will be limited to cases where a single heteroatom is directly bonded via a covalent bond to the aromatic, hexagonal lattice of graphenic carbon frameworks.3. Why does selenium doping matter?
There are many possible combinations to dope and co-dope carbonaceous materials with a variety of heteroatoms. In theory, the whole Periodic Table is at our disposal.17 Consequently, before going into details concerning Se-doped carbons, a question must be asked: why does it even matter? Selenium is a truly peculiar element. From the human body's perspective, it is simultaneously an essential and toxic element, and the range between the necessary and poisonous amount is very narrow. The recommended selenium intake for adults is 55 μg a day, while the toxic limit is 800 μg a day.18 When it comes to its environmental abundance, Se is considered a trace element in the Earth's crust with an average abundance of ca. 0.05 ppm.19 Nevertheless, it is still more abundant than the noble metals of Pt, Pd and Au. Sulfide minerals associated with copper provide most of the produced selenium and thus Se is obtained mainly as a byproduct of copper refining. Because selenium is photoconductive, it is often classified as an energy-critical element necessary for photovoltaic device production.From a chemical point of view, Se is most similar to sulfur. They share the same oxidation states (i.e., −2, 0, +2, +4, and +6) and functional group types (Fig. 1).20 In fact, they are so similar that analogous compounds of S and Se often co-crystallize. Thiophene (C4H4S) and selenophene (C4H4Se) are similar compounds – both are planar aromatic structures, and the bond lengths and angles of the C–S–C motif in thiophene are very close in values to those of C–Se–C in selenophene. But despite these similarities there are some subtle differences with tremendous consequences. Generally, with respect to sulfur, Se tends to be more stable in its intermediate oxidation states but less stable in the extreme states (e.g., the high oxidation states of Se are less stable than those of sulfur). In biological systems, Se is found in compounds such as selenols, diselenides, and selenoethers. These compounds are usually more reactive than their sulfur-based counterparts, and this is due to the greater polarity and lower strength of the C–Se, N–Se, and O–Se bonds.21 Biochemically, the most relevant difference between Se and S is the higher polarizability and thus the nucleophilicity of Se, and the lower pKa of the Se–H moiety.22 While there are many chemical forms of selenium in biology, selenocysteine is the most important one, as it is part of many selenoproteins. Most Se-containing enzymes (selenozymes) utilize the nucleophilic and reducing properties of the selenolate form of selenocysteine (Sec-Se−) to perform redox reactions. Among the best-studied selenoenzymes are the glutathione peroxidases (GPx) – antioxidant enzymes that scavenge peroxides and hence protect cells from oxidative damage.
 | ||
| Fig. 1 Structures and oxidation states of S and Se compounds resulting from 2e− oxidation reactions. Arrows show the conversion pathways between oxidation states. The open circle represents carbon, and the number inside the circle represents the oxidation number of S or Se that it is attached to. Magenta X denotes a slow reaction. The [O] symbol represents a 2e− oxidant. Reproduced with permission from ref. 20. Copyright © 2016, American Chemical Society. | ||
Differences between selenium and sulfur are largely due to differences in their atomic mass. Se possesses a larger covalent radius vs. sulfur (120 vs. 105 pm).3 As Se creates weaker bonds than S, it exhibits faster bond-breaking reactions. Nearly all organic reactions involving Se are faster with respect to sulfur. In biology, redox chemistry constitutes the largest chemical difference between S and Se. Se exhibits the exceptional ability to react with oxygen and related reactive oxygen species (ROS) in a readily reversible manner.20 While sulfur and selenium are good nucleophiles that react with ROS in 2e− oxidation events, the resultant S-oxides and Se-oxides show striking differences in their chemical reactivities. Unlike S-oxides, the Se-oxides can be rapidly reduced back to their original state. As it is more polarizable, Se is a better nucleophile and reacts with ROS faster than sulfur. The propensity of selenium for rapid oxidation and then rapid reduction is due to weak π-bonding in the Se-oxide.20 In contrast to sulfur, Se can create Se-containing biomolecules that resist permanent oxidation, and the weak selenium–oxygen bonding makes Se an exceptional oxygen carrier. As highlighted above, the reaction of selenium with oxygen is highly reversible. This is due to the strong nucleophilic character of selenium (to attack electrophiles), yet this reaction can be reversed because of the simultaneous strong electrophilic character of selenium and the weakness of the Se–C bond.23 Just like sulfur, selenium forms stable chalcogenides with transition and heavy metals. This strong affinity of sulfur and heavier chalcogens for heavy metals has been utilized to remove metals from aqueous solutions by using sulfurized activated carbons.24 According to the Pearson rule, the strong affinity of sulfur (a soft base) to heavy metals (soft acids) originates from soft acid–soft base interactions. In fact, the toxicity of some heavy elements may be associated with their chemistry towards selenium. For instance, Hg toxicity originates most likely from its strong binding to selenium in selenoproteins of the glutathione peroxidase. Hg binds to the selenium sites on these proteins and totally inhibits their function, causing oxidative stress and cell damage.25 Selenium has an exceptionally high binding affinity constant towards Hg of 1022 (one million times higher than the binding affinity between mercury and S).26
The reversible and versatile chemistry of selenium in biological systems inspired research in the area of Se-based (nano)materials due to their potential applications in catalysis, biosensing, diagnosis/imaging, and disease treatment.27,28 Traditionally, Se-based compounds and polymers have been prepared as homogeneous catalysts to assist organic transformations or to mimic natural enzymes. Recently, many new functional selenium-based nanomaterials have been reported, including selenium nanoparticles and Se-based quantum dots.27 Se NPs can serve as new nutritional supplements as they exhibit some therapeutic properties. They can also be utilized as antibacterial, antiviral, and antifungal agents or even as fertilizers. The unique bioactivity of selenium grants the carbon materials modified with Se a wide range of feasible applications in heterogeneous catalysis and, more generally, in environment protection solutions.29
Numerous theoretical studies suggested that doping and co-doping graphenic structures with selenium could produce novel materials of valuable electronic, magnetic, and optical properties.30,31 For instance, Se-doped 2D carbon crystals were theoretically considered as 2D superconductors.32 A range of 2D crystals of CxSe stoichiometry (x = 4, 5, and 6) was scrutinized, and the C4Se and C5Se structures showed superconductivity. While pure, undoped graphene is chemically inactive, Se-doping increases its reactivity, making it useful in chemical applications.33 But the question remains: is it possible to substitutionally replace carbon atoms with Se in the graphene structure? Theoretical considerations show that in such a case, the Se heteroatom significantly protrudes out of the hexagonal, sp2-hybridized carbon framework plane due to its larger radius. Se is located ∼1.6 Å above the graphene plane – protruding more than sulfur in S-doped graphene.33 In summary, Se is considered a promising dopant due to its high polarizability, nucleophilicity, and large atomic radius, which causes severe structural deformation of the hexagonal carbon structure, leading to increased reactivity of the doped carbonaceous materials additionally amplified by the reversible chemistry of a selenium element itself.
4. Synthesis of Se-doped and co-doped carbon materials
Doping of carbon materials with foreign atoms is usually achieved by some kind of high-temperature treatment. Whether it is direct pyrolysis of organic matter or post-treatment with heteroatom-containing gases, thermochemical reactions are involved at some stage of doping. This approach is an obvious choice since graphenic carbon exhibits extremely low volatility and high thermochemical stability. However, while pyrolytic synthesis has been successfully used to produce many functional carbon materials, pyrolysis remains a classical black box case where reagents/precursors constitute an input, and the final functional material is an output.34 For this reason, it is somewhat difficult to control the molecular structure of heteroatom-doped carbons obtained via pyrolytic decomposition of organic matter and carbothermic reduction of inorganic reagents.In the following sections, we discuss some prominent examples concerning the synthesis of Se-doped carbonaceous materials. Special attention is paid to the type of Se sources utilized for doping, the chemical structure of the doped Se atom within the carbon matrix, and the porosity of the obtained materials (e.g., specific surface area (SSA)). Relying on knowledge concerning sulfur-doped carbons10–12 and the organic chemistry of selenium compounds, one can predict possible Se configurations in Se-doped carbonaceous materials. Fig. 2 depicts possible molecular configurations and surface functional groups for Se-modified graphenic carbon.
 | ||
| Fig. 2 Possible configurations of Se incorporated into the graphenic carbon framework. Selenium tends to occupy vacancies and edge sites. | ||
Because Se-doped carbon materials are obtained via pyrolysis or high-temperature annealing, the peak temperature treatment determines their final characteristic. For this reason, we always try to specify the values of the HTT. Moreover, since Se-content is one of the main characteristics of Se-doped carbons, we also specify how much selenium was doped in each presented case and by what analytical method the amount was assessed. This is crucial because if, for instance, X-ray photoelectron spectroscopy (XPS) is applied as a quantifying tool, then it reveals only surface content, while tools such as inductively coupled plasma/optical emission spectroscopy (ICP-OES) assess bulk content, which usually differs substantially from surface content. For the commonly used energy-dispersive X-ray spectroscopy (EDS/EDX) technique the content of elements, such as S or Se in carbonaceous powders, may vary even more than 25% depending on the fraction/area of the scrutinized sample.35
4.1. Se-doped carbon materials based on rGO
GO and rGO are characterized by a variety of topological defects and oxygen functional groups, which remain present even after the reduction process. Consequently, rGO is not the 2D crystal of graphene but a different kind of material. While a true graphene structure cannot be obtained via reduction of GO, there are various approaches to endow rGO with properties similar to those of the genuine 2D graphene crystal. For instance, charge-transfer chemical doping with d-electron-rich heteroatoms has been exercised to improve the electronic transport properties of rGO. It was found that Se can be chemisorbed onto rGO in particular locations, such as edge sites or vacancy defects.36 Se atoms were predominantly present in the form of a C–Se–C bonding configuration, and successful restoration of thermally reduced GO by atomic-level Se doping has indeed been achieved. High-resolution transmission electron microscopy (HRTEM) analysis proved that selenium atoms were introduced into the rGO structure at an atomic level. Se-doping served as n-type doping, improving the electrical conductivity of the obtained Se-rGO material, and resulting in temperature-independent carrier mobility, similar to that of theoretical graphene. This showed that the carrier mobility of rGOs can be restored to that of graphene by simple Se-doping. To obtain Se-rGO, GO was first prepared from natural graphite using the Hummers’ method. Then, a mixture of the lyophilized GO powder and selenium powder was heated at 800 °C (i.e., above the Se boiling/sublimation temperature (685 °C)). The amount of doped Se was estimated to be ca. 13 at% (by EDS) or 15 wt% (by elemental analysis). Yang and co-workers proposed synthesis of Se-doped graphenic carbons utilizing diphenyl diselenide (C6H5)2Se2 (DDSe) as a Se source.37,38 They prepared Se-doped graphene by pyrolyzing commercial graphene oxide and DDSe at 1050 °C. The formation of covalent C–Se bonds was confirmed by XPS analysis.37 Using commercial GO and carbon nanotubes (CNTs), they also produced Se-doped composite carbon materials by directly annealing the GO/CNT with DDSe at 900 °C.38 The resultant Se-doped CNT/graphene composite contained ∼1 wt% of Se (assessed by XPS) in the form of selenophene-like –C–Se–C– structures located at the carbon layer edges.A Se-doped carbonaceous material where the Se atom binds two sheets of rGO was also prepared.39 First, GO obtained via the Hummers’ method was iodinated using HI to replace the C–OH groups with C–I. Next, a coupling reaction was performed using selenourea ((NH2)2CSe) as a linker and copper oxide as a catalyst. The C–I groups underwent coupling, where C–Se–C bonds were created by linking two rGO sheets. XPS indicated the dominance of Se–C bonds over Se–O bonds and ∼0.16 at% of doped Se. The rGO possessed residual oxygen functionalities and was additionally doped with nitrogen originating from the selenourea. In fact, the final material constituted rGO co-doped with Se and N and contaminated with some residual O, Cu, and I.39 Another example of co-doped carbonaceous material is the Se/N co-doped rGO-CNT assembly obtained by heat treatment at 900 °C, where dicyandiamide was used as the N-doping source and diphenyldiselenide as the Se doping source.40 The GO was obtained via the Hummers’ method and self-assembled with partially oxidized CNTs. Se was doped in the carbon structure mainly as a –C–Se–C– entity. A ternary Se/S/N co-doped counterpart was also prepared using diphenyl disulfide as an additional sulfur source. The Se content was around 0.05 at% in both the binary and ternary doped carbons (ICP analysis).40 The heteroatom-doped carbons had similar N-doping amounts (∼5.5 at%). This co-doped rGO/CNT composite was tested as a catalyst for the oxygen reduction reaction (ORR). Utilizing GO produced via the Hummers’ method, a N/Se co-doped graphene aerogel was prepared and proposed as a binder-free electrode for capacitors.41 The GO aerogel was prepared by hydrothermal treatment, freeze-dried, and then urea and (C6H5)2Se2 were added as N and Se sources, respectively, and the mixture was heated at 800 °C. The N and Se contents in the final co-doped material were 5.89 and 2.13 at%, respectively. It was noted that N and Se elements can synergistically enhance electrical conductivity by inducing n-type doping. The co-dopants also improved the wettability of the electrode surface. The Brunauer–Emmett–Teller (BET) specific surface area of the N/Se co-doped aerogel was 337 m2 g−1. Significantly, the SBET value of the corresponding undoped material was only 199 m2 g−1. This suggests that selenium may act as some kind of activator, enhancing the specific surface area of carbonaceous materials. In fact, this observation agrees with our earlier report on the influence of chalcogens on carbonaceous material porosity.42 As we have proved, sulfur and selenium desorb from the carbon framework at temperatures >800 °C as CS2, S2, CSe2, and Se2 vapors, causing a distinct increase in microporosity.
4.2. Se-doped carbon materials based on metal–organic frameworks
Metal–organic frameworks (MOFs) are competitive precursors of heteroatom-doped porous carbon materials. MOF-derived carbons constitute promising functional materials for technologies concerning energy conversion and storage (e.g., as heterogeneous catalysts) and environmental remediation because they are characterized by well-defined nanoporosity, high surface areas, and controllable morphologies.43 Many types of MOFs can be directly converted into carbon materials through simple one-step pyrolysis. The zeolitic imidazolate framework ZIF-8 (2-methylimidazole zinc salt), which is made of Zn2+ ions connected by imidazolate linkers, is of particular interest as a precursor of nanoporous carbons. ZIF-8 is commercially available under the trade name Basolite® Z1200. It exhibits high porosity (SBET of ca. 1500 m2 g−1) and high thermal stability. ZIF-8 can be carbonized with good yield, and Zn can be removed via simple evaporation above 907 °C (boiling point of Zn). Significantly, the obtained nanoporous carbon is rich in pyridinic nitrogen doping.44 This fact is of the utmost importance because N-doping in pyridinic and pyrrolic states strongly coordinates transition metals, forming thermally stable metal ion–N4 molecular structures.45ZIF-8 was utilized to prepare a single-atom catalyst (SAC) consisting of atomically dispersed Se atoms supported on the N-doped nanoporous carbon.46 First, ZIF-8 was pyrolyzed to obtain an N-doped carbon framework. Next, selenium dioxide was mixed with the carbonaceous material and annealed at temperatures up to 1000 °C. SeO2 undergoes pyrolytic reduction, and Se species are anchored on the N-doped carbon framework. Annealing at three different temperatures of 500, 900, and 1000 °C allowed assessment of the effect of HTT on the structure and porosity. As expected, Se content decreased with the temperature increase, from 13 wt% at 500 °C to 2.1 wt% at 1000 °C (assessed by inductively coupled plasma/mass spectrometry (ICP-MS)). A temperature of 1000 °C was identified as optimal for Se/N co-doped SAC preparation because materials prepared at 500 °C contained ZnSe nanoparticles, and those prepared at 900 °C contained Se clusters instead of single atoms. At 1000 °C Se formed –C–Se–C– bonds. The extended X-ray absorption fine structure (EXAFS) spectra confirmed the existence of Se–C bonds, while no Se–Se and Se–O bonds were detected. The obtained SAC was then tested as an ORR catalyst. This same synthetic procedure (i.e., pyrolytic reduction of SeO2 by ZIF-8-derived carbon) was also used to prepare a Se-based SAC for hydrogen peroxide sensing.47
An interesting example of a Se co-doped carbocatalyst based on ZIF-8 is an electrocatalyst with atomically dispersed Fe–Se atom pairs supported on N-doped carbon.48 To produce this Se-, Fe- and N-ternary doped carbon material, a Fe-doped ZIF-8 powder was first obtained and then mixed with selenium powder and pyrolyzed at 950 °C (Fig. 3). As a result, a carbonaceous material with Fe content of 2.06 wt% (ICP-OES) and SSA of 811 m2 g−1 was obtained. Soft X-ray absorption near edge structure (XANES) analysis was conducted to elucidate the microenvironment of C and N elements in the Fe/Se/N co-doped carbon. It was concluded that the Fe ion is coordinated with three pyridinic nitrogen atoms and one Se atom in a selenofenic configuration (Fig. 4). The carbon-based material was then scrutinized as a bifunctional oxygen electrocatalyst.
 | ||
| Fig. 3 (a) Scheme of the synthesis of catalysts with atomically dispersed Fe–Se atom pairs supported on N-doped carbon. (b) HAADF-STEM, (c) HRTEM, (d) HAADF-STEM, and (e–i) EDS mapping images of this material. Reproduced with permission from ref. 48. © 2023 Wiley-VCH GmbH. | ||
 | ||
| Fig. 4 Schematic representation of graphenic carbon containing an iron ion coordinated with three pyridinic N atoms and one selenofenic Se atom.48 | ||
A similar example of a dual heteroatom catalyst was inspired by natural metalloenzymes containing Cu–Fe heteroatom sites.49 A carbon-based material that comprises adjacent Cu–N4 and Se–C3 sites (Fig. 5) was produced using ZIF-8 as a support. In this material, a single Cu ion is coordinated by four pyridinic nitrogen atoms and the neighboring Se is covalently bonded by three carbon atoms. It was hypothesized that in this configuration, selenium acts as an electron-withdrawing modulator and thus polarizes electron distribution around Cu–N4 sites and facilitates oxygen reduction reactions.
 | ||
| Fig. 5 Schematic representation of graphenic carbon containing adjacent Cu–N4 and Se–C3 sites.49 | ||
To produce this Cu–Se dual atom sites material, a Cu precursor (Cu(NO3)2·3H2O) was infused into ZIF-8 and then pyrolyzed with diphenyl diselenide at 950 °C (Fig. 6). If DDSe is not added, then a material only doped with Cu/N is obtained and can be used as a reference to study the impact of Se co-doping. The contents of Cu and Se were 1.30 and 0.80 at% (EDS) and 2.5 and 1.7 wt% (ICP-OES), respectively. The N-doping content was 5.2 at% (EDS). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images revealed homogeneous distribution of Cu–Se pairs throughout the carbon matrix (marked by circles in Fig. 6).49 The distance between adjacent Cu and Se atoms was estimated at 0.32 nm. X-ray absorption spectroscopy (XAS) analysis was utilized to decode the atomic structure of the Cu–Se pair configuration in the N-doped material. The local structural parameters of the Cu/Se/N ternary-doped carbon material were extracted by EXAFS fitting, which indicated that the structure of Se sites is in a Se–C3 configuration instead of SeC2 and/or SeC4 configurations.
 | ||
| Fig. 6 (a) Scheme of Cu–Se and Cu-doped materials’ syntheses. (b) TEM image of Cu–Se material. The inset is the selected area electron diffraction (SAED) pattern of Cu–Se material, which exhibited the poor crystallinity of the carbon frame. EDS images of (c) Cu–Se and (d) Cu counterpart material. HAADF-STEM image of (e) Cu–Se and (f) Cu counterpart. (g) The intensity profiles obtained in Region 1 and Region 2 in (e) and (f). Reproduced with permission from ref. 49. © 2023 Wiley-VCH GmbH. | ||
Another prominent example of a nature-inspired carbon-based functional material is the Mo–Se dual-atomic site catalyst (DASC is an extension of the single-atom catalysts concept) inspired by the Mo-dependent formate dehydrogenases capable of CO2 activation (where Mo is coordinated by a selenocysteine).50 Mo/Se/N co-doped carbon nanobelts were obtained in the following manner. First, bis(acetylacetonato)dioxomolybdenum(VI) (MoO2(acac)2) and ZnSe[DETA]0.5 (obtained from diethylenetriamine, Na2SeO3 and zinc acetate) were dispersed in an aqueous solution of 2-methylimidazole. This yielded MoO2(acac)2 trapped in ZIF-8 (i.e., MoO2(acac)2@ZIF-8) on the surface of ZnSe[DETA]0.5 through a ligand-exchange reaction between 2-methylimidazole and diethylenetriamine. By pyrolyzing this material at 920 °C, the Mo/Se/N ternary-doped carbon with single Mo atoms supported on the Se/N-doped matrix was obtained. The Mo and Se contents were 0.46 and 2.32 wt%, respectively (ICP-OES), and the carbon nanobelts exhibited SSA of ∼1079 m2 g−1. XAFS (comprising XANES and EXAFS) showed that a single Mo atom is coordinated by four N atoms (Mo–N4), and the single Se atom is coordinated by two C atoms (Se–C2). This material was evaluated as an electrocatalyst for CO2 reduction. By employing ZIF-8 as a starting template, an atomically dispersed Sb/Se/N tri-doped carbon was prepared.51 First, N-doped carbon and SeO2 were pyrolyzed at 1000 °C. Then, SbCl3 was added to the Se/N co-doped carbon, and the Sb/Se/N ternary-doped carbon was obtained upon a second pyrolysis at 950 °C. Significantly, the Sb/Se/N-doped carbon possessed the largest SSA (1738 m2 g−1) in comparison with Sb/N (1188 m2 g−1) and Se/N (1251 m2 g−1) co-doped carbons. The Sb and Se loadings of Sb/Se/N co-doped carbon were 0.37 and 3.92 wt% (ICP-OES), respectively. The XAS analysis indicated that the Sb single atom is coordinated into the SbNxC4−x structure, and the Se atom is coordinated with two carbon atoms. In another approach, a DASC with Co–Nx and Se–C dual sites was prepared by pyrolyzing (950 °C) a mixture of ZIF-derived Co–N–C carbonaceous material and SeO2.52
Besides Zn-containing ZIF-8, cobalt-based ZIF-67 (i.e., 2-methylimidazole cobalt salt) was utilized to produce selenium-containing carbon-based materials.53 ZIF-67 particles were first deposited on a pomelo peel and were then converted into Se-containing carbonaceous material by high-temperature selenization with Se powder (1000 °C). N and Se doping in the form of N–C and Se–C bonding was achieved. The prepared material retained the original 3D structure of the utilized pomelo peel and exhibited an SBET of 326 m2 g−1. Consequently, a free-standing 3D micro–mesoporous material with Co and CoSe (nano)particles supported on N/Se co-doped biomass-derived carbon was produced. A very interesting example is the utilization of a zinc-hexamethylenetetramine (Zn-HMT) framework to produce micro–mesoporous Se/N co-doped carbon nanosheets with 10 at% of N and 2 at% of Se and SSA of 376 m2 g−1 by an oxidation-selenization process.54 The synthesis and structure of the N/Se co-doped material are schematically illustrated in Fig. 7. First, N-doped carbon was prepared from Zn-HMT by carbonization at 900 °C. Secondly, the obtained carbon was partially oxidized in nitric acid to graft oxygen functional groups. The oxygen functional groups played an essential role during the selenization process. They were thermally desorbed at high annealing temperatures and substituted by the Se atoms. The selenization was performed at 600 °C by using Se powder. The final Se/N co-doped carbon was dispersed in CS2 to remove elemental Se not bonded with the carbon scaffold (in many synthetic approaches to Se-doped carbons, CS2 is used to remove unreacted Se). XPS indicated that Se atoms are covalently incorporated into the carbon framework to form C–Se–C bonds. Additionally, supposedly no N–Se bonds were formed. Based on the X-ray diffraction (XRD) analysis, the authors concluded that the Se atoms enlarge the interlayer distance of carbon nanosheets. However, this is a somewhat controversial claim as the obtained carbonaceous materials were highly disordered, and the broadening and shift of the (002) reflection in the XRD patterns can be caused by many highly diverse factors.
 | ||
| Fig. 7 (a) Scheme of N/Se co-doped carbon material preparation; (b) SEM, (c) AFM, (d) TEM, and (e) HRTEM images; (f) the HAADF-STEM image of a typical N/Se co-doped nanosheet and (i–iv) the corresponding elemental mapping. The lower part of the figure presents a schematic model of N and Se dopants in the N/Se co-doped nanosheets. Adapted with permission from ref. 54. © 2021 Elsevier B.V. | ||
4.3. Se-doped carbon materials based on synthetic polymers
Biomass and fossil fuels are considered the main feedstock for carbonaceous material production. However, unlike substrates of natural origin, synthetic polymers yield high-purity, tunable chemical compositions, and a variety of macromolecular and self-assembled (nano)structures.55 Synthetic polymers offer many possible morphologies, including spheres, fibers, foams, monoliths, films, and membranes. Moreover, the utilization of synthetic polymers as feedstock for new functional carbon materials might diminish the problem of plastic waste.Polyacrylonitrile (PAN, (CH2CH–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)n), a thermoplastic polymer, is commonly used to obtain N-doped carbons because it provides a high level of N-doping, high carbonization yield and it is additionally a graphitizable precursor yielding carbons of ordered crystalline structure. For these reasons, PAN is the reagent of choice to produce commercial carbon fibers. PAN has been utilized to produce Se and N co-doped carbon nanofibers with hierarchical porous structure as a free-standing cathode for Li–Se batteries.56 PAN and polymethylmethacrylate (PMMA) were utilized as N-doped carbon source and pore-forming agent, respectively. PAN-PMMA nanofiber films were produced by electrospinning followed by pre-oxidation at 280 °C. The pre-oxidation stage is routinely used to prepare carbon fibers from PAN. During the pre-oxidation, PMMA decomposes, forming a porous structure within the nanofiber film. Next, Se powder was deposited on the pre-oxidized PAN fabric and annealed at 600 °C. During annealing, Se vapor reacts with unsaturated bonds of the pre-oxidized scaffold, creating Se–C covalent bonds. However, the exceptionally high Se content of 33 wt% (assessed by thermogravimetric analysis) shows that the obtained material was, in fact, Se-impregnated carbon rather than Se-doped carbon. The final material was a composite of N-doped carbon fibers impregnated with amorphous Se, where part of the Se was covalently bonded to the carbon support. Utilizing PAN, ternary Se, S, and N co-doped carbon materials were also produced.57 To obtain simultaneous Se and S doping, selenium sulfide was used as a selenization and sulfurization precursor. The ternary doped carbon with N, S, and Se contents of 4.9, 1.31, and 0.33 at% (XPS), respectively, was produced by pyrolysis at 1000 °C. A high SBET of 827 m2 g−1 and large pore volume of nearly 1.0 cm3 g−1 were created due to vulcanization processes accompanied by the release of H2S and H2Se vapors. Both S and Se were doped into the carbon matrix, creating C–S and C–Se covalent bonds. The material was evaluated as an anode for alkali metal-ion batteries.
N)n), a thermoplastic polymer, is commonly used to obtain N-doped carbons because it provides a high level of N-doping, high carbonization yield and it is additionally a graphitizable precursor yielding carbons of ordered crystalline structure. For these reasons, PAN is the reagent of choice to produce commercial carbon fibers. PAN has been utilized to produce Se and N co-doped carbon nanofibers with hierarchical porous structure as a free-standing cathode for Li–Se batteries.56 PAN and polymethylmethacrylate (PMMA) were utilized as N-doped carbon source and pore-forming agent, respectively. PAN-PMMA nanofiber films were produced by electrospinning followed by pre-oxidation at 280 °C. The pre-oxidation stage is routinely used to prepare carbon fibers from PAN. During the pre-oxidation, PMMA decomposes, forming a porous structure within the nanofiber film. Next, Se powder was deposited on the pre-oxidized PAN fabric and annealed at 600 °C. During annealing, Se vapor reacts with unsaturated bonds of the pre-oxidized scaffold, creating Se–C covalent bonds. However, the exceptionally high Se content of 33 wt% (assessed by thermogravimetric analysis) shows that the obtained material was, in fact, Se-impregnated carbon rather than Se-doped carbon. The final material was a composite of N-doped carbon fibers impregnated with amorphous Se, where part of the Se was covalently bonded to the carbon support. Utilizing PAN, ternary Se, S, and N co-doped carbon materials were also produced.57 To obtain simultaneous Se and S doping, selenium sulfide was used as a selenization and sulfurization precursor. The ternary doped carbon with N, S, and Se contents of 4.9, 1.31, and 0.33 at% (XPS), respectively, was produced by pyrolysis at 1000 °C. A high SBET of 827 m2 g−1 and large pore volume of nearly 1.0 cm3 g−1 were created due to vulcanization processes accompanied by the release of H2S and H2Se vapors. Both S and Se were doped into the carbon matrix, creating C–S and C–Se covalent bonds. The material was evaluated as an anode for alkali metal-ion batteries.
Next to PAN, polypyrrole (PPy) is also a popular synthetic polymer used to produce N-doped carbonaceous materials. A Se and N co-doped carbon was prepared by annealing a mixture of benzyl diselenide and N-doped carbon derived from PPy.58 Upon annealing at 900 °C, a carbon material of SBET = 619 m2 g−1 and Se and N content of 1.53 and 3.98 at% (XPS), respectively, was obtained. Various configurations of Se and N doping were considered, including those where Se and N atoms substitute carbon atoms within the graphene layer. In such cases, the Se dopant protrudes from the hexagonal planar structure due to its large size. In a similar approach, the PPy and Se powder mixture was pyrolyzed at relatively low temperatures of 350–550 °C to produce a Se/N-doped char.59 XPS indicated that Se was doped mainly as a C–Se–C structural motif and that Se doping promotes the transformation of pyrrole N into graphitic (i.e., tertiary) and pyridinic N. However, the very high Se content (∼27 wt% (EDS)) and low pyrolysis temperature may suggest that some portion of Se was present as amorphous selenium impregnated into the porous structure of the charred PPy.
A N/Se co-doped, ordered, macroporous carbon with an inverse opal structure was prepared by employing polydopamine-coated polystyrene spheres, where the spheres act as a macropore (100 nm) template.60 A mixture of coated polystyrene spheres and SeO2 powder was subjected to pyrolysis in the 600–1000 °C temperature range. The scheme of the synthesis and the structure and chemical composition of the N/Se co-doped carbon obtained at 900 °C are presented in Fig. 8. This material was scrutinized as a metal-free catalyst for photovoltaic devices as it exhibited the highest surface area of 1066 m2 g−1 and the largest pore volume of 0.72 cm3 g−1 in comparison with counterparts prepared at other pyrolysis temperatures (i.e., 600, 700, 800 and 1000 °C). This carbon contained 1.95 at% of N and 0.53 at% of Se (XPS). C–Se bonds in selenophene configurations were identified. A synchrotron XAS analysis was applied to decode the chemical structure of N and Se doping. Se K-edge hard XANES spectroscopy revealed the valence state of Se atoms in the carbon framework – they are positively charged, and their valence state is between 0 and +4 (ca. +1.7).
 | ||
| Fig. 8 Scheme of the N/Se co-doped ordered porous carbon synthesis and TEM images (a–c) and elemental mappings (d) of N/Se co-doped carbon prepared at 900 °C. Adapted with permission from ref. 60. Copyright © 2023, American Chemical Society. | ||
A series of Se-doped microporous carbon gels was obtained via polycondensation of resorcinol and 2-formylselenophene followed by pyrolysis at 600–1200 °C.42 A schematic representation of the synthesis is depicted in Fig. 9. Pyrolysis at temperatures >800 °C resulted in a sharp decrease in the Se-content (from 2 at% for 600 °C to 0.6 at% for 1200 °C) accompanied by a significant increase in microporosity (corresponding SBET increase from 675 to 940 m2 g−1). This extensive enhancement of microporosity is caused by thermal desorption of chalcogens such as sulfur and selenium from the doped carbonaceous materials via corresponding chalcogen and dichalcogenide vapors (S2, Se2, CS2, CSe2). In fact, even if commercial carbon black was annealed with selenium compounds at 1000 °C, its surface area increased from 1567 to 1762 m2 g−1, confirming the activating properties of heavy chalcogens.61
 | ||
| Fig. 9 Scheme of the polycondensation reaction of resorcinol and 2-formylselenophene to yield a cross-linked resin and then a Se-doped microporous carbon upon pyrolysis. Adapted with permission from ref. 42. © 2018 Elsevier Inc. | ||
Besides common polymers, polymerized ionic liquids (ILs) were also employed to produce Se-doped carbonaceous solids. Using such atypical reagents, hierarchically porous N-doped carbon membranes containing atomically dispersed Se were produced.62Fig. 10a–e depicts the synthesis and structure of the intermediates and the final Se/N co-doped carbonaceous material. KSeCN was utilized to prepare one of the ILs bearing SeCN− anion (poly(1-carboxymethyl-3-vinylimidazolium)selenocyanate). A mixture of two polymerized ILs was dried as a thin layer and immersed in an aqueous NH3 solution to create a porous membrane (Fig. 10b and c). Vacuum pyrolysis at different temperatures (800, 900, and 1000 °C) yielded the final Se/N co-doped carbon membranes of several cm2 in size (Fig. 10f). A cross-section SEM image of the carbonaceous material revealed a 3D interconnected porous system (Fig. 10g). STEM-HAADF analysis showed homogeneously distributed Se atoms, which occur exclusively in a single-atom state. As pyrolysis temperature was increased from 800 to 1000 °C, the Se content dropped from 5.90 to 3.23 wt% (as revealed by XPS). Simultaneously, the SBET and total pore volume values grew from 258 to 450 m2 g−1 and from 0.13 to 0.17 m3 g−1 (for 800 vs. 1000 °C). The increase in porosity was due to the development of micropores. These results are in line with our previous observations that pyrolysis of Se-doped carbons at temperatures >800 °C enhances microporosity due to the thermal desorption of chalcogens.42 According to XAS analysis, Se atoms were located at the edges of graphenic domains in hexatomic carbon ring structures in the form of selenabenzene configurations (i.e., a heterobenzene in which a Se atom replaces a C atom of the benzene ring). It was additionally concluded that in such a chemical state, the outermost orbitals of Se overlap with C atoms, which introduces a positive charge on Se atoms. Consequently, the doped Se atoms carry a positive charge with a valence state between 0 and +4.
 | ||
| Fig. 10 (a)–(e) Scheme of the Se/N co-doped carbon preparation and structures of the intermediates. (f)–(h) A photograph, a cross-section SEM image, and the HRTEM image of Se/N co-doped carbon upon pyrolysis at 1000 °C, respectively. (i)–(l) STEM image and the corresponding elemental mapping. Reproduced with permission from ref. 62. © 2019 Wiley-VCH GmbH. | ||
A specific polymerization/surface grafting technique was designed to produce templated Se-doped macroporous carbons.63 Chloromethylated SiO2 nanoparticles (SiO2–CH2Cl serving as a template) were surface-coated with DDSe by a Friedel–Crafts reaction utilizing dimethoxymethane as a cross-linker (Fig. 11). This material was pyrolyzed at relatively low temperatures of only 300, 500, or 700 °C. The Se content decreased with increases in temperature, from 37 wt% for 300 °C to 10 wt% for 700 °C (XPS). This was accompanied by a gradual increase in microporosity and hence surface area, which reached 557 m2 g−1 upon HTT of 700 °C. It is interesting to note that the carbon produced at 700 °C contained as much as 13 wt% of oxygen. The Se-doped carbon was studied as a sodium-ion battery (SIB) anode.
 | ||
| Fig. 11 Schematic illustration for the preparation of Se-doped templated porous carbon. Reproduced with permission from ref. 63. Copyright 2023, The Royal Society of Chemistry. | ||
A nitrogen-rich 1,3,5-triformylbenzene-tris(4-aminophenyl)benzene polymer was obtained via polymerization of a tris(4-aminophenyl)amine and a triformaldehyde and doped with iron and sulfur during carbonization.64 Then, annealing with Se powder produced Fe, S, Se, and N co-doped carbon. The Se doping enhanced microporosity yielding material of the highest SSA (i.e., 637 m2 g−1) in respect to the counterparts without Se. The multi-doped carbon contained 0.98% of Se and as much as 5.56% of O (at%, XPS). The multi-doped carbon exhibited enhanced catalytic activity as electrocatalyst in both a liquid and flexible zinc–air batteries (ZABs).
4.4. Se-doped carbon materials via mechanochemistry
Solid-state reactions permitted or sustained by the mechanical force of milling or grinding (i.e., mechanochemistry) have been recently rediscovered due to the search for cleaner synthetic methodologies.65 Ball-milling is the most common technique used in the field of mechanochemistry because it assures an enclosed reaction environment with well-defined synthesis parameters. No wonder mechanochemistry conquered the field of new functional materials synthesis. As could be expected, it also entered the field of heteroatom-doped carbons. By milling natural graphite and selenium powder, edge-selenated graphene nanoplatelets were prepared (Fig. 12).66 After ball-milling, the product was washed with CS2 and aqueous KOH solution to remove unreacted Se. TEM analysis revealed that Se is doped into the final material as single atoms in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Se and C–Se–C configurations at the edges of the graphene nanoplatelets. Theoretical considerations hinted that the Se atom binds to the graphene edge as a single-coordinated Se-dopant (>CSe). A Se-doped carbonaceous material with 5.57 at% of Se (EDS) and SSA of 106 m2 g−1 was created using this approach. It is essential to recall here that besides the desired dopants such as Se or N, carbonaceous materials always contain oxygen. The obtained Se-doped graphene nanoplatelets indeed contained ca. 10 wt% of oxygen (as determined by elemental analysis).
Se and C–Se–C configurations at the edges of the graphene nanoplatelets. Theoretical considerations hinted that the Se atom binds to the graphene edge as a single-coordinated Se-dopant (>CSe). A Se-doped carbonaceous material with 5.57 at% of Se (EDS) and SSA of 106 m2 g−1 was created using this approach. It is essential to recall here that besides the desired dopants such as Se or N, carbonaceous materials always contain oxygen. The obtained Se-doped graphene nanoplatelets indeed contained ca. 10 wt% of oxygen (as determined by elemental analysis).
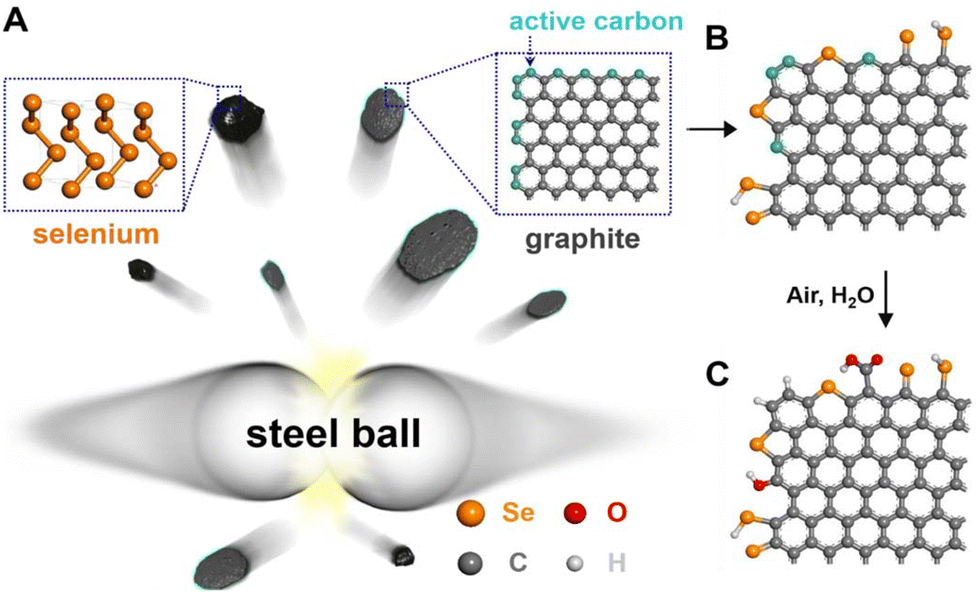 | ||
| Fig. 12 Schematic representation of ball-milling the mixture of graphite and Se powder to produce edge-selenated graphene nanoplatelets. Reproduced with permission from ref. 66. Copyright © 2016, The Authors. | ||
Similarly, Se-doped graphene was produced by ball-milling a graphene oxide (synthesized by Hummers’ method) and selenium powder, followed by high-temperature annealing in the 800–1000 °C temperature range.67 Depending on the annealing temperature, the final Se-doped graphenes contained 2.19, 1.73, and 1.65 at% of Se for 800, 900, and 1000 °C, respectively (XPS). One must note, however, that if ball-milling is followed by additional high-temperature treatment, then the final carbon structure is produced via thermally induced reactions rather than mechanochemistry. In such cases, ball-milling is applied to obtain highly homogeneous mixtures. Ball-milling followed by pyrolysis was also employed to prepare a whole family of metal-free Se/B/P/N tetra-doped carbon catalysts.68 Sucrose, selenium powder, tetraphenylphosphonium tetraphenylborate (C48H40BP), and NH4Cl were utilized as substrates and sources of heteroatoms. The quaternary-doped carbon produced upon pyrolysis at 1050 °C possessed an SBET of 1024 m2 g−1 and was further scrutinized for electroreduction of CO2.
4.5. Se-doped carbon materials from fine chemicals via the Edisonian approach
Laboratory-scale synthesis of new functional materials, including carbon-based heterogeneous catalysts, usually employs fine chemicals of high purity and hence high cost. This is justified because the key aspect of fundamental research is to establish the basic principles of synthesis-properties-performance of new materials while the production cost is less relevant. Consequently, many researchers in the field of carbon functional materials exercise the Edisonian approach, i.e., the trial-and-error method to design materials with new properties. This method usually comprises optimization via mixing diverse fine chemicals and pyrolyzing them at varied conditions. Recently, this approach is often accompanied by systematic theoretical calculations. Exploiting this approach, many different Se/Fe/N ternary-doped carbon catalysts have been produced.69–71 A Fe/Se dual-atomic-site catalyst was prepared from a mixture of melamine, L-alanine, SeO2, and iron trichloride via pyrolysis at 900 °C (Fig. 13).69 The catalytic material, denoted as Fe1Se1-NC, contained 1.8% of Fe (measured by ICP-OES) and exhibited an SSA of 422 m2 g−1. XAS, including XANES and EXAFS analyses, indicated the presence of iron in molecular unsymmetrical coordination Fe–N5 moiety (i.e., one Fe ion coordinated by five nitrogen atoms) with an adjacent selenium atom bonded to two carbon atoms (Fig. 13). | ||
| Fig. 13 Scheme of the DASCs preparation and local electronic states of the catalysts. (A), Se K-edge Fourier-transformed EXAFS spectra of Fe1Se1-NC (inset shows the XPS spectra of Se 3d). (B), Fitting curves of the EXAFS of Se SACs of Fe1Se1-NC in the R-space and K-space (inset of B). (C), The experimental Fe K-edge XANES spectra of Fe1Se1-NC, Fe1-NC (catalyst without Se-doping), and the references (Fe foil and Fe2O3). (D), Fe K-edge Fourier transformed EXAFS spectra in the R space. (E) and (F), Fitting curves of the EXAFS of Fe SACs of Fe1Se1-NC in the R-space and K-space. Adapted with permission from ref. 69. © 2022 Elsevier B.V. | ||
Some research suggests that Se-doping can modulate the type of chemical configuration of nitrogen doping. Se doping may enhance the total N content and preferential formation of pyridinic N sites.71 Such a phenomenon was reported for a catalyst consisting of iron carbide nanoparticles encapsulated in Se/N co-doped CNTs. To obtain this material, a mixture of FeSO4, elemental selenium, and melamine was pyrolyzed at 800 °C. The Se doping reduces the Fe3C particle size, improves their homogeneous dispersion, and induces a low oxidation state of Fe sites in the Se/N-CNT-Fe3C nanocomposite. The material was characterized by SSA of 525 m2 g−1 and Se- and N-contents of 0.43 and 9.0 at% (XPS), respectively.
Molten salts are often utilized to prepare porous carbonaceous materials. The addition of salts improves the formation of a nanoporous structure. In fact, NH4Cl is often utilized in the pyrolytic synthesis of carbons as a porogenic agent. In an interesting approach, a mixture of KCl/LiCl, glucose, and Na2SeO3 was carbonized at 700 °C to produce Se-doped carbon with a 3D framework and a hierarchical porosity (SSA of 446 m2 g−1).72 The interlayer spacing of graphenic layers in the produced hard carbon was 0.394 nm. The authors interpreted this as a consequence of the large Se atom size that expands the spacing. This interpretation is quite common in articles concerning Se-doped carbonaceous materials. In another approach, pyrolysis of Se and 1,4,5,8-naphthalenetetracarboxylic dianhydride at 600 °C yielded Se-doped carbon with 8.5 wt% Se content (EDX) and SBET = 117.5 m2 g−1.73 Se was doped in the carbon frameworks via C–Se bonds, and the doping generated extensive mesoporosity.
4.6. Se-doped carbon materials based on biochar
In contrast to fossil fuels, biomass is characterized by renewability and low cost due to sustainable and extensive availability and accessibility. Consequently, biomass became one of the main feedstocks for new functional carbon material preparation.74 Biomass is divided into plant and animal biomass. Plant biomass is mainly composed of cellulose, hemicellulose, and lignin, while animal biomass is composed of proteins, polysaccharides (e.g., chitosan and chitin), and minerals (Fig. 14). | ||
| Fig. 14 Typical animal and plant biomass precursors for biomass-derived carbonaceous material preparation. Reproduced with permission from ref. 74. © 2023 The Authors. Advanced Science published by Wiley-VCH GmbH. | ||
Yu's research group proposed a whole family of Se-containing carbon-based materials as catalysts (for the transfer of oxygen from oxidants to the desired products), taking advantage of the excellent oxygen-carrier feature of Se.75–78 Reduction of selenium with NaBH4 in ethanol generates NaHSe in situ, which can be applied for selenization of carbohydrates and biomass. NaHSe is a strong selenization agent used to replace some of the oxygen in carbohydrates to produce their selenized counterparts. Pyrolysis of the selenized carbohydrates at 500–550 °C yields Se-doped carbons (chars designed as Se/C), which can be successfully synthesized on a kilogram scale (Fig. 15). The final Se/C catalysts possess a highly disordered carbonaceous structure. Utilizing NaHSe selenization of various carbohydrates and biomass (e.g., glucose, fructose, sucrose, maltose, potato starch, cotton, and straw), a whole range of carbonaceous materials was prepared via pyrolysis at ∼500 °C and then probed for heavy metal removal from wastewater.79
 | ||
| Fig. 15 Fabrication of the Se/C carbonaceous catalysts via glucose selenization and pyrolysis. Adapted with permission from ref. 75. Copyright 2018, The Royal Society of Chemistry. | ||
Se and N co-doped carbon nanosheets with a micro-, meso-, macroporous hierarchical structure, SBET of up to 645 m2 g−1 and up to 1.31 wt% Se content (determined by ICP-MS) were obtained by pyrolyzing SeO2, chitosan, and NH4Cl at 1000 °C (Fig. 16).80 XPS revealed the presence of C–Se–C units. The XANES spectrum indicated that SeO2 was transformed by pyrolysis into a low valence state Se (∼+1.5). The EXAFS and soft XANES indicated that the Se atom is located at the graphene edge within the 6-membered ring. The atomic structure of the Se/N co-doped nanosheets comprises –Se–C–N– sites on the edge of graphene sheets (Fig. 16). Significantly, the proposed route could be applied to obtain other chalcogen-doped carbon nanosheets, such as Te- and Se/Te co-doped carbons.
 | ||
| Fig. 16 Scheme of the Se/N co-doped nanosheets preparation and their molecular structure. The Se atom occupies the edge on a 6-membered ring of graphene. XPS spectra of (a) Se 3d and (b) N 1s. (c) Se K-edge XANES spectra and fitted average oxidation states of Se in the insert. (d) Se K-edge EXAFS spectra. EXAFS fitting curves in (e) R space and (f) q space. (g) Wavelet transform of Se/N co-doped nanosheets (top) and Se-foil (bottom). (h) N K-edge and (i) C K-edge XANES spectra. (j) Atomic structure of Se/N co-doped nanosheets (Se red, N blue, C black). Adapted with permission from ref. 80. © 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Tree bark was also used to produce Se-doped mesoporous biochar.81 Spruce bark and SeO2 were hydrothermally treated with H3PO4 at 150 °C. Next, the material was subjected to activation with CO2 at 800 °C, producing Se-doped biochar with SSA of 1300 m2 g−1 and 2% of Se (EDS). In another study, plant leaves were utilized to prepare Se/N co-doped biochar as a metal-free catalyst for aldehyde oxidation.82 A remarkably interesting approach to Se-doped biochar was proposed by Shi et al.83 They took advantage of the biocompatibility and bioactivity of a selenium element and employed a Se-reducing bacterium (Bacillus megaterium) and NH4Cl (nitrogen precursor) to synthesize Se/N co-doped biochar (Fig. 17). The bacteria accumulated Se nanoparticles and were used as the carbon and Se source. The bacteria/NH4Cl mixture was freeze-dried and pyrolyzed at 900 °C. Se was doped in the carbon framework as atomically dispersed C–Se–C configurations. The Se/N co-doped biochar possessed SSA of 287 m2 g−1 and N and Se doping levels of 5.51 and 1.79 wt% (combustion elemental analysis and ICP-MS). Significantly, it was suggested that the Se-doping facilitates the formation of graphitic N, which was the dominant nitrogen component (55.6%). The Se/N co-doped biochar was studied as a metal-free catalyst for the oxidative removal of pollutants (Fig. 17).
 | ||
| Fig. 17 The proposed mechanism of the Se/N co-doped biochar synthesis and catalytic action. Reproduced with permission from ref. 83. © 2021 Published by Elsevier B.V. | ||
4.7. Selenium/carbon composites and Se-doped carbon quantum dots
This review report focuses on Se-doped carbonaceous materials where Se is bound covalently to the graphenic framework in a well-defined manner. However, there is a whole spectrum of carbonaceous materials where part of the Se can be doped as single atoms infused into the graphenic structure, while some part is only chemically grafted on the surface or physisorbed and a further part is just simply impregnated into pores. It is well documented that heteroatoms and heteroaromatic functional groups on the carbon surface efficiently stabilize supported metal particles.84 The surface of carbonaceous materials can be modified with selenium through non-covalent interactions, and such modified carbons can be utilized as support for noble metal catalytic nanoparticles. For instance, Vulcan XC-72 was functionalized with triphenylphosphine selenide non-covalently coupled to the surface of the oxidized carbon black.85,86 Such Se-functionalized carbon black was utilized as support to achieve high dispersion of PtRu or PtSn NPs with narrow size distribution. Nemati et al. utilized Se/N dual-doped carbons as support for Ag and Pd NPs.87,88 Besides surface functionalization, there is an extensive number of examples of selenium/carbon composites where Se occurs in its elemental form. Porous carbons can bear large amounts of Se, while Se has the advantages of electric (semi)conductivity and high stability in acidic solutions. Se-impregnated porous carbons are studied as sorbents for heavy metals,89 in Na–Se and K–Se batteries90,91 and as new advanced synthetic enzymes.92Some Se-doped carbon-based materials have been prepared via the hydrothermal method.93 In fact, hydrothermal treatment at relatively low temperatures of ca. 60–200 °C is the method of choice to prepare Se-doped carbon quantum dots (CQDs). Heteroatom-doped CQDs are characterized by a size of less than 10 nm and exhibit biocompatibility and low/no cytotoxicity.94–96 They have been proposed for applications concerning bioimaging/diagnosis, sensing, disease monitoring, and catalysis. Heteroatom doping emerged as an effective way to adjust the fluorescent properties of CQDs. One must, however, note that CQDs are a very different class of material with respect to porous carbonaceous materials. First, their unique size determines their peculiar characteristics. Secondly, they are produced at very low temperatures with respect to traditional carbonization processes, and hence the chemical structure of CQDs is different from graphenic carbons obtained by pyrolysis. Se-doped CQDs have been routinely prepared by hydrothermal treatment of selenocysteine at temperatures as low as 60 °C.97–99 The resultant water-soluble dots exhibited good free radical-scavenging capability to limit harmful levels of ROS in cells. Consequently, as Se–CQDs have strong antioxidant properties, they can be used as therapeutics for ameliorating secondary injuries. Interestingly, Se–CQDs have been proposed even as a fertilizer for improving Se delivery to plants.100 Nevertheless, due to their unique size and structure, heteroatom-doped CQDs are beyond the scope of this review, and readers are referred to excellent review papers devoted exclusively to this specific topic.94–96
5. Applications of Se-doped carbon materials
5.1. Se-doped carbon materials as electrocatalysts
N-doped carbon materials were the first metal-free, carbon-based electrocatalysts.101 The origin of their (electro)catalytic activity is usually elucidated by the difference in N and C electronegativity, which disrupts the electroneutrality of graphenic materials. This, in turn, creates charged sites for the adsorption and activation of small molecules such as oxygen, hydrogen, or carbon dioxide. In the case of selenium doping, the origin of (electro)catalytic activity must differ. Se and carbon exhibit the same electronegativity (2.55 in the Pauling scale), and therefore the huge distortion in C–C bond lengths upon Se doping, as well as selenium soft nucleophilicity, might be the reasons for enhanced catalytic activity.Metal–air batteries combine the advantages of conventional batteries and fuel cells. In fact, catalysts active in ORR for alkaline fuel cells can be successfully applied in zinc–air batteries. Aqueous (e.g., Zn–air, Al–air, and Mg–air) and nonaqueous (e.g., Li–air, Na–air, and K–air) metal–air batteries exhibit a theoretical energy density of about 3–30 times greater than that of lithium-ion batteries (LIBs).104 Nevertheless, while non-precious metal and metal-free carbocatalysts are indeed very competitive catalysts of ORR in alkaline media, they are still somewhat less active in acidic solutions when compared with Pt.
Huang et al. are pioneers in the synthesis of Se-doped carbon-based electrocatalysts.37,38 They proposed Se-doped rGO and Se-doped rGO/CNT composites (containing selenophene-like structures) as metal-free ORR catalysts in an alkaline environment.37,38 They considered that the modified spin density is the dominant factor regulating ORR activity in the Se-doped carbon catalysts. In the KOH solution, the Se-doped rGO/CNT catalyst showed catalytic activity comparable to commercial Pt/C catalysts, and the ORR occurred via a 4e− pathway. It was hypothesized that due to its large size, the Se dopant causes high strains, which may reshape charge localization and thus facilitate chemisorption of the O2 molecule on the catalyst surface. In addition, Se has high polarizability, and the valence lone electron pairs of selenium may interact with oxygen facilitating its reduction. The obtained Se-doped carbon containing only ca. 1 wt% of Se possessed activity comparable to that of N-doped carbon with up to 8 wt% of nitrogen38 Similarly, N-doped rGO–CNT was additionally co-doped with S and/or Se to prepare ORR catalysts with high activity in acidic solutions.40 Se-doping granted stronger ORR activity than S-doping, and in an acidic solution, the catalyst showed higher stability than the Pt/C catalyst. However, it must be noted that the Se-doped carbon catalyst was heavily contaminated with cobalt and iron because CoCl2 and FeCl2 were used during its synthesis. In fact, despite the acid leaching, the Se content was on the same level as the Co content and much lower than the Fe content (0.05 vs. 0.22 at%).40 Consequently, it is likely that the transition metal residues determined the catalyst activity. The problem of incidental metal impurities is indeed crucial because impurities may determine the activity of seemingly metal-free catalysts.105 It is especially concerning because many carbocatalysts are prepared from GO obtained via Hummers’ method, which involves transition metals. Similarly, metal-free catalysts derived from ZIF-8 can be heavily contaminated with Zn. For instance, Se-doped rGO prepared from GO was studied as a metal-free single-atom site catalyst of ORR in an alkaline medium.39 Unlike the Se-free rGO, the Se-rGO catalyst catalyzed the ORR via a direct 4e− pathway, and the Se atom linking two rGO sheets was identified as the catalytically active center (i.e., the O2 molecule is adsorbed on the Se site of the rGO–Se–rGO, where Se is a positive charge site in that C–Se–C configuration). However, the Se-doped rGO was contaminated with copper. Another example of a metal-free single Se atom catalyst for ORR is a ZIF-8-derived SAC, where Se single atoms are supported on N-doped carbon.46 In this Se/N co-doped single-atom catalyst (with Se content of ∼2 wt%), the Se atom served as an active site catalyzing the ORR via the direct 4e− pathway. The SAC showed activity and stability better than commercial Pt/C (in alkaline media). The catalyst was utilized as a cathodic material for an alkaline zinc-air battery, and its output power density outperformed that of the battery using a Pt/C cathode (176.9 vs. 115.4 mW cm−2). The battery also exhibited a high open circuit voltage (OCV) of 1.45 V. Fig. 18 depicts the electrochemical characteristics of the metal-free single Se atom catalysts prepared at various pyrolysis temperatures (500, 900, 1000 °C) and of the Zn–air battery with the catalyst prepared at 1000 °C and utilized as the cathodic material (with respect to a Pt/C commercial catalyst).
 | ||
| Fig. 18 (a) ORR curves at 1600 rpm in 0.1 M KOH. (b) Comparison of diffusion-limiting current (Jd) and half-wave potential (E1/2). (c) Tafel plots. (d) ORR curves of the Se/N co-doped catalyst. Inset are K–L plots. (e) Electron transfer number (n) and H2O2% of the S/N co-doped and Pt/C catalysts. (f) ORR curves before and after 5000 cycles. Inset is HAADF-STEM image after 5000 CV cycles. (g) Activity comparison of SAC Se/N catalyst with representative catalysts. (h) Discharge curves of Se/N doped carbon and Pt/C catalyst-based Zn–air batteries. The inset shows the LED screen. (i) Power density plots of the assembled Zn–air batteries. Reproduced with permission from ref. 46. © 2021 Wiley-VCH GmbH. | ||
A density functional theory (DFT) analysis with proposed models of the Se/N co-doped catalyst was performed to deconvolute the role of selenium doping in boosting the ORR catalytic activity (Fig. 19a). The doped Se atom induces electron delocalization (Fig. 19b), and this can modulate the adsorption energy of the ORR intermediates (e.g., OOH*, O*, and OH*). The obtained Gibbs free energy diagram (Fig. 19c) indicated that the rate-determination step of the ORR catalyzed by this material is the transition from O* to OH* at U = 0 V with the energy barrier (ΔG) of 0.23 eV (for a 4e− ORR pathway in an alkaline environment). It was additionally demonstrated that catalysts with single-atom Se sites outperformed the corresponding N-doped carbon without Se and a material containing selenium clusters instead of single atoms (Fig. 18a and 19c). Consequently, it was concluded that selenium single atoms with modulated local electronic configurations boost the ORR catalytic activity.
 | ||
| Fig. 19 (a) The optimized structures of the Se/N co-doped SAC and after adsorption of OOH*, O*, and OH* species. (b) Differential charge density for the SAC catalyst. Yellow and blue regions represent electron accumulation and depletion, respectively. (c) Free energy diagram of the N-doped carbon, Se cluster, and single atom Se/N co-doped SAC at U = 0 V. Reproduced with permission from ref. 46. © 2021 Wiley-VCH GmbH. | ||
One of the drawbacks of a Zn–air battery is the deposition of zinc oxides on the cathode resulting in blockage of the active sites and the oxygen diffusion pathway, thus leading to cell degeneration. ZnO originates from the anodic discharge product (i.e., zincate ions Zn(OH)42−), and deposits itself on the air electrode upon supersaturation. Se-doped, carbon-based cathodes were proposed to alleviate this issue.58 Doping N-doped carbon nanosheets with Se produced significant electron cloud delocalization from the Se atom in the carbon framework, and this, in turn, blocked Zn(OH)42− access and helped to prevent ZnO poisoning of the air cathode during battery operation. This assured a long battery life, reaching 780 cycles. Se doping not only prevented cathode poisoning but also yielded high ORR activity exceeding that of the Pt/C catalyst. The Zn–air battery assembled with the Se-doped carbon cathode exhibited a high open-circuit potential of 1.49 V and a power density of 154.45 mW cm−2. In fact, the Se-doped carbon functioned as a bifunctional catalyst as it also showed high activity in the anodic oxygen evolution reaction (OER). DFT simulations showed that Se acts as an electron-withdrawing dopant in the carbon matrix, producing electron cloud delocalization around the doped area. Enhanced electron cloud density of the adjacent carbon atoms impedes Zn(OH)42− access to the cathode surface. Se-doping not only enhances the activity of the surrounding carbon atoms, but the Se atom also acts as an active site itself.
Se-doped carbocatalysts were also studied as cathode catalysts for nonaqueous Li–O2 batteries.61 The Se-doped carbon cathode based on commercial carbon black exhibited a discharge capacity of up to 3222 mA h g−1, much higher than the corresponding undoped carbon material, i.e., BP 2000 carbon black (1953 mA h g−1). Se-doped activated carbon prepared from paper cups and Se powder was proposed as a metal-free, bifunctional (ORR/OER) air cathode electrocatalyst for the Li–O2 battery.106 The Se-doped catalyst achieved high catalytic activity (KOH solution) as compared with the corresponding B- or N-doped carbons. Interestingly, the Se-doped carbonaceous material contained 7.38% of Se and as much as 35.59% of oxygen (at%, XPS). A coin cell was fabricated using a metallic Li as an anode and the Se-doped carbon as the air-breathing cathode and showed an OCV of 3.14 V and a high discharge capacity of 1618 mA h g−1 at a current density of 50 A g−1. The Se-doped carbon enhances the ORR/OER kinetics by facilitating the reversible formation of the Li2O2/Li2O and the adsorption and reduction of O2 (Fig. 20). Se atoms alter the electronic structure of the carbon scaffold and thus act as catalytic centers. Besides SAC and DASC electrocatalysts, metal selenides supported on Se-doped porous carbons were also proposed as cathodes for metal–air batteries. For instance, a 3D self-supporting Co-CoSe@NSeC-biochar composite cathode was proposed for Li–O2 batteries.53 The cathode acted as a bifunctional catalyst showing excellent ORR/OER activity (including high capacity and good cycling stability). The activity was enhanced by the highly active Co/CoSe heterojunction. Consequently, the Se-doping into the biochar was less relevant.
 | ||
| Fig. 20 Scheme of the bifunctional ORR/OER catalysis and the reversible Li2O formation on the surface of the Se-doped carbon electrode obtained from disposable paper cups. Reproduced with permission from ref. 106. Copyright © 2023, The Royal Society of Chemistry. | ||
SACs possess many advantages, and the mononuclear nature of the catalytically active sites puts them somewhere between homogeneous and heterogeneous catalysts with 100% utilization of the doped active element. However, it was soon realized that catalysts with a single active center might be unable to drive complex, multielectron transfer processes. Some multielectron processes cannot be accommodated by a single atom site. Processes such as OER usually involve bimetallic sites. Consequently, the SAC concept was quickly expanded to dual-atomic-site catalysts. Indeed, in reality, catalytic centers are never single atoms, as their surrounding/support is equally important. In fact, many metalloenzymes responsible for crucial life-sustaining processes exhibit multinuclear catalytic centers. For example, cytochrome c oxidase contains N-coordinated Cu and Fe heteroatom sites (N3–Cu/Fe–N4) and transfers electrons for direct reduction of O2 to water. The so-called metal–nitrogen–carbon (M–N–C) catalysts based on N-doped porous carbons with N-coordinated metal atoms are a prominent example of nature-inspired single-atom catalysts.107 In such materials, the catalytic center constitutes metal ions such as Fe, Co, Mn, Cu, etc., chelated by a few (usually four, and thus the common M–N4–C designation) pyridinic or pyrrolic nitrogens. These M–N–C materials are versatile (electro)catalysts active not only in ORR and OER but also in other processes such as the CO2 reduction reaction (CO2RR). However, the activity and versatility of M–N4–C catalysts can be further modulated by tuning the local coordination environment around the symmetrical M–N4 center to break the symmetry of charge distribution and hence optimize the adsorption strength of intermediates involved in catalytic processes. Indeed, Se-doping was employed to modulate the local coordination environment of the symmetrical Cu–N4 center to improve its activity in the ORR.49 To this end, extensive theoretical analysis was performed to assess the potential benefits of Se-doping (Fig. 21). Besides Se, S and P doping around the Cu–N4 center was also simulated for comparison (Fig. 21a). Se yielded stronger polarized charge distribution in asymmetric Cu–Se atomic pairs with respect to the corresponding S and P dopants. Se breaks the inherently symmetric charge around the Cu–N4 center (Fig. 21b and c). The distance between the Cu/Se atomic pair was also scrutinized, and the medium Cu–Se distance of 3.64 Å exhibited the lowest theoretical overpotential, and thus the best 4e− ORR activity. The theoretical activity of the catalysts with different Se–Cu distances was determined by the initial transformation of O2* to OOH* (Fig. 21d and e). During the O2 reduction process, the oxygen molecule first adsorbs on the Cu atom and one electron is transferred to O2 to form the OOH*–Cu–N4 (Fig. 21f). After the release of OH−, OOH*–Cu–N4 accepts another electron and transforms into an O*–Se–C2 intermediate. The O*–Se–C2 gains one electron, and the resultant OH* returns to the Cu site to form OH*–Cu–N4. The whole ORR cycle is completed when OH*–Cu–N4 acquires the last electron. In Cu/Se DASCs, the Se dopant acts as an electron-withdrawing polarizer, which tunes the electron distribution around the Cu–N4 site and thus facilitates the second oxygen reduction step, in which the oxygen-bearing intermediate of the OOH*–Cu–N4 complex transforms to an O*–Se1–C2 intermediate.
 | ||
| Fig. 21 DFT calculations of the ORR activity on Cu–Se DASC. (a) Scheme of theoretical calculations to search for a synergetic-catalysis DASC. (b) Top view of the differential charge densities of Cu–Se. The blue and yellow surfaces represent electron depletion and electron accumulation, respectively. (c) Bader charge analysis of Cu–P, Cu–Se, and Cu–S DASCs. (d) ORR free-energy mapping at 1.23 V for the possible Cu–Se pairs with different distances. (e) ORR overpotential on different Cu–Se pairs and initial Cu–N4. (f) The calculated ORR mechanisms for the optimal Cu–Se DASC. C: gray; N: blue; Cu: orange; Se: yellow; H: white; O: red. Reproduced with permission from ref. 49. © 2023 Wiley-VCH GmbH. | ||
When employed as a cathode in an alkaline Zn–air battery (ZAB), the Se and Cu dual-atomic-site catalyst provided working parameters significantly better than the Pt/C catalyst (i.e., a maximum power density of 206.5 mW cm−2 with discharge specific capacity of 809.3 mA h g−1 at 10 mA cm−2). An operando XAS showed that Se sites enable the transformation from OOH*–Cu–N4 to the O*–Se–C2 intermediate (Fig. 22).
 | ||
| Fig. 22 Operando XAS reveals the synergistic catalysis of the cathodic Cu–Se site in ZAB. (a) Illustration of operando XAS set up under realistic device working conditions. C: gray; N: blue; Cu: orange; Se: yellow; H: white; O: red. (b) Polarization and power density curves of Cu–Se and Pt/C-based batteries. (c) Comparison of the Pmax and specific capacity of ZAB assembled by Cu–Se with other reported electrocatalysts. (d) The operando Cu K-edge XANES spectra at different voltages under ZAB working conditions. The inset shows the magnified adsorption-edge region. (e) The difference in location of the main peak and the Cu–N/O coordination number, and the intensity difference of the infrared signals at 918 and 1055 cm−1, change with applied potentials for the Cu–Se catalyst during the ORR process. The potentials presented in operando XAS and in situ SRIR spectra were referenced to Zn and RHE, respectively. (f) The fitting curves recorded at the Cu K-edge of the Cu–Se catalyst at the initial state and different applied voltages (0.4 V and 1.0 V vs. Zn) under ZAB working conditions. Reproduced with permission from ref. 49. © 2023 Wiley-VCH GmbH. | ||
Rechargeable ZABs gained considerable attention as competitive energy storage devices; nevertheless, their performance is temperature-dependent as it declines significantly below 0 °C. This is caused by the slower kinetics of ORR (occurring at the air cathode during discharging) and OER (occurring at the air cathode during charging) at low temperatures. DASCs can perform better as bifunctional ORR/OER catalysts in comparison with SACs because dual catalytic sites can alter the reaction routes and thus lead to fast kinetics. An N-doped carbon containing atomically dispersed Fe–Se atom pairs was prepared utilizing ZIF-8 as a support and tested as a bifunctional oxygen catalyst for ZABs.48 The additional Se doping yielded higher activity in comparison with Fe-based SACs. The open circuit voltage (OCV) and the maximum power density of the rechargeable ZAB assembled using the Fe–Se/NC material as the air cathode catalyst were 1.47 V and 135 mW cm−2 at 25 °C, better than for a ZAB with Pt/C + Ir/C catalyst (Fig. 23a). The ZAB exhibited stable charge–discharge of 200 h (1090 cycles) at 20 mA cm−2 at 25 °C (6.9 times that of a ZAB with Pt/C and Ir/C catalysts of ORR and OER, respectively) Fig. 23c. At −40 °C, the OCV is still up to 1.44 V, and the peak power density is 71 mW cm−2 (Fig. 23a and d). The ZAB could operate for 133 h (725 cycles) even at 5 mA cm−2 at −40 °C. XANES revealed that the Fe ion is coordinated with three pyridinic N and one selenofenic Se. The band energy of the Fe 3d orbital matches with that of the Se 4p and N 2p orbitals, resulting in an optimized electronic redistribution around the Fe site. The high charge density around the iron ion sites facilitates O2 adsorption and thus favors high ORR activity. The Fe site coordinated by N and Se exhibits the lowest ORR overpotential in comparison with the Fe site coordinated only by N or with the Se site coordinated by N. This is because the ternary Se/N/Fe catalyst exhibits the strongest O2 adsorption energy and the shortest Fe–O bond (1.93 Å). The Fe–Se atomic pair with hybridized p–d orbitals leads to asymmetrical polarized charge distributions, thus optimizing the adsorption energies of the oxygen intermediates during ORR and OER processes.
 | ||
| Fig. 23 The electrochemical performance of ZABs–Fe–Se/NC. (a) Discharging polarization curves and the corresponding power density plots at 25 and −40 °C. (b) Discharging plateaus with current densities from 1 to 50 mA cm−2 of ZABs–Fe–Se/NC and Pt/C + Ir/C at 25 °C. (c) Galvanostatic cycling of ZABs–Fe–Se/NC and ZABs–Pt/C + Ir/C at 20 mA cm−2 at 25 °C. (d) OCVs of ZABs–Fe–Se/NC and Pt/C + Ir/C at −40 °C. (e) Discharging plateaus with current densities from 1 to 10 mA cm−2. (f) Capacity, (g) galvanostatic cycling, and (h) the corresponding round-trip efficiency decay of ZABs–Fe–Se/NC and ZABs–Pt/C + Ir/C at 1 mA cm−2 at −40 °C. (h) Comparison of the round-trip efficiency decay. (i) Galvanostatic cycling at 5 mA cm−2 of ZABs–Fe–Se/NC at −40 °C. (j) Comparison with reported low-temperature ZABs. The values of coordinates (m, n, o) in the graph represent the cycle time, current density, and operating temperature, respectively. (k) Optical images of LED screens powered by ZABs–Fe–Se/NC under (i) puncturing and (ii) cutting. Reproduced with permission from ref. 48. © 2023 Wiley-VCH GmbH. | ||
To enhance the catalytic performance of carbocatalysts, they are usually co-doped with transition metals. However, the main-group metals and semimetals can also be utilized to this end. In addition, the p-orbital valence electrons of atomically dispersed main-group metals can be further modulated by co-doping with non-metallic dopants. A solid-state flexible ZAB was assembled utilizing Sb/Se/N co-doped carbon as a cathodic catalyst for ORR, and its performance was evaluated at −40 °C.51 An OCV of 1.33 V, peak power density of 54.1 mW cm−2, and a rate discharge operation of 44 h were achieved. The parameters were much better than those obtained for 20% Pt/C-based, and Sb/N doped carbon-based cathodes. DFT simulations revealed that the Se doping in the form of SeC2 moiety functions as a long-range regulator of the electronic structure of the SbN2C2 catalytic center. The adsorption energy of the active Sb site can be regulated to an optimal value via a change in the Se–N distance. The Uonset of the SbN2C2-1-Se2 structure increases to 0.795 eV if the Se–N distance = 7.4 Å, and the rate-determining step switches from *OH → H2O to *O → *OH. This is due to the Se doping, which weakens the adsorption ability of SbN2C2 by long-range electron delocalization (Fig. 24). By plotting Uonsetvs. the Se–N distance, a volcano-type relationship was obtained, where Uoneset reaches an optimal value at a Se–N distance of 7.4 Å (Fig. 24).
 | ||
| Fig. 24 DFT calculations on the long-range regulation mechanism. (a) The structure diagram and the distance between Se and N of SbN2C2-1-Sen (n = 1–5). (b) Free energy diagram of SbN2C2 and SbN2C2-1-Se2, RDS represents the rate-determining step. (c) The RDS of different structures and the free energy diagram of intermediates *OH and *OOH. The dotted line represents ΔGSbN2C2-1. The blue and green background represents the RDS of the first three and latter two structures, respectively. (d) The Uonset of SbN2C2-1-Sen (n = 1–5). (e) The PDOS of SbN2C2-1-Sb and SbN2C2-1-Se2-Sb. (e) The PDOS of SbN2C2-1-Sb and SbN2C2-1-Se2-Sb. (f) Suborbital PDOS of the O and Se orbitals combined with the bonding state shown by the crystal orbital Hamilton population (COHP); light gray means the density of states is the bond state, and dark gray is the anti-bond state. Reproduced with permission from ref. 51. Copyright © 2023, American Chemical Society. | ||
A rich family of Se-doped carbonaceous electrocatalysts additionally co-doped with Fe and N was prepared and scrutinized as catalysts for the ORR.69 High activity was observed for both single-atom Se-NC and dual-atom Fe/Se-NC catalysts in alkaline and acid electrolytes. However, while the Se SAC exhibited remarkable activity for ORR in an alkaline solution, it performed somewhat worse in acid. It was proposed that the active site of the DASC comprises adjacent Fe–N5 (iron ion coordinated by five N atoms) and SeC2 structures. DFT analysis demonstrated that the Se-doping to the Fe-NC catalyst lowers (optimizes) the adsorption energy of the ORR intermediates (*O and *OH). Moreover, the co-existence of the Fe–N5/SeC2 dual site is beneficial for the *OH desorption (the modeling and ORR mechanism on the diatomic Fe/Se-NC catalyst is presented in Fig. 25). Significantly, 57Fe Mössbauer spectroscopy indicated that the Se doping increases the content of low-spin Fe3+ polarization configuration. This low-spin Fe3+ configuration is supposed to be favorable for optimizing the adsorption strength of the ORR intermediates. Se co-doped DASCs were also evaluated in polymer electrolyte fuel cells. The DASC catalyst with Co–Nx and Se–C dual sites showed high ORR activity and, when employed as a cathodic catalyst in a H2–O2 PEFC, the cell delivered a peak power density of 297 mW cm−2.52 Se-doping also improves the stability of cathodic catalysts in PEFC. Se-doped Fe–N–C catalyst obtained from ZIF-8 showed better stability in a H2–O2 PEFC than the corresponding S-doped and the Fe–N–C catalyst without any chalcogen doping.108 However, the S-doped catalyst exhibited higher initial activity in respect to the Se-doping.
 | ||
| Fig. 25 (A), Free-energy diagram for different Fe-centered moieties. (B), The Bader charge and spin density of Fe1Se1-NC and Fe1-NC. C, Top and side views of the 3D differential charge density distributions for the three intermediate reaction steps, and the iso-surface value is 0.005 eV Å−3. (D) and (E), The projected density of states of Fe1-NC and Fe1Se1-NC. (F), The proposed ORR mechanism for Fe1Se1-NC. Reproduced with permission from ref. 69. © 2022 Elsevier B.V. | ||
Se-doped carbon-based catalysts have also been applied in energy conversion devices such as direct liquid fuel cells. Hydrazine fuel cells (HzFCs) exhibit many advantages over hydrogen fuel cells, such as the greater energy density of hydrazine vs. hydrogen and its much safer and easier handling due to its liquid state. The only byproducts of the hydrazine oxidation reaction (HzOR) in an alkaline solution are nitrogen and water (N2H4 + 4OH− → N2 + 4H2O + 4e−). One of the issues hampering practical applications of HzFCs is the sluggish kinetics of the HzOR. Consequently, electrocatalysts are required for the anodic HzOR. Platinum is usually utilized for the HzOR, but cheaper and more abundant substitutes are necessary to lower the cost of this technology. A free-standing hierarchically porous carbon membrane containing single-atom Se sites was demonstrated to be an effective catalyst for HzOR in practical O2-HzFC.62 When utilized as an anode in HzFCs, the Se-doped carbonaceous catalyst exhibited catalytic activity superior to commercial Pt/C (Fig. 26). The OCV and maximum power density of the operating O2-HzFC were 0.91 V and 182.2 mW cm−2, respectively. These parameters were not only superior to that of Pt/C-based HzFC but also among the best performances ever reported for O2-HzFC. Significantly, doping carbon with atomically dispersed selenium yielded outstanding enhancement in the electrochemically active surface area, which boosted catalytic activity toward HzOR. The superior catalytic activity was attributed to the positively charged Se sites on which the HzOR proceeded via four dehydrogenation steps (N2H4 → N2H3 → N2H2 → N2H → N2).
 | ||
| Fig. 26 (a) Schematic diagram of O2-HzFC. (b) Polarization and power density plots for O2-HzFCs at 80 °C with Se-doped and Pt/C catalysts, respectively. (c) Long-term stability of the O2-HzFC with the Se-doped catalyst. Reproduced with permission from ref. 62. © 2019 Wiley–VCH GmbH. | ||
Se or S-modified Cu and N co-doped carbons were utilized as cathode catalysts for microbial fuel cells.109 The S or Se doping optimized the electronic structure of the Cu–Nx active sites and hence improved the ORR activity of the Cu–N–C catalyst. In addition, the Cu2S and Cu2Se nanoparticles provided antibacterial properties, thus preventing biofilm formation on cathodes. This is important because it shows that chalcogen nanoparticles are formed during chalcogen doping of transition metal containing carbons.
Inspired by natural molybdenum-dependent enzymes, a synthetic, carbon-based Mo–Se DASC electroreduced CO2 to CO with FECO >90% over a potential window of −0.4 to −1.0 V vs. RHE.50 Mo was the catalytic center directly interacting with CO2, and the Se atom adjacent to Mo functioned as a modulator of its electronic structure through a long-range electron delocalization. When the distance between Se and Mo atoms = 6.0 Å, a strong electronic hybridization occurs as electrons can be transferred from the Mo to the Se atom. The long-range electron delocalization reduces the d-electron center of Mo and hence weakens CO* adsorption. This, in turn, protects Mo from poisoning via strong CO adsorption. Se co-doping improves the selectivity of the neighboring Mo for CO2 adsorption and suppresses the competitive HER. However, the influence of Se on neighboring Mo vanishes gradually with increasing Se–Mo distance. For Se–Mo distances ≥8.5 Å, the electron hybridization between Mo and Se atoms drops substantially. But despite this, the Se atoms far from Mo (>8.5 Å) can still improve the catalytic activity of the Mo–Se DASC by repelling H2O molecules from the catalyst surface and hence accelerating CO2 transport to Mo centers. The performance of the Mo–Se DASC for CO2 electroreduction was evaluated in a gas diffusion electrode (GDE)-based flow cell (Fig. 27). The Mo/Se-based GDE exhibited high CO selectivity with FECO >95% in a constant potential test. At −0.6 V and −0.9 V, the CO partial current density (jCO) exceeded 100 and 200 mA cm−2, respectively.
 | ||
| Fig. 27 Performance evaluation of the Mo–Se DASC in a GDE-based flow-cell. (a) Scheme of the GDE-based flow-cell. (b) FECO of the Mo–Se DASC in the constant potential flow-cell test. (c) jCO in the constant potential flow-cell test. Reproduced with permission from ref. 50. © 2022 Wiley-VCH GmbH. | ||
Se and N co-doped carbon nanosheets with a hierarchical micro-meso-macroporous structure were analyzed as CO2RR electrocatalysts.80 When compared with the corresponding material without Se doping, the partial current density of CO was enhanced 11-fold alongside 90% selectivity for CO production at a low potential of −0.6 V (vs. RHE). The catalytic center constitutes a Se atom in a 6-membered ring of graphene edge with neighboring pyridinic nitrogen (–Se–C–N– edge configuration). DFT calculations showed that Se doping lowers the free energy barrier of CO2RR and inhibits the competitive HER. The Se-doped catalyst exhibited stronger CO2 than hydrogen adsorption, assuring high selectivity. In contrast, the N-doped carbon without Se co-doping exhibited stronger hydrogen adsorption. The Se and N co-doped carbon nanosheets showed outstanding performance in the conversion of CO2 to CO, comparable even to platinum and gold catalysts. Significantly, it was demonstrated that the maximum FECO increased with increasing Se content in the catalyst. In another approach, a series of metal-free Se, B, P, and N tetra-doped carbocatalysts have been prepared at various pyrolysis temperatures and evaluated in CO2RR.68 The tetra-doped catalyst prepared at 1050 °C reduced CO2 to CO with FEco of up to 96.2% (at −0.5 V) and with H2 as the only byproduct. The high catalytic activity was attributed to extensive porosity providing exposed active sites and to the optimal graphitization degree. The synergy of simultaneous Se, B, P, and N co-doping produces charge and spin redistribution/delocalization, and the asymmetric electron redistribution yields improved CO2RR performance when compared with the ternary (B/P/N), binary (Se/N), and single doping (N) of the carbonaceous catalyst. Besides metal-free catalysts, an iron carbide catalyst encapsulated in N-doped CNT was also studied for electrocatalytic CO2RR.71 When this material was additionally doped with Se, its surface gained aerophilic and basic (i.e., electron-rich) properties. The aerophilic surface increased the affinity for CO2. This was due to the overall enhancement of nitrogen content and preferential formation of pyridinic N upon Se doping. DFT models showed that the electron-rich surface created by the abundant pyridinic N species and more negatively charged Fe sites facilitate electron transfer from the catalyst surface to the anti-bonding orbitals of the acidic CO2 molecule. The Se/N-Fe3C-CNT-based catalyst exhibited excellent CO selectivity with FECO of 92% at −0.5 V (vs. RHE).
Se doping at the edges was identified as the source of enhanced electrocatalytic activity. The graphitic structure permitted rapid transfer of electrons from the external circuit to the adsorbed I−/I3− ions, improving the overall photovoltaic performance.
Qiu et al. proposed Se-doped rGO as a CE material for DSSCs.67 The catalyst was produced via high-temperature annealing, and the temperature allowed tuning of the density of defects (Fig. 28). The material prepared at an annealing temperature of 900 °C with Se content of 1.73 at% showed high catalytic activity for I3− reduction with a high PCE of 8.42% (under AM 1.5G illumination), much higher than for the reference Pt-based CE (7.88%). DFT calculations showed that the high catalytic activity of Se-doped rGO originates from Se-doping, which increases the electron-donating capabilities of the material. This, in turn, facilitates electron transfer to the iodine adsorbed on the surface of the catalyst. Significantly, it was highlighted that the catalytic activity of the Se-doped rGO for I3− reduction was diminished by the oxygen-containing functional groups. Oxygen-containing groups hinder the electron-donating properties of the catalyst because they influence the charge distribution and destroy the conjugated π-system of rGO. This is one of only a few examples highlighting the importance of the impact of chemisorbed oxygen on the catalytic properties of carbocatalysts. As oxygen functional groups are omnipresent on carbon-based material surfaces, their impact should not be neglected.14
 | ||
| Fig. 28 (a) The preparation of Se-doped rGO. (b) Operating principles of DSSC and the mechanism of the I3− reduction over the CE based on Se-doped rGO. Reproduced with permission from ref. 67. © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Song et al. performed extensive theoretical analysis concerning dually doped graphene as a catalyst for iodine reduction.113 They showed that graphene dually doped with N and Se exhibited enhanced catalytic activity due to the improved transfer of electrons to iodine species. Qiu's research group continued their search for precious metal-free CEs for DSSCs and confirmed that theoretical assumption.60 They prepared a N and Se co-doped porous carbon with an ordered macroporous structure. This material exhibited high electrocatalytic activity toward the triiodide reduction reaction, outperforming the single N-doped carbon (without Se). Under AM 1.5G illumination, the DSSC assembled with the N/Se co-doped catalyst delivered PCE of 9.79%, outperforming the Pt-based reference CE (7.75%). The DSSC with the dually doped N/Se catalyst exhibited a 33% increase in PCE compared with the device with a single N-doped carbon catalyst. This is strong evidence of the advantage of dual-doping to improve electrocatalytic activity in I3− reduction. DFT calculations indicated that the high activity of the N/Se co-doped porous carbon is due to the synergy between N and Se dopants creating active sites for iodine adsorption (Fig. 29). The molecular configuration of ac-QuaN-Se3-C2 (Fig. 29g) yields optimal adsorption energy of the intermediate I atom (Ead,I) of 0.67 eV, showing that the C2 atom adjacent to the quaternary N and Se configurations at the armchair edge of graphene is the most active site for iodine adsorption and hence the major contributor to the catalytic activity toward I3− reduction. In addition, the high surface area and ordered open porosity increase the exposure and accessibility of the active sites to the electrolyte.
 | ||
| Fig. 29 DFT calculated adsorption energy of the I atom on various models. (a) Reaction pathway of I2 reduction over the NSeC model. (b) Graphene slab as the adsorption model. Adsorption models with (c) quaternary N and (d) Se at the armchair edge. Charge-transfer analysis of (e) ac-QuaN and (f) ac-Se3-QuaN. (g) Adsorption model with ac-Se3-QuaN configuration. (h) Ead,I value as a function of models with heteroatoms at armchair edges. Adsorption models with (i) quaternary N and (j) Se at the zigzag edge. (k) Adsorption model with zz-Se3-QuaN configuration. (l) Ead,I values as a function of models with heteroatoms at zigzag edges. Reproduced with permission from ref. 60. Copyright © 2023, American Chemical Society. | ||
5.2. Se-doped carbon materials as electrodes for alkali metal-ion batteries and electrochemical capacitors
Alkali metal-ion batteries comprise the commercially successful lithium-ion batteries and their alternatives: sodium-ion batteries (SIBs) and potassium-ion batteries (PIBs).114 LIBs are a crucial commercial energy storage technology in portable electronics, electric vehicles (trucks included), and grid-scale facilities. However, the uneven geographical distribution of lithium accelerated research on Na-ion and K-ion batteries as competitive replacements for LIBs. The development of SIBs and PIBs is hindered by a shortage of high-performance anode materials. In the case of LIBs, graphite (in the spherical form) remains the critical anode material. Li intercalates into graphite easily, forming a stable LiC6 intercalation compound. Potassium can also intercalate into graphite quite easily, forming KC8 (but the sluggish kinetics and the drastic volume expansion due to the large K+ radius cause poor rate capability and capacity deterioration). In contrast, the intercalation capacity of graphite anodes for SIBs is exceptionally low, as only high-stage graphite intercalation compounds, such as NaC48, NaC64, and NaC80, have been reported. Graphite anodes are not appropriate for Na+ intercalation and thus for high energy density SIBs. The so-called hard carbons, i.e., disordered carbonaceous materials obtained from non-graphitizing precursors, are attractive anode materials for SIBs and PIBs with enhanced capacity (nevertheless, the ion storage mechanism for Na+ has not yet been clearly understood).115 Doping hard carbons with heteroatoms is a promising strategy to improve their performance as anode materials for SIBs and PIBs. This is often associated with the enlargement of graphenic carbon lattice parameters upon insertion of heteroatoms. Foreign atoms also provide a profusion of active sites for Na+ storage through fast pseudo-capacitive reactions, which improves both the capacity and rate performance.Fu et al. prepared sulfur, selenium, and nitrogen co-doped hard carbon as the anode material for SIBs and PIBs.57 This anode material exhibited high Na+ storage performance with a capacity of 210.4 mA h g−1 at 1 A g−1 after 1000 cycles (and a capacity of 125.9 mA h g−1 at a current density of up to 10 A g−1). When utilized in a PIB, it exhibited a capacity of 207 mA h g−1 at 0.1 A g−1 after 450 cycles and good capacity retention of 143.5 mA h g−1 after 1100 cycles at 1 A g−1. The capacitive charge storage grants fast rate performance. It was concluded that the reaction kinetics and ion storage performances are improved by the active sites and surface defects induced by the S, Se, and N co-doping. This same research group also reported a Se-doped carbon anode for LIBs.73 They demonstrated that the Se-doping introduced a profusion of defects which yielded an anode material with excellent rate capability and a high specific capacity of 450 mA h g−1 after 580 cycles at the current density of 500 mA g−1. Heteroatom doping is indeed utilized not only to prepare anode materials for SIBs but also to improve the performance of existing LIBs. Doping carbons with heteroatoms such as N, Se, S, or P can modulate Li ion adsorption and diffusion by expanding the interlayer spacing of carbon materials and improving adsorption energy.72 When selenium-doped rGOs were evaluated for LIBs, they showed better Li storage performance than their undoped counterparts.36 This was attributed to the improved electrical conductivity. Se atoms were homogeneously introduced on the entire rGO surface, leading to improved electrical transport properties caused by the n-type doping. Se-rGOs showed higher rate performance and increased capacity (reversible capacity of 570 mA h g−1 at a current density of 1 C) when compared with Se-free rGO (310 mA h g−1).
Se/N co-doped carbon nanosheets were scrutinized as anode materials for SIBs, and their performance was compared with corresponding N-doped nanosheets without Se.54 Because high-rate performance and cyclic stability constitute the two critical parameters determining the practical application of an anode material, these parameters were studied in detail. The N/Se co-doped carbon exhibited higher reversible capacity, better cycling stability, and improved rate performance and capacity retention (at a high rate) compared with the studied Se-free counterparts. The N/Se co-doped anode delivered a high specific capacity of 302 mA h g−1 at 0.1 A g−1 and an enhanced rate capability of 123 mA h g−1 even at 10 A g−1 (Fig. 30). Fig. 30 also depicts a comparison of the Se/N co-doped carbon performance with undoped, N/S, and N/P co-doped carbon materials reported earlier in the literature as anodic materials for SIBs. The comparative analysis revealed the superior performance of the anode upon Se doping. It was concluded that the Se dopant acts as a strong electron-withdrawing center for Na storage when compared with C and N sites (the Se atom constitutes an active site for Na+ adsorption, thus improving ion storage capacity). In addition, Se enlarges the interlayer distance of carbon sheets to create more active sites for ion storage and enhance the Na+ diffusion kinetics. The additional N co-doping was responsible for improved conductivity and wettability of the anode material. Se-doping to porous hard carbons allows for SIBs with an excellent rate performance. For instance, Se-doped macroporous carbon showed large capacity and fast Na storage capability with a high reversible capacity of 335 mA h g−1 at 0.1 A g−1.63 Significantly, the capacity was still 251 mA h g−1 under a large current density of 5 A g−1, showing an ultrafast Na+ storage process. The Se-doped carbon exhibited surface-dominated ion storage behaviors owing to the selenium-assisted capacitive redox reactions.
 | ||
| Fig. 30 Electrochemical performance of three analyzed carbonaceous materials (N-doped, N/O co-doped, and N/Se co-doped): (a) cycling performance at 0.1 A g−1; (b) rate capability; (c) rate capacity retention at different current densities; (d) comparison of Na+ storage capability with other anode materials reported in the literature; (e) long-term cycling performance of N/Se co-doped carbon at 5 A g−1. Reproduced with permission from ref. 54. © 2021 Elsevier B.V. | ||
Alkali metal–selenium batteries such as lithium–selenium (Li–Se), sodium–selenium (Na–Se), and potassium–selenium (K–Se) batteries are considered an exciting alternative to the alkali metal–sulfur batteries. Various carbon materials were loaded with elemental selenium to prepare cathodes for such batteries. However, in such cathodes, the carbon is only a host for amorphous Se, and the selenium loadings are very high – for instance, around 30–60 wt%.56,91 We do not portray such systems within the scope of this review as they concern Se-impregnated and not Se-doped carbon materials.
Heteroatom doping of carbonaceous materials has also been utilized to improve their performance as electrode materials for electrochemical capacitors (supercapacitors). Supercapacitors constitute the most important electric energy storage technologies next to batteries. They store energy via a charge storage mechanism, which includes a non-faradaic electric double-layer capacitance and a faradaic pseudocapacitance (both mechanisms coexist in these capacitors).116 Supercapacitors based on unmodified carbon electrodes suffer from low charge storage capacity. Doping with heteroatoms creates additional pseudocapacitance via surface redox reactions.117 Heteroatoms can also improve the wettability of the electrode surface. N and Se co-doped graphene aerogels (NSe-GA) were proposed as binder-free electrodes for supercapacitors.41 The produced electrode exhibited a high specific capacity of 302.9 F g−1 at a current density of 1 A g−1 – higher than that of solely Se-doped or N-doped aerogels. Carbon-based supercapacitors usually show poor energy density (lower than 20 W h kg−1). However, the symmetric supercapacitor constructed utilizing this N/Se co-doped aerogel exhibited an energy density of 26.3 W h kg−1 at a power density of 900 W kg−1 (1 M Na2SO4 electrolyte), which is higher than for carbon-based supercapacitors presented recently in the literature (Fig. 31). Fig. 31 demonstrates the practical performance of the capacitor by lighting an LED device. The doped carbon possessed a SSA of only 337 m2 g−1; however, the N and Se contents were relatively high at ca. 5.9 and 2.1 at%, respectively. The N and Se atoms were considered to synergistically improve the electrical conductivity caused by the n-type doping. In addition, co-doping improves electrode wettability and thus the capacitance performance. The high polarizability of Se enhances interactions between Se and the surrounding electrolyte and hence improves the charge-transfer between the electrode surface and electrolyte. Se-doping produces strain in the graphenic structure, which facilitates the adsorption of ions from the electrolyte. The excellent electrical performance was due to the highly reversible faradaic redox reactions of Se-doping, which provided large pseudocapacitance.
 | ||
| Fig. 31 Electrochemical performance of the NSe-GA//NSe-GA symmetric supercapacitor in 1 M Na2SO4 electrolyte. (A) CV curves in different voltage ranges at the scan rate of 20 mV s−1. (B) GCD curves at various current densities. (C) Specific capacitance as a function of the current density and the inset shows the application of NSe-GA//NSe-GA symmetric supercapacitor. (D) Ragone plots of the NSe-GA//NSe-GA symmetric supercapacitor and other previously reported carbon-based symmetrical supercapacitors. Reproduced with permission from ref. 41. © 2020 Wiley-VCH GmbH. | ||
5.3. Se-doped carbon materials for pollutant neutralization/recycling and detection
Heteroatom-doped carbons are commonly scrutinized as efficient materials for pollutant removal and neutralization.118,119 In this regard doping with chalcogens is especially attractive because they exhibit a high affinity to transition and heavy metals. While sulfur is a well-known example of a p-block element with a strong affinity to such metals, selenium's affinity to these metals is truly remarkable. For instance, the affinity of mercury for Se is ca. 106 greater than for sulfur, with an association constant of 1045 (vs. 1039 for sulfur).120 HgSe solubility in water is also extremely low, with a constant of 1.0 × 10−59 (vs. 10−52 for HgS). This is because Se possesses a larger atomic radius and hence is more polarizable and nucleophilic than sulfur. It is not surprising then, that Se-modified sorbents are considered effective means for toxic metal capture. Many organoselenium polymers showed high sorption capacity for heavy metals, allowing their removal and recycling.121,122 In fact, it was demonstrated that Se-doped resins possess much higher adsorption capacity for noble metals than corresponding sulfur-doped polymers.122Biomass is a renewable and low-cost feedstock to produce Se-doped carbonaceous sorbents. Many interesting examples of biochars serving as high-performance adsorbents for metal removal and recycling can be found in the literature.79,123 Zhu et al. presented a rich family of Se-doped biochars prepared from various carbohydrates via their selenization followed by pyrolysis.79,123 The Se-doped chars were utilized to remove Pb from wastewater from a lead battery factory.79 Even a small amount of Se doping (∼0.34 wt%) significantly enhanced the Pb adsorption capacity. The Se-doped carbon adsorbed over 2.56 times more lead with respect to its undoped counterpart. Interestingly, the Se-doped chars prepared from aldoses perform better than those from ketoses (e.g., glucose vs. fructose). In addition, the sorbents’ adsorption capacity decreased with increasing molecular weight of the utilized carbohydrate (Fig. 32). Potato starch, cotton, and straw were unfavorable precursors for Pb sorbents. Se-doped char from glucose proved to be the most favorable for lead removal. Pb sorption occurred on the Se-doped carbons via strong chemisorption, and hence it was not affected by temperature (Fig. 32). In contrast, the adsorption capacity of the undoped carbon diminished severely with temperature, indicating only weak physisorption.
 | ||
| Fig. 32 Pb adsorption by the Se-doped chars prepared from different carbohydrates (aqueous solution 0.05 mol L−1 Pb, 25 °C, adsorption time of 10 h) and the effect of the adsorption temperature (0.05 mol L−1 Pb, adsorption time of 10 h). Reproduced with permission from ref. 79. Copyright © 2021, Springer Nature. | ||
A Se-doped mesoporous biochar obtained from tree bark was studied as an adsorbent for orange 16 dye and sodium diclofenac removal.81 The advantages of Se-doping were revealed by comparing the sorbent with an undoped biochar. The selenium-modified biochar possessed a higher SSA with respect to the reference (1300 vs. 1207 m2 g−1). Se was present in the biochar as highly dispersed nanoparticles bonded to the carbon framework. The oxygen functional groups of the carbon framework could interact with Se NPs to form Se-oxygen bonds, and these entities facilitated the binding of the orange 16 dye and sodium diclofenac molecules. The achieved adsorption capacities for the dye and diclofenac were 538 and 434 mg g−1 (at 25 °C), respectively. The unmatched affinity of mercury to Se has been utilized for removal of Hg0 vapors. Hg0 capture is especially challenging due to its high volatility, yet it is technologically significant for mercury emission control from coal power plants. In this regard, Se powder was utilized to prepare Se-doped Cu–N–C catalyst with atomically dispersed active sites for oxidative capture of Hg.124 The optimal catalyst with Cu/Se ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exhibited mercury removal performance with over eight-fold enhancement in its Hg0 oxidation capacity with respect to the Se-free Cu–N–C counterpart. The electron-accepting property of Se is pivotal in promoting oxidation of the adsorbed mercury formation of very stable HgSe. Similar research showed that Se-loaded activated carbon (with ca. 15.3 wt% of Se) obtained by Se powder impregnation at 300 °C exhibited an ultra-high efficiency for Hg0 capture from vapors.89 The Se-loaded carbon reached an adsorption capacity of 17.98 mg g−1 at 160 °C in a nitrogen atmosphere. Selenium captured mercury from the gas phase via HgSe formation. The Hg0 chemisorption was significantly enhanced with the increase in temperature.
:1 exhibited mercury removal performance with over eight-fold enhancement in its Hg0 oxidation capacity with respect to the Se-free Cu–N–C counterpart. The electron-accepting property of Se is pivotal in promoting oxidation of the adsorbed mercury formation of very stable HgSe. Similar research showed that Se-loaded activated carbon (with ca. 15.3 wt% of Se) obtained by Se powder impregnation at 300 °C exhibited an ultra-high efficiency for Hg0 capture from vapors.89 The Se-loaded carbon reached an adsorption capacity of 17.98 mg g−1 at 160 °C in a nitrogen atmosphere. Selenium captured mercury from the gas phase via HgSe formation. The Hg0 chemisorption was significantly enhanced with the increase in temperature.
Shi et al. proposed Se and N co-doped biochar derived from bacterium cells as a bifunctional material for adsorptive and oxidative removal of phenols from water.83 The Se co-doping not only enhanced the catalytic performance but also significantly improved the catalyst's stability. This is of the utmost importance because oxidative degradation of phenols could also damage the adsorbent. The oxidation of the catalytically active adsorbent was prevented thanks to the antioxidant properties of Se. Compared with solely Se- or N-doped reference adsorbents, the Se/N co-doped material exhibited the highest efficiency of oxidative degradation of phenol assisted by peroxymonosulfate (PMS). It was hypothesized that the Se co-doping enhanced the doping of nitrogen in the graphitic-N configuration, and this in turn facilitated the electron transfer between the carbon surface and PMS. The graphitic N affected the adjacent C site making it an active catalytic site for PMS activation. PMS would bond covalently to the carbon site to form a metastable complex with a high ability for accelerated oxidation of phenol. It was assumed that phenol degradation occurs via a non-radical mechanism comprising the direct transfer of electrons from phenol to PMS. Se-doping enhanced charge density on the carbon surface to facilitate PMS activation and generated electron cloud delocalization which helped to protect the C site from oxidative inactivation. The rapid degradation of phenol by this metal-free catalyst was indeed excellent when compared with other metal-based carbonaceous catalysts. This is significant because iron and cobalt are usually doped into carbon to produce efficient adsorbents for advanced oxidation processes. In somewhat similar research, Peng et al. employed ZIF-8 to prepare Se/N co-doped porous carbon by pyrolysis (500–900 °C), where SeO2 was utilized as the Se source.125 A range of Se/N co-doped microporous carbon catalysts were prepared to study the degradation of organic pollutants by activating PMS. Upon annealing at 900 °C, the obtained material showed the highest PMS activation capability. Ten various pollutants were successfully degraded utilizing this approach. Degradation proceeded via a non-radical pathway through the formation of an outer-sphere complex between the Se/N co-doped carbon and the adsorbed PMS. In such a complex, Se/N co-doped carbon works as an electron donor to PMS and as an electron mediator to transfer electrons from a pollutant molecule to PMS. Se doping (1.06 at%), a high content of graphitic N, enhanced graphitization, and a very high SBET (1560 m2 g−1) helped to improve the catalytic activity of this Se/N co-doped carbon.
Besides adsorption and oxidative degradation, Se-doped carbonaceous materials were also applied for sensing. For instance, a Se-doped carbonaceous material was studied as an electrochemical sensor and adsorbent for Hg(II) and Cd(II) ions.126 Square-wave anodic stripping voltammetry (utilizing a glassy carbon electrode covered with the Se-doped carbon) was applied for detection. The achieved limits of detection of Cd(II) and Hg(II) were 1.9 and 4.3 ppb, respectively. Maximum adsorption capacities were 210.81 and 205.53 mg g−1 for Hg(II) and Cd(II), respectively. It was deduced that the C–Se functionality was responsible for ion adsorption, while elemental Se played a key role in electrochemical sensing. Dong and co-workers studied Se-containing metal-free and metal-doped single and dual-atomic-site catalysts for nonenzymatic sensing of H2O2.47,70 Significantly, the designed sensors were successfully utilized for H2O2 detection in disinfectant and urine samples. For example, a ZIF-8 derived Se/N co-doped material with single Se sites exhibited high electrocatalytic activity for H2O2 reduction and thus was applied for its detection in a wide range from 0.04 to 11.1 mM, with a low detection limit (0.018 mM) and high sensitivity of ca. 404 μA mM−1 cm−2.47 This same research group also synthesized a dual-site Fe–Se catalyst based on an N-doped carbon and studied it in nonenzymatic detection of H2O2.70 The simultaneous doping of Se and Fe enhanced the electrocatalytic activity for peroxide reduction, allowing outstanding sensing properties. Thanks to the synergistic effect of Fe/Se dual doping that improved the electron transfer rate, the hydrogen peroxide could be detected in a wide linear range (0.02–13 mM) with a high sensitivity of 1508.6 μA mM−1 cm−2 and an 11.5 μM detection limit. In another interesting approach for a sensing platform, a Se-doped carbon obtained from PPy was utilized as it exhibited peroxidase-like activity 6.51-fold higher than PPy-based char without Se.59 The Se-doped PPy char catalyzed rapid oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) by H2O2. This permitted the design of a colorimetric sensing system for the detection of D-penicillamine (a therapeutic drug) with a detection limit of 60 nM. The peroxidase-like activity of the Se-doped PPy-derived char was ascribed to the C–Se–C bonds catalyzing the decomposition of H2O2 into ROS to oxidize TMB. Because D-penicillamine inhibited the peroxidase-like activity of the Se-doped char, a simple and fast colorimetric assay was proposed for its detection.
5.4. Se-doped carbons as heterogeneous catalysts in organic synthesis
The most important use of selenium in organic synthesis is selenoxide elimination – a convenient way to form CC bonds.127 It requires a selenylating agent and an oxidant to generate the selenoxide entity in situ. Significantly, CC bond formation can proceed even below 0 °C.127 This reaction is considered a green synthesis, as hydrogen peroxide can be used as the oxidant in the selenylation–deselenylation catalytic cycles. Corresponding chalcogenoxide eliminations, such as sulfoxide eliminations, occur at much higher temperatures and are 100000 times slower. This highlights the core difference between selenium and sulfur chemistry. Se is considered eco-friendly as it can be metabolized in organisms, which gives it an advantage over transition metal-based catalysts. Consequently, transition metal-free Se-based catalysis, including organoselenium catalysis, is of great significance. Selenium catalysis offers methods for synthesizing epoxides, 1,2-diols, and carbonyls via alkene oxidation. Se-catalyzed oxidation reactions employ green oxidants such as H2O2, O2, or air and can be conducted under mild conditions.128 Numerous review reports are devoted to organoselenium reagents in homogeneous catalysis of various transformations in the presence of mild oxidants where selenium acts as an oxygen carrier catalyst.129,130 Yu's research group conducted several projects on oxygen-transfer processes where Se-containing compounds/materials were used as catalysts to transfer the oxygen from oxidants to the desired products, taking advantage of the prominent oxygen carrier abilities of selenium.131 The Se–O bond is much weaker than the corresponding S–O, and this enables the transfer of oxygen from the high-valent Se-based catalytic species to the products to restart the catalysis cycle.
Heteroatom-doped carbonaceous materials constitute attractive metal-free heterogeneous catalysts for a wide range of organic transformations.132,133 Yu's group designed a new family of Se-doped carbon materials and evaluated them as heterogeneous catalysts in selenium-assisted oxygen transfer processes.29 The catalyst could be prepared on a kilogram scale by pyrolyzing selenized carbohydrates, such as glucose. They reported a breakthrough in conventional homogeneous organoselenium catalysis when they showed that a heterogeneous Se-doped carbon catalyst was more active than the homogeneous organoselenium catalysts in the epoxidation of β-ionone (yielding epoxide as the only product).75 Significantly, while homogeneous organoselenium-catalyzed oxidations of β-ionone are not selective, reactions catalyzed by the Se-doped carbon material are regiospecific due to the steric hindrance caused by the carbon support. The reaction occurred probably via a non-radical pathway, and the formation of the –Se(O)OOH moiety was proposed. However, the product yield was relatively low, and the catalytic activity of the recycled Se-doped carbon decreased significantly due to the surface degradation caused by the oxidizing conditions. Many carbohydrates were utilized for the fabrication of Se-doped carbon catalysts, and their catalytic activities were evaluated in the selective β-ionone epoxidation reaction.75 The Se-containing catalyst obtained from fructose was equally effective as the one from glucose. The catalytic activities decreased when using glycans (e.g., sucrose and chitosan) as carbon sources. Significantly, cheap biomass such as potato amylum and sawdust was also employed as a precursor to prepare Se-doped carbon catalysts for β-ionone epoxidation reactions. The catalytic activity of Se-doped carbonaceous catalysts was further improved via potassium intercalation.78 The K-intercalated catalyst was assessed in the selective epoxidation reaction of β-ionone (5,6-epoxy-β-ionone) using O2 as a mild oxidant. The enhanced porosity caused by potassium addition was crucial as small mesopores enabled the β-ionone molecules to reach the Se sites. Consequently, higher utilization of active selenium sites was possible. The selective aerobic epoxidation of β-ionone proceeded via a free radical mechanism, as presented in Fig. 33. First, oxidation of the supported selenium by O2 led to the active radical 3, while the reaction of O2 with NHPI (N-hydroxyphthalimide – an essential additive increasing the reaction yield) yielded the active radicals of HO2 and phthalimido-N-oxyl (PINO). The reaction of 3 with the HO2 radical yielded the intermediate 4, and then the intermediate 5 and the hydroxyl radical via homo-cleavage of the peroxy bond. The produced hydroxyl radicals react with 3 to yield intermediate 6, which could additionally furnish the radical 5 through the action with PINO radicals. The α,β–CC bond with electron-withdrawing carbonyl was less active than the electron-enriched endocyclic γ,δ–CC bond, the interaction of which with the carbonaceous support could yield the intermediate state moiety 7. This granted the regioselectivity of the reaction. Then, the oxygen radical could attack the electron-deficient γ-carbon of the intermediate 7 to produce the intermediate 8, and its decomposition via cleavage of the Se–O bond released the product 2 and yielded the radical species 3 to re-start the whole catalysis circle.
 | ||
| Fig. 33 Scheme of β-ionone (1) oxidation to 5,6-epoxy-β-ionone (2), and proposed mechanism of the epoxidation catalyzed by Se-doped carbon. Adapted with permission from ref. 78. Copyright 2022, The Royal Society of Chemistry. | ||
Carbon materials with sulfonic acid functional groups are well known as solid acids and are considered a promising replacement for sulfuric acid catalysts.134 In an analogous manner, selenium-doped carbon was utilized as a solid acid catalyst in the Beckmann rearrangement of ethyl 2-(2-aminothiazole-4-yl)-2-hydroxyiminoacetate.76 It was probably the first example of using Se-containing materials as a solid acid catalyst, and the unique steric hindrance of the carbon support restrained the undesired side reactions. The Beckmann rearrangement allowed the production of useful derivatives of Cefixime (an antibiotic). H2O2 functioned as an activator of the virgin Se-doped catalyst oxidizing the elemental selenium into –SeOxH moieties of strong acidity (i.e., into carbon-supported selenic acid). Besides Se-doped heterogeneous catalysts, selenium and nitrogen co-doped biochar-based catalysts were produced and shown to be active metal-free catalysts for the oxidation of aromatic aldehydes to their corresponding carboxylic acids.82 The catalyzed oxidation could be performed under solvent-free conditions using H2O2 as an oxidant. The catalytic activity of the co-doped catalyst was superior to its non-doped counterpart.
A variety of new Se-doped carbon-based catalysts is inspired by glutathione peroxidase – an antioxidant selenoenzyme, which is responsible for protecting cells from oxidative stress by activating peroxides with the assistance of glutathione. Many naturally inspired organoselenium compounds with enzyme-mimicking activity have been developed over recent decades.92,93 A Se-doped carbonaceous material prepared via the hydrothermal method showed GPx-like activity in the epoxidation of various alkenes via H2O2 activation.93 This catalysis allowed the conversion of ∼90% (with ∼75% yield; with acetonitrile used as a solvent). Styrene was selected as a model substrate for optimizing the alkene epoxidation conditions.
The mechanism of olefin epoxidation was established with the assistance of DFT calculations. It was assumed that C–Se functionality in the catalyst could exist in four different configurations as presented in Fig. 34: (i) a hexagonal ring in a graphitic structure, (ii) a pentagonal ring like in selenophene, (iii) Se bonded to the edge of graphene via a double bond, and (iv) –SeH functionality. All these Se configurations allow hydrogen atom abstraction from H2O2 by the Se atom. The epoxidation reaction proceeded through a radical pathway with only a small contribution of the non-radical pathway through the formation of –Se(O)OH species. The Se-doped catalyst activated H2O2 to ˙OOH radical via hydrogen atom abstraction. The ˙OOH attacked CC to form an epoxide.
 | ||
| Fig. 34 Representation of possible C–Se functionalities in the Se-doped carbon catalyst of alkene epoxidation. Reproduced with permission from ref. 93. Copyright 2022, The Royal Society of Chemistry. | ||
Liu et al. studied metal-free Se co-doped carbon-based catalysts obtained from biomass for ammoxidation and oxidation of alcohols with molecular oxygen.135,136 For instance, a Se/P/N ternary-doped carbonaceous material was evaluated as a metal-free catalyst for the direct ammoxidation of (hetero)aromatic alcohols to nitriles with aqueous NH3 and O2.135 To prepare the metal-free catalysts, biomass of agricultural origin (e.g., straw, corn cob, and chaff) was used as carbon feedstock and NH4Cl, SeO2, and triphenylphosphine as N, Se, and P sources. Compared with the mono- and binary-doped carbons, the Se/P/N tri-doped carbon showed the best catalytic performance with up to 93.1% conversion of benzyl alcohol to benzonitrile and with selectivity of 99.5%. It was shown that the Se and N dopants enhance the activity, while the P dopant enhances the selectivity for benzonitrile. Pyrrole N, pyridine N, and Se–C species were identified as active sites. Se-doped and co-doped carbonaceous materials were also utilized as supports for noble metal NPs. For instance, Ag NPs supported on Se/N co-doped mesoporous carbon catalyzed the hydrogenation of a series of nitroaromatics to corresponding amines.87 Similarly, the Pd NPs supported on the Se/N dually doped carbon support catalyzed the Heck cross-coupling reaction (C–C bond formation via Heck–Mizoroki cross-coupling reaction).88
6. Challenges and outlook
Heteroatom-doped carbonaceous materials exhibit many advantages over unmodified carbons, and this in turn widens the range of their future applications as advanced functional materials. Nitrogen doping has so far dominated this research field, and much less attention has been paid to dopants such as heavy chalcogens. Selenium constitutes a peculiar carbon dopant due to its unique chemical properties, including nucleophilicity and moderate affinity to oxygen, yet extreme affinity to heavy metals. Se-doped carbonaceous materials represent promising heterogeneous catalysts for electro- and thermocatalytic processes, including a broad range of electrochemical reduction processes such as oxygen, carbon dioxide, iodine, cobalt, or hydrazine reduction reactions, and organic syntheses involving oxygen transfer. The weak selenium–oxygen bonding makes the reaction of Se with oxygen highly reversible and hence Se is an excellent oxygen carrier. In the case of heterogeneous (electro)catalysis, the highly polarizable and valence electron-rich Se atom (4s24p4) proved to be an efficient long-range modulator of the d- or p-electron structure of transition metal and main-group semimetal single-atom catalytic centers. Doping Se into chars makes the resultant hard carbons attractive materials for alkali-ion batteries and electrochemical capacitors. The synthesis of Se-doped carbons comprises pyrolysis of carbon and selenium-bearing feedstocks. Selenium powder, followed by selenium dioxide, and diphenyl diselenide have been identified as the most commonly utilized Se dopant sources (Table S1†). This shows that Se and SeO2 powders are the most convenient and the cheapest Se-sources for doping carbonaceous materials. It is important to note that to boost the performance of the final material, Se doping is often accompanied by nitrogen co-doping and additional co-doping with transition metals – mostly Fe. By employing simple pyrolysis, a wide range of selenium-doped and co-doped materials with varying Se contents and surface areas has been prepared and then evaluated in practical applications. The pyrolytic temperatures vary in a very wide range of ca. 300 up to 1110 °C yielding Se-doped carbons with SSAs of up to 1700 m2 g−1. Nevertheless, there are still some challenges to be overcome before chalcogen-doped carbonaceous materials find their way to commercial devices such as fuel cells, batteries, or photovoltaic cells. The main trouble spots we have managed to pinpoint include:• Discrimination between substitutional doping and an undefined presence of the Se dopant is often too elusive in scientific reports.
• Abuse of the synergy term is widespread. The complex impact of carbon doping/co-doping on its catalytic properties is usually elucidated by using elusive expressions such as: synergistically enhanced performance.
• When metal-free catalysts are studied, incidental contamination with traces of metals should be borne in mind, especially since Se possesses extraordinary affinity to transition and heavy metals.
• As chalcogens exhibit high affinity to transition metals, for Se-doped carbons additionally co-doped with these metals selenide particles may be preferentially formed instead of single-atom sites with Se atom neighboring transition metal atom. Consequently, obtaining SACs and DASCs without selenide NPs might be challenging.
• While Se doping does modify fundamental properties of carbonaceous materials, the omnipresence of the lightest chalcogen, i.e., oxygen, on the carbon surface affects their physicochemical characteristics, especially where surface chemistry is concerned (e.g., catalysis or pseudo-capacitance).
• While pyrolysis is a straightforward approach for preparing heteroatom-doped carbons, it does not allow us to obtain doped carbons with a well-defined heteroatom configuration. In addition, the very wide range of peak pyrolysis temperatures applied (300–1110 °C) yields Se-doped carbons of extremely varied characteristics, making systematic comparison challenging.
• While HTT allows control of the Se doping level, it simultaneously causes other serious modifications, such as microporosity enhancement via chalcogen desorption. Consequently, in some cases, the amount of doped Se might be less important than the resultant nanoporosity.
• Se content assessment is more challenging than quantification of the more commonly doped heteroatoms such as N or S. EDS and XPS are commonly used, but these techniques are not accurate enough in the particular case of carbonaceous samples of heterogeneous nature. Consequently, the accuracy of such techniques is limited to several or even to several dozen percent.
• Graphitization of carbonaceous scaffold can be significantly affected by Se-doping either by increasing amorphicity (via formation of micropores upon thermal desorption of Se) or via prevention of metal-catalyzed graphitization due to blockage of metal carbides formation (i.e., Se prevents dissolution of elemental carbon in transition metals).
Perusing through reports on Se-doped carbons, some misinterpretations of the observed effects can also be found. For instance, the interlayer d-spacing value for the Se-doped partly graphitized carbons is always higher than that of graphite (0.335 nm), and this is often attributed to the large size of Se atoms. This is an erroneous interpretation because chars, especially biochars, constitute non-graphitizing carbons, and they yield only turbostratic carbons with interlayer space of ∼0.344 nm, even after pyrolysis at high temperatures >2000 °C. Consequently, the interlayer space in (bio)chars cannot be assigned to the presence of large heteroatoms. In fact, carbonaceous materials obtained via pyrolytic decomposition of biomass are never graphitic as they possess the highly disordered structure of the so-called hard carbons. And finally, while there is a profusion of papers considering how Se-doping influences the electronic structure of graphenic carbons, there are not many papers elucidating other crucial aspects, such as how Se-doping affects the formation of porosity or how it enhances or hinders graphitization of the carbonaceous matrix. In fact, in this regard, Se-doping is very similar to sulfur-doping, as S-doping has the same effect on microporosity enhancement with the increase of pyrolysis temperature. Nevertheless, despite some challenges, there is significant and increasing evidence that Se-doped and co-doped carbons are attractive, functional materials for the implementation of green chemistry ideas as well as sustainable and renewable technological transition.
List of abbreviations
| BET | Brunauer–Emmett–Teller |
| Bpy | 2,2′-Bipyridine |
| CE | Counter electrode |
| CNT | Carbon nanotube |
| CO2RR | CO2 reduction reaction |
| CRR | Cobalt reduction reaction |
| CQD | Carbon quantum dot |
| DASC | Dual-atomic-site catalyst |
| DDSe | Diphenyl diselenide |
| DETA | Diethylenetriamine |
| DFT | Density functional theory |
| DSSC | Dye-sensitized solar cell |
| EDS (EDX) | Energy-dispersive X-ray spectroscopy |
| EXAFS | Extended X-ray absorption fine structure |
| FECO | CO faradaic efficiency |
| GDE | Gas diffusion electrode |
| GO | Graphene oxide |
| GPx | Glutathione peroxidase |
| HAADF | High-angle annular dark field |
| HER | Hydrogen evolution reaction |
| HMT | Hexamethylenetetramine |
| (HR)TEM | (High-resolution) transmission electron microscopy |
| HTT | Heat treatment temperature |
| HzFC | Hydrazine fuel cell |
| HzOR | Hydrazine oxidation reaction |
| ICP-MS | Inductively coupled plasma mass spectrometry |
| ICP-OES | Inductively coupled plasma optical emission spectroscopy |
| IL | Ionic liquid |
| IRR | Iodine reduction reaction |
| LIB | Lithium-ion battery |
| M–N–C | Metal–nitrogen–carbon |
| MOF | Metal–organic framework |
| NP | Nanoparticle |
| OCV | Open circuit voltage |
| OER | Oxygen evolution reaction |
| ORR | Oxygen reduction reaction |
| PAN | Polyacrylonitrile |
| PBA | Poly(n-butyl acrylate) |
| PCE | Power conversion efficiency |
| PEFC | Polymer electrolyte fuel cell |
| PIB | Potassium-ion battery |
| PINO | Phthalimido-N-oxyl |
| PMMA | Polymethylmethacrylate |
| PMS | Peroxymonosulfate |
| PPy | Polypyrrole |
| rGO | Reduced graphene oxide |
| RHE | Reversible hydrogen electrode |
| ROS | Reactive oxygen species |
| SAC | Single-atom catalyst |
| SEM | Scanning electron microscopy |
| SIB | Sodium-ion battery |
| SSA | Specific surface area |
| STEM | Scanning transmission electron microscopy |
| TMB | 3,3′,5,5′-Tetramethylbenzidine |
| XAFS | X-ray absorption fine structure spectroscopy |
| XANES | X-ray absorption near-edge structure |
| XAS | X-ray absorption spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
| ZAB | Zinc–air battery |
| ZIF | Zeolitic imidazolate framework |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Centre, Poland, grant number 2020/37/B/ST5/01688.References
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS
.
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC
- C. Hu, R. Paul, Q. Dai and L. Dai, Chem. Soc. Rev., 2021, 50, 11785–11843 RSC
- B. Wu, H. Meng, D. M. Morales, F. Zeng, J. Zhu, B. Wang, M. Risch, Z. J. Xu and T. Petit, Adv. Funct. Mater., 2022, 32, 2204137 CrossRef CAS
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC
- J. Zhang, G. Chen, K. Mullen and X. Feng, Adv. Mater., 2018, 30, 1800528 CrossRef PubMed
- E. J. Askins, M. R. Zoric, M. Li, Z. Luo, K. Amine and K. D. Glusac, Nat. Commun., 2021, 12, 3288 CrossRef CAS PubMed
- S. Zhao, D. W. Wang, R. Amal and L. Dai, Adv. Mater., 2019, 31, 1801526 CrossRef PubMed
- T. J. Bandosz and T.-Z. Ren, Carbon, 2017, 118, 561–577 CrossRef CAS
- S. Shaheen Shah, S. M. Abu Nayem, N. Sultana, A. J. Saleh Ahammad and M. Abdul Aziz, ChemSusChem, 2022, 15, 202101282 CrossRef PubMed
- W. Kiciński, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef
- P. A. Denis, ACS Omega, 2022, 7, 45935–45961 CrossRef CAS PubMed
- M. Jerigova, M. Odziomek and N. Lopez-Salas, ACS Omega, 2022, 7, 11544–11554 CrossRef CAS PubMed
- P. Lisowski, J. C. Colmenares, O. Mašek, W. Lisowski, D. Lisovytskiy, A. Kamińska and D. Łomot, ACS Sustainable Chem. Eng., 2017, 5, 6274–6287 CrossRef CAS
- J. C. Colmenares, R. S. Varma and P. Lisowski, Green Chem., 2016, 18, 5736–5750 RSC
- I. A. Pasti, A. Jovanovic, A. S. Dobrota, S. V. Mentus, B. Johansson and N. V. Skorodumova, Phys. Chem. Chem. Phys., 2018, 20, 858–865 RSC
- M. Kieliszek, Molecules, 2019, 24, 1298 CrossRef CAS PubMed
- L. L. Stillings , Selenium, chap. Q of K.J. Schulz, J.H. DeYoung, Jr., R.R. Seal II, and D.C. Bradley, eds., Critical mineral resources of the United States-Economic and environmental geology and prospects for future supply: U.S. Geological Survey Professional Paper 1802, 2017 p. Q1–Q55.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed
- J. Meyer, J.-M. Moulis, J. Gaillard and M. Lutz, Adv. Inorg. Chem., 1992, 38, 73–115 CrossRef CAS
- L. A. Wessjohann, A. Schneider, M. Abbas and W. Brandt, Biol. Chem., 2007, 388, 997–1006 CrossRef CAS PubMed
- M. J. Maroney and R. J. Hondal, Free Radical Biol. Med., 2018, 127, 228–237 CrossRef CAS PubMed
- K. A. Krishnan and T. S. Anirudhan, Ind. Eng. Chem. Res., 2002, 41, 5085–5093 CrossRef CAS
- H. A. Spiller, Clin. Toxicol., 2018, 56, 313–326 CrossRef CAS PubMed
- S. Ahmed, J. Brockgreitens, K. Xu and A. Abbas, Adv. Funct. Mater., 2017, 27, 1606572 CrossRef
- Y. Huang, E. Su, J. Ren and X. Qu, Nano Today, 2021, 38, 101205 CrossRef CAS
- T. M. Sakr, M. Korany and K. V. Katti, J. Drug Delivery Sci. Technol., 2018, 46, 223–233 CrossRef CAS
- X. Xiao, Z. Shao and L. Yu, Chin. Chem. Lett., 2021, 32, 2933–2938 CrossRef CAS
- Y. Safaei Ardakani and M. Moradi, Eur. Phys. J. B, 2020, 93, 99 CrossRef CAS
- P. A. Denis and F. Iribarne, Chem. Phys. Lett., 2016, 658, 152–157 CrossRef CAS
- P. Kong, X. Zhang, J. Wang, W. Tian, Y. Ni, B. Sun, H. Wang, H. Wang, Y. P. Feng and Y. Chen, Phys. Rev. B, 2023, 107, 184115 CrossRef CAS
- P. A. Denis, ChemPhysChem, 2014, 15, 3994–4000 CrossRef CAS PubMed
- Y. Huang, Y. Chen, M. Xu, T. Asset, P. Tieu, A. Gili, D. Kulkarni, V. De Andrade, F. De Carlo, H. S. Barnard, A. Doran, D. Y. Parkinson, X. Pan, P. Atanassov and I. V. Zenyuk, Mater. Today, 2021, 47, 53–68 CrossRef CAS
- D. E. Newbury and N. W. Ritchie, Scanning, 2013, 35, 141–168 CrossRef CAS PubMed
- Y. S. Yun, G. Yoon, M. Park, S. Y. Cho, H.-D. Lim, H. Kim, Y. W. Park, B. H. Kim, K. Kang and H.-J. Jin, NPG Asia Mater., 2016, 8, 338 CrossRef
- Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205–211 CrossRef CAS PubMed
- Z. Jin, H. Nie, Z. Yang, J. Zhang, Z. Liu, X. Xu and S. Huang, Nanoscale, 2012, 4, 6455–6460 RSC
- S. Pal, S. Bawari, T. Veettil Vineesh, N. Shyaga and T. N. Narayanan, ACS Appl. Energy Mater., 2019, 2, 3624–3632 CrossRef CAS
- C. H. Choi, M. W. Chung, Y. J. Jun and S. I. Woo, RSC Adv., 2013, 3, 12417 RSC
- J. Chen, C. Lin, M. Zhang, T. Jin and Y. Qian, ChemElectroChem, 2020, 7, 3311–3318 CrossRef CAS
- W. Kiciński, B. Budner, M. Norek, M. Szala, M. Polański and S. Dyjak, Microporous Mesoporous Mater., 2018, 272, 260–264 CrossRef
- J. Wang, Y. Wang, H. Hu, Q. Yang and J. Cai, Nanoscale, 2020, 12, 4238–4268 RSC
- H. Wang, L. R. Grabstanowicz, H. M. Barkholtz, D. Rebollar, Z. B. Kaiser, D. Zhao, B.-H. Chen and D.-J. Liu, ACS Energy Lett., 2019, 4, 2500–2507 CrossRef CAS
- S. Wei, A. Li, J. C. Liu, Z. Li, W. Chen, Y. Gong, Q. Zhang, W. C. Cheong, Y. Wang, L. Zheng, H. Xiao, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, Nat. Nanotechnol., 2018, 13, 856–861 CrossRef CAS PubMed
- H. Hu, J. Wang, B. Cui, X. Zheng, J. Lin, Y. Deng and X. Han, Angew. Chem., Int. Ed., 2022, 61, 202114441 CrossRef PubMed
- C. Qi, W. Wang and Y. Dong, Anal. Bioanal. Chem., 2023, 415, 5391–5401 CrossRef CAS PubMed
- Y. Wang, J. Wu, S. Tang, J. Yang, C. Ye, J. Chen, Y. Lei and D. Wang, Angew. Chem., Int. Ed., 2023, 62, 202219191 CrossRef PubMed
- Z. Sun, H. Zhang, L. Cao, X. Liu, D. Wu, X. Shen, X. Zhang, Z. Chen, S. Ru, X. Zhu, Z. Xia, Q. Luo, F. Xu and T. Yao, Angew. Chem., Int. Ed., 2023, 62, 202217719 CrossRef PubMed
- K. Sun, K. Yu, J. Fang, Z. Zhuang, X. Tan, Y. Wu, L. Zeng, Z. Zhuang, Y. Pan and C. Chen, Adv. Mater., 2022, 34, 2206478 CrossRef CAS PubMed
- Z. Niu, Z. Lu, Z. Qiao, M. Xing, L. Han, S. Wang and D. Cao, ACS Catal., 2023, 13, 7122–7131 CrossRef CAS
- J. Lian, J. Zhao, X. Wang and Q. Bai, Carbon, 2023, 213, 118257 CrossRef CAS
- H. Liang, L. Jia, F. Chen, S. Jing and P. Tsiakaras, Appl. Catal., B, 2022, 317, 121698 CrossRef CAS
- B. Yang, S. Liu, Y. V. Fedoseeva, A. V. Okotrub, A. A. Makarova, X. Jia and J. Zhou, J. Power Sources, 2021, 493, 229700 CrossRef CAS
- H. Wang, Y. Shao, S. Mei, Y. Lu, M. Zhang, J. K. Sun, K. Matyjaszewski, M. Antonietti and J. Yuan, Chem. Rev., 2020, 120, 9363–9419 CrossRef CAS PubMed
- J. Zhou, M. Chen, T. Wang, S. Li, Q. Zhang, M. Zhang, H. Xu, J. Liu, J. Liang, J. Zhu and X. Duan, iScience, 2020, 23, 100919 CrossRef CAS PubMed
- Y. Liu, H. Dai, Y. An, L. Fu, Q. An and Y. Wu, J. Mater. Chem. A, 2020, 8, 14993–15001 RSC
- S. Liu, M. Wang, T. Qian, J. Liu and C. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 20056–20063 CrossRef CAS PubMed
- M. Zhou, X. Zhang, Y. Huang and P. Feng, Sens. Actuators, B, 2024, 399, 134779 CrossRef CAS
- Y. Ma, X. Meng, K. Li, L. Zhang, Y. Du, X. Cai and J. Qiu, ACS Catal., 2023, 13, 1290–1298 CrossRef CAS
- Z. Qian, R. Guo, Y. Ma, C. Li, L. Du, Y. Wang, C. Du, H. Huo and G. Yin, Chem. Eng. Sci., 2020, 214, 115413 CrossRef CAS
- T. Wang, Q. Wang, Y. Wang, Y. Da, W. Zhou, Y. Shao, D. Li, S. Zhan, J. Yuan and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 13466–13471 CrossRef CAS PubMed
- M. Zhang, M. Ning, K. Xiong, Z. Duan, X. Yang and Z. Li, Phys. Chem. Chem. Phys., 2023, 25, 7213–7222 RSC
- Y. Wu, M. Jing, J. Li, W. Deng, M. Yi, Z. Chen, M. Yang, J. Wu, X. Xu, Y. Bai, X. Zou, T. Wu and X. Wang, Chin. Chem. Lett., 2023, 109269 CrossRef
- J. L. Do and T. Friscic, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS
- M. J. Ju, I. Y. Jeon, H. M. Kim, J. I. Choi, S. M. Jung, J. M. Seo, I. T. Choi, S. H. Kang, H. S. Kim, M. J. Noh, J. J. Lee, H. Y. Jeong, H. K. Kim, Y. H. Kim and J. B. Baek, Sci. Adv., 2016, 2, 1501459 CrossRef PubMed
- X. Meng, C. Yu, X. Song, J. Iocozzia, J. Hong, M. Rager, H. Jin, S. Wang, L. Huang, J. Qiu and Z. Lin, Angew. Chem., Int. Ed., 2018, 57, 4682–4686 CrossRef CAS PubMed
- Y. Sun, K. Zhao, X. Deng, M. Zhang, X. Wang and W. Wang, J. Environ. Chem. Eng., 2023, 11, 110435 CrossRef CAS
- Z. Chen, X. Su, J. Ding, N. Yang, W. Zuo, Q. He, Z. Wei, Q. Zhang, J. Huang and Y. Zhai, Appl. Catal., B, 2022, 308, 121206 CrossRef CAS
- C. Qi, Y. Luo and Y. Dong, Appl. Surf. Sci., 2023, 637, 157900 CrossRef CAS
- J. Yan, H. Ma, J. Ni, J. Ma, J. Xu, J. Qi, S. Zhu and L. Lu, J. Colloid Interface Sci., 2023, 648, 558–566 CrossRef CAS PubMed
- X. Cai, Y. Xu, F. Mo, F. Kong, L. Fan, Y. Tan, G. Zhang, S. Chu, W. Chu, S. Tao and L. Song, ACS Appl. Mater. Interfaces, 2023, 15, 29204–29213 CrossRef CAS PubMed
- W. Zhou, Y. Liu, H. Dai, X. Yuan, Y. Peng, W. Huang, L. Fu, Y. Zhu, Y. Wu and X. Wang, J. Solid State Electrochem., 2021, 25, 457–464 CrossRef CAS
- H. He, R. Zhang, P. Zhang, P. Wang, N. Chen, B. Qian, L. Zhang, J. Yu and B. Dai, Adv. Sci., 2023, 10, 2205557 CrossRef CAS
- Y. Yang, X. Fan, H. Cao, S. Chu, X. Zhang, Q. Xu and L. Yu, Catal. Sci. Technol., 2018, 8, 5017–5023 RSC
- S. Chu, H. Cao, T. Chen, Y. Shi and L. Yu, Catal. Commun., 2019, 129, 105730 CrossRef
- H. Cao, Y. Yang, X. Chen, J. Liu, C. Chen, S. Yuan and L. Yu, Chin. Chem. Lett., 2020, 31, 1887–1889 CrossRef CAS
- P. Li, Z. Qi, L. Yu and H. Zhou, Catal. Sci. Technol., 2022, 12, 2241–2247 RSC
- Z. Zhu, S. Sun and X. Jing, Chem. Pap., 2022, 76, 401–408 CrossRef CAS
- B. Zhang, J. Zhang, F. Zhang, L. Zheng, G. Mo, B. Han and G. Yang, Adv. Funct. Mater., 2020, 30, 1906194 CrossRef CAS
- G. S. dos Reis, J. Thivet, E. Laisné, V. Srivastava, A. Grimm, E. C. Lima, D. Bergna, T. Hu, M. Naushad and U. Lassi, Chem. Eng. Sci., 2023, 281, 119129 CrossRef CAS
- N. Abedian-Dehaghani, S. Sadjadi and M. M. Heravi, J. Mol. Struct., 2022, 1264, 133237 CrossRef CAS
- K. Zhang, X. Min, T. Zhang, M. Xie, M. Si, L. Chai and Y. Shi, J. Hazard. Mater., 2021, 413, 125294 CrossRef CAS PubMed
- Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS
- R. Wang, H. Da, H. Wang, S. Ji and Z. Tian, J. Power Sources, 2013, 233, 326–330 CrossRef CAS
- H. Wang and Q. Ma, J. Solid State Electrochem., 2015, 19, 355–360 CrossRef CAS
- F. Sabour, F. Nemati and Y. Rangraz, Appl. Organomet. Chem., 2022, 36, 6716 CrossRef
- A. Samandi, F. Nemati, M. S. Mirhosseyni and S. Sabaqian, Appl. Organomet. Chem., 2023, 37, 6988 CrossRef
- Y. Liu, C. Ma, J. Zhou, L. Zhu, L. Cao and J. Yang, Environ. Sci. Pollut. Res., 2022, 29, 69450–69461 CrossRef CAS PubMed
- L. Wu, S. Guo, H. Yue, H. Li, W. Li, C. Yao, P. Li, W. Fa, B. Song, K. Li, B. Zhou, Q. Yu, Y. Xu, C. Yang, Z. Zheng and Y. Gao, Batteries, 2023, 9, 143 CrossRef CAS
- J. K. Kim and Y. C. Kang, ACS Nano, 2020, 14, 13203–13216 CrossRef CAS PubMed
- Y. Huang, C. Liu, F. Pu, Z. Liu, J. Ren and X. Qu, Chem. Commun., 2017, 53, 3082–3085 RSC
- S. Maity and B. B. Dhar, Catal. Sci. Technol., 2022, 12, 1296–1312 RSC
- X. Kou, S. Jiang, S. J. Park and L. Y. Meng, Dalton Trans., 2020, 49, 6915–6938 RSC
- F. Li, D. Yang and H. Xu, Chem. – Eur. J., 2019, 25, 1165–1176 CrossRef CAS PubMed
- S. Miao, K. Liang, J. Zhu, B. Yang, D. Zhao and B. Kong, Nano Today, 2020, 33, 100879 CrossRef CAS
- F. Li, T. Li, C. Sun, J. Xia, Y. Jiao and H. Xu, Angew. Chem., Int. Ed., 2017, 56, 9910–9914 CrossRef CAS PubMed
- Z. T. Rosenkrans, T. Sun, D. Jiang, W. Chen, T. E. Barnhart, Z. Zhang, C. A. Ferreira, X. Wang, J. W. Engle, P. Huang and W. Cai, Adv. Sci., 2020, 7, 2000420 CrossRef CAS PubMed
- B. Bai, S. Qi, K. Yang, X. Yu, R. Jian, T. Zhang, D. Wang, H. Meng, Y. Zhao, Y. Xia, H. Xu, G. Yu and Z. Chen, Small, 2023, 19, 2300217 CrossRef CAS PubMed
- J. Li, Z. Wang, Y. Zhang, X. Cao, F. Lian and S. Gu, Environ. Sci.: Nano, 2023, 10, 866–878 RSC
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 RSC
- W. T. Borden, R. Hoffmann, T. Stuyver and B. Chen, J. Am. Chem. Soc., 2017, 139, 9010–9018 CrossRef CAS
- A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS PubMed
- Y. Li and J. Lu, ACS Energy Lett., 2017, 2, 1370–1377 CrossRef CAS
- W. Kiciński and S. Dyjak, Carbon, 2020, 168, 748–845 CrossRef
- V. Sankar Devi and P. Elumalai, New J. Chem., 2023, 47, 2189–2201 RSC
- Z. Shi, W. Yang, Y. Gu, T. Liao and Z. Sun, Adv. Sci., 2020, 7, 2001069 CrossRef CAS
- W. Liu, H. He, Q. Liu, X. Wan and J. Shui, Particuology, 2024, 89, 99–108 CrossRef CAS
- B.-L. Lai, H.-X. Liao, S.-Q. Zhou, H.-X. Wei, A.-Y. Li, N. Li and Z.-Q. Liu, J. Environ. Chem. Eng., 2023, 11, 111076 CrossRef CAS
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed
- A. B. Munoz-Garcia, I. Benesperi, G. Boschloo, J. J. Concepcion, J. H. Delcamp, E. A. Gibson, G. J. Meyer, M. Pavone, H. Pettersson, A. Hagfeldt and M. Freitag, Chem. Soc. Rev., 2021, 50, 12450–12550 RSC
- Y. Hou, D. Wang, X. H. Yang, W. Q. Fang, B. Zhang, H. F. Wang, G. Z. Lu, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 1583 CrossRef PubMed
- Y. Zhong, H. Xu, Q. Zhang, X. Song and C. Hao, ChemistrySelect, 2022, 7, 202200380 CrossRef
- L. F. Zhao, Z. Hu, W. H. Lai, Y. Tao, J. Peng, Z. C. Miao, Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2021, 11, 2002704 CrossRef CAS
- Y. Huang, Y. Wang, P. Bai and Y. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 38441–38449 CrossRef CAS PubMed
- Y. Wang, L. Zhang, H. Hou, W. Xu, G. Duan, S. He, K. Liu and S. Jiang, J. Mater. Sci., 2021, 56, 173–200 CrossRef CAS
- Y. Zheng, K. Chen, K. Jiang, F. Zhang, G. Zhu and H. Xu, J. Energy Storage, 2022, 56, 105995 CrossRef
- Z. Y. Choong, K. A. Lin, G. Lisak, T. T. Lim and W. D. Oh, J. Hazard. Mater., 2022, 426, 128077 CrossRef CAS PubMed
- G. R. Paixão, N. G. Camparotto, G. d. V. Brião, R. d. L. Oliveira, J. C. Colmenares, P. Prediger and M. G. A. Vieira, Chem. Eng. Res. Des., 2022, 187, 225–239 CrossRef
- D. Dyrssen and M. Wedborg, Water, Air, Soil Pollut., 1991, 56, 507–519 CrossRef
- X. Jing, C. Chen, X. Deng, X. Zhang, D. Wei and L. Yu, Appl. Organomet. Chem., 2018, 32, 4332 CrossRef
- Z. Qian, Y. Zhang, X. Pan, N. Li, J. Zhu and X. Zhu, React. Funct. Polym., 2019, 142, 223–230 CrossRef CAS
- Z. Zhu, W. Wang, F. Zhang and J. Liu, Mater. Lett., 2020, 261, 127011 CrossRef CAS
- L. Shen, Z. Xiong, Z. Du, H. Wang, W. Li, Z. Zhang, H. Wu, C. Zhou, Y. Zhou, G. Tang, J. Liu and H. Yang, Sep. Purif. Technol., 2024, 334, 126078 CrossRef CAS
- Y. Chai, H. Dai, P. Zhan, Z. Liu, Z. Huang, C. Tan, F. Hu, X. Xu and X. Peng, J. Hazard. Mater., 2023, 452, 131202 CrossRef CAS PubMed
- S. Maity, S. Deshmukh, S. S. Roy and B. B. Dhar, ChemElectroChem, 2022, 10, 202201044 CrossRef
- A. Madabeni, S. Zucchelli, P. A. Nogara, J. B. T. Rocha and L. Orian, J. Org. Chem., 2022, 87, 11766–11775 CrossRef CAS PubMed
- H. Cao, R. Qian and L. Yu, Catal. Sci. Technol., 2020, 10, 3113–3121 RSC
- F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073–1091 RSC
- X. Xiao, C. Guan, J. Xu, W. Fu and L. Yu, Green Chem., 2021, 23, 4647–4655 RSC
- C. Chen, Y. Cao, X. Wu, Y. Cai, J. Liu, L. Xu, K. Ding and L. Yu, Chin. Chem. Lett., 2020, 31, 1078–1082 CrossRef CAS
- R. L. Oliveira, K. Nicinski, M. Pisarek, A. Kaminska, A. Thomas, G. Pasternak and J. C. Colmenares, ChemCatChem, 2022, 14, 202200787 CrossRef
- S. S. Shang and S. Gao, ChemCatChem, 2019, 11, 3730–3744 CrossRef CAS
- A. Mahajan and P. Gupta, Environ. Chem. Lett., 2020, 18, 299–314 CrossRef CAS
- W. Wang, L. Ma, H. Wang, X. Jiang, Z.-H. He, K. Wang, Y. Yang, L. Li and Z.-T. Liu, ACS Appl. Nano Mater., 2023, 6, 15193–15203 CrossRef CAS
- W. Wang, T. Bao, H. Wang, X. Jiang, Z.-H. He, K. Wang, Y. Yang, L. Li and Z.-T. Liu, Mol. Catal., 2023, 541, 113112 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04804g |
| This journal is © The Royal Society of Chemistry 2024 |