
Direct electrochemical synthesis of arenesulfonyl fluorides from nitroarenes: a dramatic ionic liquid effect†
Xianqiang
Kong
 *a,
Qianwen
Liu
a,
Yiyi
Chen
a,
Wei
Wang
a,
Hong-Fa
Chen
b,
Wenjie
Wang
a,
Shuangquan
Zhang
a,
Xiaohui
Chen
*a and
Zhong-Yan
Cao
*b
*a,
Qianwen
Liu
a,
Yiyi
Chen
a,
Wei
Wang
a,
Hong-Fa
Chen
b,
Wenjie
Wang
a,
Shuangquan
Zhang
a,
Xiaohui
Chen
*a and
Zhong-Yan
Cao
*b
aSchool of Chemical Engineering and Materials, Changzhou Institute of Technology, No. 666 Liaohe Road, Changzhou 213032, China. E-mail: kongxq@czu.cn; chenxh@czu.com
bCollege of Chemistry and Molecular Sciences, Henan University, Kaifeng 475004, P. R. China. E-mail: zycao@henu.edu.cn
First published on 1st February 2024
Abstract
A practical electrochemical strategy for the direct synthesis of arenesulfonyl fluorides from industrial feedstock nitroarenes is described. The key to success lies in using a cheap ionic liquid N-methylimidazolium p-toluenesulfonate ([Mim]TolSO3) as an effective additive and electrolyte to facilitate the selective reduction of nitroarenes to the corresponding aniline intermediate, promoting the desired fluorosulfonylation with broad functional group tolerance under mild conditions.
Introduction
With the emergence of new applications for fluorinating reagents,118F radiolabeling agents,2 and other fluorinated compounds for chemical biology research,3 interest in the synthesis of sulfonyl fluorides, including arenesulfonyl fluorides, has dramatically increased recently.4 Aryl sulfonyl fluorides are typically synthesized by chloride–fluoride exchange reactions of the corresponding arenesulfonyl chlorides,5 oxidative fluorination of sulfur-containing substrates (such as sodium sulfinic,6 thiols,7 sulfonyl hydrazides,8 disulfides,7b,9 and sulfonates or sulfonic acids10) or by converting various prefunctionalized substrates such as aryl (pseudo)halides,11 aryl boronic acids,12 aryl diazonium salts,13 and others.14 Despite these advances, the development of practical, mild methods for accessing arenesulfonyl fluorides from inexpensive, widely available industrial feedstocks remains highly desirable.On the other hand, nitroarenes are inexpensive and abundant industrial chemical feedstocks that can be easily accessed by the electrophilic aromatic nitration of arenes.15 The reported methods for transforming nitroarenes into arenesulfonyl fluorides often require multiple steps, which include the conversion of nitroarenes to the corresponding diazo salts by reduction/derivatization first, and the subsequent transformation with an SO2 surrogate and fluorinating reagents enables the delivery of arenesulfonyl fluorides (Scheme 1a).13 Although these reliable multistep methods are widely used in organic synthesis, they have some drawbacks, including the need for tedious separation procedures and the generation of a lot of chemical waste.13 Therefore, a strategy for the direct denitrative fluorosulfonylation of nitroarenes to afford arenesulfonyl fluorides would be highly desirable in terms of step economy, environmental friendliness, cost, and speed. However, to the best of our knowledge, the use of nitroarenes for the direct synthesis of arenesulfonyl fluorides remains unexplored.
 | ||
| Scheme 1 Synthesis of arenesulfonyl fluorides from nitroarenes. | ||
Electrochemistry, which has the advantages of being green and sustainable, has received considerable attention from researchers in academia and industry in the last decade.16 In this scenario, coupled with our previous success in electrosynthesis17 and the reductive dialkylation of nitroarenes,18 we envisioned that the use of electrochemistry tools to reduce nitroarenes in situ might provide a meaningful strategy to realize the anticipated denitrative fluorosulfonylation by overcoming the previous methods’ tedious reduction and pre-functionalization. Nevertheless, it is nontrivial to achieve this goal, as precedent examples have identified that depending on the specific reduction conditions, the intermediates for the reduction of nitroarenes might be completely different and complicated.19 For example, the reduction of nitrobenzene could result in nitrosobenzene, N-phenyl hydroxylamine, azoxybenzene, azobenzene, 1,2-diphenylhydrazine, or PhNH2.18,19 To this end, a prerequisite for the desired fluorosulfonylation lies in choosing suitable conditions that enable the selective reduction of nitroarenes to be realized in a controllable way. In addition, such reduction conditions should be compatible with the subsequent fluorosulfonylation process. Herein, by successfully overcoming the aforementioned challenges, we wish to report our preliminary results on the electrochemical denitrative fluorosulfonylation of nitroarenes (Scheme 1b). As detailed below, our strategy enables the preparation of arenesulfonyl fluorides with high efficiency and shows broad functional group tolerance.
Results and discussion
To find the optimal conditions for the desired denitrative fluorosulfonylation of nitroarenes, 4-nitrotoluene (1a) was chosen as the model substrate. After extensive screening of various reaction parameters, we were pleased to notice that the desired product 3a could be facilely isolated with 72% yield using commercially available 1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) (DABSO, 2) as an SO2 surrogate, Selectfluor as a fluorinating reagent in an undivided cell with a graphite anode and a glassy carbon (GC) cathode (Table 1). As shown in entry 1, the reaction for 6 h at room temperature in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of N-methylimidazolium p-toluenesulfonate ([Mim]TolSO3) and MeOH containing tert-butyl nitrite (tBuONO) as an additive at a constant current of 12 mA gave the best result. During the whole process, the voltage of the cell was 2.6–3.0 V. The essential dual role (as an electrolyte and a key additive for the selective reduction of nitroarenes) of [Mim]TolSO3 will be detailed later. In comparison, replacing MeOH with MeCN, DMF, EtOH, CF3CH2OH or (CF3)2CHOH led to a lower or trace yield (entries 2–4). The replacement of [Mim]TolSO3 with various alternative ionic liquids or common Brønsted acids (such as TsOH, H2SO4, or HCl) also decreased the yield (entries 5 and 6). This indicates that both the cation and anion parts of (Mim)TolSO3 play very important roles for achieving good results. Other electrode combinations—RVC(+)/RVC(−) (RVC = reticulated vitreous carbon), Pt(+)/GC(−), and GC(+)/GC(−)—also gave similar yields (entry 7), as did alternative SO2 surrogates (entry 8) and fluoride sources (entry 9). In addition, reducing or increasing the current lowered the yield (entry 10). Under an air atmosphere, the yield was only 43% (entry 11). The control experiment showed that tBuONO played a vital role in this transformation (entry 12), as did the electric current and [Mim]TolSO3 (entry 13); none of the desired product was obtained in their absence.
:1 (v/v) mixture of N-methylimidazolium p-toluenesulfonate ([Mim]TolSO3) and MeOH containing tert-butyl nitrite (tBuONO) as an additive at a constant current of 12 mA gave the best result. During the whole process, the voltage of the cell was 2.6–3.0 V. The essential dual role (as an electrolyte and a key additive for the selective reduction of nitroarenes) of [Mim]TolSO3 will be detailed later. In comparison, replacing MeOH with MeCN, DMF, EtOH, CF3CH2OH or (CF3)2CHOH led to a lower or trace yield (entries 2–4). The replacement of [Mim]TolSO3 with various alternative ionic liquids or common Brønsted acids (such as TsOH, H2SO4, or HCl) also decreased the yield (entries 5 and 6). This indicates that both the cation and anion parts of (Mim)TolSO3 play very important roles for achieving good results. Other electrode combinations—RVC(+)/RVC(−) (RVC = reticulated vitreous carbon), Pt(+)/GC(−), and GC(+)/GC(−)—also gave similar yields (entry 7), as did alternative SO2 surrogates (entry 8) and fluoride sources (entry 9). In addition, reducing or increasing the current lowered the yield (entry 10). Under an air atmosphere, the yield was only 43% (entry 11). The control experiment showed that tBuONO played a vital role in this transformation (entry 12), as did the electric current and [Mim]TolSO3 (entry 13); none of the desired product was obtained in their absence.
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield of 3ab (%) |
| a Standard conditions: graphite (C, 0.8 × 0.2 cm2) anode, glassy carbon (GC, 0.8 × 0.2 cm2) cathode, 12 mA, 1a (0.30 mmol), 2 (0.20 mmol), Selectfluor (0.60 mmol), tBuONO (0.90 mmol), [Mim]TolSO3 (0.5 mL), MeOH (4 mL), room temperature (rt), N2 atmosphere, and 6 h. b Isolated yields. NFSI: N-fluorobenzenesulfonamide. [Bmim]: 1-butyl-3-methylimidazolium hexafluorophosphate. | ||
| 1 | None | 72 |
| 2 | CH3CN instead of MeOH | 32 |
| 3 | DMF instead of MeOH | 42 |
| 4 | EtOH, CF3CH2OH, (CF3)2CHOH instead of MeOH | 65/trace/trace |
| 5 | [Bmim]Br, [Bmim]PF6, [Bmim]OAc, [Mim]Cl, [Mim]OAc, or [Mim]HSO4 instead of [Mim]TolSO3 | Trace/8/13/0/0/13 |
| 6 | H2SO4, HCl, TsOH instead of [Mim]TolSO3 | 0/0/0 |
| 7 | RVC(+)/RVC(−), Pt(+)/GC(−), or GC(+)/GC(−) instead of C(+)/GC(−) | 64/66/70 |
| 8 | K2S2O5, CF3SO2Na, or Na2S2O5 instead of DABSO | 8/0/43 |
| 9 | Et3N·3HF, NFSI, KHF2 or KBF4 instead of Selectfluor | 54/56/40/15 |
| 10 | 10 or 15 mA instead of 12 mA | 69/53 |
| 11 | Air instead of N2 | 43 |
| 12 | No tBuONO | 0 |
| 13 | No electric current or [Mim]TolSO3 | 0 |
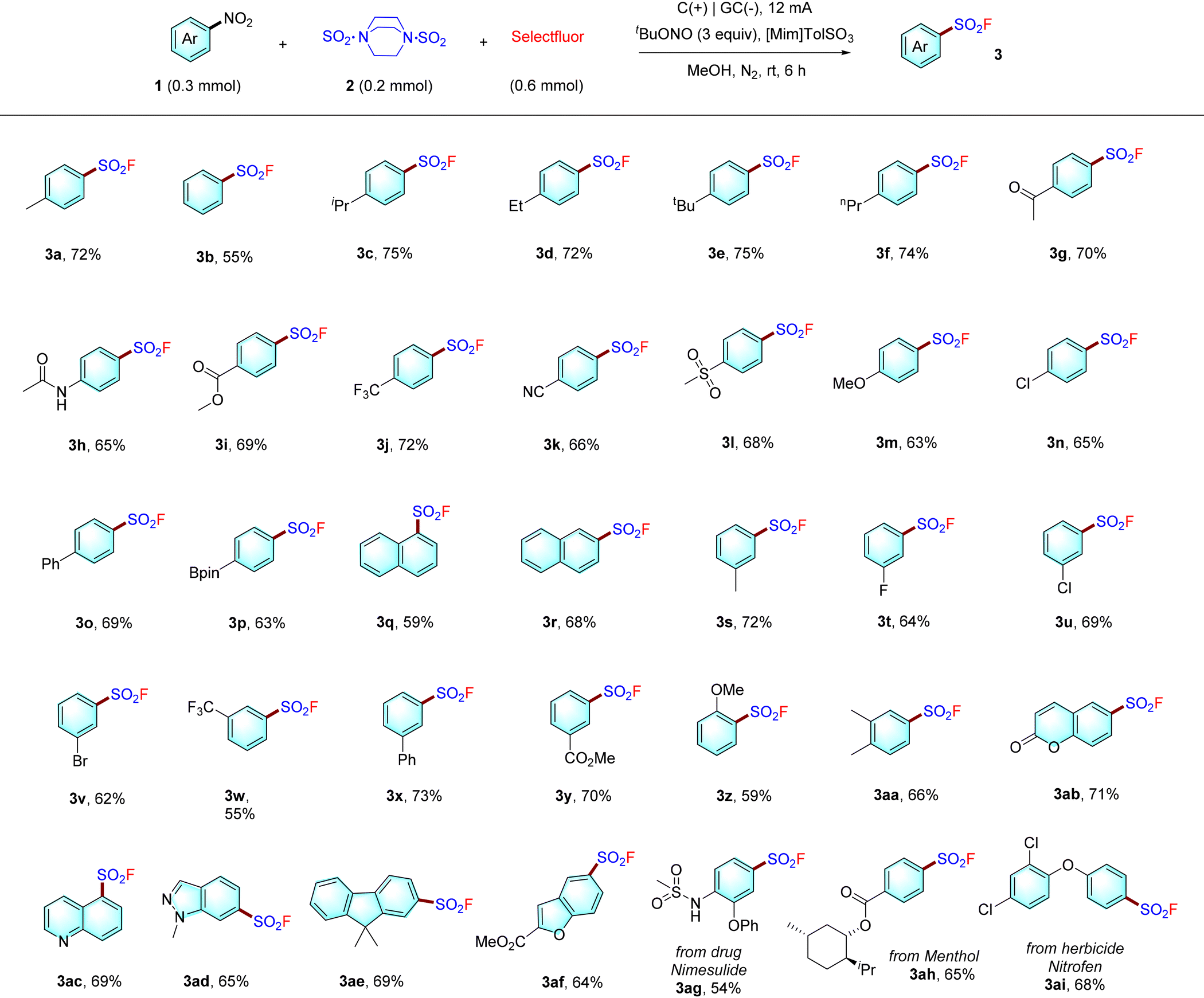
With the optimal conditions in hand, we carefully evaluated various nitroarene substrates 1 (Table 2). Nitroarenes with alkyl groups (1a–1f), electron-withdrawing substituents (1g–1l, 1n), methoxy (1m), phenyl (1o), or a readily transformable bis(pinacolato)diboron substituent (1p) at the para position of the phenyl ring were successfully converted to the desired products 3a–3p in moderate to good yields (63–75%). Two naphthalene-based nitro compounds underwent fluorosulfonylation to give the desired products 3q and 3r in moderate yields. In addition, nitro compounds with a meta methyl (3s), halogen (3t–3v), trifluoromethyl (3w), phenyl (3x), or ester (3y) substituent were tolerated. We also evaluated ortho-methoxy substrates, which gave 3z under the standard conditions, albeit with moderate yield (59%). 3,5-Dimethyl substituted substrate was also tolerated (3aa). Furthermore, heteroarenesulfonyl fluorides with a coumarin, quinoline, indazole, fluorene or benzofuran core (3ab–3ad, 3af) were also accessed in reasonable yields. Finally, to our delight, sulfonyl fluorides derived from the pharmaceutical nimesulide, the natural product menthol, and the pesticide nitrofen could also be obtained (3ag–3ai), highlighting the good functional group tolerance of our strategy. It is noteworthy that denitrative arenes can often by detected in these cases as the major byproduct.
| a For details, see the ESI.† |
|---|
|
To further demonstrate the green metrics of our method, Table 3 summarizes the green metrics of our method with the known protocols reported by Liu et al.13a,b and Weng,13c and product 3a has been chosen as a model example. For the four major green chemistry metrics we analysed, that is the E-factor, atom economy (AE), reaction mass efficiency (RME), and process mass intensity (PMI), lower values of the E-factor and the PMI, as well as higher values of RME, were observed for our protocol (for details, please see Part 5, ESI†). These indicate less waste is formed and a smaller total mass of reagents is required for our protocol along with better atom efficiency.20
This novel electrochemical method could be performed on a gram scale. For example, the reaction of 9 mmol of 1a with DABSO 2 and Selectfluor gave the desired product 3a in 70% isolated yield under slightly modified conditions (Scheme 2).
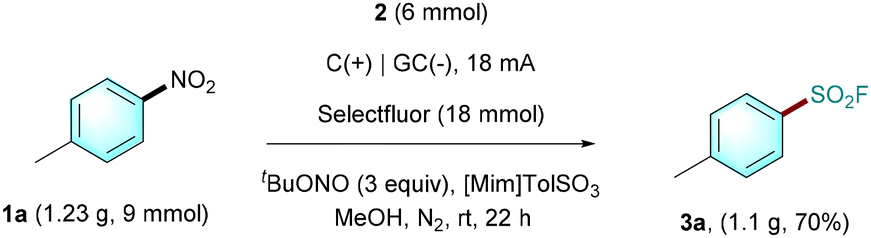 | ||
| Scheme 2 Gram-scale reaction. | ||
To gain insight into the mechanism of this denitrative fluorosulfonylation of nitroarenes, a series of control experiments were conducted (Scheme 3). (1) First of all, the addition of 2 equiv. of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) to the model reaction only led to a trace amount of 3a (Scheme 3a). (2) Second, a radical clock experiment involving 2-nitro-1,1-biphenyl 4 only afforded dibenzo[b,d]thiophene 5,5-dioxide 5 in 81% yield, and sulfonyl fluoride product 6 could not be detected (Scheme 3b). These results suggest that an aryl radical might be generated during this transformation. (3) To identify the key intermediate for the transformation, a variety of plausible intermediates, such as nitrosobenzene 7, N-phenyl hydroxylamine 8, azoxybenzene 9, azobenzene 10, 1,2-diphenylhydrazine 11, or PhNH212, were tested under the optimal conditions (Scheme 3c). It turns out that only the use of nitrosobenzene 7 or PhNH212b enables the delivery of a comparable yield of 3b. In addition, no 3b could be detected in the absence of electricity using either 7 or 12b as the substrate. This indicates that either 7 or 12b might serve as the reactive species for our fluorosulfonylation. (4) To give a deep understanding of the role of [Mim]TolSO3, cyclic voltammetry (CV) studies were conducted.19c–l As shown in Scheme 3d, while no signal was observed from 0 to −2.0 V for 1a, only one irreversible reduction peak at −1.2 V was observed in the presence of [Mim]TolSO3.19k This is different from the previous work of Zhang,19i who reported that two obvious reduction peaks were observed for the reduction of PhNO2 in the presence of nBuNHSO4. (5) To further identify the key role of [Mim]TolSO3, an exhaustive electrolysis experiment was performed. As shown in Scheme 3e, it was noticed that the presence of [Mim]TolSO3 was crucial for the selective reduction of the nitro group. While the presence of [Mim]TolSO3 enabled us to isolate intermediate 12a with 83% yield, no 12a was detected when the reaction was carried out without [Mim]TolSO3. Furthermore, while the use of [Mim]HSO4 could deliver 12a with 21% yield, the presence of other acids such as [Mim]Cl, [Mim]OAc, H2SO4, HCl or TsOH instead of [Mim]TolSO3 only led to trace 12a. These results not only demonstrate that such an ionic liquid is essential for promoting the selective reduction of nitroarenes to the corresponding aniline intermediate, but also indicate that aniline might serve as the key intermediate for the transformation.
 | ||
| Scheme 3 Mechanistic studies (for details, please see the ESI†). | ||
On the basis of the above-described results and previous reports,11–15,19 two plausible mechanisms have been proposed (Fig. 1). Initially, under the assistance of [Mim]TolSO3, nitroarenes 1 could accept six electrons at the cathode, along with protons, to form aniline Bvia ArNO intermediate A. The aniline B could react with tBuONO to afford aryl diazo salt C, which undergoes single-electron reduction on the cathode, to form aromatic radical D with the elimination of N2.19i The aryl radical D could be rapidly captured by SO2 to form the corresponding aryl sulfonyl intermediate E. After this, intermediate E could be converted to desired arenesulfonyl fluoride 3via rapid radical fluorination by Selectfluor.13a,21 Alternatively, intermediate E could combine with radical cation F (formed via the oxidation of DABCO at the cathode), giving rise to the species G.22 The subsequent nucleophilic substitution with BF4− (the anion of Selectfluor) gives rise to the observed 3. This is consistent with the fact that the use of KBF4 as a fluoro source also enables the production of product 3a, albeit with low yield (Table 1, entry 8).
 | ||
| Fig. 1 Two plausible mechanisms. | ||
Conclusions
In summary, we have developed the first electrochemical fluorosulfonylation of nitroarenes. This practical, efficient method affords arenesulfonyl fluorides in moderate to good yields and shows good functional group tolerance. The method, which utilizes inexpensive electrodes and simple ionic liquid as a mediator to help the selective reduction of nitroarenes and does not require additional oxidants or reducing agents, can be expected to be a valuable addition to the toolkit for the preparation of aromatic hydrocarbon sulfonyl fluorides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (22102012, 22202021, 22272011, 22201062, and 22372015) and the National Natural Science Foundation of Henan Province (222300420111) is gratefully acknowledged.References
- (a) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed; (b) C. J. Smedley, A. S. Barrow, C. Spiteri, M.-C. Giel, P. Sharma and J. E. Moses, Chem. – Eur. J., 2017, 23, 9990–9995 CrossRef CAS PubMed; (c) M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS PubMed.
- L. Matesic, N. A. Wyatt, B. H. Fraser, M. P. Roberts, T. Q. Pham and I. Greguric, J. Org. Chem., 2013, 78, 11262–11270 CrossRef CAS PubMed.
- (a) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC; (b) D. A. Shannon, C. Gu, C. J. McLaughlin, M. Kaiser, R. A. L. van der Hoorn and E. Weerapana, ChemBioChem, 2012, 13, 2327–2330 CrossRef CAS PubMed; (c) N. P. Grimster, S. Connelly, A. Baranczak, J. Dong, L. B. Krasnova, K. B. Sharpless, E. T. Powers, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 5656–5668 CrossRef CAS PubMed; (d) E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed.
- (a) T. Zhong, Z. Chen, J. Yi, G. Lu and J. Weng, Chin. Chem. Lett., 2021, 32, 2736–2750 CrossRef CAS; (b) F. He, Y. Li and J. Wu, Org. Chem. Front., 2022, 9, 5299–5305 RSC; (c) T. S.-B. Lou and M. C. Willis, Nat. Rev. Chem., 2022, 6, 146–162 CrossRef CAS PubMed.
- (a) W. Davies and J. H. Dick, J. Chem. Soc., 1932, 483–486 RSC; (b) T. A. Bianchi and L. A. Cate, J. Org. Chem., 1977, 42, 2031–2032 CrossRef CAS; (c) C. Patel, E. André-Joyaux, J. A. Leitch, X. M. Irujo-Labalde, F. Ibba, J. Struijs, M. A. Ellwanger, R. Paton, D. L. Browne, G. Pupo, S. Aldridge, M. A. Hayward and V. Gouverneur, Science, 2023, 381, 302–306 CrossRef CAS PubMed.
- (a) M. Kulka, J. Am. Chem. Soc., 1950, 72, 1215–1218 CrossRef CAS; (b) L. Zhang, X. Cheng and Q.-L. Zhou, Chin. J. Chem., 2022, 40, 1687–1692 CrossRef CAS; (c) B. J. Thomson, S. R. Khasnavis, E. C. Grigorian, R. Krishnan, T. D. Yassa, K. Lee, G. M. Sammis and N. D. Ball, Chem. Commun., 2023, 59, 555–558 RSC.
- (a) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080–1084 CrossRef CAS PubMed; (b) G. Laudadio, A. d. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. de Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS PubMed.
- L. Tang, Y. Yang, L. Wen, X. Yang and Z. Wang, Green Chem., 2016, 18, 1224–1228 RSC.
- (a) M. Kirihara, S. Naito, Y. Ishizuka, H. Hanai and T. Noguchi, Tetrahedron Lett., 2011, 52, 3086–3089 CrossRef CAS; (b) M. Kirihara, S. Naito, Y. Nishimura, Y. Ishizuka, T. Iwai, H. Takeuchi, T. Ogata, H. Hanai, Y. Kinoshita, M. Kishida, K. Yamazaki, T. Noguchi and S. Yamashoji, Tetrahedron, 2014, 70, 2464–2471 CrossRef CAS.
- (a) Y. Jiang, N. S. Alharbi, B. Sun and H.-L. Qin, RSC Adv., 2019, 9, 13863–13867 RSC; (b) M. Perez-Palau and J. Cornella, Eur. J. Org. Chem., 2020, 2497–2500 CrossRef CAS.
- (a) A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233–1237 RSC; (b) A. L. Tribby, I. Rodriguez, S. Shariffudin and N. D. Ball, J. Org. Chem., 2017, 82, 2294–2299 CrossRef CAS PubMed; (c) T. S.-B. Lou, Y. Kawamata, T. Ewing, G. A. Correa-Otero, M. R. Collins and P. S. Baran, Angew. Chem., Int. Ed., 2022, 61, e202208080 CrossRef CAS PubMed.
- (a) Y. Chen and M. C. Willis, Chem. Sci., 2017, 8, 3249–3253 RSC; (b) P. K. T. Lo, Y. Chen and M. C. Willis, ACS Catal., 2019, 9, 10668–10673 CrossRef CAS; (c) M. Magre and J. Cornella, J. Am. Chem. Soc., 2021, 143, 21497–21502 CrossRef CAS PubMed.
- (a) Y. Liu, D. Yu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Org. Lett., 2020, 22, 2281–2286 CrossRef CAS PubMed; (b) Q. Lin, Z. Ma, C. Zheng, X.-J. Hu, Y. Guo, Q.-Y. Chen and C. Liu, Chin. J. Chem., 2020, 38, 1107–1110 CrossRef CAS; (c) T. Zhong, M.-K. Pang, Z.-D. Chen, B. Zhang, J. Weng and G. Lu, Org. Lett., 2020, 22, 3072–3078 CrossRef CAS PubMed.
- (a) J. Kwon and B. M. Kim, Org. Lett., 2019, 21, 428–433 CrossRef CAS PubMed; (b) C. Lee, N. D. Ball and G. M. Sammis, Chem. Commun., 2019, 55, 14753–14756 RSC; (c) X. Kong, Y. Chen, Q. Liu, W. Wang, S. Zhang, Q. Zhang, X. Chen, Y.-Q. Xu and Z.-Y. Cao, Org. Lett., 2023, 25, 581–586 CrossRef CAS PubMed; (d) L. Shan, Z. Ma, C. Ou, Y. Cai, Y. Ma, Y. Guo, X. Ma and C. Liu, Org. Biomol. Chem., 2023, 21, 3789–3793 RSC; (e) Y. Ma, Q. Pan, C. Ou, Y. Cai, X. Ma and C. Liu, Org. Biomol. Chem., 2023, 21, 7597–7601 RSC. For other recent examples for the synthesis of sulfonyl fluorides, please see: (f) D. Chen, X. Nie, Q. Feng, Y. Zhang, Y. Wang, Q. Wang, L. Huang, S. Huang and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 27271–27276 CrossRef CAS PubMed; (g) N. L. Frye, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202115593 CrossRef CAS PubMed; (h) P. Wang, H. Zhang, M. Zhao, S. Ji, L. Lin, N. Yang, X. Nie, J. Song and S. Liao, Angew. Chem., Int. Ed., 2022, 61, e202207684 CrossRef CAS PubMed; (i) W. Zhang, H. Li, X. Li, Z. Zou, M. Huang, J. Liu, X. Wang, S. Ni, Y. Pan and Y. A. Wang, Nat. Commun., 2022, 13, 3515 CrossRef CAS PubMed; (j) R. Xu, T. Xu, M. Yang, T. Cao and S. Liao, Nat. Commun., 2019, 10, 3752 CrossRef PubMed; (k) J. F. Erchinger, R. Hoogesteger, R. Laskar, S. Dutta, C. Hümpel, D. Rana, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 2364–2374 CrossRef CAS PubMed; (l) H. Zhang, X. Sun, C. Ma, C. Li, Y. Ni, Y. Yu, Y.-Q. Xu, S.-F. Ni and Z.-Y. Cao, ACS Catal., 2024 DOI:10.1021/acscatal.3c05154. For a recent review, please see: (m) Y. Zheng, W. Lu, T. Ma and S. Huang, Org. Chem. Front., 2024, 11, 217–235 RSC.
- (a) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 CrossRef; (b) G. Booth, Nitro Compounds, Aromatic, Wiley-VCH, New York, 2000 Search PubMed.
- For selected reviews, please see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (c) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (d) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (e) J. C. Siu, N. K. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (f) Y. Yu, P. Guo, J.-S. Zhong, Y. Yuan and K.-Y. Ye, Org. Chem. Front., 2020, 7, 131–135 RSC; (g) C. Ma, P. Fang, Z. Liu, S. Xu, K. Xu, X. Cheng, A. Lei, H. Xu, C. C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS PubMed; (h) C. Xu, A.-W. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C.-C. Zeng, CCS Chem., 2021, 4, 1120–1152 Search PubMed; (i) Z. Tan, H. Zhang, K. Xu and C.-C. Zeng, Sci. China: Chem., 2024, 67, 450–470 CrossRef CAS; (j) H. Zhou, H.-T. Tan and W.-M. He, Chin. J. Catal., 2023, 46, 4–10 CrossRef CAS.
- (a) X. Kong, Y. Liu, L. Lin, Q. Chen and B. Xu, Green Chem., 2019, 21, 3796–3801 RSC; (b) X. Kong, Y. Wang, Y. Chen, X. Chen, L. Lin and Z.-Y. Cao, Org. Chem. Front., 2022, 9, 1288–1294 RSC; (c) X. Kong, Y. Chen, X. Chen, Z. Lu, W. Wang, S.-F. Ni and Z.-Y. Cao, Org. Lett., 2022, 24, 2137–2142 CrossRef CAS PubMed; (d) X. Kong, X. Chen, Y. Chen and Z.-Y. Cao, J. Org. Chem., 2022, 87, 7013–7021 CrossRef CAS PubMed; (e) X. Chen, N.-Z. Wang, Y.-M. Cheng, X. Kong and Z.-Y. Cao, Synthesis, 2023, 2833–2842 CAS; (f) X. Kong, Y. Chen, X. Chen, C. Ma, M. Chen, W. Wang, Y.-Q. Xu, S.-F. Ni and Z.-Y. Cao, Nat. Commun., 2023, 14, 6933 CrossRef CAS PubMed.
- H.-D. He, Z.-K. Zhang, H.-B. Tang, Y.-Q. Xu, X.-B. Xu, Z.-Y. Cao, H. Xu and Y. Li, Org. Chem. Front., 2022, 9, 4875–4881 RSC.
- (a) H.-U. Blaser, Science, 2006, 313, 312–313 CrossRef CAS PubMed; (b) H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS. For studies on the electrochemical reduction of nitroarenes, please see: (c) F. Z. Haber, Z. Elektrochem. Angew. Phys. Chem., 1898, 5, 235 Search PubMed; (d) J. Marquez and D. Pletcher, J. Appl. Electrochem., 1980, 10, 567–573 CrossRef CAS; (e) D. S. Silvester, A. J. Wain, L. Aldous, C. Hardacre and R. G. Compton, J. Electroanal. Chem., 2006, 596, 131–140 CrossRef CAS; (f) S. Won, W. Kim and H. Kim, Bull. Korean Chem. Soc., 2006, 27, 195–196 CrossRef CAS; (g) Y.-Z. Liu, M.-Y. Lin, L.-P. Xiao, K. Zhang and J.-X. Lu, Chin. J. Chem., 2008, 26, 1168–1172 CrossRef CAS; (h) J.-L. Fan, W.-L. Ye, R. Wang, L.-Q. Xu and X.-Q. Wu, J. Electrochem., 2009, 15, 260 CAS; (i) S. Wang, X. Xia, Q. Chen, K. Li, X. Xiao and F. Chen, ACS Appl. Mater. Interfaces, 2024, 16(4), 5158–5167 CrossRef CAS PubMed; (j) P. Du, J. L. Brosmer and D. G. Peters, Org. Lett., 2011, 13, 4072–4075 CrossRef CAS PubMed; (k) A. A. Sokolov, M. A. Syroeshkin, V. N. Solkan, T. V. Shebunina, R. S. Begunov, L. V. Mikhal'chenko, M. Y. Leonova and V. P. Gultyai, Chem. Bull., 2014, 63, 372–380 CAS; (l) L. Du, B. Zhang, S. Ji, H. Cai and H. Zhang, Sci. China: Chem., 2023, 66, 534–539 CrossRef CAS; (m) S. Ji, L. Zhao, B. Miao, M. Xue, T. Pan, Z. Shao, X. Zhou, A. Fu and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304434 CrossRef CAS PubMed.
- (a) K. V. Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2 DOI:10.1186/1860-5397-2-3; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC; (c) R. A. Sheldon, Chem. Commun., 2008, 44, 3352–3356 RSC; (d) C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15(4), 912–917 CrossRef CAS.
- Y. Liu, H. Wu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Angew. Chem., Int. Ed., 2017, 56, 15432–15435 CrossRef CAS PubMed.
- D. Louvel, A. Chelagha, J. Rouillon, P.-A. Payard, L. Khrouz, C. Monnereau and A. Tlili, Chem. – Eur. J., 2021, 27, 8704–8708 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04528e |
| This journal is © The Royal Society of Chemistry 2024 |