
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating catalytic activity of a modified flavin analogue via judicially positioned metal ion toward aerobic sulphoxidation†
M. S. S. Vinod Mouli and
Ashutosh Kumar Mishra *
*
Department of Chemistry, Indian Institute of Technology-Hyderabad, Kandi-502284, India. E-mail: akm@chy.iith.ac.in
First published on 1st February 2022
Abstract
This manuscript describes the synthesis, spectroscopic and crystallographic characterization of a cadmium complex of 10-propoylisoalloxazine-7-carboxylic acid (Flc-Cd). Catalytic activity of Flc-Cd towards aerobic sulphoxidation reaction was investigated in the presence of L-ascorbic acid as the reducing agent. Notably the neutral un-metalated flavin analogue did not show any significant catalytic activity. The design strategy for Flc provides a close proximity of the metal centre to the flavin core without compromising the catalytic site thereby assisting the product formation when compared to unmetallated Flc. Minor enantioselectivity is also observed in cases where unsymmetrical sulphides were used; indicative of the possible involvement of chiral L-ascorbic acid in the intermediate formation.
1. Introduction
Developing novel bioinspired constructs continues to be at the forefront of research investigations, as they not only offer modulation of functional aspects of the molecule by subtle changes around the core chemical structure but could also provide an insight into the associated biological phenomena.1 In this context, naturally occurring flavins have enthralled researchers due to their excellent photoreceptor and redox properties. The flavin moiety as a cofactor in flavoenzymes is well documented to play a key role in numerous chemical transformations, involving one or two electron redox processes.2 For instance, mammalian liver containing flavin dependant monooxygenase can metabolise xenobiotic substrates having soft nucleophilic sites such as amines, sulphides, and phosphites via the formation of an enzyme bound hydroperoxyflavin intermediate (as shown in Scheme 1a);3 or yeast flavoenzyme assisted protein folding via disulphide bond formation in endoplasmic reticulum4 or the involvement of flavin dependent DNA-photolyase in photoinduced DNA repair mechanism.5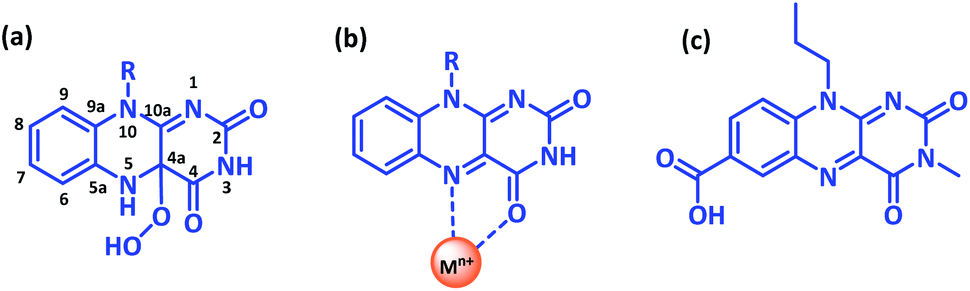 | ||
| Scheme 1 Representation of the chemical structure of flavin moiety displaying (a) C4a-hydroperoxyflavin intermediate, (b) primary metal coordination site and (c) 10-propoylisoalloxazine-7-carboxylic acid (Flc). | ||
Notably, a significant loss in monooxygenase activity was observed for isolated flavins analogues as compared to flavin in enzyme environment suggesting the crucial role of the noncovalent interactions operating between the apoprotein and flavin cofactor in regulating the functional activity and the substrate specificity in the natural system.6 While its challenging to investigate the noncovalent interactions within the biological framework, a suitable chemical model is highly desirable to design and develop its biomimetic analogue. Contrary to the natural counterpart, most of the synthetic flavin analogues are designed around charged flavinium core skeleton either as N5-alkylisoalloxaziniums or 5-alkylalloxaziniums or as 1,10-bridged alloxaziniums7 and reports pertaining to neutral flavin are scarce.8
Recently, short peptide conjugates covalently linked with the neutral flavin analogues was reported to stabilize the hydroperoxyflavin intermediate via intramolecular interactions, thereby assisting the aerobic oxygenation reactions;8a–d another example employs carboxy functionality within the neutral flavin core to stabilise the flavin semiquinone radical state and the hydroperoxyflavin intermediate formed during the thioanisole oxidation reaction.8e Alternatively, proximity of the metal ion can also influence the functional behaviour of the flavin entity by modulating the redox properties. Though direct coordination with flavin is not yet reported in natural system, close proximity of metal centre affecting the enzymatic transformations is invariably reported for iron–sulphur flavoenzymes, cytochrome P450 reductase, xanthine reductase etc.9 Model flavin analogues have exhibited the C4–O and N5 of flavin as the preferred coordination site (Scheme 1b),10 thereby blocking the active site for monooxygenase like function. However, a significant change in the redox activity of the metalated flavins was reported on photoexcitation suggestive of excellent photocatalytic potential.11
With this background, we intend to design metalated-flavin analogue without compromising on the C4–O catalytic site. Introducing a free carboxy functionality at corner of the aromatic flavin skeleton could provide direct access to the coordinating metal without interfering the reactive centre. Herein, we report the synthesis and characterization of 3-methyl, 10-propoylisoalloxazine-7-carboxylic acid (Flc) (Scheme 1c) and its complexation with the cadmium ion (Flc-Cd). Single crystal X-ray diffraction technique was used to elucidate the structure of Flc-Cd complex. The catalytic efficiency of Flc-Cd was investigated towards aerobic sulphoxidation reactions in presence of L-ascorbic acid as reducing agent.
2. Results and discussion
Synthesis and characterization
The synthesis of 10-propoylisoalloxazine-7-carboxylic acid (Flc) was performed following the slightly modified procedure12 and shown in Scheme 2. Experimental details for the synthetic procedure followed is given in the ESI.† The synthetic procedure starts with the acid catalysed esterification of 4-chloro-3-nitrobenzoic acid in methanolic solution, followed by nucleophilic aromatic substitution with propyl amine using sodium acetate as base to give 2 (methyl 3-nitro-4-(propylamino)benzoate). Reduction of nitro group in 2 with NaBH4, Pd/C catalyst in methanol, followed by condensation with alloxan monohydrate in presence of boric acid in acetic acid/methanol mixture results in methylated flavin methyl ester 3; which is further methylated at N3 position followed by deprotection of the carboxy group results in the final compound Flc obtained as yellow solid. Complex formation for the synthesis of catalyst Flc-Cd was performed by refluxing the methanolic solution of Flc with cadmium acetate dihydrate for four hours resulting in the formation of Flc-Cd as orange solid which is filtered, washed and dried. Both the Flc and the metalated complex Flc-Cd was characterized by usual spectroscopic analysis as reported in ESI.†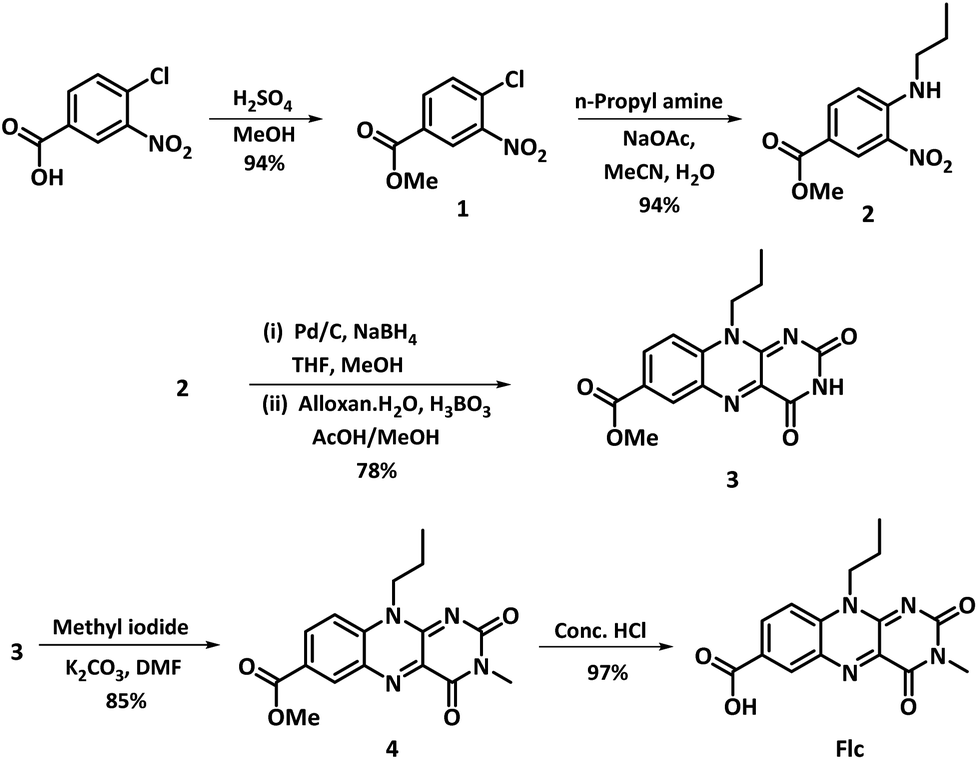 | ||
| Scheme 2 Synthetic scheme for the preparation of (Flc). | ||
Comparative NMR analysis was performed in deuterated DMSO using Flc as standard, to better understand the structural aspect of Flc-Cd catalyst as shown in the ESI (Fig. S1†). A preliminary analysis of the recorded and spectra reveals similar chemical shift values for N10-propyl and N3-methyl protons, suggesting absence of any nuclear interaction, possibly due to distantly placed metal ion. However, subtle de-shielding effect was observed for the Flc aromatic protons (H6 and H8) on cadmium coordination indicative of weak metal ion interaction. Overall, the NMR studies suggests the absence of any significant interaction of the metal ion with the flavin core skeleton.
The electronic state of the flavin moiety was also evaluated to ascertain the effect of cadmium coordination by recording the absorption and emission spectra in DMSO as shown in the ESI (Fig. S2†). The absorption spectra of Flc displays three bands with wavelength maxima at 284, 334 and 432 nm which can be attributed to the typical π–π* transition assigned to isoalloxazine ring.13 Similarly, emission spectra also reveal insignificant changes on cadmium coordination when excited at 432 nm wavelength which show band maxima at around 519 nm, typically assigned to emission from isoalloxazine ring. Subtle changes in the absorption and emission spectra for Flc-Cd and Flc suggest that metal coordination does not alter the electronic energy state of the isoalloxazine ring to a significant level.
A definite proof for the structural organization and the coordination behaviour of Flc-Cd catalyst comes from the single crystal X-ray diffraction studies. Suitable crystal for diffraction studies was grown by mixing methanolic solution of Flc and cadmium acetate in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and kept for slow evaporation to form yellow crystals. Crystal analysis of Flc-Cd reveals triclinic system with P
:1 ratio and kept for slow evaporation to form yellow crystals. Crystal analysis of Flc-Cd reveals triclinic system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, where each cadmium ion is coordinated to carboxyl oxygen from two different Flc moieties, water molecules acquiring axial positions and methanol molecules occupying the two opposite equatorial positions. The angle between any two coordinated oxygen atoms with cadmium in centre is approximately 90° (±1°); and the distance between the cadmium and coordinate oxygen falls within 2.21 to 2.31 Å. This arrangement reveals a prefect octahedral geometry around the cadmium centre in a mononuclear dimer arrangement as shown in Fig. 1.
space group, where each cadmium ion is coordinated to carboxyl oxygen from two different Flc moieties, water molecules acquiring axial positions and methanol molecules occupying the two opposite equatorial positions. The angle between any two coordinated oxygen atoms with cadmium in centre is approximately 90° (±1°); and the distance between the cadmium and coordinate oxygen falls within 2.21 to 2.31 Å. This arrangement reveals a prefect octahedral geometry around the cadmium centre in a mononuclear dimer arrangement as shown in Fig. 1.
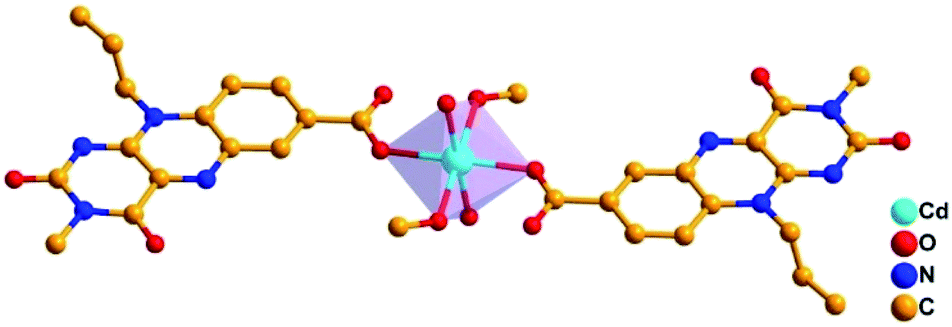 | ||
| Fig. 1 Pictorial representation of the geometry around cadmium ion in Flc-Cd. | ||
A closer inspection of the crystal lattice along the b-axis reveals perfectly aligned Flc moieties through π–π stacking interactions; where the distance between the centroid of the two isoalloxazine rings are 3.507 Å and 3.522 Å as shown in the ESI (Fig. S3†). The crystal lattice is further stabilized by the hydrogen bonding interaction between the N1-nitrogen and C2–O oxygen of Flc with the coordinated water molecule with H-bonding distance between the N1![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) H–O and C2–OH–O are 2.60 and 1.99 Å respectively. Interestingly, the preliminary success of our design strategy was quite evident, as the Flc moiety displayed monodentate coordination behaviour via carboxyl-oxygen; leaving the primary coordination site (N5, C4–O) free for the substrate access during the catalytic activity.
H–O and C2–OH–O are 2.60 and 1.99 Å respectively. Interestingly, the preliminary success of our design strategy was quite evident, as the Flc moiety displayed monodentate coordination behaviour via carboxyl-oxygen; leaving the primary coordination site (N5, C4–O) free for the substrate access during the catalytic activity.
Catalytic activity of Flc-Cd towards aerobic sulphoxidation
A preliminary investigation to explore the catalytic efficiency of Flc-Cd catalyst towards aerobic sulphoxidation reactions, was performed using thioanisole as substrate under slightly modified reported procedure.14 It was observed that in presence of 10 mol% of Flc-Cd catalyst, one equivalent of L-ascorbic acid (AA) as reductant and molecular oxygen in 2:1 TFE–water solvent system at room temperature, thioanisole was converted to methyl phenyl sulfoxide with 70% yield in 24 hours as shown in Table 1, entry 4. Under identical conditions, the reaction did not proceed in absence of the Flc-Cd catalyst or with Flc alone or cadmium acetate dihydrate alone or (Table 1, entries 1–3). Unexpectedly other reducing agents such as hydrazine hydrate and formic acid/TEA, which have been frequently reported for similar flavin catalytic reactions,8a,15 failed to give significant yield even in presence of Flc-Cd catalyst as shown in Table 1, entries 7 and 8. This might be indicative of important role played by L-ascorbic acid in deciding the course of reaction.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Reducing agent | Oxidant | Yieldb (%) |
| a Conditions: substrate (0.194 mmol), AA = L-ascorbic acid (1 eq.), catalyst loading of 10 mol% used for entry 3–8, and 20 mol% for entry 2.b Yield determined by HPLC. | ||||
| 1 | — | AA | O2 | Trace |
| 2 | Flc | AA | O2 | Trace |
| 3 | Cd(OAc)2·2H2O | AA | O2 | Trace |
| 4 | Flc-Cd | AA | O2 | 70 |
| 5 | Flc-Cd | — | O2 | Trace |
| 6 | Flc-Cd | — | H2O2 (2 eq.) | Trace |
| 7 | Flc-Cd | Hydrazine hydrate (4 eq.) | O2 | Trace |
| 8 | Flc-Cd | Formic acid/TEA (8:2) (10 eq.) |
O2 | Trace |

While 10 mol% of Flc-Cd catalyst was found to be optimum for the catalytic activity in 2:1 TFE/water system as indicated by data presented in Table 2, entries 1–4; significant decrease in reaction yield was observed for reaction in TFE or water alone as solvent (Table 2, entries 5–7). All further reactions were performed with the above-mentioned optimized reaction conditions.
With this information we decided to proceed further to explore the substrate scope of the Flc-Cd catalyst using a wide range of sulphides as substrate as reported in Table 3. It was observed that with simple thioanisole or thioanisole with electron donating substituent gives good to moderate catalytic yield; however drastic decrease in the catalytic yield was observed for the thioanisole derivatives with electron withdrawing groups (Table 3, entries 1–9). Furthermore, replacing methyl substitution from thioanisole with phenyl group as in diphenylsulphide, results in poor yield of the product; slightly better yield was obtained for dibenzylsulphide and bis(furan-2-ylmethyl)sulphane.

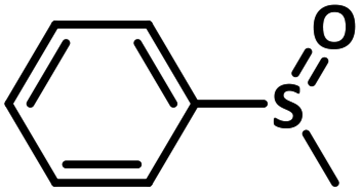
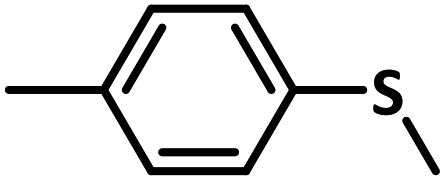
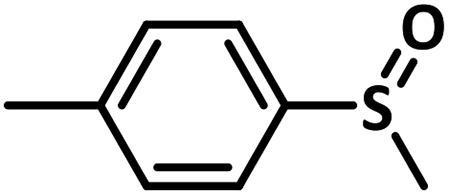
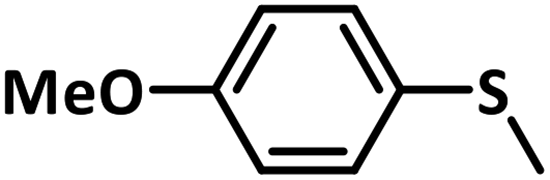
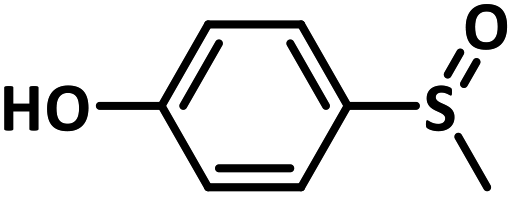
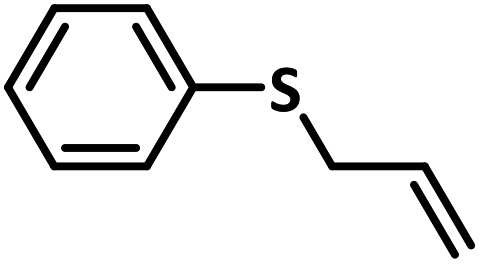
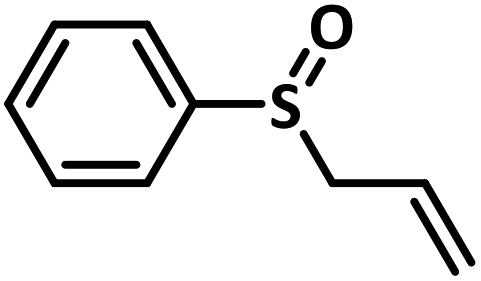
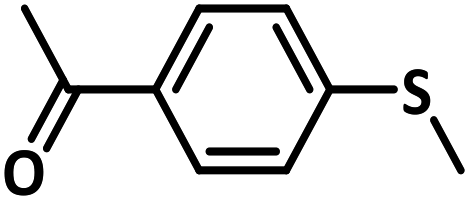
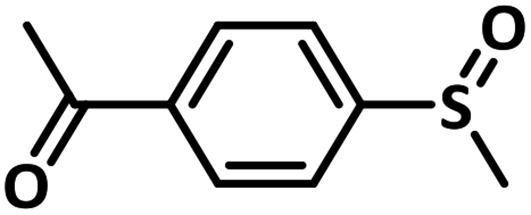
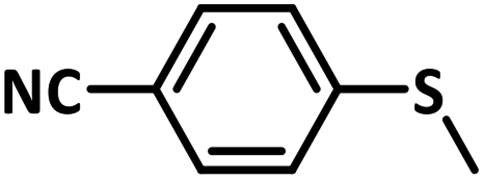
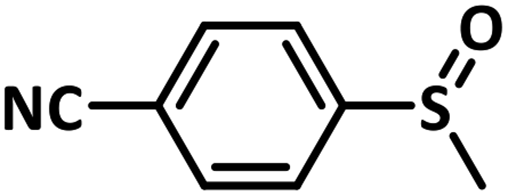
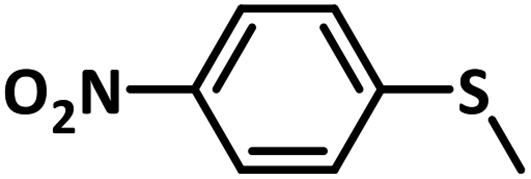
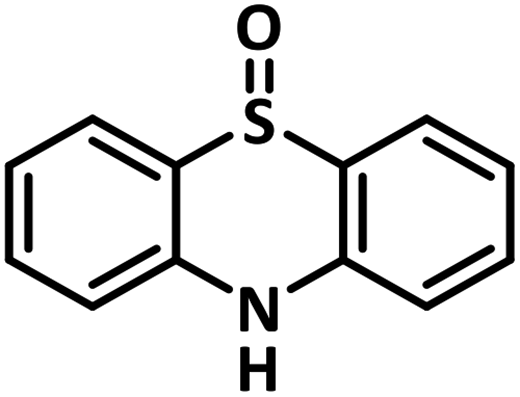
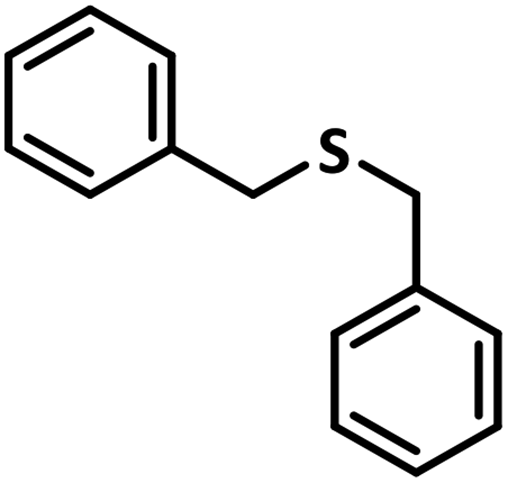
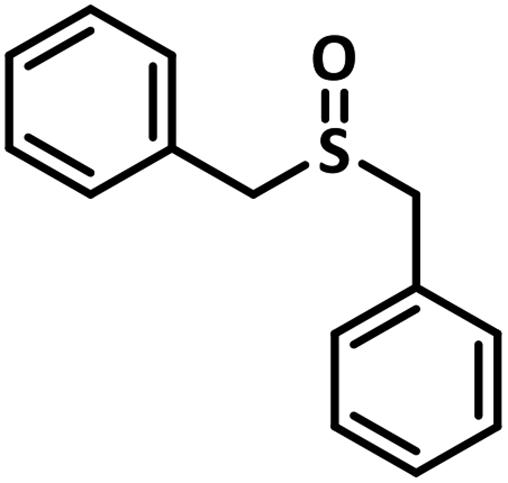
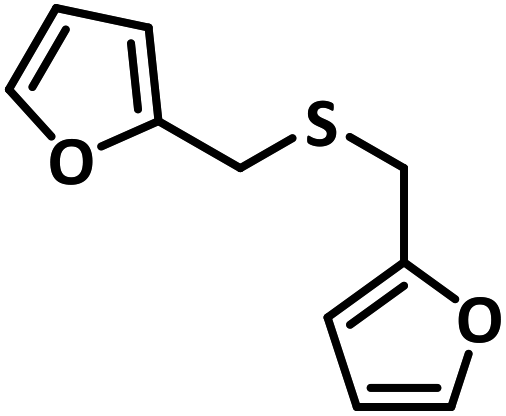
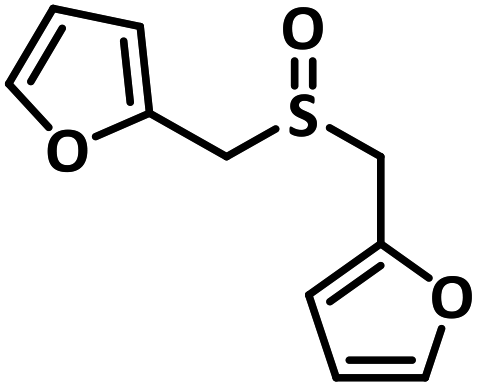
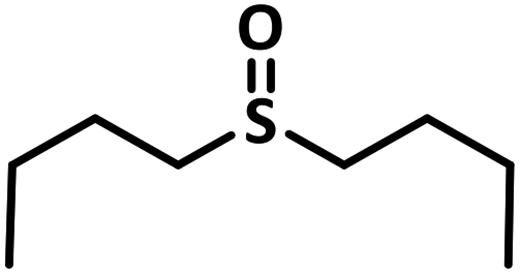
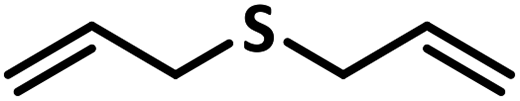
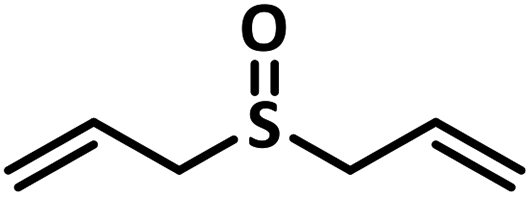
Excellent yield was obtained for aliphatic sulphides including the one with allylic group. In this context, nucleophilic attack of sulphur atom on the electrophilic oxygen of the C4a-hydroperoxyflavin intermediate was reported to be the key step in flavin catalysed sulphoxidation reaction16 and this observation supports the involvement of 4a-hydroperoxyflavin as key intermediate in this reaction as well.
Interestingly, the unsymmetrical sulphide in the present study when checked for enantioselectivity through NMR using R-binol as chiral shift reagent17 reveals enantiomeric excess in the range of 9 to 26% as shown in Table 4. Considering the achiral nature of the Flc-Cd catalyst, the plausible explanation for the observed enantioselectivity is the involvement of chiral L-ascorbic acid in stabilizing the intermediate formation. This assumption also gain support from the loss of catalytic activity in presence of other reducing agents as mentioned above. Earlier reports suggest that ascorbic acid can interact with cadmium through its O3-oxygen.18 Based on our observation and literature report, a possible intermediate state is shown in Fig. 2.
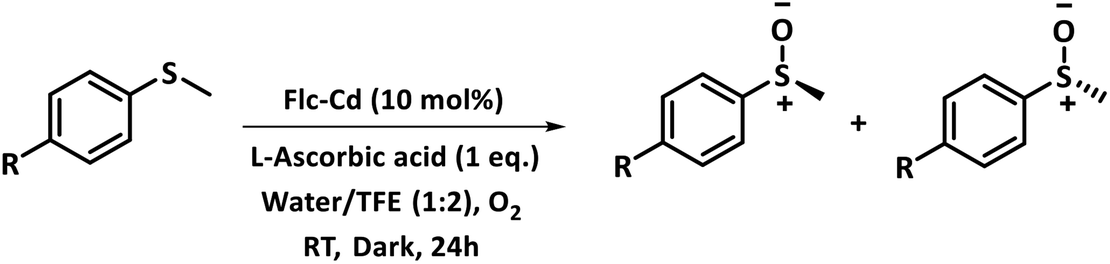
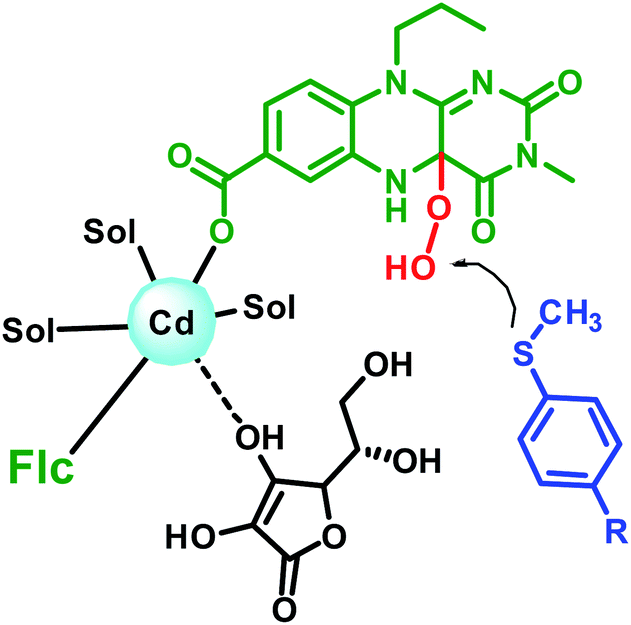 | ||
| Fig. 2 Schematic representation of the possible arrangement of the reaction components in the intermediate formation. | ||
3. Conclusion
In conclusion, we report the design and synthesis of a cadmium complex of a modified flavin analogue and investigated its catalytic activity toward aerobic sulphoxidation reaction. Our design strategy include introducing a carboxyl-group at C7 position of the flavin moiety to facilitate the metal coordination without compromising the access to the catalytic site. Spectroscopic and crystallographic studies was performed to elucidate the structural aspect of the catalyst. Catalytic activity was investigated with a range of sulphides in presence of L-ascorbic acid and molecular oxygen, displaying moderate to good yield, when compared with flavin alone. Interestingly subtle amount of enantioselectivity was also observed indicating possibly involvement of L-ascorbic acid in the intermediate formation. Further work is currently in progress to develop highly enantioselective catalyst using our modified metalated flavin moiety.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MSSVM thanks MHRD for scholarship. AKM thanks Science and Engineering Research Board (SERB) India for the research funding (CRG/2018/001645). Both the authors thanks IIT-Hyderabad for providing the necessary infrastructure and the instrumentation facilities. This manuscript is dedicated to Prof. Frederick D. Lewis on his 78th birthday.References
- (a) G. M. Whitesides, Interface Focus, 2015, 5, 20150031 CrossRef PubMed; (b) V. Artero, Nat. Energy, 2017, 2, 17131 CrossRef CAS; (c) C. Sanchez, H. Arribart and M. M. G. Guille, Nat. Mater., 2005, 4, 277–288 CrossRef CAS PubMed.
- (a) R. Hille, S. Miller and B. Palfey, Oxidase, dehydrogenase and related systems, De Gruyter, Berlin, Boston, 2012, vol. 1 Search PubMed; (b) C. T. Walsh and T. A. Wencewicz, Nat. Prod. Rep., 2013, 30, 175–200 RSC; (c) L. De Colibus and A. Mattevi, Curr. Opin. Struct. Biol., 2006, 16, 722–728 CrossRef CAS PubMed; (d) W. Kaim, B. Schwederski, O. Heilmann and F. M. Hornung, Coord. Chem. Rev., 1999, 182, 323–342 CrossRef.
- (a) W. J. H. van Berkel, N. M. Kamerbeek and M. W. Fraaije, J. Biotechnol., 2006, 124, 670–689 CrossRef CAS PubMed; (b) M. M. E. Huijbers, S. Montersino, A. H. Westphal, D. Tischler and W. J. H. van Berkel, Arch. Biochem. Biophys., 2014, 544, 2–17 CrossRef CAS PubMed; (c) S. K. Krueger and D. E. Williams, Pharmacol. Ther., 2005, 106, 357–387 CrossRef CAS PubMed.
- J. K. Suh, L. L. Poulsen, D. M. Ziegler and J. D. Robertus, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2687–2691 CrossRef CAS PubMed.
- (a) A. Sancar, Chem. Rev., 2003, 103, 2203–2238 CrossRef CAS PubMed; (b) M. Zhang, L. Wang and D. Zhong, Photochem. Photobiol., 2017, 93, 78–92 CrossRef CAS PubMed.
- Representative recent examples for the comparison between flavoenzyme and isolated cofactor: (a) P. D. Clayman and T. K. Hyster, J. Am. Chem. Soc., 2020, 142, 15673–15677 CrossRef CAS PubMed; (b) X. Huang, B. Wang, Y. Wang, G. Jiang, J. Feng and H. Zhao, Nature, 2020, 584, 69–74 CrossRef CAS PubMed; (c) B. A. Sandoval, P. D. Clayman, D. G. Oblinsky, S. Oh, Y. Nakano, M. Bird, G. D. Scholes and T. K. Hyster, J. Am. Chem. Soc., 2021, 143, 1735–1739 CrossRef CAS PubMed.
- (a) A. Rehpenn, A. Walter and G. Storch, Synthesis, 2021, 53, 2583–2593 CrossRef CAS; (b) H. Iida, Y. Imada and S. I. Murahashi, Org. Biomol. Chem., 2015, 13, 7599–7613 RSC; (c) R. Cibulka, Eur. J. Org. Chem., 2015, 2015, 915–932 CrossRef CAS; (d) G. deGonzalo and M. W. Fraaije, ChemCatChem, 2013, 5, 403–415 CrossRef CAS; (e) J. Zelenka, E. Svobodová, J. Tarábek, I. Hoskovcová, V. Boguschová, S. Bailly, M. Sikorski, J. Roithová and R. Cibulka, Org. Lett., 2018, 21, 114–119 CrossRef PubMed; (f) T. Sakai, T. Kumoi, T. Ishikawa, T. Nitta and H. Iida, Org. Biomol. Chem., 2018, 16, 3999–4007 RSC; (g) T. Hartman and R. Cibulka, Org. Lett., 2016, 18, 3710–3713 CrossRef CAS PubMed; (h) S.-I. Murahashi, D. Zhang, H. Iida, T. Miyawaki, M. Uenaka, K. Murano and K. Meguro, Chem. Commun., 2014, 50, 10295–10298 RSC; (i) Y. Imada, H. Iida, S.-I. Murahashi and T. Naota, Angew. Chem., Int. Ed., 2005, 44, 1704–1706 CrossRef CAS PubMed.
- (a) Y. Arakawa, K. Yamanomoto, H. Kita, K. Minagawa, M. Tanaka, N. Haraguchi, S. Itsuno and Y. Imada, Chem. Sci., 2017, 8, 5468–5475 RSC; (b) Y. Arakawa, N. Takechi, K. Yamanomoto, K. Minagawa and Y. Imada, Tetrahedron Lett., 2021, 73, 153107 CrossRef CAS; (c) T. Akiyama, F. Simeno, M. Murakami and F. Yoneda, J. Am. Chem. Soc., 1992, 114, 6613–6620 CrossRef CAS; (d) K. Yamanomoto, H. Kita, Y. Arakawa, K. Minagawa and Y. Imada, Chimia, 2018, 72, 866–869 CrossRef CAS PubMed; (e) Y. Arakawa, K. Minagawa and Y. Imada, Polym. J., 2018, 50, 941–949 CrossRef CAS; (f) Y. Imada, T. Kitagawa, T. Ohno, H. Iida and T. Naota, Org. Lett., 2010, 12, 32–35 CrossRef CAS PubMed.
- (a) M. Iijima, J. Ohnuki, T. Sato, M. Sugishima and M. Takano, Sci. Rep., 2019, 9, 9341 CrossRef PubMed; (b) M. A. Vanoni, Open Biol., 2021, 11, 210010 CrossRef CAS PubMed; (c) C. Enroth, B. T. Eger, K. Okamoto, T. Nishino, T. Nishino and E. F. Pai, Proc. Natl. Acad. Sci., 2000, 97, 10723–10728 CrossRef CAS PubMed.
- (a) W. Kaim, B. Schwederski, O. Heilmann and F. M. Hornung, Coord. Chem. Rev., 1999, 182, 323–342 CrossRef; (b) S. Miyazaki, K. Ohkubo, T. Kojima and S. Fukuzumi, Angew. Chem., Int. Ed., 2007, 46, 905–908 CrossRef CAS PubMed.
- (a) B. Mühldorf and R. Wolf, Chem. Commun., 2015, 51, 8425–8428 RSC; (b) S. Fukuzumi and T. Kojima, JBIC, J. Biol. Inorg. Chem., 2008, 13, 321–333 CrossRef CAS PubMed.
- (a) Y. L. Xu, H. Y. Lin, X. Ruan, S. G. Yang, G. F. Hao, W. C. Yang and G. F. Yang, Eur. J. Med. Chem., 2015, 92, 427–438 CrossRef CAS PubMed; (b) A. Mugford, E. J. Trybulski, A. T. Vu, G. T. Whiteside and P. E. Mahaney, J. Med. Chem., 2009, 52, 5703–5711 CrossRef PubMed; (c) T. R. G. Edwards and D. Gani, Tetrahedron, 1990, 46, 935–956 CrossRef CAS.
- (a) A. Kobayashi, K. Ohbayashi, R. Aoki, H.-C. Chang and M. Kato, Dalton Trans., 2011, 40, 3484–3489 RSC; (b) S. Salzmann, V. Martinez-Junza, B. Zorn, S. E. Braslavsky, M. Mansurova, C. M. Marian and W. Gärtner, J. Phys. Chem. A, 2009, 113, 9365–9375 CrossRef CAS PubMed; (c) M. Insinska-Rak, E. Sikorska, J. L. Bourdelande, I. V. Khmelinskii, W. Prukala, K. Dobek, J. Karolczak, I. F. Machado, L. F. V. Ferreira, A. Komasa, D. R. Worrall and M. Sikorski, J. Mol. Struct., 2006, 783, 184–190 CrossRef CAS; (d) S. Salzmann, J. Tatchen and C. M. Marian, J. Photochem. Photobiol., A, 2008, 198, 221–231 CrossRef CAS; (e) A. Weigel, A. L. Dobryakov, M. Veiga and J. L. P. Lustres, J. Phys. Chem. A, 2008, 112, 12054–12065 CrossRef CAS PubMed.
- Y. Imada, T. Kitagawa, H.-K. Wang, N. Komiya and T. Naota, Tetrahedron Lett., 2013, 54(7), 621–624 CrossRef CAS.
- S.-I. Murahashi, D. Zhang, H. Iida, T. Miyawaki, M. Uenaka, K. Murano and K. Meguro, Chem. Commun., 2014, 50, 10295–10298 RSC.
- S. Oae, K. Asada and T. Yoshimura, Tetrahedron Lett., 1983, 24, 1265–1268 CrossRef CAS.
- S. Schoumacker, O. Hamelin, S. Téti, J. Pécaut and M. Fontecave, J. Org. Chem., 2005, 70, 301–308 CrossRef CAS PubMed.
- (a) B. Zumreoglukaran, Coord. Chem. Rev., 2006, 250(17–18), 2295–2307 CrossRef; (b) H. A. Tajmir-Riahi, J. Inorg. Biochem., 1991, 42(1), 47–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The experimental details including synthetic procedure and characterization, crystallographic details etc. CCDC 2106178. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra06558k |
| This journal is © The Royal Society of Chemistry 2022 |