
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogenation properties of lithium and sodium hydride – closo-borate, [B10H10]2− and [B12H12]2−, composites†
Steffen R. H.
Jensen
a,
Mark
Paskevicius
 a,
Bjarne R. S.
Hansen
a,
Anders S.
Jakobsen
a,
Kasper T.
Møller
a,
James L.
White
b,
Mark D.
Allendorf
b,
Vitalie
Stavila
*b,
Jørgen
Skibsted
c and
Torben R.
Jensen
*a
a,
Bjarne R. S.
Hansen
a,
Anders S.
Jakobsen
a,
Kasper T.
Møller
a,
James L.
White
b,
Mark D.
Allendorf
b,
Vitalie
Stavila
*b,
Jørgen
Skibsted
c and
Torben R.
Jensen
*a
aCenter for Materials Crystallography, Interdisciplinary Nanoscience Center (iNANO) and Department of Chemistry, Aarhus University, Langelandsgade 140, 8000 Aarhus C, Denmark. E-mail: trj@chem.au.dk
bChemistry, Combustion, and Materials Center, Sandia National Laboratories, Livermore, California 94551, USA. E-mail: vnstavi@sandia.gov
cDepartment of Chemistry and Interdisciplinary Nanoscience Center (iNANO), Aarhus University, Langelandsgade 140, 8000 Aarhus C, Denmark
First published on 4th June 2018
Abstract
The hydrogen absorption properties of metal closo-borate/metal hydride composites, M2B10H10–8MH and M2B12H12–10MH, M = Li or Na, are studied under high hydrogen pressures to understand the formation mechanism of metal borohydrides. The hydrogen storage properties of the composites have been investigated by in situ synchrotron radiation powder X-ray diffraction at p(H2) = 400 bar and by ex situ hydrogen absorption measurements at p(H2) = 526 to 998 bar. The in situ experiments reveal the formation of crystalline intermediates before metal borohydrides (MBH4) are formed. On the contrary, the M2B12H12–10MH (M = Li and Na) systems show no formation of the metal borohydride at T = 400 °C and p(H2) = 537 to 970 bar. 11B MAS NMR of the M2B10H10–8MH composites reveal that the molar ratio of LiBH4 or NaBH4 and the remaining B species is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.63 and 1:0.21, respectively. Solution and solid-state 11B NMR spectra reveal new intermediates with a B:H ratio close to 1:1. Our results indicate that the M2B10H10 (M = Li, Na) salts display a higher reactivity towards hydrogen in the presence of metal hydrides compared to the corresponding [B12H12]2− composites, which represents an important step towards understanding the factors that determine the stability and reversibility of high hydrogen capacity metal borohydrides for hydrogen storage.
:0.63 and 1:0.21, respectively. Solution and solid-state 11B NMR spectra reveal new intermediates with a B:H ratio close to 1:1. Our results indicate that the M2B10H10 (M = Li, Na) salts display a higher reactivity towards hydrogen in the presence of metal hydrides compared to the corresponding [B12H12]2− composites, which represents an important step towards understanding the factors that determine the stability and reversibility of high hydrogen capacity metal borohydrides for hydrogen storage.
Introduction
The human consumption of energy has increased over the past decades, and has mainly been covered by burning fossil fuels at an increasing rate, which has led to an increase in CO2 levels in the atmosphere.1,2 Despite an abundance of available renewable energy from sun and wind, these energy sources are intermittent and fluctuate over time and geography. Thus, energy storage is required to compensate for the variability in renewable energy fluxes, and hydrogen is a promising energy carrier with the highest gravimetric energy density among all known compounds (ρm = 120 MJ kg−1).3 However, compressed hydrogen gas has a low volumetric energy density (ρV = 8.5 MJ L−1). The volumetric density can be improved by storing hydrogen in a solid, for example in reversible metal hydrides.3–5 Metal borohydrides have been proposed as solid-state hydrogen storage media due to their high hydrogen content and potential reversibility.6–9 The challenge of using metal borohydrides as hydrogen carriers is that they often possess high hydrogen release temperatures, coupled with harsh conditions needed for hydrogenation.10–14 These issues have generated interest in the synthesis and investigation of hydrogen storage properties of bi- and tri-metallic main group/transition metal borohydrides with tunable hydrogen desorption temperatures.15–21 Another approach has been to stabilize unstable borohydrides e.g. Fe(BH4)2 and Co(BH4)2, using ammonia.22 Reactive hydride composites have also been used to thermodynamically destabilize metal borohydrides by altering the decomposition mechanism, e.g. LiBH4–MgH2, or by changing the morphology of the sample, e.g. NaBH4–KBH4, where the solid is transformed into a liquid.23,24 Finally, several materials have been studied as catalysts for hydrogen uptake and release, e.g., transition metals and their halides and oxides.25–27 However, to the best of our knowledge, an efficient catalyst for breaking and forming B–H bonds has not yet been discovered.Polyhydro-closo-polyborates (also known as closo-borates), BnHn2− (n = 6–12), are anions comprised of boron atoms with solely terminal hydrogen in closed polyhedral clusters. These compounds can be prepared through a variety of solid-state and solution synthetic approaches.28 Higher closo-borates, such as dodecahydro-closo-dodecaborates ([B12H12]2− salts) and decahydro-closo-decaborates ([B10H10]2− salts) are often assumed to be intermediates in the mechanism for hydrogen release and uptake in metal borohydrides, M(BH4)x, and their relatively high stability may retard the reversibility of these reactions.28–31 The formation of higher closo-borates during decomposition of metal borohydrides may occur in a reaction between the metal borohydride and transient diborane (B2H6). The formation of either Li2B10H10 or Li2B12H12, depending on temperature, has clearly been observed by mechano-chemical treatment of LiBH4 in a diborane atmosphere.32 The formation of Li2B12H12 is also observed for the reactive hydride composite, LiBH4–MgH2–Al, which shows decreasing reversible hydrogen content commensurate with increasing amounts of Li2B12H12 during cycling of hydrogen release and uptake.30 In a similar manner, magnesium borohydride, Mg(BH4)2, decomposes under vacuum at T ≈ 200 °C and forms arachno-Mg(B3H8)2, which can absorb hydrogen at more moderate conditions, e.g., p(H2) = 120 bar and T = 250 °C, in contrast to the closo-borates.33
Hydrogen release and absorption reactions in boron-based hydrides remain not fully understood. Density functional theory (DFT) calculations have been used to predict reaction enthalpies for multiple chemical reactions that reversibly store hydrogen and to identify promising reactions with large storage capacities and relevant thermodynamic properties.34,35 Experiments have revealed that LiBH4 decomposes into LiH and B, with Li2B12H12 as an intermediate phase. Theoretical work has suggested formation of (LiBH4)n, n ≤ 12, nanoclusters, which decompose into mixed LinBn clusters via a series of intermediate clusters of LinBnHm, (m ≤ 4n).36 A variety of other intermediates have also been suggested, with the [B12H12]2− as the most stable, which is considered to hamper hydrogen uptake reactions.37,38 Experimental determination of structure and composition of intermediates is often hampered by poor crystallinity and difficulties in preparation of phase-pure samples.
Recently, M2B12H12–10MH composites were hydrogenated at p(H2) = 1000 bar and 500 °C for 72 hours forming the corresponding metal borohydrides, MBH4, M = Li, Na or K.39 This has prompted the present hydrogenation study of closo-polyborate-containing composites, M2B10H10–8MH and M2B12H12–10MH, M = Li or Na, using less harsh conditions in custom-made hydrogenation equipment designed to monitor the hydrogen uptake.40 In doing so, the reaction pathway and kinetic issues associated with the hydrogenation of closo-borate containing samples can be assessed. It is expected that this information will provide insights into the “boron-sink” closo-borates in borohydride-based hydrogen storage systems. The mechanism for hydrogen uptake is investigated using in situ synchrotron radiation powder X-ray diffraction (SR-PXD), ex situ powder X-ray diffraction (PXD), Fourier transform infrared spectroscopy (FT-IR), as well as solution and solid-state 11B NMR and 23Na magic-angle spinning (MAS) NMR.
Experimental
Sample preparation
Lithium hydride, LiH (Sigma-Aldrich, 95%), and sodium hydride, NaH (Sigma-Aldrich, 95%), were used as supplied. Anhydrous Li2B10H10 and Na2B10H10 were synthesized from decaborane (B10H14, Katchem), dimethyl sulfide, liquid ammonia and the respective hydroxides MOH, using a method described elsewhere.41 Li2B12H12·4H2O and Na2B12H12·xH2O (Katchem) were heated to 245 °C under dynamic vacuum for 12 hours to remove coordinated water. The closo-borates M2B10H10 or M2B12H12 were mixed with metal hydrides MH, M = Li or Na, in the molar ratio 1:8 or 1:10, respectively, to form a composite with a M:B ratio of 1:1. The mixtures were then manually ground for 3 minutes. These samples are denoted Li2B10-A, Li2B12-A, Na2B10-A and Na2B12-A. Other samples were prepared with either 3 minutes of manual grinding (Li2B12-As and Na2B12-As) or 15 minutes of ball-milling (ball to powder ratio of 25:1) (Li2B10-Ab, Na2B10-Ab, Li2B10-Ab and Na2B12-Ab). All handling of the chemicals was performed in an argon-filled glovebox with p(O2, H2O) < 1 ppm.
The closo-borate-hydride composites (Li2B10-A, Li2B12-A, Na2B10-A and Na2B12-A) were hydrogenated in custom-made equipment by loading the mixed powders (80–200 mg) into a steel container, which is closed with a filter and placed in a specialized sample cell.42 High pressures (HP) are generated using a metal hydride hydrogen compressor, i.e. heating a commercial AB5 hydrogen storage alloy with the composition MmNi4.35Co0.8Al0.05 where Mm is a mixture of rare earth metals, primarily La and Ce. The pressure generated at room temperature, p(H2) ∼ 400 bar, was increased to 526–547 bar during heating to either ∼300 or ∼400 °C (ΔT/Δt = 5 °C min−1) and was then kept isothermal for 24 hours. The high-pressure hydrogenation experiments were performed at 400 °C under hydrogen pressures of 970 to 998 bar in a stainless steel reactor. An overview of the hydrogen-treated samples is given in Table 1.
| Name | Ratio M2B10/12H10/12:MH, M = Li or Na |
Synthesis, hydrogenation of sample | T [°C] | p(H2)start [bar] | p(H2)end [bar] | Δm/m [wt% H2] | Time [h] | Products |
|---|---|---|---|---|---|---|---|---|
| a Vessel had a ∼4 bar h−1 leak, leading to a large decrease in pressure over the course of the experiment. | ||||||||
| Li2B10-B | 1:8 |
Li2B10-A | 307 | 526 | 516 | 2.9 | 24 | LiBH4 + LiH + 1 |
| Li2B10-C | 1:8 |
Li2B10-Ab | 400 | 998 | 773a | — | 48 | LiBH4 + LiH |
| Li2B12-B | 1:10 |
Li2B12-A | 402 | 547 | 540 | 2.2 | 24 | Li2B12H12 + LiH |
| Li2B12-C | 1:10 |
Li2B12-Ab | 400 | 970 | 955 | — | 48 | Li2B12H12 + LiH |
| Li2B12-D | 1:10 |
Li2B12-As | 400 | 970 | 955 | — | 48 | Li2B12H12 + LiH |
| Na2B10-B | 1:8 |
Na2B10-A | 289 | 534 | 527 | 3.2 | 24 | NaBH4 + NaH + 2 |
| Na2B10-C | 1:8 |
Na2B10-Ab | 400 | 998 | 773a | — | 48 | NaBH4 + NaH |
| Na2B12-B | 1:10 |
Na2B12-A | 401 | 536 | 533 | 1.5 | 24 | Na2B12H12 + NaH + 3 + 4 |
| Na2B12-C | 1:10 |
Na2B12-Ab | 400 | 970 | 955 | — | 48 | Na2B12H12 + NaH |
| Na2B12-D | 1:10 |
Na2B12-As | 400 | 970 | 955 | — | 48 | Na2B12H12 + NaH |
Characterization
Results and discussion
Hydrogenation of metal decahydro-closo-decaborate M2B10H10-8MH compositions
 | ||
| Fig. 1 Normalized powder X-ray diffraction patterns of Li2B10H10–8LiH after manually grinding (Li2B10-A) and after hydrogenation (Li2B10-B) at T = 307 °C and p(H2) = 526 bar for 24 hours (λ = 1.540593 Å). Symbols: ■ o-LiBH4, ● Li2B10H10, ▲ LiH, and compound 1. The dotted pattern is that of pure LiBH4, overlaid for comparison. | ||
The hydrogenation of Li2B10H10–8LiH was further studied by in situ SR-PXD at high hydrogen pressure, p(H2) = 407 bar, and isothermal conditions, T = 300 °C (Fig. 2). The PXD signatures of the reactants, Li2B10H10 and LiH, were observed at RT, after ∼6 hours the unidentified compound 1 emerged in accordance with ex situ PXD (Fig. 1 and Table S1, ESI†), and reflections from Li2B10H10 completely disappeared after ∼9 hours. This agrees with the induction period observed in Fig. S1 (ESI†). As Bragg reflections from Li2B10H10 disappear, the reflections from 1 become more pronounced. After ∼11 hours the sample is cooled and crystallization of h-LiBH4 (h-hexagonal) is observed (Tmelt = 280 °C), which transforms into the ambient o-LiBH4 polymorph (o-orthorhombic).12,43 This indicates that 1 may be an intermediate in the formation of LiBH4.
 | ||
| Fig. 2 In situ high pressure synchrotron powder X-ray diffraction patterns of the hydrogenation of Li2B10H10–8LiH heated from RT to 300 °C, at p(H2) = 407 bar (ΔT/Δt = 10 °C min−1, λ = 0.20720 Å). The sample was kept isothermal at T = 300 °C for ∼10 hours. The dashed line indicates the temperature profile. Symbols: ■ o-LiBH4, ⊠ h-LiBH4, ● Li2B10H10, ▲ LiH, and compound 1. | ||
The 11B MAS NMR spectrum of Li2B10-B is dominated by the narrow centerband resonance at −41.1 ppm from LiBH4 (Fig. 3a). In addition, a somewhat broader centerband is observed at −23 ppm, which is ascribed to boron in compound 1, since the resonance does not match with the chemical shift reported for Li2B10H10 at −28.8 ppm.44 All spinning sidebands for the central and satellite transitions are observed for LiBH4 whereas only a part of the spinning sideband manifold from the satellite transitions is observed for compound 1. Thus, the total intensity for the 11B central and satellite transitions for LiBH4 is obtained as the sum of intensities for the centerband and all spinning sidebands, whereas the central-transition intensity for compound 1 is obtained from the intensities for the centerband and first-order spinning sidebands after an intensity correction to these peaks from the contribution from the satellite transitions. The central-transition intensity for LiBH4 is obtained as 4/10 of the total intensity, which holds for a spin 3/2 nucleus, and comparison of this value with the central transition intensity for 1 reveals that the molar ratio for boron in LiBH4 and 1 is 1:0.63, which corresponds to an uptake of 2.8 wt% H2, matching well with the observed hydrogenation, 2.9 wt% H2.44 This indicates that the absorbed hydrogen only goes toward producing LiBH4 as the B:H ratio in the unknown compound 1 is 1:1, consistent with a higher borate, which is also reflected in its characteristic IR mode and 11B chemical shift. Thus, 11B MAS NMR reveals that all Li2B10H10 (−0.9 ppm and −28.8 ppm) is consumed during hydrogenation and that LiBH4 and 1 are the only reaction products, besides a minor amount of a BO3 species (∼20 ppm), corresponding to 3.5% of the boron in the sample. Solution-state 11B NMR spectra of Li2B10-B dissolved in THF obtained without and with 1H decoupling (Fig. S4, ESI†) allow identification of resonances at −14.0 ppm (1JBH = 142 Hz), −15.6 ppm (1JBH = 120 Hz), −16.0 ppm (1JBH = 120 Hz), −16.7 ppm (1JBH = 120 Hz), −18.0 ppm (1JBH = 150 Hz) and −20.7 ppm (1JBH = 142 Hz) in addition to the main peak at −41.8 ppm from the [BH4]− units. All resonances in the −14.0 to −20.7 ppm region show doublets in the 1H-coupled spectra, demonstrating that they originate from boron sites which are directly bonded to one H atom. The resonance and 1JBH coupling at −15.6 ppm is in agreement with the 11B NMR data reported for [B12H12]2−,45 matching the impurity of Li2B12H12 in the solid-state 11B NMR spectrum (Fig. 3a). The remaining resonances, and, in particular, the peak at −20.7 ppm, may originate from the unknown compound 1 and thereby suggest that this phase contains boron sites directly bonded to a single H atom. Moreover, comparison of the 11B chemical shifts with those reported for relevant borate species (Table S4, ESI†) strongly suggests that compound 1 contains closo-borate units.
 | ||
| Fig. 3 11B MAS NMR spectra (16.45 T, νR = 15.0 kHz) of the hydrogenated samples (a) Li2B10-B and (b) Na2B10-B. The centerbands for LiBH4 and NaBH4 are cut-off at 1/2 and 1/50 of their total heights in (a) and (b), respectively. The asterisk indicates the second-order quadrupole lineshape from a minor impurity of BO3 species (identified for both samples), most likely produced prior to the NMR experiments, whereas the open circle in (a) identifies a centerband at −15.1 ppm (0.7% of the boron in the sample) from a small amount of Li2B12H12 in the sample. | ||
The formation of LiBH4 is also confirmed by FT-IR (Fig. S3, ESI†). The IR spectrum of Li2B10-A has a single large B–H stretching mode at 2500 cm−1 with minor modes at lower wavenumbers consistent with metal closo-borates.46,47 For Li2B10-B, the B–H stretching mode at 2500 cm−1 has almost disappeared while B–H stretching modes appear around 2300 cm−1, which correspond to those of LiBH4.48,49
This work suggests that 1 has a lithium–boron ratio in between Li2B10H10 and LiBH4, i.e. 0.2 < Li/B < 1, and a hydrogen–boron ratio close to 1:1. A hydrogen uptake of 2.9 wt% H2 was measured, which corresponds to a sample composition of 10Li–10B–23.6H. After hydrogenation, the sample contains LiBH4 and LiH and, therefore, also some metal borates richer in boron and hydrogen than lithium. However, the spectroscopy and diffraction data do not unambiguously identify any such compounds.
PXD of Na2B10-B reveals Bragg reflections from NaBH4 along with an unidentified compound, denoted 2, and some unreacted NaH (Fig. 4). PXD of the high-pressure sample, Na2B10-C, reveals that NaBH4 is the major crystalline product, along with 2 (Fig. S6, ESI†). The new compound 2 has reflections at low Bragg angles 2θ < 7° (d > 13 Å) (Table S2, ESI†), which can be indexed with a relatively large monoclinic unit cell, a = 12.7068(5), b = 23.3384(7), c = 16.8802(8) Å, β = 105.456(4)°, V = 4824.9(5) Å3.
 | ||
| Fig. 4 Normalized powder X-ray diffraction patterns of Na2B10H10–8NaH after manually grinding (Na2B10-A) and after hydrogenation (Na2B10-B) at T = 289 °C and p(H2) = 534 bar for 24 hours (λ = 1.54056 Å). Symbols: □ NaBH4, ○ NaH, △ LT-Na2B10H10, and compound 2. The dotted pattern is that of pure NaBH4, overlaid for comparison. | ||
The formation of NaBH4 is further confirmed by IR spectroscopy (Fig. S7, ESI†). The two B–H stretching bands at ∼2500 cm−1 from Na2B10H10 in Na2B10-A are replaced by a single broad band at ∼2400 cm−1 in Na2B10-B, indicating that a different B–H containing cluster is formed, possibly a closo-, arachno-, or nido-borate, which could potentially be ascribed to the unidentified compound 2. Finally, the two stretching modes between 2350–2250 cm−1 are assigned to NaBH4.
The hydrogenation of the composite Na2B10H10–8NaH was investigated by HP in situ SR-PXD and the diffraction data is presented in Fig. 5. Initially, Bragg reflections from the reactants, Na2B10H10 and NaH, are observed followed by the well-known α- to β-Na2B10H10 polymorphic transition at T ∼ 130 °C.50 After 20 min at T = 302 °C and p(H2) ∼ 440 bar (t = 60 min), Bragg reflections from 2 are observed, whereas NaBH4 is observed after approximately 220 minutes at T = 250 °C during cooling. This reaction time agrees well with the ∼4 hours induction time observed in ex situ hydrogen absorption in Fig. S5 (ESI†). Thus, 2 may represent an intermediate in the formation of NaBH4 from the Na2B10H10–8NaH composite.
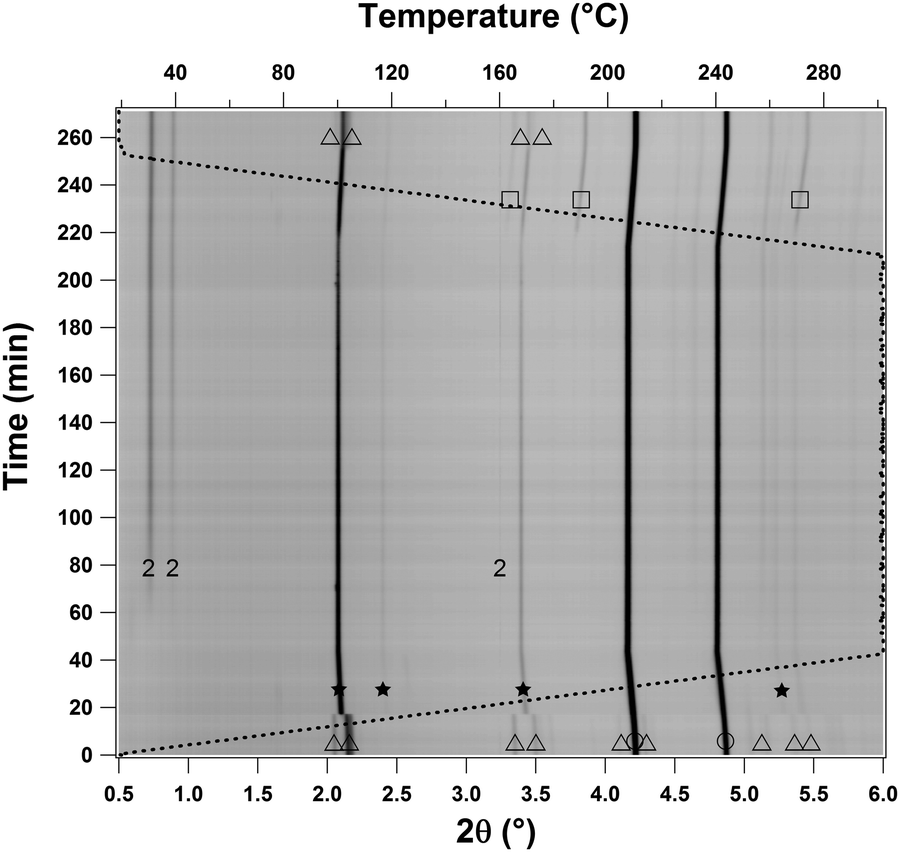 | ||
| Fig. 5 In situ high pressure synchrotron powder X-ray diffraction patterns of the hydrogenation of Na2B10H10–8NaH heated from RT to 302 °C, at p(H2) = 440 bar (ΔT/Δt = 10 °C min−1, λ = 0.20720 Å). The sample was kept isothermal at T = 302 °C for ∼3 hours. The dashed line indicates the temperature profile. Symbols: □ NaBH4, ○ NaH, △ LT-Na2B10H10, ★ HT-Na2B10H10, and compound 2. | ||
The 11B MAS NMR spectrum of Na2B10-B (Fig. 3b) reveals at least two different boron environments, with the dominating resonance at −41.9 ppm originating from NaBH4.51 The second broad resonance is centered at −28 ppm and includes a sharp low-intensity component at −30 ppm. The main component of the resonance at −28 ppm is ascribed to 2 while the sharp resonance at −30 ppm is ascribed to a small amount of the Na2B10H10 starting material.44 The molar ratio for boron in NaBH4 and 2 of 1:0.21 and is derived from the intensities of the central and satellite transitions for NaBH4 and the central transition for the −28 ppm resonance.
The 23Na MAS NMR spectrum of the Na2B10-B sample (Fig. 6a) is dominated by resonances from NaBH4 (−7.9 ppm) and NaH (18.2 ppm).52 Spectral integration, considering the centerband intensities only, reveals that NaBH4 and NaH are present in a 1:0.16 molar ratio. The vertical expansion of the spectrum (Fig. 6a) shows at least four additional resonances, i.e., a second-order quadrupolar lineshape with singularities at 12.2 and 11.1 ppm, a broadened resonance at 2.7 ppm, a shoulder to the NaBH4 centerband at −2.8 ppm and a resonance at −17.9 ppm. These additional resonances constitute 8.3% of the total intensity in the centerband region and thereby represent smaller amounts of other Na containing compounds, possibly 2.
 | ||
| Fig. 6 23Na MAS NMR spectra (16.45 T, νR = 15.0 kHz) of the hydrogenated samples (a) Na2B10-B and (b) Na2B12-B. The spectra illustrate the spectral region for the central transitions and they are shown on normalized as well as expanded intensity scales. | ||
Solution 11B NMR spectra of Na2B10-B dissolved in THF and obtained with and without 1H decoupling (Fig. 7) show resonances at −1.7 ppm (1JBH = 134 Hz), −17.5 ppm (1JBH = 125 Hz), −20.7 ppm (1JBH = 125 Hz), −21.3 ppm (1JBH = 125 Hz) and −29.9 ppm (1JBH = 119 Hz), all corresponding to boron bonded to one H atom, and a main peak at −41.8 ppm from the [BH4]− units. The peaks at −1.7 ppm and −29.9 ppm exhibit a 1:4 intensity ratio and are ascribed to the two distinct B sites in [B10H10]2−, in accordance with 11B NMR data from literature.53 Thus, the resonances at −17.5 ppm, −20.7 ppm, and −21.3 ppm are assigned to 2 and is in accord with data reported for [B11H11]2− (Table S4, ESI†), which is a closo-borate with terminal hydrogens only, i.e., B–H units producing doublets in liquid state NMR spectra.
 | ||
| Fig. 7 Solution 11B NMR spectra (14.1 T) of Na2B10-B dissolved in THF obtained (a) without and (b) with 1H decoupling. | ||
Thus, the measured hydrogen uptake of 3.2 wt% H2 corresponds to a sample composition of 10Na–10B–29.3H, and Na NMR reveals a sample composition of NaBH4–NaH 1:0.16 and that Na2B11H11 (denoted 2) accounts for <8.3% of the sodium in the sample. Boron NMR reveals a ratio between NaBH4–Na2B11H11 of 1:0.21. The products besides NaBH4 and Na2B11H11 are difficult to determine and may be a mixture of different borates.
Hydrogenation of metal dodecahydro-closo-dodecaborate M2B12H12–10MH compositions
 | ||
Fig. 8 Normalized powder X-ray diffraction patterns of Li2B12H12–10LiH after manually grinding (Li2B12-A) and after hydrogenation (Li2B12-B) at T = 402 °C and p(H2) = 546 bar for 24 hours (λ = 1.54056 Å). Symbols: ![[hexagon open, flat-side down]](https://www.rsc.org/images/entities/char_e1c0.gif) LT-Li2B12H12, ▲ LiH, and LT-Li2B12H12, ▲ LiH, and  Li2O. Li2O. | ||
This finding is further supported by the 11B MAS NMR spectrum of Li2B12-B (Fig. S10a, ESI†) that almost exclusively shows a single resonance peak at −15.2 ppm, in agreement with the reported chemical shift for Li2B12H12.44
 | ||
| Fig. 9 Normalized powder X-ray diffraction patterns of Na2B12H12–10NaH after manually grinding (Na2B12-A) and after hydrogenation (Na2B12-B) at T = 401 °C and p(H2) = 537 bar for 24 hours (λ = 1.54056 Å). Symbols: ◇ LT-Na2B12H12, ○ NaH, and compound 3. | ||
The change in structures is investigated by IR spectroscopy (Fig. S13, ESI†). The B–H stretching bands at ∼2478 cm−1 from Na2B12H12 in Na2B12-A are shifted slightly to a lower value of 2462 cm−1 in Na2B12-B, which may be ascribed to 3 along with a new bending mode at ∼1650 cm−1.
The formation of 3 is studied by HP in situ SR-PXD (Fig. 10). Initially, Bragg reflections from Na2B12H12 and NaH are present, and, during heating, the polymorphic transition of α- to β-Na2B12H12 is observed at ∼275 °C.54 In addition, an unidentified compound denoted 4 is observed. No changes in the diffraction pattern are observed at the isothermal temperature, but Bragg reflections from compound 4 disappear at T ∼ 210 °C during cooling. At the same time, Bragg reflections from compound 3 appear, which indicates a phase transition of 4 into 3. Reflections from compounds 3 and 4 are listed in Table S3 (ESI†) with the d-spacing and relative intensities. Indexing suggests an orthorhombic unit cell for 3: a = 6.9114(6), b = 12.877(2), c = 14.296(1) Å, V = 1272.2(5) Å3, and an orthorhombic unit cell for 4: a = 6.3383(2), b = 10.5223(8), c = 14.8283(7) Å and V = 987.4(2) Å3.
 | ||
| Fig. 10 In situ high pressure synchrotron powder X-ray diffraction patterns of the hydrogenation of Na2B12H12–10NaH heated from RT to 407 °C, at p(H2) = 417 bar (ΔT/Δt = 10 °C min−1, λ = 0.20720 Å). The sample was kept isothermal at T = 407 °C for ∼3 hours. The dashed line indicates the temperature profile. Symbols: ◇ LT-Na2B12H12, ◆ HT-Na2B12H12, ○ NaH, compound 3, and compound 4. | ||
The 11B MAS NMR spectrum of Na2B12-B (Fig. S10b, ESI†), exhibits a narrow resonance (full width at half maximum (FWHM) = 0.7 ppm) at −15.7 ppm, in accordance with the 11B chemical shift reported for Na2B12H12, which may suggest that 3 has boron coordination similar to that of Na2B12H12.44 PXD clearly shows the formation of a new crystalline product after hydrogenation at high temperature, whereas NMR results only indicate the presence of [B12H12]2−-containing compounds. A previous study has shown that annealing Li2B12H12 under hydrogen pressure can result in the formation of additional Bragg diffraction peaks, indicating that other compounds may form at high temperature.55 Compound 3 appears to consist of [B12H12]2− anions as suggested by NMR.
The 23Na MAS NMR spectrum of Na2B12-B (Fig. 6b) is dominated by a resonance at 18.2 ppm from NaH. In addition, minor peaks are observed at 32.1 ppm, 11.9 ppm, 2.3 ppm, −11.4 ppm, −28.8 ppm and −38 ppm. A 23Na MAS NMR spectrum of Na2B12H12 reveals that the resonance at −11.4 ppm originates from this compound. Moreover, the peaks at −28.8 ppm and −38 ppm are most likely the low-frequency part (singularity and edge) of a second-order quadrupolar lineshape. However, the resonances at 32.1 ppm, 11.9 ppm, 2.3 ppm, −28.8 ppm, and −38 ppm are not assigned and thus they may include contributions from the unidentified compound 3. Spectral integration shows that the four resonances correspond to 10.7% of the total 23Na centerband intensity.
The Na2B12-B sample absorbed 1.5 wt% H2, which corresponds to a sample composition of 12Na–12B–28.4H. Solid state 11B MAS NMR show that 3 has similar chemical environment as [B12H12]2− and diffraction reveal that 4 transforms to 3 upon cooling.
Comparison of the investigated closo-borate composites
Comparison of the reactivity of the Li and Na closo-borate composites, Li2B10H10–8LiH and Na2B10H10–8NaH, reveals several important differences. For instance, the Na composites have faster kinetics for hydrogenation than the corresponding Li samples, e.g., Li2B10-A absorbs hydrogen slowly for the first 6 hours, then ∼5× faster for 8 hours and then again slower whereas Na2B10-A does not absorb hydrogen for the first 3 hours then absorbs quickly for 12 h, and then more slowly. From a kinetic point of view, the shorter induction period for the sodium system may be associated with faster nucleation and growth of intermediate compounds prior to formation of NaBH4 as compared to similar reactions for the analogous lithium composites. This is well documented by solid-state 11B NMR, which reveals sample compositions of LiBH4–1 (1:0.63) for Li2B10H10–8LiH and NaBH4–2 (1:0.21) for Na2B10H10–8NaH after 24 h. In situ HP SR-PXD experiments reveal the formation of NaBH4 after 4 hours of Na2B10B10–8NaH, whereas LiBH4 forms after 10 hours. A major difference between composites based on M2B10H10 and M2B12H12 is their reactivity; those based on M2B10H10 more readily form the corresponding metal borohydride, MBH4, and intermediate compounds. In contrast, only unidentified compounds are observed in the Na2B12H12–10NaH composite, whereas no reaction is observed for Li2B12H12–10LiH under the conditions used in this study. Four different intermediate compounds have been observed during hydrogenation reactions in this investigation. However, they are different than intermediates predicted theoretically for the decomposition of LiBH4 and to those observed experimentally.36,56
In a previous study, Li2B12H12–10LiH and Na2B12H12–10NaH were fully converted to the respective metal borohydrides, LiBH4 and NaBH4, under harsher conditions, i.e. T = 500 °C, p(H2) = 1.0 kbar for 72 h.39 Milder conditions are used in this study, i.e. T ∼ 400 °C, p(H2) ∼ 998 bar for 24 h for hydrogenation of M2B10H10–8MH and T ∼ 400 °C, p(H2) ∼ 970 bar for 48 h for M2B12H12–10MH, which proves insufficient to form the respective metal borohydride. The M2B10H10 composites are clearly more reactive and take up hydrogen at milder conditions compared to the M2B12H12 composites, possibly due to the lower thermodynamic stability of the [B10H10]2− anion, which allows B–B bond breaking to occur under milder reaction conditions.
Conclusions
The composites M2B12H12–10MH and M2B10H10–8MH (M = Li and Na) have been reacted with hydrogen at elevated pressures and temperatures and studied by X-ray diffraction both PXD and in situ SR-PXD and FT-IR, 23Na and 11B NMR spectroscopy. Both the in situ and ex situ characterization results show that the M2B10H10–8MH composites react with hydrogen gas to form the respective metal borohydride, MBH4, at T ∼ 300 °C and p(H2) > 500 bar. These conditions were deliberately selected to obtain partial hydrogenation of the samples in order to focus on the mechanism of hydrogen uptake. The relatively mild conditions used in this investigation did not allow formation of MBH4 by hydrogenation of the M2B12H12–10MH composites: no hydrogen absorption was detected for Li2B12H12–10LiH, but new intermediates were observed for Na2B12H12–10NaH. Generally, the sodium-containing composites are observed to be more reactive towards hydrogen compared to the lithium analogues under similar conditions of temperature and H2 pressure, and M2B10H10–8MH are more reactive than M2B12H12–10MH. The high stability of the [B12H12]2− anions is associated with the pseudoaromatic bonding in dodecahydro-closo-dodecaborate cages and lack of chemically distinct and more reactive apical boron atoms within the closo-borate polyhedra that the decahydro-closo-decaborate cages contain.The conditions for hydrogenation used in this study are clearly above the thermodynamic limit needed to form the corresponding metal borohydrides, LiBH4 or NaBH4, for the composites containing [B10H10]2−. Therefore, the observed differences in the length of the induction periods prior to hydrogen absorption, which occur at different rates, are assigned to kinetic constraints. Remarkably, four different intermediate compounds have been observed in this study for the hydrogenation of the composites. The formation of intermediate compounds appears to be the rate-limiting step of the reaction, with the slowest kinetics observed for the M2B12H12 compounds. This suggest that the molecular mechanism for hydrogen absorption is different for the two closo-borate cages, [B10H10]2− and [B12H12]2−.
This study not only demonstrates that lithium and sodium M2B10H10 salts can be hydrogenated into the corresponding metal borohydrides, but it also reveals the presence of, at least, four B–H intermediates, which are different to those previously observed experimentally or suggested based on theoretical calculations. Additional investigations are needed to clarify composition, structure, and properties of these intermediate compounds. The analysis of the available solid-state and solution 11B NMR data indicates that these intermediates are not salts of [B3H8]−, [B10H10]2− or [B12H12]2− anions. We hypothesize that these could be other closo-polyborates, e.g., [B9H9]2− or [B11H11]2− salts, or oligomers resulting from [B10H10]2− polymerization.29 For example, closo-decaborate salts are known to form dimeric anions, such as various isomers of [B20H18]4− and [B20H18]2− anions.57
A detailed understanding of the reactivity of these important compounds may allow further tailoring of the reaction mechanisms of hydrogen storage reactions. Further investigations should focus on identifying reaction pathways in the dehydrogenation of metal borohydrides that avoid the formation of stable [B12H12]2− anions, and favoring formation of intermediate B–H species that can be cycled under more reasonable conditions of hydrogen pressure and temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Danish National Research Foundation is thanked for funding to the Center for Materials Crystallography (CMC, DNRF93). Furthermore, we acknowledge financial support from The Danish Council for Independent Research for a DFF Mobility grant (no. 1325-00072) and the research project HyNanoBorN (DFF – 4181-00462). The beam line P02.1, PETRA III, DESY, Germany is acknowledged for providing beam time to conduct the in situ experiments. The use of the facilities at the Instrument Centre for Solid-State NMR Spectroscopy, Aarhus University, sponsored by the Danish Natural Science Research Councils and the Carlsberg Foundation is acknowledged. The authors gratefully acknowledge research support from the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office through the Hydrogen Storage Materials Advanced Research Consortium (HyMARC). Sandia National Laboratories is a multi-mission laboratory managed by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analyses. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.References
- The Relentless Rise of Carbon Dioxide, http://climate.nasa.gov/climate_resources/24/, accessed October 2nd, 2017.
- U. S. EIA, International Energy Outlook, 2016, http://www.eia.gov/forecasts/ieo/pdf/0484(2016).pdf.
- M. B. Ley, L. H. Jepsen, Y.-S. Lee, Y. W. Cho, J. M. Bellosta von Colbe, M. Dornheim, M. Rokni, J. O. Jensen, M. Sloth, Y. Filinchuk, J. E. Jørgensen, F. Besenbacher and T. R. Jensen, Mater. Today, 2014, 17, 122–128 CrossRef.
- L. H. Jepsen, M. B. Ley, Y.-S. Lee, Y. W. Cho, M. Dornheim, J. O. Jensen, Y. Filinchuk, J. E. Jørgensen, F. Besenbacher and T. R. Jensen, Mater. Today, 2014, 17, 129–135 CrossRef.
- K. T. Møller, T. R. Jensen, E. Akiba and H.-W. Li, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- H.-W. Li, Y. Yan, S. Orimo, A. Züttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef.
- J. Puszkiel, S. Garroni, C. Milanese, F. Gennari, T. Klassen, M. Dornheim and C. Pistidda, Inorganics, 2017, 5 CrossRef.
- G. Moussa, R. Moury, U. B. Demirci, T. Şener and P. Miele, Int. J. Energy Res., 2013, 37, 825–842 CrossRef.
- M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Cerny, D. B. Ravnsbaek, Y. Filinchuk, M. Dornheim, F. Besenbacher and T. R. Jensen, Chem. Soc. Rev., 2017, 46, 1565–1634 RSC.
- G. Severa, E. Rönnebro and C. M. Jensen, Chem. Commun., 2010, 46, 421–423 RSC.
- O. Friedrichs, J. W. Kim, A. Remhof, F. Buchter, A. Borgschulte, D. Wallacher, Y. W. Cho, M. Fichtner, K. H. Oh and A. Züttel, Phys. Chem. Chem. Phys., 2009, 11, 1515–1520 RSC.
- M. Paskevicius, M. B. Ley, D. A. Sheppard, T. R. Jensen and C. E. Buckley, Phys. Chem. Chem. Phys., 2013, 15, 19774–19789 RSC.
- P. Martelli, R. Caputo, A. Remhof, P. Mauron, A. Borgschulte and A. Züttel, J. Phys. Chem. C, 2010, 114, 7173–7177 Search PubMed.
- Q. Lai, M. Paskevicius, D. A. Sheppard, C. E. Buckley, A. W. Thornton, M. R. Hill, Q. Gu, J. Mao, Z. Huang, H. K. Liu, Z. Guo, A. Banerjee, S. Chakraborty, R. Ahuja and K. F. Aguey-Zinsou, ChemSusChem, 2015, 8, 2789–2825 CrossRef PubMed.
- T. D. Humphries, M. B. Ley, C. Frommen, K. T. Munroe, T. R. Jensen and B. C. Hauback, J. Mater. Chem. A, 2015, 3, 691–698 Search PubMed.
- P. Schouwink, M. B. Ley, A. Tissot, H. Hagemann, T. R. Jensen, L. Smrcok and R. Cerny, Nat. Commun., 2014, 5, 5706–5715 CrossRef PubMed.
- M. B. Ley, M. Paskevicius, P. Schouwink, B. Richter, D. A. Sheppard, C. E. Buckley and T. R. Jensen, Dalton Trans., 2014, 43, 13333–13342 RSC.
- P. Schouwink, V. D’Anna, M. B. Ley, L. M. Lawson Daku, B. Richter, T. R. Jensen, H. Hagemann and R. Černý, J. Phys. Chem. C, 2012, 116, 10829–10840 Search PubMed.
- I. Lindemann, R. Domenech Ferrer, L. Dunsch, Y. Filinchuk, R. Cerny, H. Hagemann, V. D'Anna, L. M. Lawson Daku, L. Schultz and O. Gutfleisch, Chem. – Eur. J., 2010, 16, 8707–8712 CrossRef PubMed.
- R. Černý, P. Schouwink, Y. Sadikin, K. Stare, L. Smrcok, B. Richter and T. R. Jensen, Inorg. Chem., 2013, 52, 9941–9947 CrossRef PubMed.
- R. Černý and P. Schouwink, Acta Crystallogr., Sect. B: Struct. Sci., 2015, 71, 619–640 CrossRef PubMed.
- E. Roedern and T. R. Jensen, Inorg. Chem., 2015, 54, 10477–10482 CrossRef PubMed.
- S. R. H. Jensen, L. H. Jepsen, J. Skibsted and T. R. Jensen, J. Phys. Chem. C, 2015, 119, 27919–27929 Search PubMed.
- J. J. Vajo and S. L. Skeith, J. Phys. Chem. B, 2005, 109, 3719–3722 CrossRef PubMed.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef.
- C. J. Webb, J. Phys. Chem. Solids, 2015, 84, 96–106 CrossRef.
- J. Wang, H.-W. Li and P. Chen, MRS Bull., 2013, 38, 480–487 CrossRef.
- B. R. S. Hansen, M. Paskevicius, H.-W. Li, E. Akiba and T. R. Jensen, Coord. Chem. Rev., 2016, 323, 60–70 CrossRef.
- Y. Yan, A. Remhof, D. Rentsch and A. Züttel, Chem. Commun., 2015, 51, 700–702 RSC.
- B. R. Hansen, D. B. Ravnsbaek, J. Skibsted and T. R. Jensen, Phys. Chem. Chem. Phys., 2014, 16, 8970–8980 RSC.
- V. Stavila, J.-H. Her, W. Zhou, S.-J. Hwang, C. Kim, L. A. M. Ottley and T. J. Udovic, J. Solid State Chem., 2010, 183, 1133–1140 CrossRef.
- O. Friedrichs, A. Remhof, S. J. Hwang and A. Züttel, Chem. Mater., 2010, 22, 3265–3268 CrossRef.
- M. Chong, A. Karkamkar, T. Autrey, S. Orimo, S. Jalisatgi and C. M. Jensen, Chem. Commun., 2011, 47, 1330–1332 RSC.
- K. C. Kim, A. D. Kulkarni, J. K. Johnson and D. S. Sholl, Phys. Chem. Chem. Phys., 2011, 13, 7218–7229 RSC.
- S. V. Alapati, J. Karl Johnson and D. S. Sholl, Phys. Chem. Chem. Phys., 2007, 9, 1438–1452 RSC.
- Z. Q. Huang, W. C. Chen, F. C. Chuang, E. H. Majzoub and V. Ozolins, Sci. Rep., 2016, 6, 26056 CrossRef PubMed.
- V. Ozolins, E. H. Majzoub and C. Wolverton, J. Am. Chem. Soc., 2009, 131, 230–237 CrossRef PubMed.
- Y. Zhang, E. Majzoub, V. Ozoliņš and C. Wolverton, J. Phys. Chem. C, 2012, 116, 10522–10528 Search PubMed.
- J. L. White, R. J. Newhouse, J. Z. Zhang, T. J. Udovic and V. Stavila, J. Phys. Chem. C, 2016, 120, 25725–25731 Search PubMed.
- B. R. S. Hansen, K. T. Møller, M. Paskevicius, A.-C. Dippel, P. Walter, C. J. Webb, C. Pistidda, N. Bergemann, M. Dornheim, T. Klassen, J.-E. Jørgensen and T. R. Jensen, J. Appl. Crystallogr., 2015, 48, 1234–1241 Search PubMed.
- E. L. Muetterties, J. H. Balthis, Y. T. Chia, W. H. Knoth and H. C. Muller, Inorg. Chem., 1964, 3, 444–451 CrossRef.
- M. Paskevicius, D. A. Sheppard and C. E. Buckley, J. Am. Chem. Soc., 2010, 132, 5077–5083 CrossRef PubMed.
- H. I. Schlesinger and H. C. Brown, J. Am. Chem. Soc., 1940, 62, 3429–3435 CrossRef.
- L. He, H.-W. Li and E. Akiba, Energies, 2015, 8, 12429–12438 CrossRef.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier Ltd, 2nd edn, 1997 Search PubMed.
- M. Sharma, D. Sethio, V. D'Anna and H. Hagemann, Int. J. Hydrogen Energy, 2015, 40, 12721–12726 CrossRef.
- E. L. Muetterties, R. E. Merrifield, H. C. Miller, W. H. Knoth and J. R. Downing, J. Am. Chem. Soc., 1962, 84, 2506–2508 CrossRef.
- H. Hagemann, http://www.unige.ch/sciences/chifi/?ftirdb.html.
- V. D'Anna, A. Spyratou, M. Sharma and H. Hagemann, Spectrochim. Acta, Part A, 2014, 128, 902–906 CrossRef PubMed.
- T. J. Udovic, M. Matsuo, W. S. Tang, H. Wu, V. Stavila, A. V. Soloninin, R. V. Skoryunov, O. A. Babanova, A. V. Skripov, J. J. Rush, A. Unemoto, H. Takamura and S. Orimo, Adv. Mater., 2014, 26, 7622–7626 CrossRef PubMed.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831–1836 RSC.
- R. Tabeta, M. Aida and H. Saitô, Bull. Chem. Soc. Jpn., 1986, 59, 1957–1966 CrossRef.
- N. N. Greenwood and J. H. Morris, Proc. Chem. Soc., 1963, 338–340, 10.1039/PS9630000325.
- N. Verdal, J.-H. Her, V. Stavila, A. V. Soloninin, O. A. Babanova, A. V. Skripov, T. J. Udovic and J. J. Rush, J. Solid State Chem., 2014, 212, 81–91 CrossRef.
- M. P. Pitt, M. Paskevicius, D. H. Brown, D. A. Sheppard and C. E. Buckley, J. Am. Chem. Soc., 2013, 135, 6930–6941 CrossRef PubMed.
- L. Mosegaard, B. Møller, J.-E. Jørgensen, U. Bösenberg, M. Dornheim, J. C. Hanson, Y. Cerenius, G. Walker, H. J. Jakobsen, F. Besenbacher and T. R. Jensen, J. Alloys Compd., 2007, 446–447, 301–305 CrossRef.
- M. F. Hawthorne, K. Shelly and F. Li, Chem. Commun., 2002, 547–554 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp07776a |
| This journal is © the Owner Societies 2018 |