
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminum and Fenton reaction: how can the reaction be modulated by speciation? A computational study using citrate as a test case
Jon I.
Mujika
 a,
Gabriele
Dalla Torre
ab and
Xabier
Lopez
*a
a,
Gabriele
Dalla Torre
ab and
Xabier
Lopez
*a
aKimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, and Donostia International Physics Center (DIPC), P.K. 1072, 20080 Donostia, Euskadi, Spain. E-mail: xabier.lopez@ehu.es
bUCBIO/REQUIMTE, Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, s/n, Porto, Portugal
First published on 17th May 2018
Abstract
The pro-oxidant ability of aluminum is behind many of the potential toxic effects of this exogenous element in the human organism. Although the overall process is still far from being understood at the molecular level, the well known ability of aluminum to promote the Fenton reaction is mediated through the formation of stable aluminum–superoxide radical complexes. However, the properties of metal complexes are highly influenced by the speciation of the metal. In this paper, we investigate the effect that speciation could have on the pro-oxidant activity of aluminum. We choose citrate as a test case, because it is the main low-molecular-mass chelator of aluminum in blood serum, forming very stable aluminum–citrate complexes. The influence of citrate in the interaction of aluminum with the superoxide radical is investigated, determining how the formation of aluminum–citrate complexes affects the promotion of the Fenton reaction. The results indicate that citrate increases the stability of the aluminum–superoxide complexes through the formation of ternary compounds, and that the Fenton reaction is even more favorable when aluminum is chelated to citrate. Nevertheless, our results demonstrate that overall, citrate may prevent the pro-oxidant activity of aluminum: on one hand, in an excess of citrate, the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 aluminum–citrate complexes is expected. On the other hand, the chelation of iron by citrate makes the reduction of iron thermodynamically unfavorable. In summary, the results suggest that citrate can have both a promotion and protective role, depending on subtle factors, such as initial concentration, non-equilibrium behavior and the exchange rate of ligands in the first shell of the metals.
:2 aluminum–citrate complexes is expected. On the other hand, the chelation of iron by citrate makes the reduction of iron thermodynamically unfavorable. In summary, the results suggest that citrate can have both a promotion and protective role, depending on subtle factors, such as initial concentration, non-equilibrium behavior and the exchange rate of ligands in the first shell of the metals.
Introduction
Aluminum is the third most abundant element in the Earth's crust, but complex geochemical cycles prevent its solubilization from the lithosphere,1–3 keeping this abundant metal out of essential biological cycles. Nevertheless, in the last century, aluminum has been extracted on a massive scale from the soil due to its favorable properties (easy to manipulate, cheap and good physicochemical properties) and a myriad of applications (food additives, pharmaceuticals, Al-containers, etc.). As a consequence, aluminum is nowadays present in the biosphere, and humans are highly exposed to this metal.4,5 Unfortunately, this is not without consequences, and this exposure to aluminum has been linked to various diseases,6–9 starting from early evidence of dialysis encephalopathy or osteodistrophy in patients with renal failure under dialysis treatment,10,11 to more recent evidence linking aluminum to several neurodegenerative diseases.6,8 The high charge and small volume of aluminum make this metal a strong Lewis acid, with high affinity towards oxygen-containing and negatively-charged functional groups, such as phosphates or carboxylic groups. Therefore, aluminum has the potential ability to form strong interactions with important biomolecules such as proteins, phospholipids, ATP, NADH, RNA, DNA, etc.12The pro-oxidant ability of aluminum is one of the main deleterious effects of aluminum, observed in both in vitro and in vivo experiments.13,14 This is a feature not expected for an element without a redox capability.15,16 It was first hypothesized13,15 that this pro-oxidant activity was due to the formation of a strong aluminum–superoxide complex, which leads to an increase of the lifetime of the superoxide radical species. In fact, Fukuzumi et al. reported a linear relationship between the strength of the metal–superoxide interaction and the oxidant activity of the metal.17–19 Based on this hypothesis, we demonstrated20 the possibility of the formation of an aluminum–superoxide complex in solution, departing from various aluminum hydrolytic species. Further calculations confirmed that these aluminum–superoxide complexes may increase oxidative stress in biological environments by acting as promoting agents of the Fenton reaction21 (see Fig. 1).
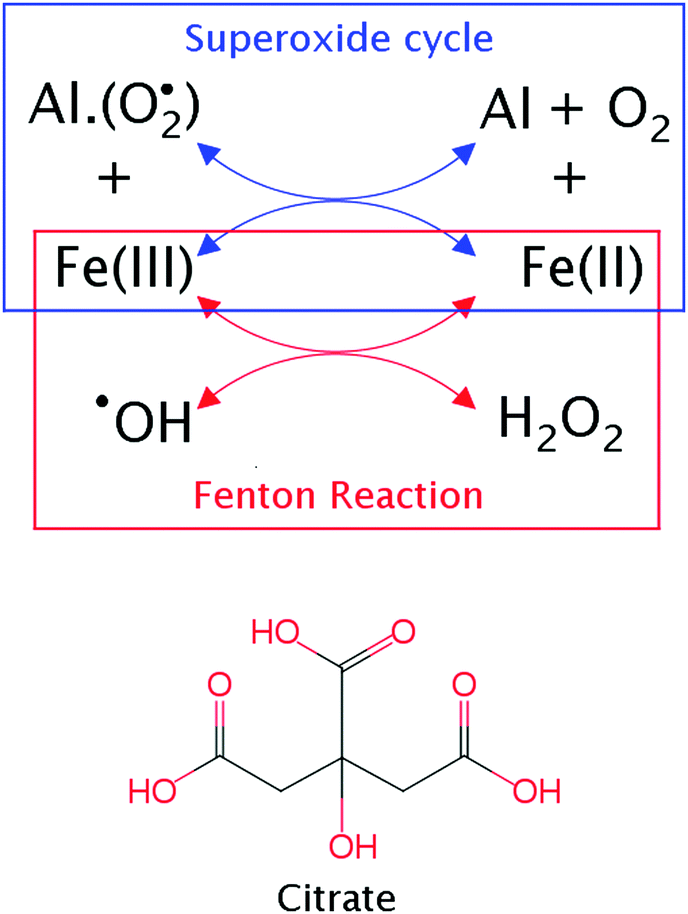 | ||
| Fig. 1 Above, a possible route for the pro-oxidant activity of aluminum by the promotion of the Fenton reaction. Below, the molecular structure of citrate. | ||
However, most of the aluminum present in the human organism is not free in solution, but forms stable complexes with low and high molecular mass biomolecules, and this could have a strong effect on the pro-oxidant activity of aluminum. Many attempts have been made to identify the molecules interacting with aluminum in biological systems.22–25 However, due to the complex chemistry of aluminum, its low total concentration, and the high risk of sample-contamination, the study of the speciation of aluminum is a complex task.24 Nowadays, it is accepted that 90% of the aluminum found in the blood serum is bound to serum transferrin protein, while citrate is the main low molecular weight chelator.26 Further, citrate has been identified as the main chelator of aluminum in brain extracellular fluid.6 This behavior is not surprising as the molecular composition of citrate with three carboxylic groups and an alcohol group (shown in Fig. 1) provides high affinity towards Al(III). Interestingly, in previous studies, it was demonstrated that the formation of ternary complexes with citrate can enable a protective role with respect to some of the deleterious effects of aluminum.27,28
In the present study, our goal is to analyze how the interaction with citrate could alter (i) the possibility of the formation of aluminum–superoxide complexes and (ii) the possibility of reducing Fe(III) to Fe(II), promoting the Fenton reaction. To do so, we evaluate the binding energies of Al(III) to superoxide and citrate, and the reaction free energy of iron reduction from Fe(III) to Fe(II) in the presence of aluminum–superoxide radical binary species and aluminum–citrate–superoxide ternary species. To be consistent, we also consider the changes in the corresponding energies when citrate is bound to iron. The study describes a complex scenario that ultimately depends on the relative concentrations of each species. However, the results point in general to the protective role of citrate with respect to the pro-oxidant ability of aluminum, especially in high-citrate concentration regimes.
Methodology
All geometries were optimized in solution at two levels of theory using the B3LYP functional29–32 and 6-31++g(d,p) basis set: B3LYP/6-31++g(d,p) and B3LYP-D3(BJ)/6-31++g(d,p). In the latter level of theory, the D3 version of Grimme's dispersion with Becke–Johnson damping is included.33 To confirm that the optimized structures were real minima on the potential energy surfaces, frequency calculations were carried out at the same level of theory. All structures showed positive force constants for all the normal modes of vibration. The frequencies were then used to evaluate the zero-point vibrational energy (ZPVE) and the thermal (T = 298 K) vibrational corrections to the enthalpies and Gibbs free energies within the harmonic oscillator approximation. To calculate the entropy, the different contributions to the partition function were evaluated using the standard statistical mechanics expressions in the canonical ensemble and the harmonic oscillator and rigid rotor approximation. The solvent effect was introduced by using the self-consistent reaction field (SCRF) method with the polarized continuum model (PCM), using the integral equation formalism variant (IEFPCM).34Once the geometries were optimized at the two levels of theory stated above, the electronic energies were refined using the larger basis set 6-311++g(3df,2p) by two distinct single-point energy calculations: (a) using the same functional as that in the geometry optimization or (b) using the wB97XD hybrid functional with the dispersion correction included in it.35 Thus, for each reaction, a set of four different energies are presented (energy evaluation//geometry optimization): (i) B3LYP/TZ//B3LYP/DZ (ΔGB3LYPaq), (ii) wB97XD/TZ//B3LYP/DZ (ΔGB3,wB97aq), (iii) B3LYP-D3/TZ//B3LYP-D3/DZ (ΔGB3–D3aq), and (iv) wB97XD/TZ//B3LYP-D3/DZ (ΔGB3–D3,wB97aq), where TZ = 6-311++g(3df,2p) and DZ = 6-31++g(d,p). All these calculations were carried out using the Gaussian 0936 and 1637 packages.
As in our previous study, we considered different hydrated models for the Al–Citr complex, where different numbers of water and hydroxide molecules were included. Departing from each of these models, the substitution reaction of a water molecule (eqn (1)) or a hydroxide (eqn (2)) by the superoxide to form an Al–Citr–O2˙ complex was studied:
| [AlCitr(H2O)n(OH)m](aq)−(1+m) + [O2˙(H2O)2](aq)− → [AlCitr(H2O)n−1(OH)mO2˙](aq)−(2+m) + [H2O(H2O)2](aq) | (1) |
| [AlCitr(H2O)n(OH)m](aq)−(1+m) + [O2˙(H2O)2](aq)− → [AlCitr(H2O)n(OH)m−1O2˙](aq)−(1+m) + [OH(H2O)2](aq)− | (2) |
The free energies in solution corresponding to these two reactions are calculated as:
| ΔGaq = Gaq(AlCitr(H2O)n−1(OH)mO2˙) + Gaq(H2O(H2O)2) − Gaq(AlCitr(H2O)n(OH)m) − Gaq(O2˙(H2O)2) | (3) |
| ΔGaq = Gaq(Al·Citr·(H2O)n·(OH)m−1·O2˙) + Gaq(OH·(H2O)2) − Gaq(Al·Citr·(H2O)n·(OH)m) − Gaq(O2˙·(H2O)2) | (4) |
The ΔGaq values were computed at the four levels of theory described above. All these values are presented in Tables 1–3, but since the overall trends are maintained, only the ΔGB3–D3aq values are discussed throughout the body text.
| ΔGB3LYPaq | ΔGB3,wB97aq | ΔGB3–D3aq | ΔGB3–D3,wB97aq | |
|---|---|---|---|---|
| [Al(H2O)6]3+ + [O2˙]− → [Al(O2˙)(H2O)5]2+ + H2O | −24.7 | −25.9 | −19.5 | −19.9 |
| [Al(OH)(H2O)5]2+ + [O2˙]− → [Al(O2˙)(OH)(H2O)4]1+ + H2O | −24.6 | −24.1 | −17.9 | −18.3 |
| [Al(OH)2(H2O)4]1+ + [O2˙]− → Al(O2˙)(OH)2(H2O)3 + H2O | −18.7 | −18.6 | −17.6 | −17.4 |
| Al(OH)3(H2O)2 + [O2˙]− → [Al(O2˙)(OH)3(H2O)]1− + H2O | −16.4 | −17.4 | −8.5 | −8.6 |
| [Al(OH)4]1− + [O2˙]− → [Al(O2˙)(OH)3]1− + OH− | 31.1 | 30.5 | 21.5 | 22.1 |
| ΔGB3LYPaq | ΔGB3,wB97aq | ΔGB3–D3aq | ΔGB3–D3,wB97aq | |
|---|---|---|---|---|
| [AlCitr(H2O)3(H2O)10]1− + [O2˙]− → [AlCitr(H2O)2(O2˙)(H2O)10]2− + H2O | −7.2 | −5.5 | −4.0 | −5.2 |
| [AlCitr(H2O)2(OH)(H2O)10]2− + [O2˙]− → [AlCitr(H2O)(OH)(O2˙)(H2O)10]3− + H2O | −4.4 | −4.2 | −3.9 | −4.5 |
| [AlCitr(H2O)(OH)2(H2O)10]3− + [O2˙]− → [AlCitr(OH)2(O2˙)(H2O)10]4− + H2O | 4.1 | 7.4 | 6.2 | 6.6 |
| [AlCitr(H2O)2(OH)(H2O)10]2− + [O2˙]− → [AlCitr(H2O)2(O2˙)(H2O)10]2− + OH− | 15.5 | 16.3 | 16.5 | 17.5 |
| [AlCitr(H2O)(OH)2(H2O)10]3− + [O2˙]− → [AlCitr(H2O)(OH)(O2˙)(H2O)10]3− + OH− | 16.6 | 16.1 | 14.0 | 14.8 |
| [AlCitr(OH)3(H2O)10]4− + [O2˙]− → [AlCitr(OH)2(O2˙)(H2O)10]4− + OH− | 3.2 | 5.2 | 4.1 | 5.2 |
| ΔGB3LYPaq | ΔGB3,wB97aq | ΔGB3–D3aq | ΔGB3–D3,wB97aq | |
|---|---|---|---|---|
| a Data taken from ref. 21. | ||||
| Absence of citrate and aluminum | ||||
| [FeW6]3+ + [O2˙]− → [FeW6]2+ + O2 | −54.7 | −57.8 | −50.0 | −55.7 |
| Absence of citratea | ||||
| [FeW6]3+ + [AlW5(O2˙)]2+ → [FeW6]2+ + [AlW5(O2)]3+ | −19.6 | −16.7 | −9.2 | −12.7 |
| [FeW6]3+ + [AlW5(O2˙)]2+ + H2O → [FeW6]2+ + [AlW6]3+ + O2 | −30.0 | −31.9 | −30.5 | −35.8 |
| [FeW6]3+ + [AlOHW4(O2˙)]+ + H2O → [FeW6]2+ + [AlOHW5]2+ + O2 | −30.1 | −33.7 | −32.1 | −37.4 |
| [FeW6]3+ + Al(OH)2W3(O2˙) + H2O → [FeW6]2+ + [Al(OH)2W4 ]+ + O2 | −1.6 | −3.9 | −32.4 | −38.4 |
| Citrate interacting only with aluminum | ||||
| [FeW6]3+ + [AlCitrW2(O2˙)]2− → [FeW6]2+ + [AlCitrW2(O2)]− | −10.2 | −4.8 | −4.9 | −2.6 |
| [FeW6]3+ + [AlCitrW2(O2˙)]2− + H2O → [FeW6]2+ + [AlCitrW3]− + O2 | −37.9 | −30.6 | −36.1 | −41.2 |
| [FeW6]3+ + [AlCitr(OH)W(O2˙)]3− + H2O → [FeW6]2+ + [AlCitr(OH)W2]2− + O2 | −40.6 | −31.8 | −36.5 | −42.1 |
| Citrate interacting only with iron | ||||
| [FeCitrW3]− + [AlW5(O2˙)]2+ → [FeCitrW3]2− + [AlW5(O2)]3+ | 28.2 | 28.7 | 34.0 | 30.2 |
| [FeCitrW3]− + [AlW5(O2˙)]2+ + H2O → [FeCitrW3]2− + [AlW6]3+ + O2 | 17.8 | 26.0 | 12.7 | 7.0 |
| [FeCitrW3]− + [Al(OH)W4(O2˙)]+ + H2O → [FeCitrW3]2− + [Al(OH)W5]2+ + O2 | 17.7 | 24.2 | 11.1 | 5.5 |
| Citrate interacting with both aluminum and iron | ||||
| [FeCitrW3]− + [AlCitrW2(O2˙)]2− → [FeCitrW3]2− + [AlCitrW2(O2)]− | 37.7 | 40.7 | 38.3 | 40.3 |
| [FeCitrW3]− + [AlCitrW2(O2˙)]2− + H2O → [FeCitrW3]2− + [AlCitrW3]− + O2 | 10.0 | 14.9 | 7.0 | 1.7 |
| [FeCitrW3]− + [AlCitr(OH)W(O2˙)]3− + H2O → [FeCitrW3]2− + [AlCitr(OH)W2]2− + O2 | 7.3 | 13.7 | 6.6 | 0.7 |
For all the aluminum containing structures, we also calculated delocalization indices (DIs). These are a measure of the covariance between the population of two atoms A and B and, consequently, a measure of the number of electrons simultaneously fluctuating between these atoms,38,39

Results and discussion
Incorporation of the O2 superoxide radical
In our previous work, we investigated the incorporation of O2˙− into the first coordination shell of aluminum hydrolytic species.20 In particular, the displacement of either a water molecule or a hydroxide from the first coordination shell was analyzed by evaluating the ΔGaq of one of the next reactions:| [Al(H2O)n(OH)m]3−m + O2˙− → [Al(H2O)n−1(OH)mO2˙]2−m + H2O | (5) |
| [Al(H2O)n(OH)m]3−m + O2˙− → [Al(H2O)n(OH)m−1O2˙]3−m + OH− | (6) |
Now, we will analyze whether aluminum can form a stable complex with the superoxide radical anion in the presence of citrate, a ligand composed of three carboxylic groups and an alcohol group (see Fig. 1). Previously, pKa values of −14.5, −8.0, 0.6 and 3.6 were computed for the four titratable groups of an aluminum-bound citrate molecule,43 which are in accordance with the two known experimental values, the two highest ones: 2.3 and 3.6.23 Therefore, we assume that under physiological conditions, the Al(III)-bound citrate is fully deprotonated. Note that the pKa values of free citrate in solution (2.9, 4.3, 5.6 and 11.6/14.4) suggest a different protonation state, with the alcohol group protonated.43 Different coordination modes between citrate and Al(III) were also compared,43 concluding that citrate interacts in a tridentate manner with aluminum.
Different solvation models were considered for the aluminum–citrate complexes, varying in the number of water/hydroxide molecules. In all of them, aluminum presents an octahedral arrangement, with three of the six first coordination shell positions occupied by citrate, and three of them available for water or hydroxide molecules. All possible combinations were taken into account, giving rise to a total of four structures (see Fig. 2). In addition, our previous calculations demonstrated the importance of including the second solvation sphere explicitly in order to obtain reliable solvation energies,44 therefore, ten water molecules were placed in the second coordination shell, mainly around the water/hydroxide molecules. All these structures are shown in Fig. 2.
 | ||
| Fig. 2 Density functional theory structures optimized at the B3LYP/6-31++g(d,p) level of theory. On the left, initial Al–Citr complexes with different numbers of H2O/OH molecules (according to eqn (1) and (2)). On the right, Al–Citr–O2˙ complexes formed by the substitution of either a H2O or a OH molecule by O2˙. | ||
We found that the substitution of a water molecule by the superoxide radical anion is thermodynamically favorable in solution, when at least two water molecules are found in the first coordination shell. Thus, the formation of [AlCitr(H2O)2(O2˙)(H2O)10]2− and [AlCitr(H2O)(OH)(O2˙)(H2O)10]3− shows ΔGaq values of −4.0 and −3.9 kcal mol−1, respectively. In contrast, the ΔGaq for the formation of [AlCitr(OH)2(O2˙)(H2O)10]4− is positive, 6.2 kcal mol−1. Thus, the amount of negatively charged groups in the first coordination shell influences the final stability of the aluminum–citrate–superoxide ternary complexes.
On the other hand, none of the substitutions of a hydroxide anion by the superoxide radical is thermodynamically favorable. The formation of [AlCitr(H2O)2(O2˙)(H2O)10]2− and [AlCitr(H2O)(OH)(O2˙)(H2O)10]3− species show values of 16.5 and 14.0 kcal mol−1, respectively. However, departing from the [AlCitr(OH)3(H2O)10]4− species, the formation of the [AlCitr(OH)2(O2˙)(H2O)10]4− complex is less endoergic, with a ΔGaq value of 4.1 kcal mol−1.
In summary, as in the case of the absence of citrate, the interaction of the superoxide with aluminum can lead to a thermodynamically favorable species, with negative formation energies, only in the case of the substitution of water molecules in the first-coordination shell around aluminum, whereas the substitution of hydroxides is highly unfavorable. As expected, the presence of other negatively-charged groups in the first-coordination shell of aluminum leads to the less favorable formation of an aluminum–superoxide complex, both in binary and ternary compounds. In this sense, the presence of citrate, with a total charge of 4−, has a sizable effect on the substitution reactions. The addition of a more negative charge by the incorporation of a superoxide radical is energetically less favorable than that in the absence of citrate. For instance, in the absence of citrate, the exchange of a water molecule with the superoxide shows a value of ΔGaq of −19.5 kcal mol−1, while the same reaction in the presence of citrate shows a value of −4.0 kcal mol−1. However, although the binding of aluminum to citrate makes less favorable the interaction of the metal with the superoxide, the formation of a ternary aluminum–citrate–superoxide complex is still thermodynamically favorable, and therefore a ternary complex of this kind could also take part in the promotion of the Fenton reaction. In the next section, we evaluate how the inclusion of citrate influences the thermodynamics of the resultant redox reaction.
Fenton reaction
Previously,21 it was shown that an aluminum–superoxide complex could thermodynamically promote the Fenton reaction by reducing Fe(III) (sextuplet spin state) to Fe(II) (pentuplet spin state) through the following redox reaction:| Fe3+ + [AlO2˙]2+ → Fe2+ + Al3+ + O2 | (7) |
Taking into account the most stable [AlCitrO2˙] ternary complex ([AlCitr(H2O)2(O2˙)(H2O)10]2−) as a reference, the ΔGaq value of reaction 7 is −36.1 kcal mol−1, thus, it is 6 kcal mol−1 more stable than in the absence of citrate. A slightly larger exothermicity is gained (ΔG value of −36.5 kcal mol−1) when the [AlCitr(H2O)(OH)(O2˙)(H2O)10]3− complex is taken as the reference. Thus, the formation of a ternary aluminum–citrate–superoxide complex favors the redox reaction that reduces Fe(III) to Fe(II). In this sense, the presence of citrate would enhance the ability of aluminum to promote the Fenton reaction. The reason for this behavior is that the presence of a highly-negative charged citrate in the coordination shell of aluminum makes the loss of an electron in the superoxide, and therefore a reduction of its negative charge, more likely.
However, citrate is not only a good chelator for aluminum in biological systems, but also a good chelator of Fe(III). In fact, the binding free energy of citrate to Fe(III) is −133 kcal mol−1, 10 kcal mol−1 more stable than the one for aluminum. Therefore, we also investigated the possibility of iron reduction with complexes in which iron is also chelated to citrate. We consider two possibilities: (i) reduction of Fe(III)–citrate to Fe(II)–citrate by an aluminum–superoxide binary species and (ii) the reduction of Fe(III)–citrate to Fe(II)–citrate by an aluminum–citrate–superoxide ternary complex. In both cases and for all the complexes considered, the redox reaction is thermodynamically unfavorable. For instance, in the case that both iron and aluminum are chelated by citrate, the ΔGaq value of the redox reaction 7 is +7.0 kcal mol−1 when the [AlCitr(H2O)2(O2˙)(H2O)10]2− complex is taken as the reference, and +6.6 kcal mol−1 with [AlCitr(H2O)(OH)(O2˙)(H2O)10]3−. Therefore, the high stabilization of Fe(III) by citrate is a dominant factor with respect to the loss of an electron by the superoxide and stabilization of the aluminum–citrate complex. Thus, the chelation of iron by citrate has a protective effect with respect to the generation of Fe(II), and it promotes the Fenton reaction.
Overall, from our calculations, a complex picture emerges of the role of citrate in the thermodynamic promotion/inhibition of aluminum pro-oxidant activity, which is summarized in the scheme of Fig. 3. Aluminum hydrolytic species can form stable complexes with the superoxide, leading to stable binary aluminum–superoxide and ternary aluminum–citrate–superoxide complexes. Both types of complex have the ability to reduce Fe(III) to Fe(II) from a thermodynamic point of view. Moreover, the presence of citrate in ternary complexes promotes the loss of an electron from the superoxide, thus, increasing the iron-reduction ability of the aluminum ternary complexes with respect to the binary ones. However, if iron is also chelated to citrate, the possibility of iron reduction is compromised, leading in all cases to endothermic redox reactions. Finally, we should also take into account that in an excess of citrate and on formation of the [Al–Citr2] complex, in which all the coordination positions of aluminum are occupied by citrate, there would be no possibility of stabilization of superoxide by aluminum, and therefore, no possibility to reduce iron from the thermodynamic point of view.
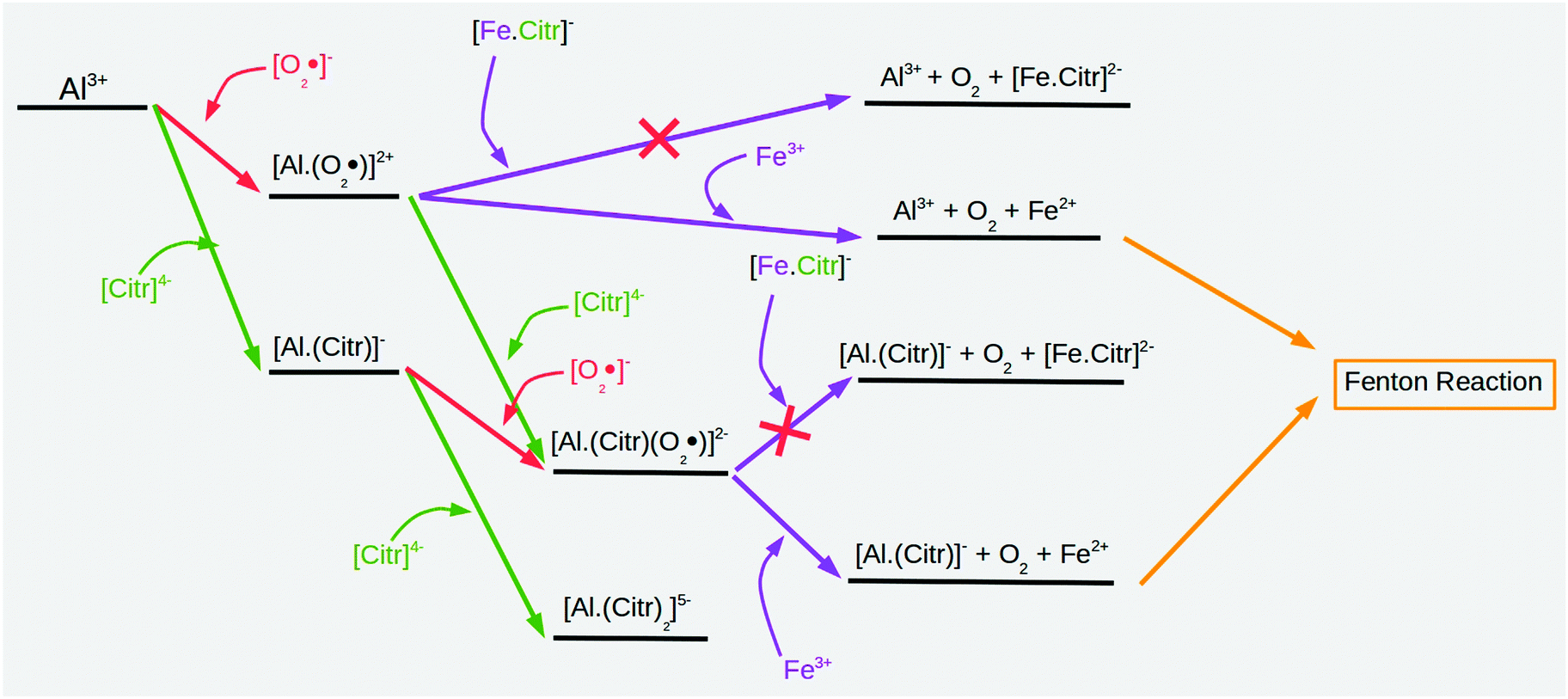 | ||
| Fig. 3 Schematic representation of how citrate modulates the promotion of the Fenton reaction by Al(III). | ||
Therefore, a complex overall picture emerges from these calculations, in which depending on the type of complex formed (binary/ternary), and the relative concentrations of citrate/aluminum/iron, one could observe the promotion or inhibition of the pro-oxidant activity of aluminum. In addition, the reactions of oligonuclear and/or mixed hydroxo complexes of Al(III) can be extremely slow, resulting in long-lived non-equilibrium states of Al(III)–ligand complexes,26,45 which, in the present context, could imply that aluminum could exert its pro-oxidant activity from the ternary species characterized in this work, even under conditions in which there is an excess of citrate.
Delocalization indices and ligand affinity
The effects of different functional groups considered herein (i.e. H2O, OH−, O2−, and the alkoxide and carboxylate groups of citrate) on the Al–O interactions are analyzed by calculating the delocalization indices for all the Al–O bonds in 17 structures: five aluminum hydrolytic species, five binary aluminum–superoxide complexes, four binary aluminum–citrate complexes and three ternary aluminum–superoxide–citrate complexes. Within each family of compounds, the complexes differ by the number of H2O/OH− ligands in the first solvation shell of aluminum. Most of the structures correspond to hexacoordinated species, but there are also examples of structures penta ([Al(H2O)2(OH)3]0) and tetra-coordinated ([Al(OH)4]−). In all cases, the structures consider a double layer of explicit waters around aluminum, embedded in an implicit solvation model. The results are summarized in Fig. 4 and in Table 4. | ||
| Fig. 4 The delocalization indices for the Al–O bonds of various aluminum hydrolytic species, aluminum–superoxide and aluminum–citrate binary complexes and ternary aluminum–superoxide–citrate complexes with different numbers of H2O/OH filling the first-coordination shell of aluminum. The DIs are classified according to the different functional groups: hydroxide (Al–OH−), alkoxide (Al–CO−), acetate (Al–CO2−), superoxide (Al–O2−) and water (Al–OH2). In the case where, for a given structure, several Al–O bonds with the same functional groups are present, the average value is provided in the figure. | ||
 referring to the O atom interacting with Al(III)
referring to the O atom interacting with Al(III)
| Aluminum hydrolytic species | ||||||
|---|---|---|---|---|---|---|
| [Al(H2O)6]3+ | 0.1614 (W) | 0.1536 (W) | 0.1383 (W) | 0.1560 (W) | 0.1507 (W) | 0.1545 (W) |
| [Al(H2O)5(OH)]2+ | 0.1476 (W) | 0.1386 (W) | 0.1454 (W) | 0.1385 (W) | 0.1378 (W) | 0.2160 (OH) |
| [Al(H2O)4(OH)2]+ | 0.1269 (W) | 0.1240 (W) | 0.1130 (W) | 0.1238 (W) | 0.2075 (OH) | 0.2428 (OH) |
| [Al(H2O)2(OH)3]0 | 0.1094 (W) | 0.1081 (W) | 0.2707 (OH) | 0.2395 (OH) | 0.2709 (OH) | |
| [Al(OH)4]− | 0.2693 (OH) | 0.2694 (OH) | 0.2393 (OH) | 0.2583 (OH) | ||
| Aluminum–superoxide complexes | ρ O2˙ |
|
||||||
|---|---|---|---|---|---|---|---|---|
| [Al(H2O)5(O2˙)]2+ | 0.1561 (W) | 0.1533 (W) | 0.1459 (W) | 0.1453 (W) | 0.1515 (W) | 0.1715 (SO) | 0.6723 | 0.3164 |
| [Al(H2O)4(OH)(O2˙)]+ | 0.1326 (W) | 0.1344 (W) | 0.1478 (W) | 0.1444 (W) | 0.2265 (OH) | 0.1500 (SO) | 0.6577 | 0.3251 |
| [Al(H2O)3(OH)2(O2˙)]0 | 0.1217 (W) | 0.1161 (W) | 0.1320 (W) | 0.2197 (OH) | 0.2123 (OH) | 0.1384 (SO) | 0.6103 | 0.3828 |
| [Al(H2O)(OH)3(O2˙)]− | 0.1148 (W) | 0.2589 (OH) | 0.2539 (OH) | 0.2336 (OH) | 0.1344 (SO) | 0.6024 | 0.3880 | |
| [Al(OH)3(O2˙)]2− | 0.2869 (OH) | 0.2771 (OH) | 0.2860 (OH) | 0.1774 (SO) | 0.6322 | 0.3599 | ||

| Aluminum–citrate complexes | ||||||
|---|---|---|---|---|---|---|
| [Al(H2O)3(Cit)]− | 0.2232 (CA) | 0.1645 (CCC) | 0.1596 (CCT) | 0.1240 (W) | 0.1480 (W) | 0.1333 (W) |
| [Al(H2O)2(OH)(Cit)]2− | 0.2134 (CA) | 0.1410 (CCC) | 0.1417 (CCT) | 0.1072 (W) | 0.1097 (W) | 0.2428 (OH) |
| [Al(H2O)(OH)2(Cit)]3− | 0.1947 (CA) | 0.1266 (CCC) | 0.1305 (CCT) | 0.0936 (W) | 0.1894 (OH) | 0.2197 (OH) |
| [Al(OH)3(Cit)]4− | 0.1845 (CA) | 0.1125 (CCC) | 0.1059 (CCT) | 0.1930 (OH) | 0.1996 (OH) | 0.1593 (OH) |
| Aluminum–superoxide–citrate complexes | ρ O2˙ |
|
||||||
|---|---|---|---|---|---|---|---|---|
| [Al(H2O)2(O2˙)(Cit)]2− | 0.2159 (CA) | 0.1610 (CCC) | 0.1574 (CCT) | 0.1244 (W) | 0.1348 (W) | 0.1613 (SO) | 0.5814 | 0.4012 |
| [Al(H2O)(OH)(O2˙)(Cit)]3− | 0.1985 (CA) | 0.1455 (CCC) | 0.1455 (CCT) | 0.1111 (W) | 0.2211 (OH) | 0.1326 (SO) | 0.5496 | 0.4310 |
| [Al(OH)2(O2˙)(Cit)]4− | 0.1872 (CA) | 0.1318 (CCC) | 0.1263 (CCT) | 0.2199 (OH) | 0.1827 (OH) | 0.1102 (SO) | 0.5306 | 0.4484 |

Delocalization indices (DIs) are a measure of the degree of electron sharing between two atoms (see the Methodology section). Although Al–O bonds are mainly electrostatic in nature, there are also important dative interactions46 from the lone pair of the oxygens to the formally vacant 3s and 3p orbitals of Al(III). For functional groups/ligands of similar charge, like in the case of hydroxide, alkoxide, carboxylate and superoxide, the analysis of DIs can help in the rationalization of the specific aluminum-binding affinities of the different oxygen donors. Among the 17 structures analyzed, we find a consistent pattern with the DI decreasing in the following order: Al–OH− >Al–CO− > Al–COO− > Al–O2˙− > Al–H2O. There are several aspects to highlight in the following trends, which allows for a clear rationalization of the ligand affinities described in the previous sections. The DI for Al–O2˙− bonds are the lowest among the charged ligands/groups but higher than that for water. This is in agreement with the favorable substitution of a water molecule by superoxide, and the unfavorable substitution of a charged ligand like hydroxide characterized in our previous work20 and shown in Table 1.
On the other hand, the highest Al–O delocalization indices correspond to the interaction of aluminum with hydroxides, with alkoxide having similar but smaller values. This is in agreement with the fact that hydrogen being less electronegative than carbon, makes OH− a better Lewis base than alkoxide. However, the DIs for carboxylate groups are substantially lower, due to the resonance of the –COO− moiety, which leads to a less dative oxygen. Finally, considering the superoxide, the higher electronegativity of oxygen compared to carbon and hydrogen makes O2− the poorest Lewis base among the charged functional groups/ligands of the present work.
We should also remark that the strong Al–OH− interaction characterized in our structures is in agreement with the inherent stability of hydroxides at the first solvation layer around aluminum.47 Moreover, the presence of hydroxides has a sizable effect on the strength of the rest of the Al–O bonds with other ligands and functional groups, leading to a weakening of the rest of the Al–O bonds, and therefore, a lowering of their DIs. This is also in agreement with the known fact that the presence of hydroxides leads to a depleting of the toxicity of aluminum, by hindering the direct interaction of aluminum with bioligands. This leads to a low toxicity of aluminum at neutral pH values at the limit of chemical equilibrium, i.e., when all the aluminum is in the [Al(OH)]4− form. However, as Exley et al.26 has established, one should always bear in mind that non-equilibrium aluminum species could be highly relevant in the biological effects of this metal.
Finally, when we compare similar structures in the presence or in the absence of citrate, we encounter the fact that the presence of citrate leads consistently to lower DIs for aluminum–superoxide bonds, in agreement with the lower affinity for superoxide displayed by aluminum–citrate complexes with respect to aluminum hydrolytic species. However, the DI of superoxide is still higher than the ones of water, and therefore the displacement of a water molecule by a O2˙− remains favorable. This decrease in aluminum–superoxide interaction in the presence of citrate also explains the fact that the aluminum–citrate–superoxide ternary complex is a better reductant than an aluminum–superoxide binary complex, since the loss of an electron and therefore the loss of an aluminum–superoxide interaction is energetically less unfavorable for the former than for the latter.
Similar conclusions can be reached based on the analysis of the Mulliken spin densities (ρ) at the two oxygen atoms of the O2˙ molecule. It should be pointed out that the spin densities computed based on the Mulliken atom partition are physically unsound, but they can be useful to determine qualitatively where the radical character is located and to describe trends in similar structures. The spin densities at the two oxygen atoms of O2˙ are shown in Table 4. For instance, the ρ values at the two oxygen atoms of O2˙ in the [Al(H2O)5(O2˙)]2+ structure are 0.6723 and 0.3164. So, the sum of these two values is approximately 1, but the spin density is mainly located at the O atom not interacting with Al(III). In other words, the molecule is highly polarized by the cation. However, the ρ values computed on other structures show that: (1) the inclusion of OH− radicals in the Al(III) first coordination shell leads to a small but consistent decrease of the polarization, and (2) this effect is more effective when Al(III) interacts with citrate.
Conclusions
We have presented a thorough computational study on the influence of citrate, the main low-molecular-mass chelator of aluminum, on the pro-oxidant activity of this metal. We have found that from the thermodynamic point of view, stable ternary aluminum–citrate–superoxide complexes can be formed in aqueous solution, which can in turn promote the Fenton reaction by reducing Fe(III) to Fe(II). The presence of citrate has a two-fold effect: (i) it reduces the affinity of aluminum towards the superoxide, although it is still thermodynamically favorable to form aluminum–citrate–superoxide compounds, and (ii) thermodynamically, it favors the redox reaction in which iron is reduced from Fe(III) to Fe(II). However, we also found that citrate has a protective role in two ways: (i) if iron is linked to citrate, the Fe(III) oxidation state is highly stabilized, and the reduction of iron is no longer thermodynamically favorable, irrespective of whether we depart from an aluminum–superoxide binary complex or an aluminum–superoxide–citrate ternary one; and (ii) in an excess of citrate, the formation of very stable 1:2 aluminum–citrate compounds is predicted to thermodynamically outcompete the formation of aluminum–superoxide–citrate complexes. In conclusion, we would like to remark that the use of delocalization indices, and in general of the QTAIM theory, is a powerful tool for the clear and full rationalization of the observed trends in complex stability, allowing us to establish an order of affinity of aluminum towards the different functional groups/ligands analyzed in the present work: Al–OH− >Al–CO− > Al–COO− > Al–O2˙− > Al–H2O.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Technical and human support was provided by SGI/IZO (SGIker) of UPV/EHU. Financial support comes from UPV/EHU (PES14/35), Eusko Jaurlaritza (IT588-13) and the Spanish Ministerio de Ciencia e Innovación (MINECO/FEDER) (CTQ2015-67608-P). G.D.T. acknowledges financial support from the European Commission, Horizon 2020 Research and Innovation Programme (grant agreement No. 642294 – TCCM).References
- C. Exley, J. Inorg. Biochem., 2003, 97, 1–7 CrossRef PubMed.
- C. Exley, Trends Biochem. Sci., 2009, 34, 589–593 CrossRef PubMed.
- J. Beardmore, X. Lopez, J. I. Mujika and C. Exley, Sci. Rep., 2016, 6, 30913 CrossRef PubMed.
- Z. Rengel, Biometals, 2004, 17, 669–689 CrossRef PubMed.
- C. Exley, Environ. Sci.: Processes Impacts, 2013, 15, 1807–1816 Search PubMed.
- L. Tomljenovic, J. Alzheimer's Dis., 2011, 23, 567–598 CrossRef PubMed.
- R. A. Yokel, Curr. Inorg. Chem., 2012, 2, 54–63 CrossRef.
- V. B. Gupta, S. Anitha, M. L. Hegde, L. Zecca, R. M. Garruto, R. Ravid, S. K. Shankar, R. Stein, P. Shanmugavelu and K. S. Jagannatha Rao, Cell. Mol. Life Sci., 2005, 62, 143–158 CrossRef PubMed.
- G. Crisponi, V. Nurchi, V. Bertolasi, M. Remelli and G. Faa, Coord. Chem. Rev., 2011, 256, 89–104 CrossRef.
- A. I. Arieff, Am. J. Kidney Dis., 1985, 6, 317–321 CrossRef PubMed.
- A. C. Alfrey, G. R. LeGendre and W. D. Kaehny, N. Engl. J. Med., 1976, 294, 184–188 CrossRef PubMed.
- J. I. Mujika, E. Rezabal, J. M. Mercero, F. Ruipérez, D. Costa, J. M. Ugalde and X. Lopez, Comput. Struct. Biotechnol. J., 2014, 9, e201403002 CrossRef PubMed.
- S. Kong, S. Liochev and I. Fridovich, Free Radical Biol. Med., 1992, 13, 79–81 CrossRef PubMed.
- A. Khan, J. P. Dobson and C. Exley, Free Radical Biol. Med., 2006, 40, 557–569 CrossRef PubMed.
- C. Exley, Free Radical Biol. Med., 2004, 36, 380–387 CrossRef PubMed.
- P. Zatta, T. Kiss, M. Suwalsky and G. Berthon, Coord. Chem. Rev., 2002, 228, 271–284 CrossRef.
- S. Fukuzumi and K. Ohkubo, Chem. – Eur. J., 2000, 6, 4532–4535 CrossRef.
- S. Fukuzumi, H. Ohtsu, K. Ohkubo, S. Itoh and H. Imahori, Coord. Chem. Rev., 2002, 226, 71–80 CrossRef.
- S. Fukuzumi, J. Phys. Chem., 2002, 15, 448–460 Search PubMed.
- J. Mujika, F. Ruipérez, I. Infante, J. Ugalde, C. Exley and X. Lopez, J. Phys. Chem. A, 2011, 115, 6717–6723 CrossRef PubMed.
- F. Ruipérez, J. Mujika, J. Ugalde, C. Exley and X. Lopez, J. Inorg. Biochem., 2012, 117, 118–123 CrossRef PubMed.
- G. F. Van Landeghem, M. E. De Broe and P. C. D'Haese, Clin. Biochem., 1998, 31, 385–397 CrossRef PubMed.
- A. Lakatos, F. Evanics, G. Dombi and R. Bertani, Eur. J. Inorg. Chem., 2001, 3079–3086 CrossRef.
- R. Milacic, S. Murko and J. Scancar, J. Inorg. Biochem., 2009, 103, 1504–1513 CrossRef PubMed.
- G. Berthon, Coord. Chem. Rev., 2002, 228, 319–341 CrossRef.
- J. Beardmore and C. Exley, J. Inorg. Biochem., 2009, 103, 205–209 CrossRef PubMed.
- J. Mujika, J. Ugalde and X. Lopez, J. Phys. Chem. B, 2014, 18, 1345–1349 Search PubMed.
- N. B. Luque, J. I. Mujika, E. Formoso and X. Lopez, RSC Adv., 2015, 5, 63874–63881 RSC.
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- A. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- S. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef PubMed.
- J. D. Chai and M. M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ã Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09 Revision E.0 1, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16 Revision A.03, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- X. Fradera, M. A. Austen and R. F. W. Bader, J. Phys. Chem. A, 1999, 103, 304–314 CrossRef.
- R. Bader and M. E. Stephens, J. Am. Chem. Soc., 1975, 97, 7391–7399 CrossRef.
- K. Ruedenberg, Rev. Mod. Phys., 1962, 34, 326–376 CrossRef.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- T. A. Keith, AIMAll (Version 14.11.23), 2014 Search PubMed.
- J. I. Mujika, J. M. Ugalde and X. Lopez, Phys. Chem. Chem. Phys., 2012, 14, 12465–12475 RSC.
- J. I. Mujika, J. M. Ugalde and X. Lopez, Theor. Chem. Acc., 2011, 128, 477–484 CrossRef.
- J. Beardmore, G. Rugg and C. Exley, J. Inorg. Biochem., 2007, 101, 1187–1191 CrossRef PubMed.
- A. E. Martell, R. D. Hancock, R. M. Smith and R. J. Motekaitis, Coord. Chem. Rev., 1996, 149, 311–328 CrossRef.
- S. Bogatko, J. Moens and P. Geerlings, J. Phys. Chem. A, 2010, 114, 7791–7799 CrossRef PubMed.
| This journal is © the Owner Societies 2018 |