
Ruthenium-catalyzed direct arylations with aryl chlorides
Gao-Feng
Zha
a,
Hua-Li
Qin
*a and
Eric Assen B.
Kantchev
*b
aSchool of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, 205 Luoshi Road, Wuhan, 430070, China. E-mail: qinhuali@whut.edu.cn; Fax: +86-27-8774-9300; Tel: +86-27-8774-9300
bSchool of Chemistry and Chemical Engineering, Hefei University of Technology, 193 Tunxi Road, Hefei 230009, China. E-mail: ekantchev@hfut.edu.cn; ekantchev@gmail.com; Fax: +86-551-6290-1450; Tel: +86-551-6290-1450
First published on 16th March 2016
Abstract
Aryl chlorides are readily available at lower cost than the corresponding bromides and iodides, but are much more challenging as substrates in metal-catalyzed cross-couplings. Using arenes as nucleophilic partners by means of C–H activation instead of arylorganometallics also carries significant cost and environmental advantages. Finally, Ru catalysts, characterized by lower metal cost, excellent chemoselectivity, and high activity in C–H functionalization even in “green solvents” such as water, compound the economic and environmental benefits of chelation-assisted direct arylation. The current state-of-the-art of the Ru-catalyzed direct aryl–aryl coupling with chloroarenes will be reviewed.
 Gao-Feng Zha | Gao-Feng Zha was born in Hubei Province, P. R. China. He received his B. S. and M. S. degrees in pharmaceutical engineering from Wuhan University of Technology in 2007 and 2015, respectively. He worked as a research scientist in the pharmaceutical industry from 2007 to 2013. He is currently a Ph. D. student in Prof. Hua-Li Qin's group at Wuhan University of Technology. His research is focused on the development of powerful synthetic methodologies for synthesis of bioactive molecules. |
 Hua-Li Qin | Hua-Li Qin received a B. E. in chemical engineering from Anhui University and M. S. in organic chemistry from University of Science and Technology of China in 2000 and 2004, respectively. In early 2009, he received his Ph. D. in Chemistry from Boston University under the direction of Prof. James S. Panek. After working in Singapore and China as a medicinal chemist, he started his academic career at Wuhan University of Technology focusing on new synthetic method development, bioorganic, and medicinal chemistry. In 2015, he joined The Scripps Research Institute as Visiting Professor with Nobel Laureate Prof. K. B. Sharpless. |
 Eric A. B. Kantchev | Eric A. B. Kantchev, a native of Bulgaria, has been Green Chemistry Group Leader and Professor at the School of Chemistry and Chemical Engineering, Hefei University of Technology, P. R. China since 2014. His current research interests center on combining computational and experimental approaches to design new and improve existing transition metal catalysts. After obtaining a B. S. (University of Sofia, 1996) and a Ph. D. (The Ohio State University, 2001) in organic chemistry, he undertook research roles both in academia and industry in Taiwan, Canada, and Singapore. Since 1999, he has co-authored 50 peer reviewed articles cited >2900 times. |
Introduction
The 2010 Nobel Prize in Chemistry1 for Pd-catalyzed cross-coupling2 has been the strongest testament to the astounding impact this class of reactions has had in organic synthesis. Despite many spectacular advances, research efforts in this field continue unabated.3 One industrially-important, major advance was the successful coupling of unactivated aryl chlorides by means of Pd ligated with bulky, highly electron-rich tertiary phosphanes and N-heterocyclic carbenes.4 A considerably larger number of chloroarenes are commercially available at lower cost than bromo- and iodoarenes used in the classical cross-coupling protocols. However, they are more challenging as reacting partners. In the early years of the field, only aryl chlorides bearing strongly electron withdrawing p-substituents (e.g., NO2 or CN) could be cross-coupled, typically under harsher conditions and in lower conversions than aryl bromides and iodides. The continuing efforts of catalyst design have dramatically changed this situation to the point where the choice of the halide is no longer a reactivity bottleneck.5 Herein we will give just one example: p-chloroanisole (an electronically-deactivated arene) can be coupled with pre-formed sodium phenylborate in methanol, without additional base, by 5 mol% of cyclo(chloropalladated) acetanilide ligated with a bulky N-heterocyclic carbene in 96% yield at 0–4 °C being the first example of Pd-catalyzed Suzuki–Miyaura coupling below ambient temperature.6The typical cross-coupling methodologies rely on aryl organometallic derivatives such as organoboron reagents7 to smoothly introduce the nucleophilic coupling partner into the coordination sphere of the metal. However, such derivatives are handicapped by non-trivial synthesis, low stability, and low air, moisture and functional group tolerance. In the course of cross-coupling, a stoichiometric amount of salt is generated, providing thermodynamic driving force for the process but also generating waste. These drawbacks can be circumvented by the recruitment of C–H bonds as reactive sites. In the recent years, numerous examples of C–H activation methodologies catalyzed by Pd,8 Rh,9 and Ru10 have emerged. However, coupling of aryl halides with arenes by C–H activation (“direct arylation”)11 is typically performed only with Pd and Ru (Fig. 1). The Ru(II)-catalyzed direct arylation with aryl halides, even though mechanistically distinct, has only been reviewed in the general context of Ru-catalyzed C–H activation. Herein, we will focus only on coupling of aryl chlorides due to their challenging substrate nature and economic and environmental significance as we anticipate important future breakthroughs in this area.
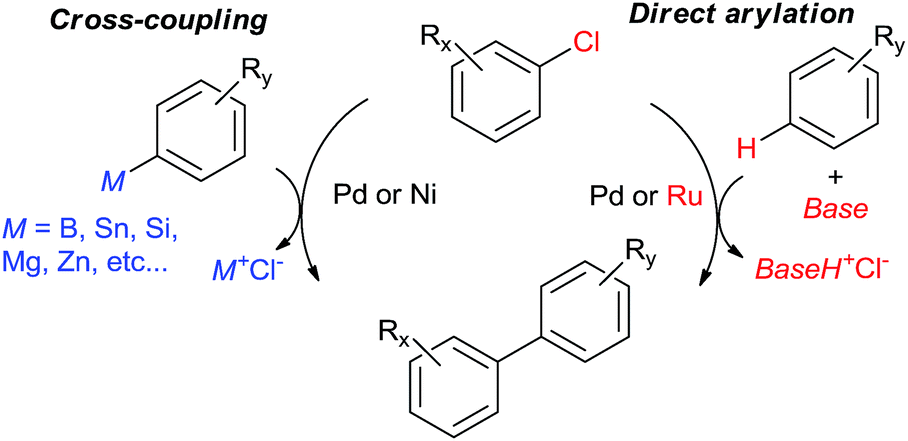 | ||
| Fig. 1 Transition metal-catalyzed directed arylation vs. cross-coupling methods for biaryl synthesis. Only metals that are most commonly used as catalysts are shown. The scope of this review is shown in red. | ||
Synthetic studies on the Ru-catalyzed direct arylation with aryl chlorides
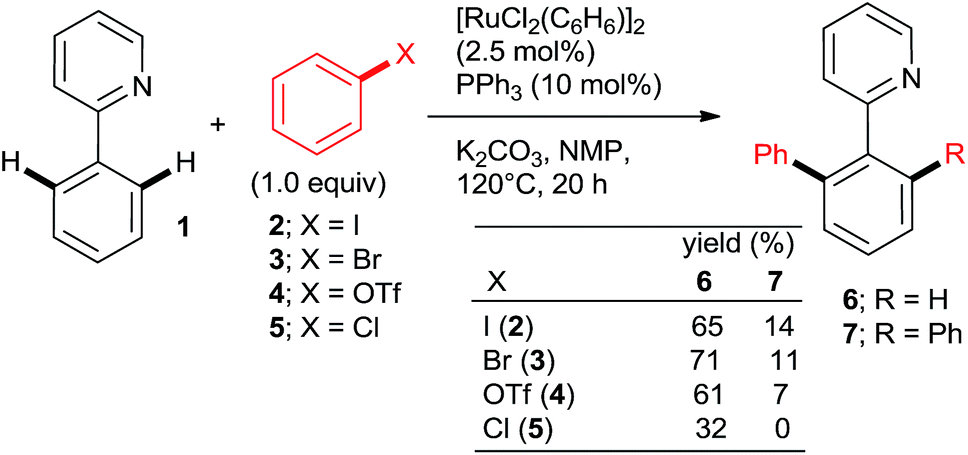 | ||
| Scheme 1 The first Ru-mediated direct arylation. | ||
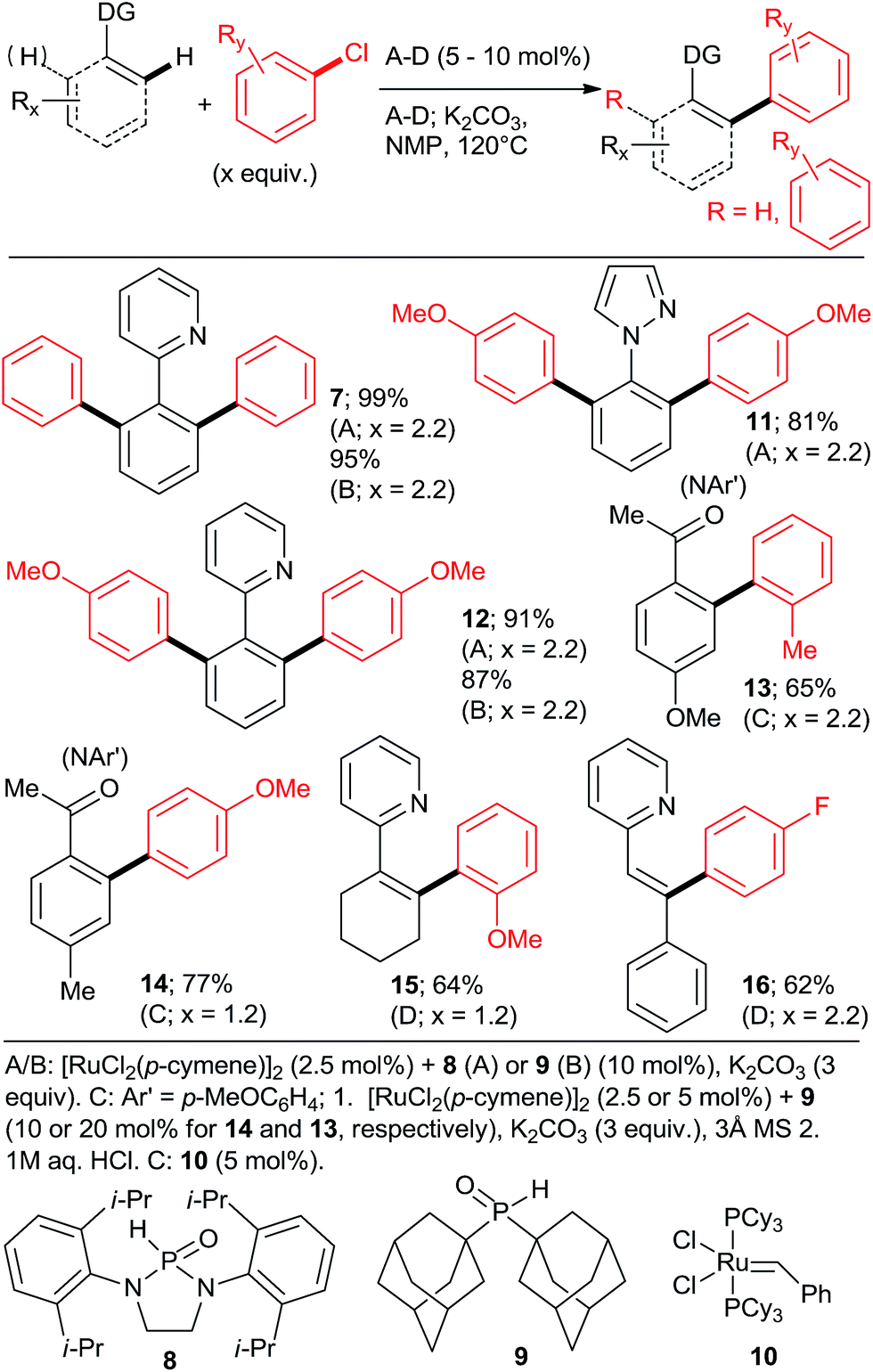 | ||
| Scheme 2 The first high yielding Ru-mediated direct arylations with unactivated aryl chlorides. | ||
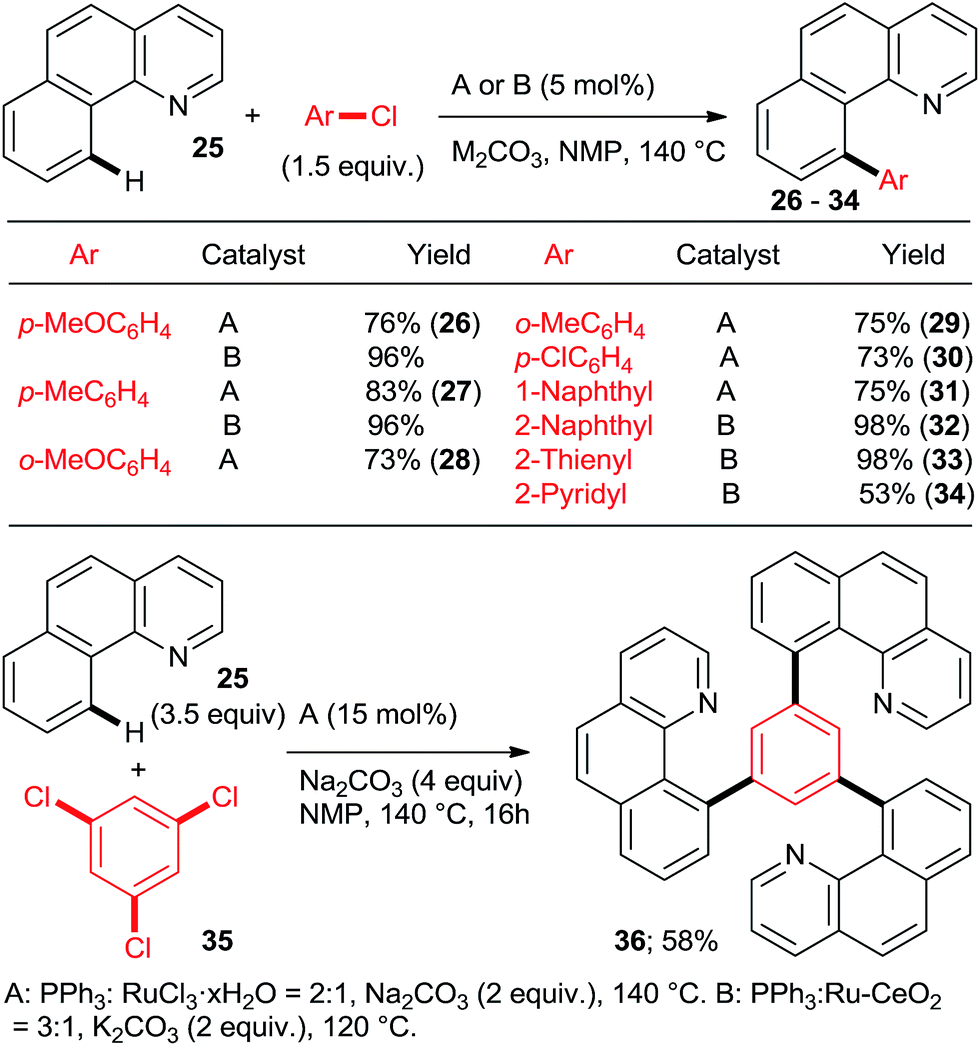
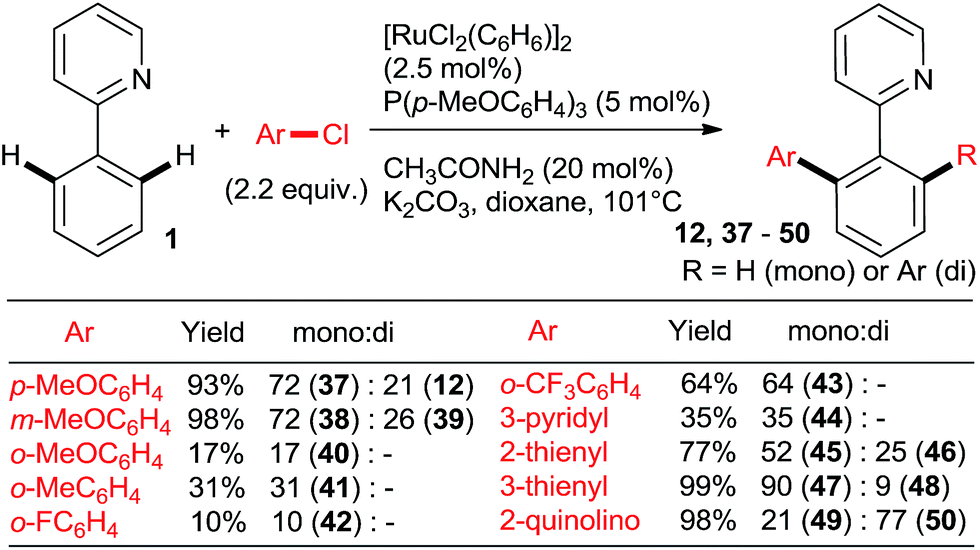
The key to success were the bulky, air-stable secondary phosphane oxides 8 and 9 used as “additives” (ligands) in conjunction with the common, commercially available, inexpensive [RuCl2(p-cymene)2] precatalyst under the Oi/Inoue conditions. The hydrolysis of the directing p-methoxyphenyl imine group gave acetophenones (e.g., 13, 14) after aqueous hydrolysis.23 Interestingly, despite the use of 2.2 equiv. of o-chlorotoluene, only the monoarylation product 14 was isolated. Subsequently, the same group found that the 2nd generation Grubbs' metathesis catalyst (10) carrying the bulky, electron-rich tricyclohexylphosphane (Cy3P) performed even slightly better in the same reaction.24 Preformed complexes by the reaction of [RuCl2(p-cymene)] and tertiary phosphanes prepared by zirconophosphanation/protonolysis by Tang, Xi et al.25 (Scheme 3, 17), and by the Diels–Alder reaction of phosphane oxides carrying 1-alkyne substituent with anthracene and subsequent reduction by Doherty and co-workers26 (KITPHOS; e.g., 18) were also competent catalysts under the Oi/Inoue conditions. These complexes were competent catalysts for the arylation of a wide range of o- and p-substituted chlorobenzenes. Particularly, the use of tertiary amino-substituted chloroarenes is noteworthy (products 20, 21, 22, 23). Exchange of the η6-complexed arene from p-cymene in 18 to a β-Ph from the phosphane in 19 had no significant effect on arylation yields. Two studies in 2010 explored in situ prepared Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PPh3 = 1:n catalysts (Scheme 4) for arylation of benzo[h]quinoline (25) with a wide range of aryl and heteroaryl chlorides. Yu's group27 utilized 5 mol% RuCl3·xH2O (the most inexpensive commercial source of Ru) and 10 mol% PPh3. The impressive 3-fold coupling of 35 with 3.5 equiv. 25 proceeded in 58% yield of 36 with 15 mol% RuCl3·xH2O and 30 mol% PPh3 at 140 °C. Wada et al.28 explored a range of solid supported catalysts, the most active of which was prepared from [RuCl2(CO)3] on CeO2 (precipitated by KOH) followed by impregnation with 3 equiv. PPh3 under atmosphere of H2. These solid phase catalysts were recyclable, exhibiting no loss of yield over 3 runs. Li et al.29 have demonstrated the coupling of an extended range of aryl- and heteroaryl chlorides, with 5 mol% of [RuCl2(p-cymene) (p-MeOC6H4)3P] and 20 mol% of acetamide as an additive in refluxing 1,4-dioxane. Under these milder conditions, even the use of 2.2 equiv. of m- and p-substituted chlorobenzenes resulted in the mono arylated product being the major in all but one case. Chloro benzenes carrying o-substituents gave only mono arylated products in low to moderate yields (Scheme 5). On the other hand the use of heterocyclic aryl chlorides was capricious: 4-chloropyridine failed to react whereas 2-chloroquinoline attained 98% conversion, with 77% yield of the di-arylated product. Surprisingly, electron withdrawing o-substituents had in most cases a deleterious effect on reactivity: coupling yield for o-chloroacetophenone was 0% and for methyl o-chlorobenzoate less than 5%.
:PPh3 = 1:n catalysts (Scheme 4) for arylation of benzo[h]quinoline (25) with a wide range of aryl and heteroaryl chlorides. Yu's group27 utilized 5 mol% RuCl3·xH2O (the most inexpensive commercial source of Ru) and 10 mol% PPh3. The impressive 3-fold coupling of 35 with 3.5 equiv. 25 proceeded in 58% yield of 36 with 15 mol% RuCl3·xH2O and 30 mol% PPh3 at 140 °C. Wada et al.28 explored a range of solid supported catalysts, the most active of which was prepared from [RuCl2(CO)3] on CeO2 (precipitated by KOH) followed by impregnation with 3 equiv. PPh3 under atmosphere of H2. These solid phase catalysts were recyclable, exhibiting no loss of yield over 3 runs. Li et al.29 have demonstrated the coupling of an extended range of aryl- and heteroaryl chlorides, with 5 mol% of [RuCl2(p-cymene) (p-MeOC6H4)3P] and 20 mol% of acetamide as an additive in refluxing 1,4-dioxane. Under these milder conditions, even the use of 2.2 equiv. of m- and p-substituted chlorobenzenes resulted in the mono arylated product being the major in all but one case. Chloro benzenes carrying o-substituents gave only mono arylated products in low to moderate yields (Scheme 5). On the other hand the use of heterocyclic aryl chlorides was capricious: 4-chloropyridine failed to react whereas 2-chloroquinoline attained 98% conversion, with 77% yield of the di-arylated product. Surprisingly, electron withdrawing o-substituents had in most cases a deleterious effect on reactivity: coupling yield for o-chloroacetophenone was 0% and for methyl o-chlorobenzoate less than 5%.
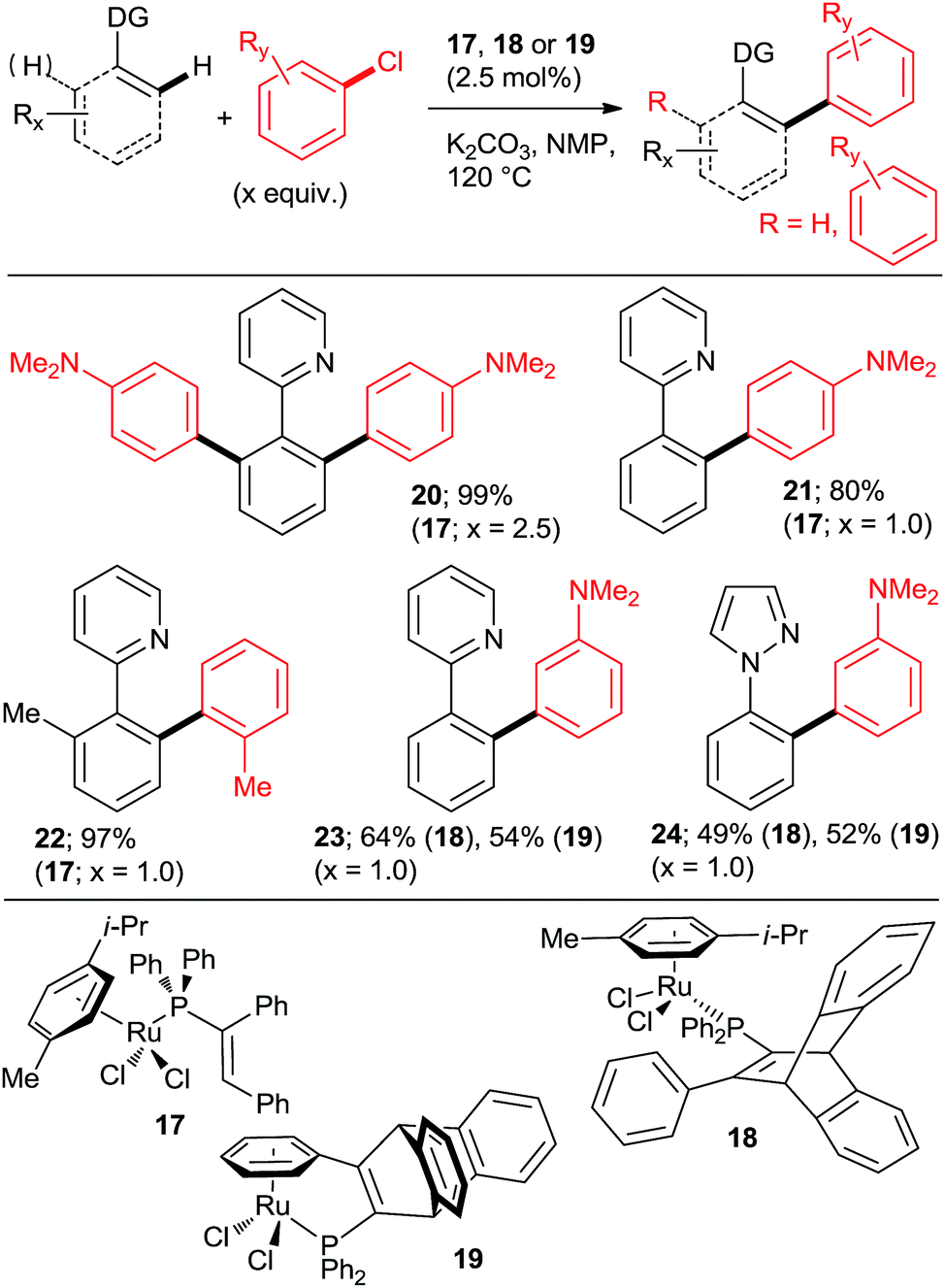 | ||
| Scheme 3 Direct arylations with unactivated aryl chlorides catalyzed by well-defined Ru complexes of bulky vinylphosphanes. | ||
 | ||
| Scheme 4 Direct arylations with unactivated aryl chlorides mediated by in situ prepared PPh3–Ru catalysts. | ||
 | ||
| Scheme 5 Selective mono-arylations of 2-chloropyridine with unactivated aryl chlorides. | ||
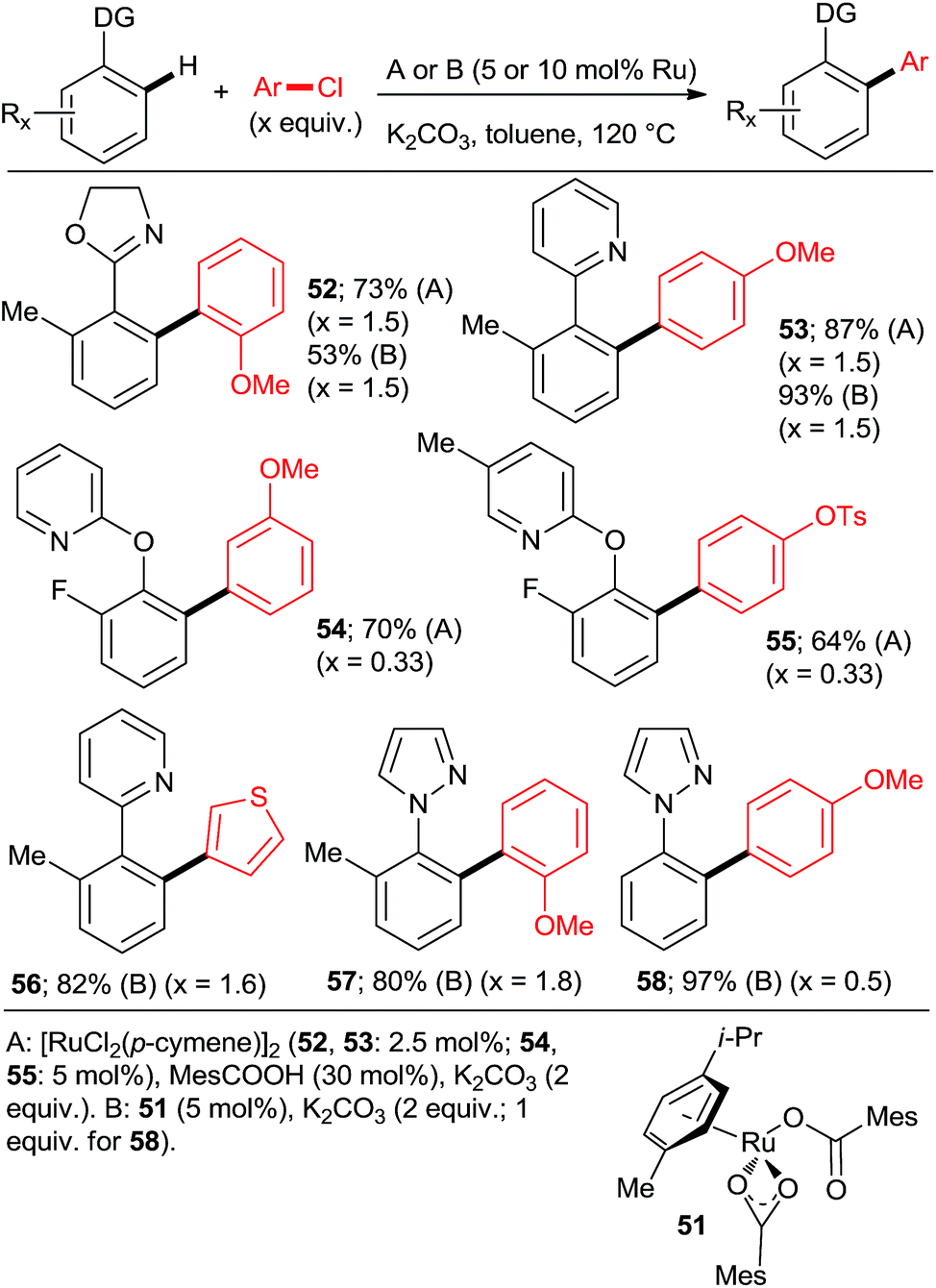
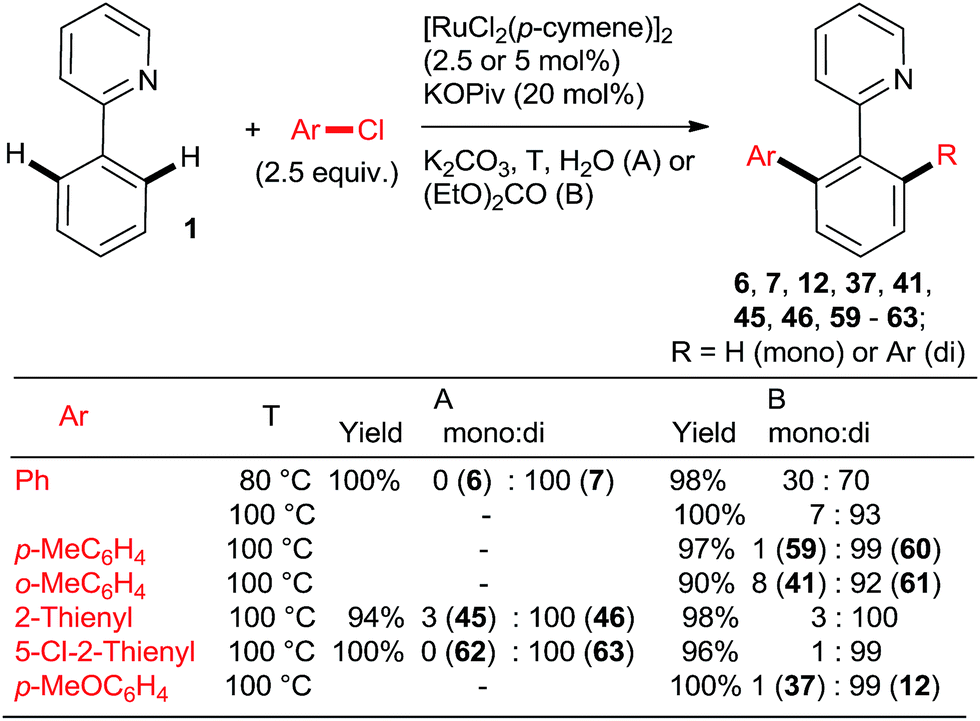
Ackermann et al. explored the couplings of a range of aryl chlorides in toluene as the solvent with a catalyst prepared either in situ from [RuCl2(p-cymene)]2 and MesCOOH (Mes = 2,4,6-trimethylphenyl)30 or from the preformed complex 5131 (Scheme 6). Overall, both catalytic protocols gave similar yields within the experimental error over a range of o-, m-, and p-substituted chlorobenzenes. In the case of the 2-aryloxypyridines, the pyridine was removed after hydrolysis of the corresponding N-methylpyridinium triflate formed after treatment with MeOTf, revealing much more synthetically useful o-arylphenols. The Ru–carboxylate catalyst tolerates Ar-OTs and Ar-F functionalities (e.g., 55). Dixneuf, Fischmeister, and co-workers performed highly efficient o,o-diarylations of a range of aryl and heteroaryl chlorides with Ru–pivalate catalysts in neat water32 or diethyl carbonate33 (Scheme 7), both of which are “geener” solvents than NMP or toluene and permit lower reaction temperatures, typically 100 °C. Importantly, it was found that in water the reactivity order of phenyl halides was reversed (5 > 3 > 2), attributed to the better solubility of chlorobenzene. Remarkably, 1,3,5-trichlorobenzene (35) underwent 3-fold arylation with 25 in refluxing water over 36 h to yield 45% of 36 (analogous to Scheme 4, bottom). The catalytic activity was slightly inferior in diethyl carbonate than water, as suggested by conversions and mono:di arylation ratios. Gimeno et al. published arylations of 2-phenylpyridine (1) with an extensive range of chloroarenes catalyzed by a ligandless Ru(II) carboxylate catalyst prepared in situ by reduction of RuCl3·xH2O (5 mol%) with Zn or H3PO2 in water containing 3 equiv. NaOAc as the base (Scheme 8).34 In contrast to the catalyst derived from [RuCl2(p-cymene)]2, the monoarylated products predominated. The reaction proceeded well also under microwave conditions. Challenging substrates such as o-chloroaniline and p-chlorophenol gave low yields of only the monoarylated product.
 | ||
| Scheme 6 Mono-arylations with unactivated aryl chlorides catalyzed by (MesCOO)2Ru catalysts. | ||
 | ||
| Scheme 7 Selective di-arylations of 2-phenylpyridine with unactivated aryl chlorides in water or diethyl carbonate. | ||
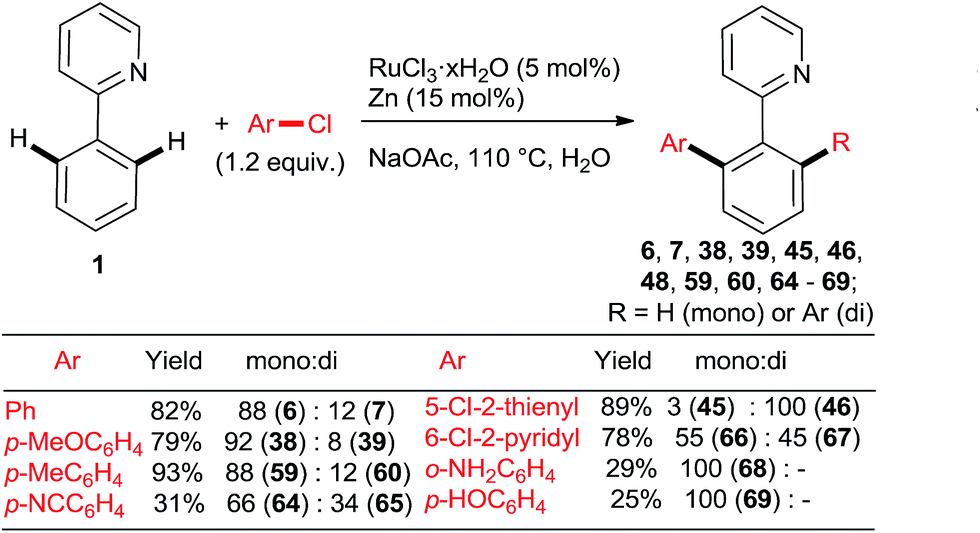 | ||
| Scheme 8 Selective di-arylations of 2-phenylpyridine with unactivated aryl chlorides in water or diethyl carbonate. | ||
:Ru = 3:1). Despite the forcing conditions, only 41% of 75 were isolated (21% with PPh3). Among the non-heteroaryl directing groups, Ackermann group investigated only acetophenone N-(p-methoxyphenyl)imines.23 The same group also demonstrated the only high-yielding examples of arylation proceeding via a 6-membered ruthenacycle in the case of 2-pyridylaryl ethers (Scheme 6, 54 and 55).30
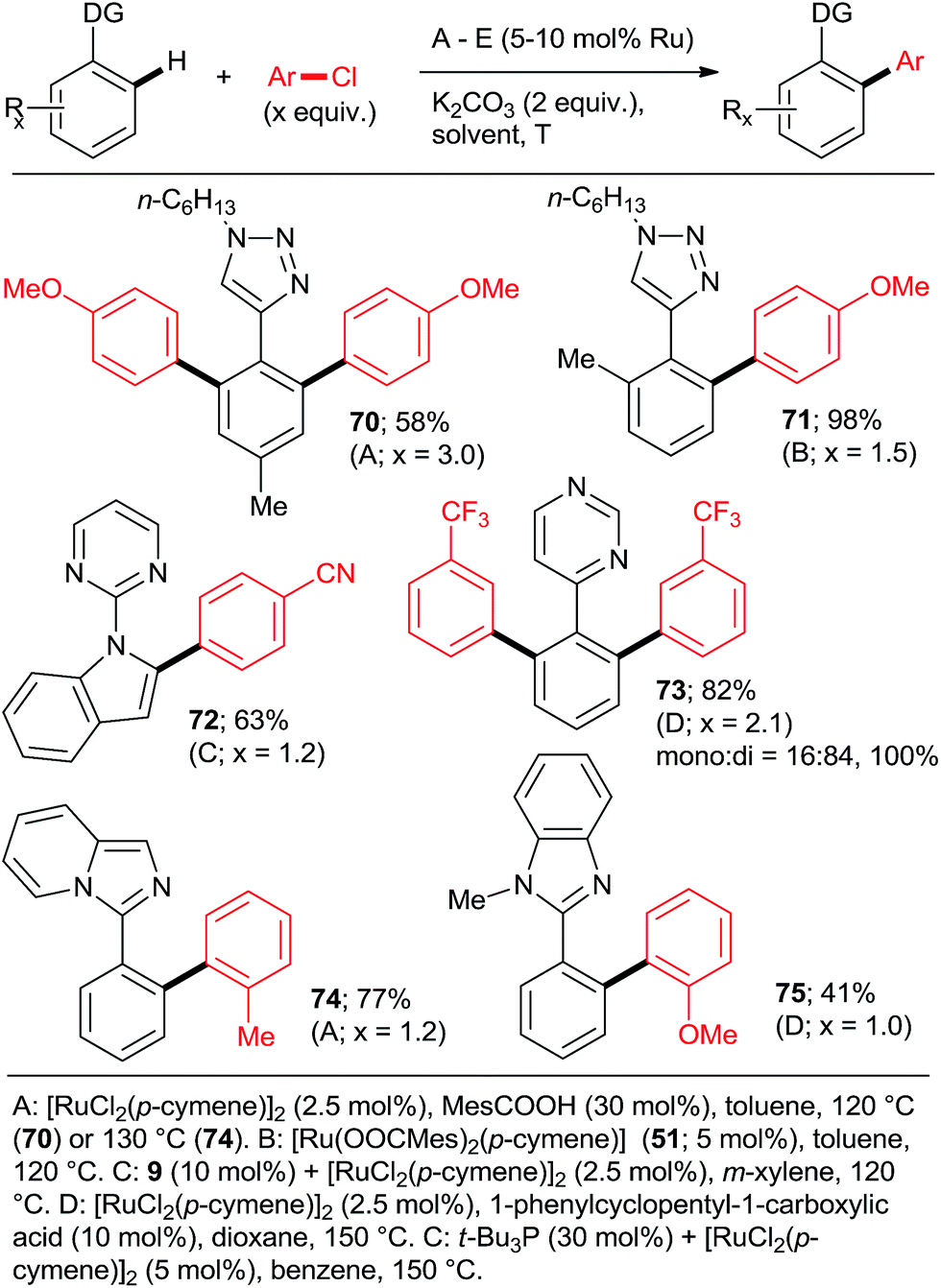 | ||
| Scheme 9 Arylations of substrates with rare directing groups. | ||
:di selectivities > 5:95 and yields close to quantitative can be achieved at reasonable catalyst loadings for reasonable reaction times only with the most highly active catalysts. One such system comprises the bulky secondary phosphane oxides 8 or 9 with [RuCl2(p-cymene)]2 (Scheme 2, products 7 and 12).22 Detailed studies by Dixneuf et al. showed that [Ru(OPiv)2(p-cymene)] (and other carboxylate analogs) is an excellent choice as a di-arylation catalyst in either water32 or diethyl carbonate33 (Scheme 7, products 37, 46, 60, 63; Scheme 10b). The analogous carboxylate catalyst derived from MesCOOH was found by Ackermann group42 to mediate diarylation of 4-phenyl-1,2,3-triazoles in good yields (58–78%). However, the yield of the mono-arylated product was not listed, nor the selectivity issue discussed. In one of the most extensive studies, Yu's group27 published a number of examples of diarylation of 2-phenylpyridine, 2-phenyloxazoline, and 1-phenylpyrazole with 20 mol% PPh3 and 10 mol% RuCl3·xH2O (40/20 mol% for the latter). However, despite the high loading yields close to quantitative were recorded only for the easiest pairs of substrates, 2-phenylpyridine and chlorobenzene or p-chloroacetophenone. For the remaining examples, yields were only good to moderate (no product was obtained for 4-chloronitrobenzene). Dixneuf et al. extended their aqueous medium-carboxylate catalyst protocol to [RuCl(p-cymene) (L)] complexes (L = anions of tropolone, kojic acid (78) or glycine) (Scheme 11, products 7, 83, and 84).36 The same group also investigated [RuH(codyl)2](BF4) (codyl = η5-cyclooctadienyl) as a ruthenium source to be combined with a number of anionic additives under the Oi/Inoue conditions.37 The highest activity was attained with potassium acetate and potassium phthalimide: conversion of 1 with 2.5 equiv. chlorobenzene reached 100% with only 1 hour at 120 °C in NMP (mono:di selectivity 6:94 and 98:2, respectively). Designer N-heterocyclic carbene–Ru complexes by Peris (79,358015) and Özdemir (81,388239) were also quite capable of delivering diarylated products (7 and 12) in high yields under the Oi/Inoue conditions.
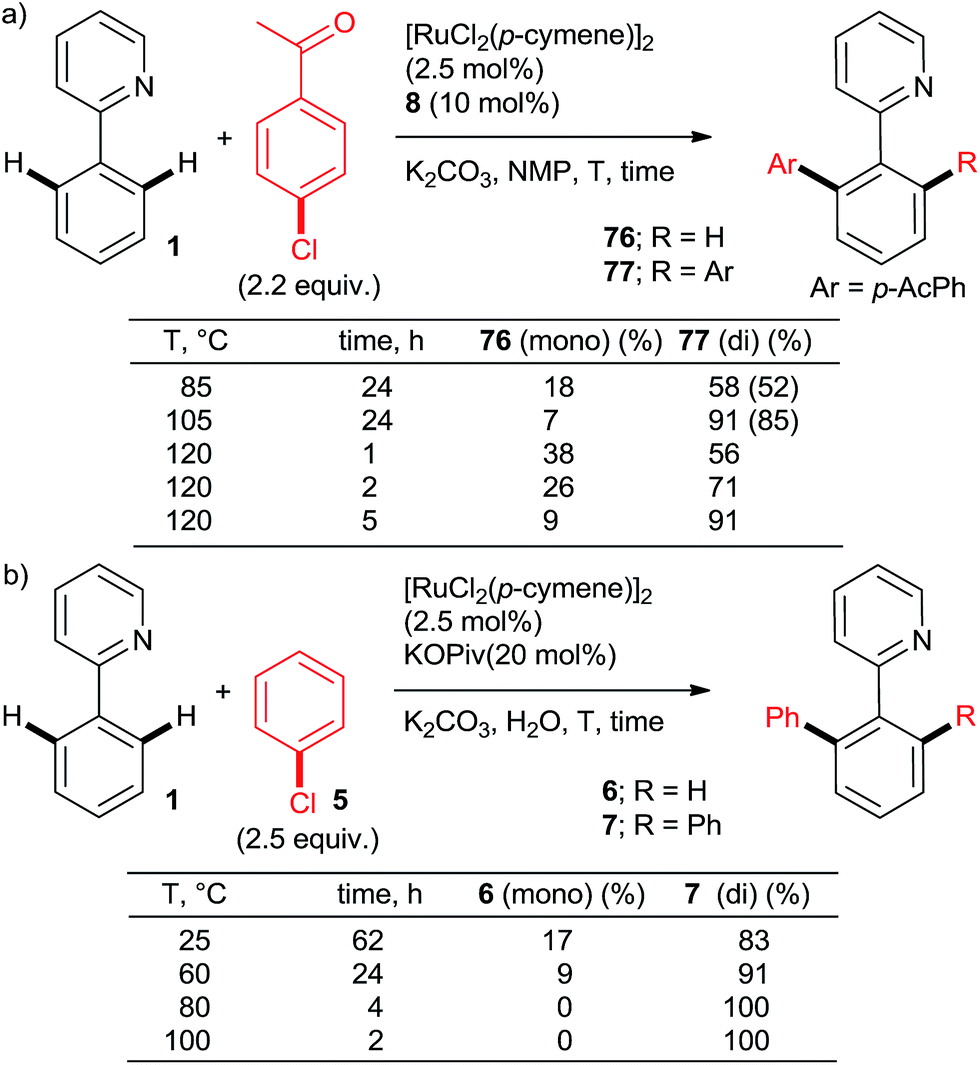 | ||
| Scheme 10 Effect of temperature and time on arylation mono:di selectivity. Isolated yields shown in brackets. | ||
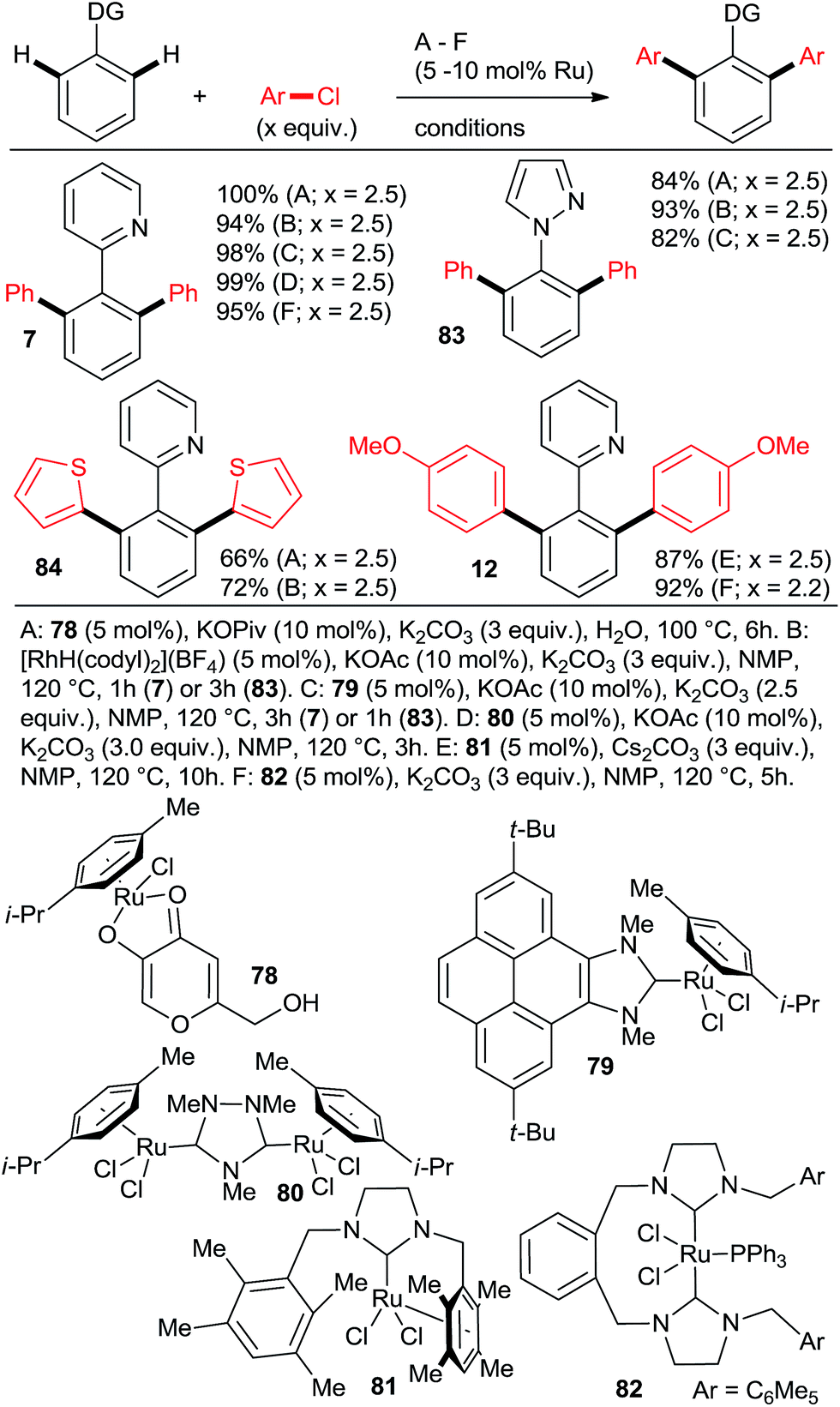 | ||
| Scheme 11 Additional catalytic diarylation protocols. | ||
Next, approaches towards mono arylation will be discussed. Having one o-substituent naturally precludes di-arylation (Scheme 2, 22; Scheme 4; Scheme 6, 52–57; Scheme 9, 72) as does the use of vinyl instead of aryl C–H activation substrate (Scheme 2, 16). Ackermann et al.24,31 and Yu et al.27 demonstrated that a m-methyl or m-methoxy substituent in 2-phenylpyridine or 1-phenylpyrazole resulted in a complete monoarylation, most likely by virtue of increasing steric hindrance unless that substituent is fluorine (Scheme 12). However, it should be noted that Ackermann's group experiments (products 87–90) were conducted with 2-fold excess of the C–H activation compound. As it can be expected, p-substituents generally do not preclude double arylation.
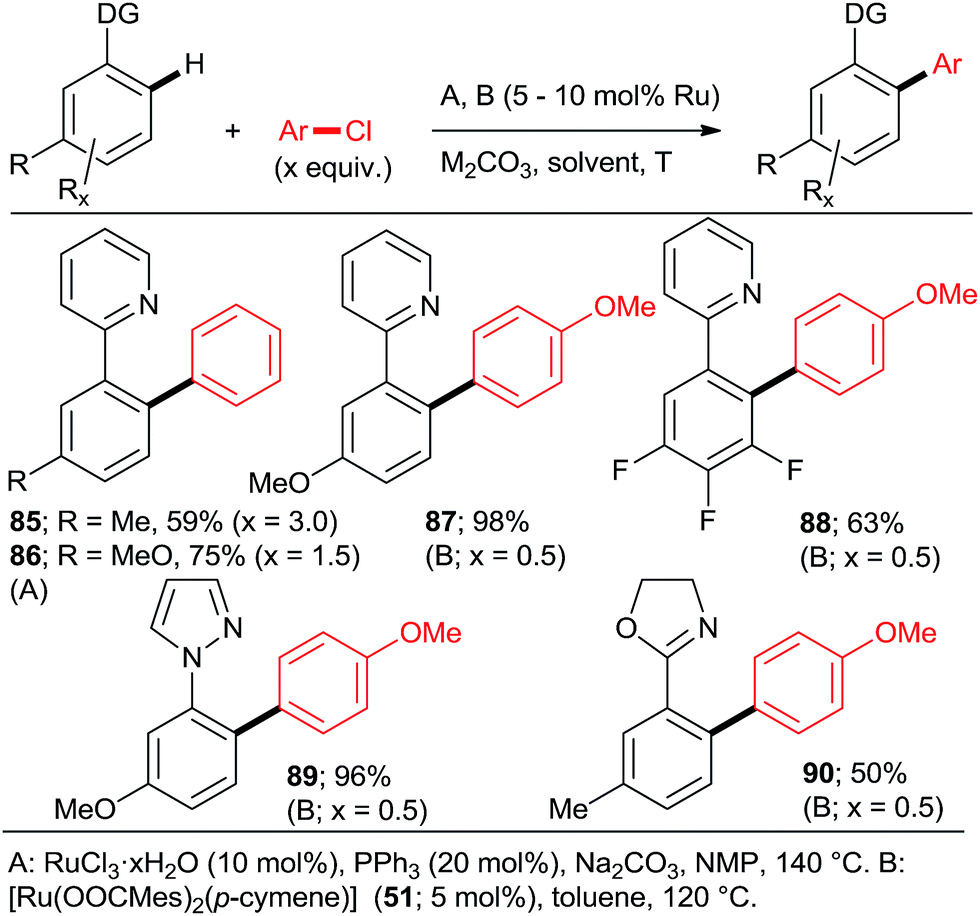 | ||
| Scheme 12 Mono arylation under the influence of the m-substituent in the C–H activation substrate. | ||
Using catalysts and/or reaction conditions to control the mono/di selectivity is a much more powerful and useful approach. As mentioned above, lower temperatures, shorter reaction times (Scheme 10), and lower catalyst loadings all result in increasing mono-arylated product percentage. However, tedious optimization of the reaction conditions may be necessary to maximize the yield of mono-arylated product. Whereas such undertaking may be justified in process chemistry/manufacturing, the time pressures during discovery/medicinal chemistry campaigns make it rather unattractive. Fortunately, a number of studies have demonstrated that synthetically useful mono-arylations can be reliably attained by using tertiary phosphanes as ligands (Schemes 3, 5 and 8). Particularly, the inexpensive PPh3 was explored by the groups of Yu27 and Dixneuf40 as a mono-directing ligand in combination with RuCl3·xH2O and [RuCl2(p-cymene)]2, respectively. The monoarylated products (Scheme 13; 37, 40, 58, 91–93) were obtained in moderate yields when the C–H activation partner was used in stoichiometric or slightly larger, amount.
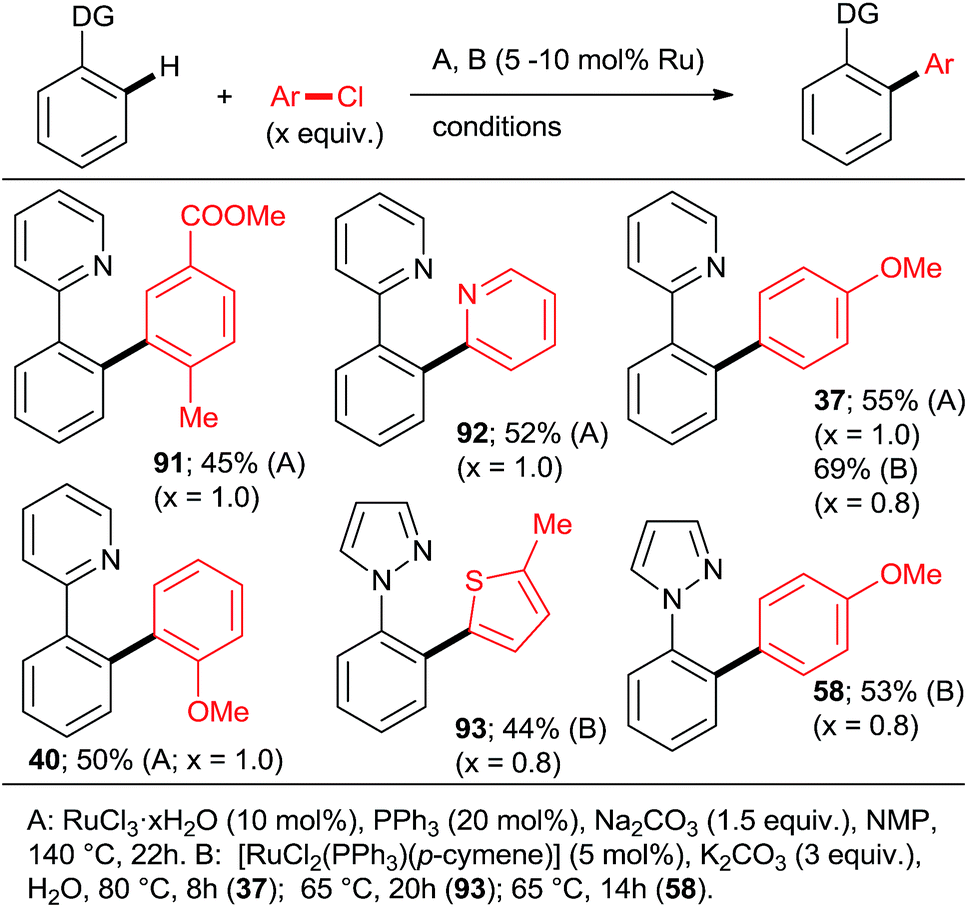 | ||
| Scheme 13 Ligand (PPh3)-controlled mono arylation. | ||
:7:77 = 73:20:7, 100% conversion). Peris' group successfully combined35 directed hydroarylation of alkenes with directed o-arylation of 2-phenylpyridine (1) catalyzed by a di-NHC-Ru complex 95 in the presence of MesCOOK, including one example with an aryl chloride (Scheme 15, product 96). Utilizing a simpler, mononuclear NHC-Ru complex (97), Peris' group also performed15 (Scheme 16) a dehydrogenation of the secondary alcohol 98 to produce much more active 4-chloroacetophenone, which underwent in situ di-o-arylation of 2-phenylpyridine (giving 77) and 1-phenylpyrazole. Unlike with bromoarenes, up to 10% of mono-arylated products were also isolated. Ackermann et al. telescoped24 (Scheme 17) direct o-arylation with 3- and 4-chloroarylketones and subsequent Ru-catalyzed hydrosilylation to 1-arylethanol TES ethers (e.g., product 99) catalyzed by the Ru(IV)–alkylidene complex 10. The resulting TES ethers could be deprotected with TBAF in situ to free secondary alcohols. Only one example of one pot Ru-catalyzed directed o-arylation and Pd-mediated direct arylation was disclosed very recently by Charette group,14 and in that case the only aryl chloride tested gave a very low yield compared to aryl bromides and iodides.
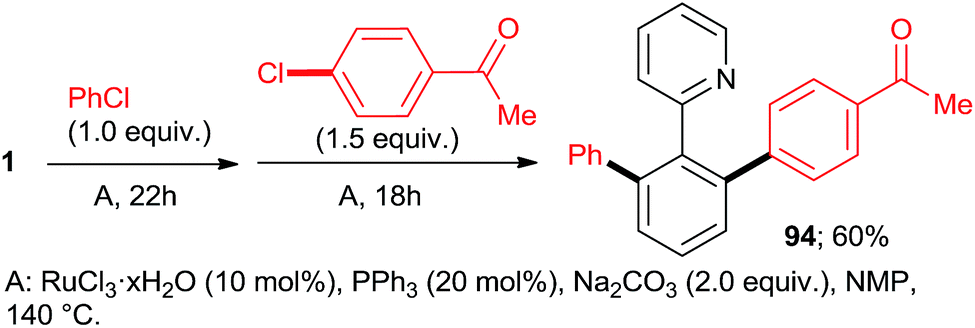 | ||
| Scheme 14 One-pot, sequential arylation. | ||
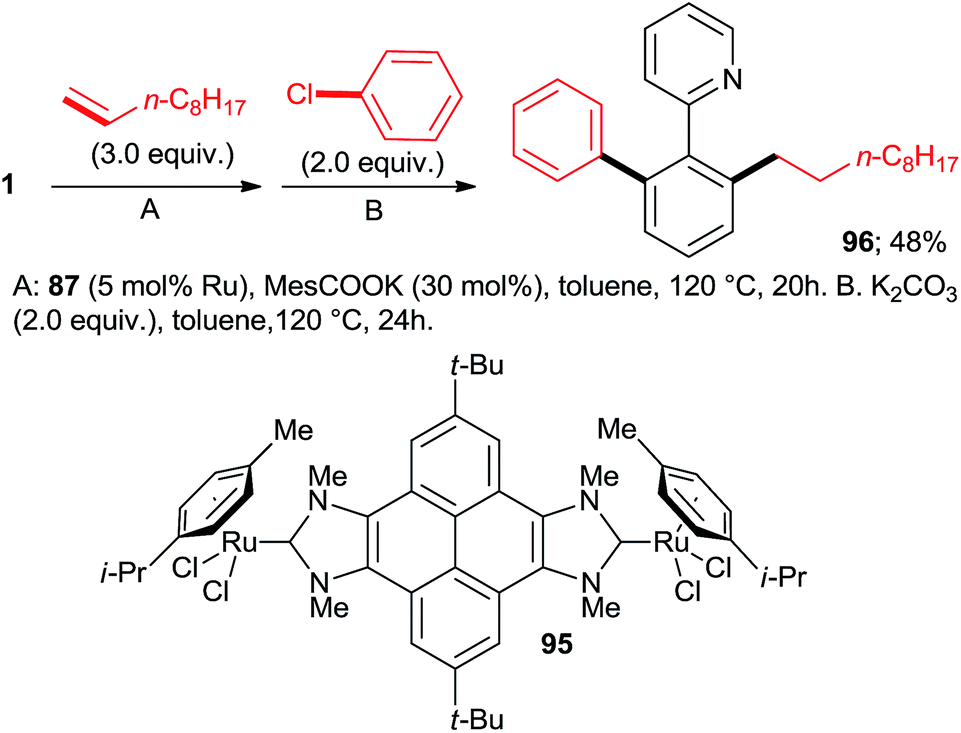 | ||
| Scheme 15 One-pot, chelation-assisted hydroarylation and arylation sequence. | ||
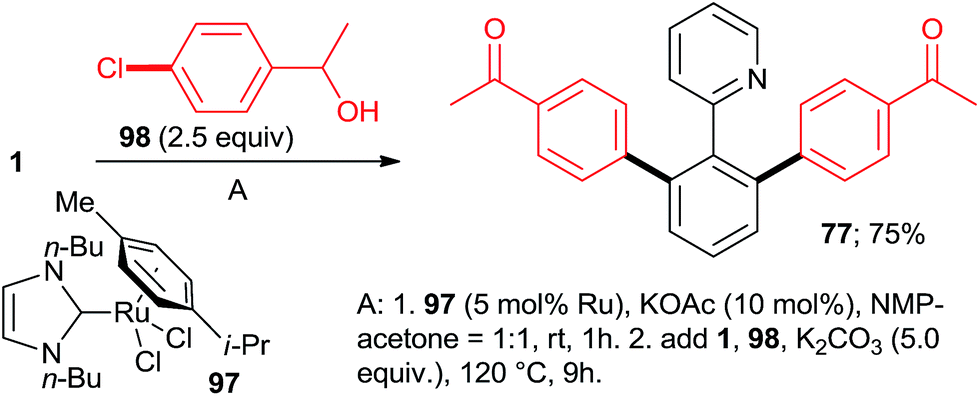 | ||
| Scheme 16 One-pot transfer dehydrogenation/directed arylation sequence. | ||
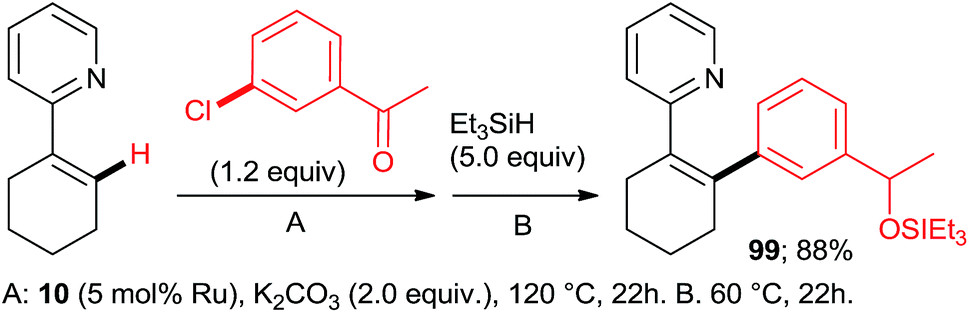 | ||
| Scheme 17 One-pot directed arylation/hydrosilylation sequence. | ||
Mechanism of the Ru-catalyzed direct arylation with unactivated aryl chlorides
The seminal Oi and Inoue paper12 proposed a catalytic cycle (Scheme 18a) consisting of an oxidative addition of the aryl chloride to Ru(II) to produce Ru(IV) followed by a C–H activation, reductive elimination and base-promoted halide removal. Notably, the C–H activation step was postulated to proceed by electrophilic aromatic substitution (SEAr) via a zwitterionic (Wheland) intermediate (100). This cycle features the same sequences of steps as the cycle of Pd-catalyzed directed arylation, which is a modification of the ubiquitous aryl–aryl cross-coupling cycle with the C–H activation taking the place of transmetalation (Scheme 18b). This initial mechanistic proposal was found to be inconsistent with stoichiometric experiments published by Ackermann's group.31 Importantly, [Ru(MesCOO)2(p-cymene)] (51) did not react with p-chloroanisole in toluene at 110 °C (Scheme 18c), ruling out oxidative addition as the first step in the cycle. On the other hand, treating 2-(4-methoxyphenyl)pyridine with 51 at 80 °C gave a neural cyclometalated Ru(p-cymene) complex with one monodentate MesCOO anion retained in the inner coordination sphere of the metal (101). Complex 101 reacted with p-chloroanisole to give 67% yield of 102 in the presence of K2CO3 at 80 °C. Both 51 and 101 were competent catalysts for direct arylation to give 102. The above results suggest that the cycle commences with C–H activation instead of C–Cl oxidative addition. This is also consistent with the observation that unreactive (e.g., o-substituted) aryl chlorides promote the dimerization (homocoupling) of the C–H activation component in synthetically useful yields.42 The recent interest in C–H activation chemistry8–10 has led to intense investigations of its mechanism. However, studies for Ru are relatively sketchy next to those for Pd and Rh. The main difference between Pd and Ru (Rh-catalyzed direct arylations are very scarce,44 hence will not be discussed here) lies in the C–H activation step. Extensive studies8 have shown that C–H activation in Pd(II)-catalyzed proceeds by a number elementary reactions in a substrate- and catalyst-dependent manner, with as well as without chelation assistance (Scheme 18d).45 Studies with Pd(OAc)2-based catalysts suggest an inner sphere (bimolecular) concerted metallation-deprotonation (CMD-SE2).46 In certain cases, carbopalladation/β-hydride elimination (“Heck-type”)47 or direct transmetalation (“carbanion arylation type”),48 are known. As a consequence, the substrate scope for Pd is much wider than Ru. Dixneuf, Jutand et al. conducted a more detailed investigation on Ru(II) dicarboxylate-catalyzed arylations of bromoarenes.49 The C–H activation was concluded to proceed by outer sphere (trimolecular) chelation-assisted CMD (CMD-SE3). In this elementary step, free carboxylate acts as a base abstracting o-H from the Ph ring in 1 after the pyridine nitrogen forms a Wheland complex with the cationic [Ru(RCOO) (p-cymene)] (96). This conclusion is consistent with the observed auto-catalytic effect of the liberated RCOOH, attributed to facilitating the dissociation of a carboxylate from the initial precatalyst, thus providing an explanation for the higher activity of this type of catalysts. Based on these studies, a revised catalytic cycle comprising CMD-SE3 C–H activation, oxidative addition, and reductive elimination has been established (Scheme 18e). The oxidative addition (giving 97) has been accepted as the rate-determining step for the whole cycle, as corroborated by numerous observations described above that aryl halides bearing electron-withdrawing groups are more active than those bearing electron-donating ones. This conclusion was made based on observations that: (1) the carboxylate-assisted cycloruthenation is facile even at room temperature50 whereas the arylation reaction as a whole is not; (2) deuterium labelling experiments point at the C–H activation being reversible.14,31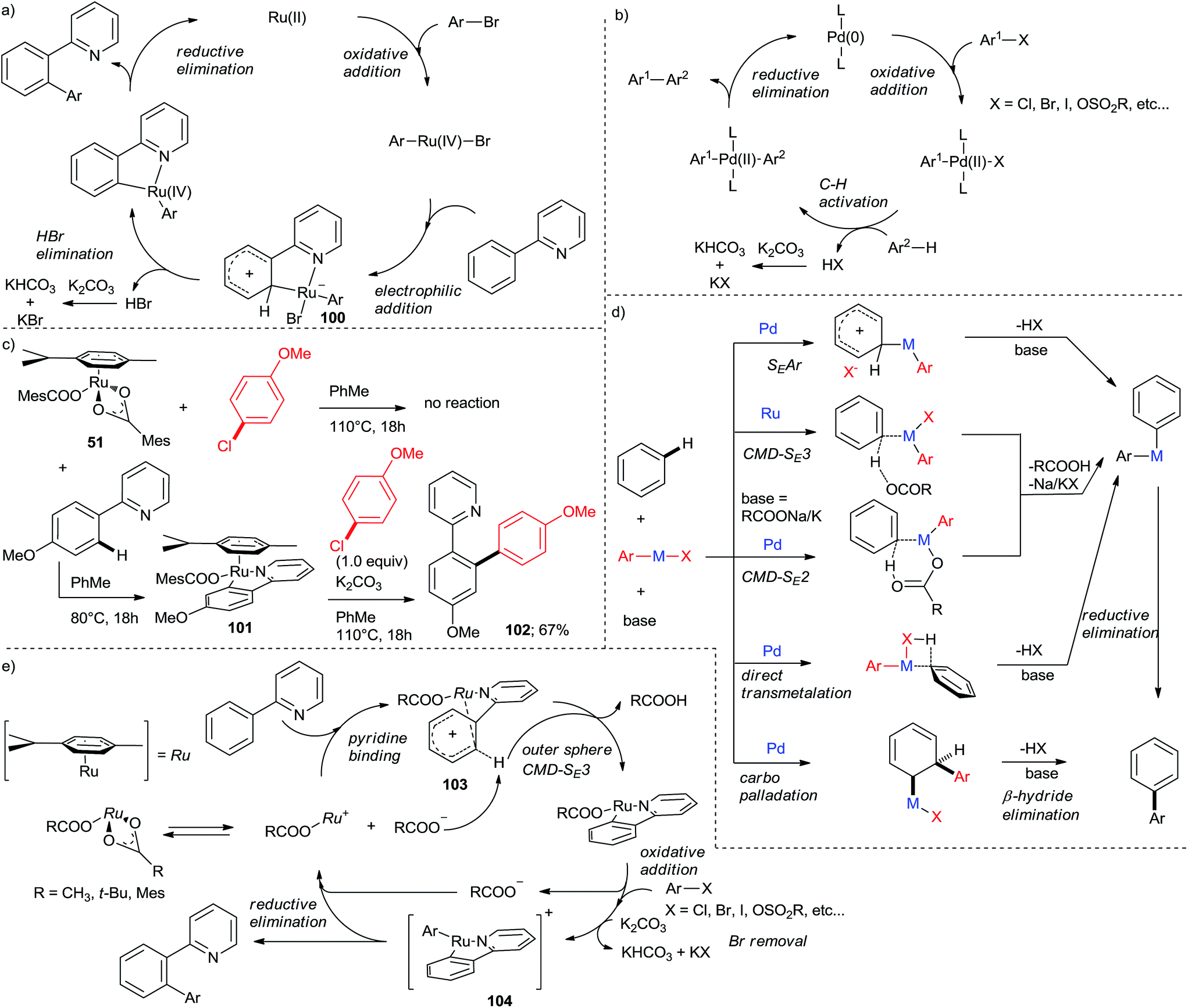 | ||
| Scheme 18 (a) Initially proposed catalytic cycle for the Ru-catalyzed chelation-assisted direct arylation. (b) Accepted catalytic cycle for the Pd-catalyzed direct arylation. (c) Stoichiometric experiments that refute the initially proposed catalytic cycle. (d) Comparison of the C–H activation mechanisms for Pd and Ru-catalyzed direct arylation. The elementary steps are shown in a simplified manner. (e) Currently accepted catalytic cycle for the Ru-catalyzed chelation-assisted direct arylation. | ||
Conclusion and future directions
Aryl chlorides are cheaper and more easily available than aryl bromides and iodides, but pose greater challenge due to the relatively low reactivity of the C–Cl bond. The use of Ru(II) catalysts has been very successful in inducing even deactivated (electron-rich) aryl chlorides to couple with arenes containing a pendant chelating substituent in the course of the direct arylation reaction.Despite the significant progress evidenced in this comprehensive mini review, a number of issues remain. Some of those are briefly discussed next: (1) even though high yields have been attained using a large number of Ru catalysts, the majority of these have come from simple aryl chlorides such as chlorobenzene, m- and p-chlorotoluenes, or m- and p-chloroanisoles. It remains to be seen whether more complex substrates such as those used in pharmaceutical or agrochemical industries, or materials sciences can be converted in similarly high yields and which catalysts would enable that. (2) The scope is even more limited on the C–H activation side and warrants further investigations. Notably, many substrates that have been successfully C–H activated in other Ru-catalyzed reactions have not been demonstrated to participate in direct arylation yet. Therefore, the issue of the C–H component reactivity may be quite complex. (3) With respect to the catalysts, the work carried out to date has shown a great diversity in Ru complexes capable of catalyzing direct arylations in good yields as well as mono:di selectivity. The “ligandless” catalysts prepared from [RuCl2(η6-arene)] complexes and anionic “additives” (such as carboxylates) have been systematically optimized. These catalysts have shown excellent levels of activity, particularly being the top choice for attaining high diarylation yields. On the other hand, large-scope, systematic exploration of the multitude of phosphane ligands available, particularly those that are bulky and highly sterically hindered, is still lacking. The situation with NHCs is similar. Both of these ligands classes are indispensable for activation of aryl chlorides in palladium catalysis. (4) More detailed mechanistic studies by both experiment and computation are overdue, particularly for phosphane- and NHC-ligated Ru catalysts. Investigating the molecular basis of the pronounced inclination of phosphane ligands to selectively catalyze mono-arylation may pave the way for improved catalyst design.
Acknowledgements
Financial support from Wuhan University of Technology (to GFZ and HLQ) and Hefei University of Technology (to EABK) is gratefully acknowledged.Notes and references
- C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2010, 51, 5062–5085 CrossRef PubMed
.
-
Metal-catalyzed cross-coupling reactions, ed. A. De Meijere and F. Diederich, John Wiley & Sons, New York, 2nd edn, 2004 Search PubMed
- H. Li, C. C. C. J. Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS
-
(a) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768–2813 CrossRef CAS
- P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497–5508 CrossRef CAS
- G.-R. Peh, E. A. B. Kantchev, J.-C. Er and J. Y. Ying, Chem.–Eur. J., 2010, 16, 4010–4017 CrossRef CAS
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC
- Recent reviews:
(a) R. K. Rit, M. R. Yadav, K. Ghosh and A. K. Sahoo, Tetrahedron, 2015, 71, 4450–4459 CrossRef CAS
- Recent reviews:
(a) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS PubMed
- Recent reviews:
(a) R. Manikandan and M. Jeganmohan, Org. Biomol. Chem., 2015, 13, 10420–10436 RSC
- Recent reviews:
(a) L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS
- S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579–2581 CrossRef CAS
-
(a) S. Rajkumar, S. Karthik and T. Gandhi, J. Org. Chem., 2015, 80, 5532–5545 CrossRef CAS
- D. S. Roman, V. Poiret, G. Pelletier and A. B. Charette, Eur. J. Org. Chem., 2015, 67–71 CrossRef
- S. Sabater, J. A. Mata and E. Peris, Organometallics, 2012, 31, 6450–6456 CrossRef CAS
- L. Ackermann and A. V. Lygin, Org. Lett., 2011, 13, 3332–3335 CrossRef CAS
- L. Ackermann, A. Althammer and R. Born, Tetrahedron, 2008, 64, 6115–6124 CrossRef CAS
- L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299–2302 CrossRef CAS
- S. Seršen, J. Kljun, F. Požgan, B. Štefane and I. Turel, Organometallics, 2013, 32, 609–616 CrossRef
- Y. Goriya and C. V. Ramana, Chem.–Eur. J., 2012, 18, 13288–13292 CrossRef CAS
- B. Štefane, J. Fabris and F. Požgan, Eur. J. Org. Chem., 2011, 3474–3481 CrossRef
- L. Ackermann, A. Althammer and R. Born, Angew. Chem., Int. Ed., 2006, 45, 2619–2622 CrossRef CAS
- L. Ackermann, Org. Lett., 2005, 7, 3123–3125 CrossRef CAS
- L. Ackermann, R. Born and P. Álvarez-Bercedo, Angew. Chem., Int. Ed., 2007, 46, 6364–6367 CrossRef CAS
- B. Yu, X. Yan, S. Wang, N. Tang and C. Xi, Organometallics, 2010, 29, 3222–3226 CrossRef CAS
- S. Doherty, J. G. Knight, C. R. Addyman, C. H. Smyth, N. A. B. Ward and R. W. Harrington, Organometallics, 2011, 30, 6010–6016 CrossRef CAS
- N. Luo and Z. Yu, Chem.–Eur. J., 2010, 16, 787–791 CrossRef CAS
- H. Miura, K. Wada, S. Hosokawa and M. Inoue, Chem.–Eur. J., 2010, 16, 4186–4189 CrossRef CAS
- J. Zhang, Q. Yang, Z. Zhu, M. L. Yuan, H. Y. Fu, X. L. Zheng, H. Chen and R. X. Li, Eur. J. Org. Chem., 2012, 6702–6706 CrossRef CAS
- L. Ackermann, E. Diers and A. Manvar, Org. Lett., 2012, 14, 1154–1157 CrossRef CAS PubMed
- L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed
- P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2010, 49, 6629–6632 CrossRef CAS
- P. Arockiam, V. Poirier, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2009, 11, 1871–1875 RSC
- L. A. Adrio, J. Gimeno and C. Vicent, Chem. Commun., 2013, 49, 8320–8322 RSC
- S. Gonell and E. Peris, ACS Catal., 2014, 4, 2811–2817 CrossRef CAS
- K. S. Singh and P. H. Dixneuf, ChemCatChem, 2013, 5, 1313–1316 CrossRef CAS
- W. Li, P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2315–2319 RSC
- S. Demir, I. Özdemir and B. Çetinkaya, J. Organomet. Chem., 2009, 694, 4025–4031 CrossRef CAS
- I. Özdemir, S. Demir, N. Gürbüz, B. Çetinkaya, L. Toupet, C. Bruneau and P. H. Dixneuf, Eur. J. Inorg. Chem., 2009, 1942–1949 CrossRef
- P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2013, 15, 67–71 RSC
- N. Dastbaravardeh, M. Schnürch and M. D. Mihovilovic, Eur. J. Org. Chem., 2013, 2878–2890 CrossRef CAS
- L. Ackermann, P. Novák, R. Vicente, V. Pirovano and H. K. Potukuchi, Synthesis, 2010, 2245–2253 CrossRef CAS
- Y. Kobayashi, M. Kashiwa, M. Sonoda, M. Kirihata and S. Tanimori, Synthesis, 2014, 46, 3185–3190 CrossRef CAS
- Some examples:
(a) T. S. S. Yanagisawa, R. Noyori and K. Itami, J. Am. Chem. Soc., 2006, 128, 11748–11749 CrossRef
- S. I. Gorelsky, Coord. Chem. Rev., 2013, 257, 153–164 CrossRef CAS
-
(a) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS
- C. Colletto, S. Islam, F. Juliá-Hernández and I. Larossa, J. Am. Chem. Soc., 2016, 138, 1677–1683 CrossRef CAS
- M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS
-
(a) I. Fabre, N. v. Wolff, G. L. Duc, E. Ferrer-Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, Chem.–Eur. J., 2013, 19, 7595–7604 CrossRef CAS
- B. Li, C. Darcel, T. Roisnel and P. H. Dixneuf, J. Organomet. Chem., 2015, 793, 200–209 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |