
Palladium supported on amino functionalized magnetic MCM-41 for catalytic hydrogenation of aqueous bromate†
Huan Chena,
Peng Zhanga,
Wenhui Tana,
Fang Jiang*a and
Rong Tangb
aKey Laboratory of Jiangsu Province for Chemical Pollution Control and Resources Reuse, School of Environmental & Biological Engineering, Nanjing University of Science and Technology, Nanjing 210094, PR China. E-mail: fjiang@njust.edu.cn; Fax: +86-25-84315352; Tel: +86-25-84311819
bJiangsu Open University, Nanjing 210036, PR China
First published on 21st August 2014
Abstract
Pd supported on amino functionalized magnetic MCM-41 was synthesized by deposition–precipitation (DP) and impregnation (IMP) methods. The catalysts were characterized by N2 adsorption, XRD, FTIR, CO chemisorption, TEM, XPS and VSM. Liquid phase catalytic hydrogenation of bromate was tested over these catalysts. The results showed that the DP catalyst had a higher activity than the IMP catalyst, due to its higher Pd dispersion. With regards to DP catalysts supported on magnetic MCM-41 with different NH2 loading amounts, a positive relationship was observed between the Pd dispersion and the initial catalytic activity. Moreover, the DP-2 catalyst (supported on magnetic MCM-41 with 0.99 mmol of NH2 per g) was completely recovered with an external magnetic field, and the catalytic activity remained unaltered even after 5 repeated cycles for the bromate reduction.
1. Introduction
Bromate is the main inorganic disinfection byproduct formed during the ozonation of bromide-containing water.1,2 Due to its high toxicity, the United States Environmental Protection Agency (USEPA) and the World Health Organization have formulated the acceptable bromate level of 0.01 mg L−1 in drinking water.3,4 Thus, many effective treatment methods have been developed to remove bromate from drinking water, such as adsorption,5 photocatalysis,6 zero-valent iron reduction,7 and recently catalytic hydrogenation.8The catalytic hydrogenation method has been identified as a promising method for the removal of many contaminants from water, such as nitrate,9 perchlorate,10 chlorophenol11 and nitrophenol.12 Noble metals have been extensively applied as catalysts in the hydrogenation process, and among them Pd is suggested as a highly active metal. Generally, the activity of supported Pd catalysts is related to the nature of active Pd, which is influenced by the property of support, method of preparation and nature of Pd precursor, etc.13,14 In this case, many researchers have focused their attention on developing catalysts with high activity by improving the surface property of catalyst support. Employing functional group, such as amine or amino, onto the surface of catalyst, is an effective approach to modify the surface and electronic properties.15 The amine or amino groups on catalyst support are well known to stabilize Pd nanoparticles against aggregation and to increase Pd dispersion.16,17 Yi et al.18 reported that an amine-functionalized silica supported Pd catalyst provided excellent reactivity and reusability in the hydrogenation of nitrobenzene. They interpreted that the phenomenon was resulted from the high Pd dispersion and the suppression of the aggregation of Pd nanoclusters during the hydrogenation process. Similarly, Mandal et al.19 investigated a Pd nanoparticles immobilized on amine-functionalized zeolite catalyst, and observed a high activity and stability in the hydrogenation reaction. Besides of the surface property of catalyst support, the preparation method of catalyst is considered to be another important impact factor on catalytic behavior. Compared to conventional impregnation (IMP) method, deposition–precipitation (DP) method is suggested to be a more appropriate method to produce catalysts with higher Pd dispersion. Hence, it is feasible to improve hydrogenation efficiency by applying this method to develop high dispersed Pd catalyst. As reported by Gopinath et al.,20 the high activity of 1 wt% Pd/ZrO2 catalyst prepared by the DP method in hydrodechlorination of chlorobenzene could be attributed to the high Pd dispersion.
Apart from the catalytic activity, another key challenge in the industrial application of hydrogenation is the reusability of catalysts. Therefore, developing catalysts which can be easily separated and recycled is highly desirable. One approach to such a problem is using magnetic materials as catalyst support. The catalyst supported on magnetic materials can be easily separated from the reaction solution by the application of an external magnetic field. For example, Guin et al.21 prepared Pd supported amine-terminated Fe3O4 and NiFe2O4 catalysts for a series of hydrogenation reactions, and found that the catalysts could be completely recoverable by magnetic separation and the efficiency of the catalyst remained unaltered after 10 repeated cycles.
In the present investigation, magnetic Pd catalysts supported on amino functionalized MCM-41 with different NH2 loading amount are developed by DP and IMP methods. A comparison is made between the activities of these catalysts on liquid phase catalytic hydrogenation of bromate. The variation in catalytic activity is interpreted based on the Pd dispersion.
2. Experimental
2.1 Catalyst preparation
The amino functionalized magMCM-41 (magMCM-41-NH2) was prepared by a post-grafting method. Typically, 2.0 g of magMCM-41 particles were suspended in 100 mL of dry toluene containing a certain amount of 3-aminopropyltrimethoxysilane (APTES). Then, the mixture was stirred under N2 flow, and refluxed at 110 °C for 12 h. Finally, the magMCM-41-NH2 sample was separated from the toluene solution by magnetic decantation, washed with ethanol and acetone for several times, and dried at 70 °C under vacuum for 12 h. Five different functionalized magMCM-41 samples were obtained by using different ratios of volume of APTES (mL) to mass of magMCM-41 (g). Samples with the ratio of 0.1, 0.25, 0.5, 1.0, and 2.0 were designated as magMCM-41N1, magMCM-41N2, magMCM-41N3, magMCM-41N4 and magMCM-41N5, respectively. The concentrations of amino group was calculated from nitrogen content obtained by elemental analyzer (CHN–O-Rapid, Heracus) were 0.83, 0.99, 1.19, 1.41 and 1.59 mmol g−1 for magMCM-41N1, magMCM-41N2, magMCM-41N3, magMCM-41N4 and magMCM-41N5, respectively. The unfunctionalized magMCM-41 was designated as magMCM-41N0.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PdCl2 = 2.5, pH 3–4). Then, the mixture was evaporated to dryness under stirring at 90 °C.
:PdCl2 = 2.5, pH 3–4). Then, the mixture was evaporated to dryness under stirring at 90 °C.All of the samples made by DP and IMP were dried overnight and calcined at 200 °C for 2 h under a N2 flow of 40 mL min−1, and reduced at 200 °C for 4 h under a H2 flow of 40 mL min−1. All catalysts were ground to pass through a 400-mesh sieve (<37 μm) to ensure negligible intraparticular diffusion before bromate reduction.23,24 The Pd contents of the obtained DP and IMP catalysts were around 2.0% determined by X-ray fluorescence (ARL-9800). The DP catalysts using magMCM-41N1, magMCM-41N2, magMCM-41N3, magMCM-41N4 and magMCM-41N5 as support were designated as DP-1, DP-2, DP-3, DP-4, DP-5, respectively. The IMP catalysts using magMCM-41N1, magMCM-41N2, magMCM-41N3, magMCM-41N4 and magMCM-41N5 as support were designated as IMP-1, IMP-2, IMP-3, IMP-4, IMP-5, respectively. The catalysts prepared by unfunctionalized MCM-41 were designated as IMP-0 and DP-0, respectively.
2.2 Catalyst characterization
BET surface areas of the catalysts were determined by N2 adsorption method on Micromeritics ASAP 2200. The samples were pretreated at 200 °C under vacuum (1.33 Pa) for 4 h. X-ray diffraction (XRD) patterns of the catalysts were obtained by a powder diffraction-meter (D/max-RA, Rigaku), operating with a Cu anode at 30 kV and 20 mA. The Fourier transform infrared spectra (FTIR) spectra were acquired in transmission mode on a Nicolet Nexus 870 FTIR spectrophotometer in the range of 4000–400 cm−1. The morphology and size of catalyst was obtained by a JEOL 2100 transmission electron microscope. X-ray photoelectron spectroscopy (XPS) was performed on a PHI 5000 VersaProbe XPS instrument using a monochromatized Al Kα excitation source (hν = 1486.6 eV). The magnetic properties of the catalysts were measured by a vibration sample magnetometry (VSM 7410).Pd dispersion was evaluated using the CO chemisorption method. Typically, 100 mg of the reduced catalyst was activated under a H2 flow of 40 mL min−1 at 200 °C for 2 h. After flushing by Ar flow (30 mL min−1) for 1 h at 200 °C, the catalyst was cooled down to room temperature. The CO chemisorption was performed with pulses of 0.5 mL. In order to calculate the metal dispersion, an adsorption stoichiometry of Pd/CO = 1 was assumed.25
2.3 Catalytic bromate reduction
Hydrogenation reduction of bromate was conducted at atmospheric pressure under 25 °C in a 250 mL three-necked flask reactor. Typically, a mixture of the catalyst (10.0 mg) and deionized water (180 mL) was placed in the reactor at first, and H2 was introduced (40 mL min−1) under stirring rate of 1000 rpm for 30 min. Then, 20 mL of 7.8 mM bromate solution was added rapidly, and the solution pH was 5.6 without adjustment. The samples were taken at the desired reaction time and the catalyst particles were filtered with a 0.45 μm membrane. Bromate and bromide concentration in the filtrate was determined by Ion Chromatography (ICS 2000, Dionex). The initial activities of the catalysts were calculated by the removal rate of the bromate within initial 7 min.As shown in Fig. 1S† (see ESI), the sum concentrations of bromate and bromide were nearly identical to the initial bromate concentrations during the reaction procedure, reflecting that bromide was the sole bromate reduction product by DP-2. The effect of mass transfer limitation under the reaction condition was evaluated. Bromate reduction over DP-2 with varied catalyst dosages and stirring rates was compared and the results were presented in Fig. 2S.† As catalyst dosage and stirring rate increased, the initial activities were nearly constant, indicating that the reaction was absence of mass transfer limitation.26,27
3. Results and discussion
3.1 Catalyst characterization
 | ||
| Fig. 1 (a) Wide-angle XRD patterns of DP-0, DP-2, IMP-0, IMP-2, magMCM-41 and (b) low-angle XRD patterns of DP-0 and amino functionalized DP catalysts. | ||
As seen from the small-angle XRD patterns in Fig. 1b, the DP-0 catalyst exhibited three diffraction peaks corresponding to (100), (110), and (200) reflections, which assigned to the hexagonal mesoporous structure. After the amino modification, the intensities of the three peaks were reduced and the positions were slightly shifted towards higher value, revealing the decrease of the structural order in amino functionalized DP catalysts. The results were consistent with the XRD patterns of MCM-41 and MCM-41-NH2 materials in previous literatures.30,31
 | ||
| Fig. 2 FTIR spectra of DP-0 and amino functionalized DP catalysts. | ||
For DP catalysts before and after amino functionalization, it was clearly observed from Table 1 that DP-2 catalyst had much higher Pd dispersion than DP-0. This is because that during the procedure of catalyst preparation, the amino groups have large affinity with Pd precursor, hence suppress the agglomeration of Pd particles.18 Furthermore, in comparison of DP catalysts with different surface concentration of amino groups, the volcano-type dependence of Pd dispersion on the NH2 loading was observed. In detail, when the concentration of amino groups in magMCM-41 increased from 0 to 0.99 mmol g−1, the Pd dispersion raised significantly from 39.40% to 84.23%. However, further increase in the concentration of amino groups decreased Pd dispersion.
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) s = ∑nidi3/∑nidi2 s = ∑nidi3/∑nidi2
| (1) |
 | ||
| Fig. 3 TEM images of (a) DP-0, (b) DP-1, (c) DP-2 (d) DP-3, (e) DP-4, (f) DP-5, (g) IMP-0 and (h) IMP-2. | ||
The average sizes of the Pd particles were calculated to be 2.80, 1.73, 1.40, 1.57, 2.33, 2.48, 5.33 and 4.98 nm for DP-0, DP-1, DP-2, DP-3, DP-4, DP-5, IMP-0 and IMP-2, respectively. The small Pd particle sizes on DP catalyst could be attributed to the effective Pd particle dispersion in the catalyst surface. Among the amino functionalized DP catalysts, the DP-2 owned the smallest Pd particle. It confirmed the result of CO chemisorption that DP-2 had the highest Pd dispersion.
 | ||
| Fig. 4 XPS spectra of N 1s and Pd 3d for DP-0, DP-2, IMP-0 and IMP-2 catalysts. | ||
The Pd 3d5/2 binding energy value at 335.1 eV in DP-0 could be attributed to the presence of metallic Pd species, while the binding energy shifted to 336.1 eV in DP-2. It indicated that there was an interaction between Pd and amino group to make Pd more stable on the surface of DP-2 catalyst, thus suppress the agglomeration of Pd particles.36 In the case of DP catalysts with different amount of amine groups, the binding energies were 336.1 eV, 336.1 eV, 336.0 eV, 335.9 eV and 335.7 eV for DP-1, DP-2, DP-3, DP-4 and DP-5, respectively (Fig 4S†). The lower Pd 3d5/2 binding energy in DP-5 related to the attenuate interaction between Pd and amino groups, associated with the phenomenon of Pd agglomeration. The result was consistent with the result of Pd particle size that DP-5 owned the largest Pd particle size among the texted catalysts. As for IMP-0 and IMP-2 catalysts, the Pd 3d5/2 binding energy was 335.4 eV, which corresponded to metallic Pd species. No significant interaction between Pd and the catalyst support was observed in IMP catalysts.
 | ||
| Fig. 5 Magnetization curves and magnetic separation pictures (insert) of DP-2 before and after bromate reduction reaction. | ||
3.2 Catalytic bromate reduction
Catalytic bromate reduction as a function of reaction time over IMP-0, IMP-2, DP-0 and DP-2 is compiled in Fig. 6. The bromate reduction efficiencies were 31.4%, 31.9%, 40.0% and 100% for IMP-0, IMP-2, DP-0 and DP-2, respectively, indicating DP catalysts presented higher catalytic activities than IMP catalysts. The difference in catalytic activities caused by catalyst preparation method was related to Pd dispersion. A higher Pd dispersion resulted in more Pd active sites and active H, thus caused higher activity.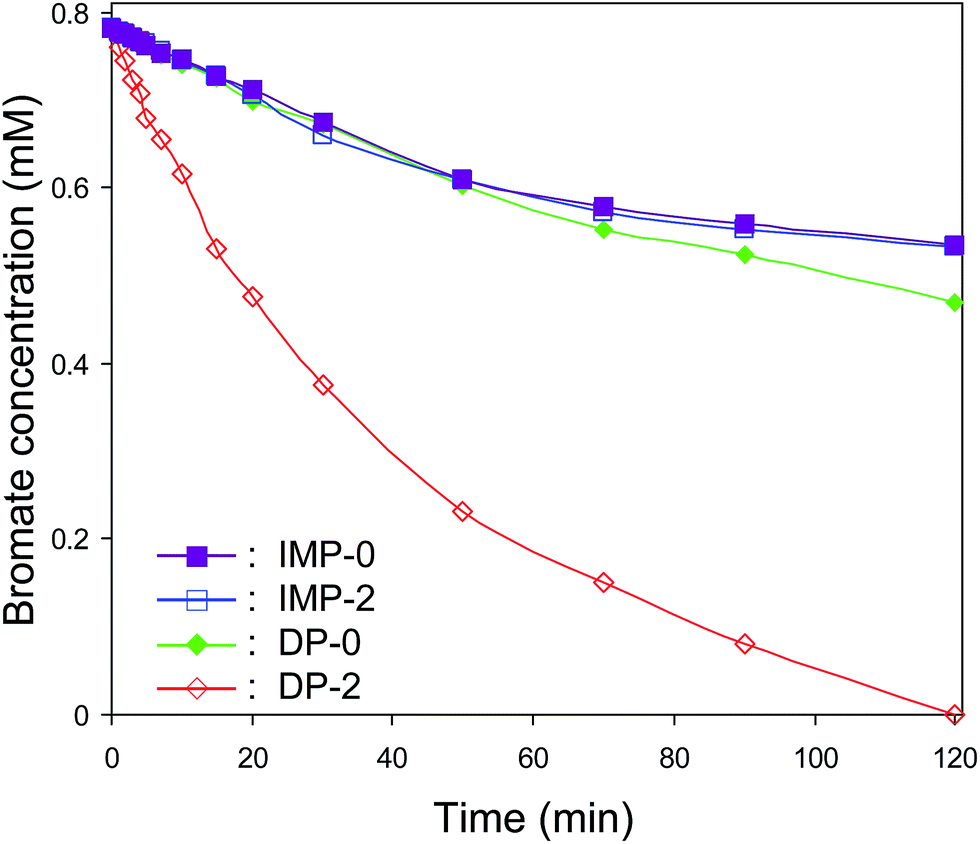 | ||
| Fig. 6 Catalytic bromate reduction over DP-0, DP-2, IMP-0 and IMP-2 catalysts. | ||
To further verify the effect of Pd dispersion, the catalytic hydrogenation of bromate over amino functionalized DP catalysts is compared in Fig. 7a. The bromate reduction efficiency increased from 40.0% to 100% when the concentration of amino groups increased from 0 (DP-0) to 0.99 mmol g−1 (DP-2), followed by a decline in activity with a further increase of NH2 loading. More information obtained by revealing the relationship of initial catalytic activity and Pd dispersion was shown in Fig. 7b. As can be seen, increasing Pd dispersion created enhanced catalytic activity.
 | ||
| Fig. 7 (a) Catalytic bromate reduction over amino functionalized DP catalysts and (b) initial catalytic activity of amino functionalized DP catalysts as a function of Pd dispersion. | ||
The reusability of catalyst is very important for the practical applications of hydrogenation system. Therefore, the reusability of DP-2 catalyst is tested by repeated five runs at the same reaction condition. The DP-2 catalyst could be efficiently recycled for bromate reduction by magnetic separation. As shown in Fig. 8, the DP-2 catalyst could be reused five times without significant loss of activity performance (<6%) in the reduction of bromate. This indicated that the properties of Pd particles on the DP-2 catalyst had little change after the recycle reactions. Minor loss of catalytic activity was also observed in DP-1, DP-3, DP-4 and DP-5 catalysts after five repeated cycles (Fig 5S†). Thus, the DP catalyst could be a promising reusable catalyst for catalytic hydrogenation of bromate.
 | ||
| Fig. 8 Reusability of DP-2 catalyst for bromate reduction. | ||
4. Conclusions
Magnetic Pd catalysts supported on amino functionalized MCM-41 were prepared by deposition–precipitation (DP) and impregnation (IMP) methods. Characterization results revealed that DP catalysts possessed higher Pd dispersion than IMP catalysts. Liquid phase catalytic hydrogenation of bromate was tested over the as-made catalysts. The DP catalyst exhibited a higher activity than IMP catalyst, due to the higher Pd dispersion of DP catalyst. In addition, the initial catalytic activity for DP catalysts with varied NH2 loading amount was also dependent on the Pd dispersion. In particular, the DP-2 catalyst could be easily recovered by magnet and the reusability was excellent for the bromate reduction. The results showed that the as-synthesized DP-2 catalyst was of great potential as an active and reusable catalyst for the catalytic reduction of bromate.Acknowledgements
The financial supports from the Natural Science Foundation of China (no. 51208257 and 51178223), the Natural Science Foundation of Jiangsu Province (SBK201240759), China Postdoctoral Science Foundation (no. 2013M541677) and the Natural Science Fund for Colleges and Universities in Jiangsu Province (12KJB610002) are gratefully acknowledged.References
- U. V. Gunten, Water Res., 2003, 37, 1469–1487 CrossRef.
- Y. Nie, C. Hu, N. Li, L. Yang and J. Qu, Appl. Catal., B, 2014, 147, 287–292 CrossRef CAS PubMed.
- H. S. Weinberg, C. A. Delcomyn and V. Unnam, Environ. Sci. Technol., 2003, 37, 3104–3110 CrossRef CAS.
- World Health Organization, Bromate in drinking-water. Background document for preparation of WHO guidelines for drinking-water quality, World Health Organization Press, Geneva, 2005 Search PubMed.
- R. Chitrakar, Y. Makita, A. Sonoda and T. Hirotsu, Appl. Clay Sci., 2011, 51, 375–379 CrossRef CAS PubMed.
- H. Noguchi, A. Nakajima, T. Watanabe and K. Hashimoto, Environ. Sci. Technol., 2003, 37, 153–157 CrossRef CAS.
- L. I. Xie and C. Shang, Environ. Sci. Technol., 2005, 39, 1092–1100 CrossRef CAS.
- H. Chen, Z. Y. Xu, H. Q. Wan, J. Z. Zheng, D. Q. Yin and S. R. Zheng, Appl. Catal., B, 2010, 96, 307–313 CrossRef CAS PubMed.
- U. Prusse and K. D. Vorlop, J. Mol. Catal. A: Chem., 2001, 173, 313–328 CrossRef CAS.
- J. Y. Liu, J. K. Choe, Z. Sasnow, C. J. Werth and T. J. Strathmann, Water Res., 2013, 47, 91–101 CrossRef CAS PubMed.
- Z. J. Wu, C. X. Sun, Y. Chai and M. H. Zhang, RSC Adv., 2011, 1, 1179–1182 RSC.
- Z. M. Wang, C. L. Xu, G. Q. Gao and X. Li, RSC Adv., 2014, 4, 13644–13651 RSC.
- R. Gopinath, N. S. Babu, J. V. Kumar, N. Lingaiah and P. S. S. Prasad, Catal. Lett., 2008, 120, 312–319 CrossRef CAS.
- Y. Li, X. Xu, P. F. Zhang, Y. T. Gong, H. R. Li and Y. Wang, RSC Adv., 2013, 3, 10973–10982 RSC.
- F. Zhang, J. Jin, X. Zhong, S. Li, J. Niu, R. Li and J. Ma, Green Chem., 2011, 13, 1238–1243 RSC.
- R. Abu-Reziq, D. Wang, M. Post and H. Alper, Chem. Mater., 2008, 20, 2544–2550 CrossRef CAS.
- A. J. Amali and R. K. Rana, Green Chem., 2009, 11, 1781–1786 RSC.
- D. K. Yi, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 2459–2461 CrossRef CAS.
- S. Mandal, D. Roy, R. V. Chaudhari and M. Sastry, Chem. Mater., 2004, 16, 3714–3724 CrossRef CAS.
- R. Gopinath, N. Lingaiah, B. Sreedhar, I. Suryanarayana, P. S. Sai Prasad and A. Obuchi, Appl. Catal., B, 2003, 46, 587–594 CrossRef CAS.
- D. Guin, B. Baruwati and S. V. Manorama, Org. Lett., 2007, 9, 1419–1421 CrossRef CAS PubMed.
- X. Chen, K. F. Lam, Q. Zhang, B. Pan, M. Arruebo and K. L. Yeung, J. Phys. Chem. C, 2009, 113, 9804–9813 CAS.
- M. A. Aramendía, V. Boráu, I. M. García, C. Jiménez, F. Lafont, A. Marinas, J. M. Marinas and F. J. Urbano, J. Mol. Catal. A: Chem., 2002, 184, 237–245 CrossRef.
- G. Yuan and M. A. Keane, Chem. Eng. Sci., 2003, 58, 257–267 CrossRef CAS.
- G. Neri, M. G. Musolino, C. Milone, D. Pietropaolo and S. Galvagno, Appl. Catal., A, 2001, 208, 307–316 CrossRef CAS.
- O. M. Ilinitch, F. P. Cuperus, L. V. Nosova and E. N. Gribov, Catal. Today, 2000, 56, 137–145 CrossRef CAS.
- P. Zhang, F. Jiang and H. Chen, Chem. Eng. J., 2013, 234, 195–202 CrossRef CAS PubMed.
- J. H. Jang and H. B. Lim, Microchem. J., 2010, 94, 148 CrossRef CAS PubMed.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- D. B. Nale, S. Rana, K. Parida and B. M. Bhanage, Appl. Catal., A, 2014, 469, 340–349 CrossRef CAS PubMed.
- T. Joseph, K. Vijay Kumar, A. V. Ramaswamy and S. B. Halligudi, Catal. Commun., 2007, 8, 629–634 CrossRef CAS PubMed.
- M. Yamaura, R. L. Camilo, L. C. Sampaio, M. A. Macêdo, M. Nakamura and H. E. Toma, J. Magn. Magn. Mater., 2004, 279, 210–217 CrossRef CAS PubMed.
- K. M. Parida and D. Rath, J. Mol. Catal. A: Chem., 2009, 310, 93–100 CrossRef CAS PubMed.
- N. S. Babu, N. Lingaiah, N. Pasha, J. V. Kumar and P. S. Prasad, Catal. Today, 2009, 141, 120–124 CrossRef CAS PubMed.
- G. Yuan and M. A. Keane, Appl. Catal., B, 2004, 52, 301–314 CrossRef CAS PubMed.
- M. Ma, Q. Zhang, D. Yin, J. Dou, H. Zhang and H. Xu, Catal. Commun., 2012, 17, 168–172 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05593d |
| This journal is © The Royal Society of Chemistry 2014 |