
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2,3-Diaryl-substituted indole based COX-2 inhibitors as leads for imaging tracer development†‡
Markus Laubeab,
Christoph Tonderaab,
Sai Kiran Sharmac,
Nicole Bechmannab,
Franz-Jacob Pietzschad,
Arne Pigorsche,
Martin Köckerlinge,
Frank Wuestc,
Jens Pietzsch*ab and
Torsten Kniess*a
aDepartment Radiopharmaceutical and Chemical Biology, Institute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, 01328 Dresden, Germany. E-mail: t.kniess@hzdr.de; j.pietzsch@hzdr.de; Tel: +493512602760 Tel: +493512602622
bDepartment of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany
cDepartment of Oncology, Cross Cancer Institute, University of Alberta, Edmonton, Alberta, Canada T6G 1Z2
dCentre for Translational Bone, Joint, and Soft Tissue Research, Medical Faculty and University Hospital, Technische Universität, 01307 Dresden, Germany
eDepartment of Inorganic Solid State Chemistry, Institute of Chemistry, University of Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany
First published on 13th August 2014
Abstract
A series of 2,3-diaryl-substituted indoles containing a fluorine or methoxy group was synthesized via Fischer indole synthesis, McMurry cyclization, or Bischler–Möhlau reaction to identify potential leads for positron emission tomography (PET) radiotracer development as well as for optical imaging. All 2,3-diaryl-substituted indoles possess autofluorescent properties with an emission maximum in a range of 443–492 nm, which is acceptable for biological studies in vitro and, in part, in vivo. The molecular structure of compounds 3a and 3j was confirmed by X-ray crystal structure analysis. COX inhibitory activity was evaluated by a fluorescence-based and enzyme immunoassay-based assay. Redox activity of all target compounds was also determined. All synthesized 2,3-diaryl-substituted indoles are inhibitors of COX-2 enzyme in the low micromolar range. Compounds 3e, 3f, 3g and 3m displayed a 30–40% inhibition of COX-2 at 0.1 μM concentration while compounds 3f and 3g also exhibited COX-1 inhibitory activity. Various compounds like 3g showed substantial antioxidative potential (RDIENE = 2.85, RHAVA = 1.98), an effect that was most measurable with methoxy-substituted compounds. With respect to PET radiotracer synthesis, OMe-containing compound 3j was selected as a promising candidate for carbon-11 labeling, and F-containing compound 3m as a lead for the development of a fluorine-18 labeled derivative.
Introduction
Since the discovery of the cyclooxygenase-2 (COX-2) isoenzyme in the early 1990s it has been well accepted that this isoform of cyclooxygenases plays an essential role in a number of various (patho)physiological processes such as chronic inflammatory diseases, neurodegenerative disorders, and cancer. Both isoforms (COX-1 and COX-2) catalyze the conversion of arachidonic acid into prostaglandin H2 (PgH2), which subsequently leads to the biosynthesis of prostaglandins and thromboxanes as mediators of a variety of important physiological functions like vasodilation, regulation of inflammatory response, and platelet aggregation. While COX-1 is constitutively expressed, COX-2 is nearly absent under normal physiological conditions. However, various inflammatory stimuli induce COX-2 expression and function. In a clinical picture, overexpression of COX-2 leads to high levels of eicosanoids resulting in acute inflammation, pain and fever. Hence, inflammation is treated by inhibition of COX-2. First generation of COX inhibitors are termed as non-steroidal anti-inflammatory drugs (NSAIDs), which target both COX-1 and COX-2. Typical examples of NSAIDs represent ibuprofen, indomethacin, and diclofenac. COX-1 inhibition through NSAIDs can cause undesirable side effects such as peptic ulcer or stomach bleeding. To overcome these drawbacks, a second generation of compounds, termed as COXIBs, was developed. COXIBs like celecoxib, valdecoxib, and rofecoxib are characterized by high COX-2/COX-1 selectivity profile. These selective COX-2 inhibitors bind competitively to the cyclooxygenase active site of COX-2, but not COX-1, and hence block COX-2-mediated prostaglandin synthesis. Later it became clear that complete inhibition of COX-2 also blocks the formation of vasodilators like prostaglandin I2 and, consequently, resulted in a shift from the COX pathway to the lipoxygenase pathway. This shift was accompanied with increased cardiovascular risk. This matter of facts demonstrates that the search of new lead structures with high COX-2 selectivity but without cardiac or other side effects is an ongoing research field.1Most COXIBs are characterized by a central five membered heterocyclic core structure (pyrrole, thiazole, oxazole, furan and imidazole) with two adjacent aromatic rings bearing a methylsulfonyl or aminosulfonyl group as COX-2 pharmacophore. This basic concept was extended for the design of new lead structures possessing a six-membered or bicyclic heterocyclic core.2,3 The indole motif is a classical pharmacophore present in the non-selective COX inhibitor indomethacin which was as core-structure for the design of various selective COX-2 inhibitors reported by Black et al. (Scheme 1).4 Later, novel N-substituted indole carboxyclic acid esters,5 and various 3,6- and 2,6-disubstituted indole derivatives were described as potent and selective COX-2 inhibitors.6–8 Particularly high potent and selective COX-2 inhibitors as 2,3-diaryl substituted indoles were reported by Hu and Guo et al.9–11 Moreover, quantitative structure–activity relationship (SAR) analysis on benzyl-substituted indoles demonstrated a correlation of the C-2 and C-3 substitution pattern with the found high COX-2 inhibitory and selectivity profile.12 More recently, Kaur et al.13 discussed various N-1 and C-3 substituted indole Schiff bases as selective COX-2 inhibitors. A selection of potent and selective COX-2 inhibitors based on indoles is depicted in Scheme 1.
 | ||
| Scheme 1 Potent COX-2 inhibitors containing an indole motif. | ||
Over the last years, COX-2 has also been associated with the development and progression of cancer. Elevated COX-2 levels are found in many human epithelium-derived malignancies. This finding correlates with aggressiveness, metastatic and invasive potential of tumors.14,15 Exemplarily, human malignant melanoma, a non-epithelial tumor that is characterized by a marked inflammatory response and high metastatic potential, has been shown to overexpress COX-2,16,17 and COX-2 was proposed as a prognostic marker in melanomata and also in various epithelial tumors.18–21 Consequently, tumor-promoting inflammation has been recognized as an emerging hallmark of cancer, and is a current therapeutic target for the development of anticancer drugs.22
Current anticancer drug design and discovery increasingly includes non-invasive molecular imaging methodologies to assess efficacy of novel drugs based on their molecular mode of action. We are interested in radionuclide-guided functional molecular imaging of COX-2 by means of positron emission tomography (PET). The use of selective COX-2 inhibitors as PET radiotracers for molecular imaging of COX-2 would provide valuable information on tumor prognosis, metastasis, or therapy response.23–25
Up to now, a number of radiotracers basing on clinically used COXIBs like celecoxib and valdecoxib have been described;26,27 as well as novel compounds with hitherto unknown pharmacological profile were synthesized, radiolabeled and evaluated in vitro and in vivo.24 Some recent studies with 18F-labeled pyrazole28 or azulene29 based COX-2 inhibitors have successfully demonstrated a specific uptake of radiolabeled COX-2 probes in inflammatory lesions as well as in xenografted tumors and gave the proof of principle of targeting COX-2 in vivo with radiotracers. However, despite of promising pre-clinical results a suitable COX-2 specific radiotracer for human use is still absent, a fact that is stimulating radiopharmaceutical research in that field.
In our efforts to develop radiolabeled COX-2 inhibitors we selected 2,3-diaryl-substituted indole as lead structure, and we recently reported on the radiosynthesis and radiopharmacological evaluation of 3-(4-[18F]fluorophenyl)-2-(4-methylsulfonylphenyl)-1H-indole as novel 18F-radiotracer for PET imaging of COX-2. Although the radiotracer displayed a promising in vitro and in vivo stability profile, no uptake into COX-2-expressing human HT-29 tumor xenografts transplanted in NMRI nu/nu mice could be observed.30 We concluded that the COX-2 inhibitory activity and selectivity profile of the compound in vivo was not suitable for radiotracer applications. However, potential of varying the C-2/C-3 substitution pattern in the indole scaffold as well as the innovative and highly effective radiosynthesis via McMurry cyclization prompted us to synthesize a comprehensive series of 2,3-diaryl-substituted indoles containing fluorine or methoxy groups as leads for the development of respective 11C- and 18F-radiolabeled PET radiotracers. The motivation of the present work was to identify from a pool of 2,3-diaryl-substituted indoles one or two candidates with nanomolar affinity towards COX-2, most favorable COX-2/COX-1 selectivity, having fluorine or methoxy substituents suitable to be replaced by fluorine-18 or carbon-11 respectively.
In this paper we describe the synthesis of thirteen 2,3-diaryl-substituted indoles. We in detail assessed their chemical and fluorescent spectroscopic characteristics. All compounds were screened for their COX-2 inhibitory activity and selectivity profile using a fluorescence-based and an enzyme immunoassay-based COX assay. Many indoles possess antioxidative properties which is an important parameter for their exerted biological activity profile. Antioxidative potential of novel indole compounds was assessed and compared with prominent indole-based antioxidant melatonin.31,32 Concluding SAR studies assisted us in the selection of suitable candidates for radiotracer development.
Results and discussion
Chemistry
The synthetic routes towards 2,3-diaryl-substituted indoles are shown in Schemes 2 and 4, both follow in general literature procedures which however have been improved and modified by us for higher yields and products purity.The synthesis of 2-sulfonylphenyl-3-phenyl-1H-indoles 3a–3g (Scheme 2) followed the synthetic strategy of Hu et al.10 involving a McMurry cyclization as the final step to build up the indole core structure. For this approach the benzophenone derivatives 1a–1d served as starting materials. Compounds 1a and 1d were obtained in 25% and 21% yield, respectively, from N-tosyl protected anthranilic acid by conversion into the corresponding acid chloride with PCl5 and subsequent reaction with fluorobenzene or anisole under Friedel–Crafts conditions followed by detosylation using a mixture of acetic acid and perchloric acid.30,33 Compounds 1b and 1c were synthesized in one step by Friedel–Crafts acylation using BCl3 to achieve selective ortho-benzoylation relative to the amino residue.34 Following this route by starting from p-toluidine and p-fluorobenzonitrile, 1b was obtained in 21% yield. Comparable to that, the yield for 1c was 23% but it should be noted that the hydroxy derivative 1c was obtained unexpectedly by using p-anisidine and p-fluorobenzonitrile for the synthesis of the methoxy derivative. Apparently under the used reaction conditions, boron trichloride caused a demethylation of p-anisidine so that the hydroxy derivative 1c was formed. Since similar but slightly milder reaction conditions were used by Dinsmore and Bergman35 for the synthesis of 2-amino-3′-fluoro-5-methoxy-benzophenone, we also applied this procedure for the synthesis of the methoxy derivative but no product was formed. We assumed that the methoxy-derivative was not directly accessible by this route and we applied the Bischler–Möhlau synthesis as shown below to get access to a 5-methoxy-substituted indole. As a second note, 1c was not stable in alkaline solution during work-up so that for isolation of 1c neutralization of the reaction mixture with NaHCO3 and extraction before column chromatography was applied. The amino substituted benzophenones 1a–1d were converted into the N-(2-benzoyl)phenyl-benzamides 2a–2g by reaction with the corresponding methylsulfonyl- or aminosulfonyl-substituted benzoyl chloride and Et3N in THF in 62–96% yield. In the final step, the benzamides 2a–2g were cyclized under McMurry conditions to yield the 2,3-diaryl-substituted indoles 3a–2g in 21–85%.
 | ||
| Scheme 2 Synthesis of 2-sulfonylphenyl substituted indoles 3a–3g by McMurry cyclization. Reagents and conditions: (a) for 1a and 1d: (i) PCl5, 50 °C, (ii) AlCl3, 80 °C (1a) and 5–10 °C (1d), (iii) HClO4/CH3COOH, 100 °C; (b) for 1b–1c: (i) BCl3, AlCl3, toluene, reflux, (ii) 2 M HCl, reflux; (c) 4-(R2O2S)C6H4COCl, NEt3, THF, rt; (d) TiCl4, Zn, THF, 65 °C. | ||
From 3-(fluorophenyl)-2-[4-(methylsulfonyl)phenyl]-1H-indole (3a) crystals suitable for X-ray structure analysis could be obtained by slow evaporation of a solution of 3a in ethyl acetate at room temperature. This unambiguously confirmed the molecular structure of the compound (Fig. 1, for detailed results including the X-ray structure analysis of the intermediate 2c see the Experimental section and the ESI‡). Interestingly, the plane of the methylsulfonyl-substituted phenyl ring in compound 3a is only slightly twisted out of the plane of the indole core (dihedral angle: 26.74°) in comparison to the fluoro-substituted phenyl ring (dihedral angle: 52.89°). This gives evidence for the preferential interaction of the electron-rich indole system with the electron-deficient methylsulfonyl-substituted phenyl ring.
 | ||
| Fig. 1 Molecular structure of compound 3a in the crystal (ORTEP plot: displacement thermal ellipsoids are drawn at 50% probability level). | ||
The synthesis of the sulfonyl acetamide derivative 3h was accomplished by the reaction of compound 3g with acetyl chloride in acetic acid at 90 °C in 70% yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of compound 3h by acetylation. Reagents and conditions: (a) CH3COCl, CH3COOH, reflux. | ||
The 3-sulfonylphenyl-2-phenyl indoles 3i–3l were synthesized by application of a Fischer indole synthesis following the synthetic strategy of Guo et al.11 (Scheme 4). For that purpose, 1-phenyl-2-sulfonylphenyl ethanones 5a–5d served as key intermediates. The methylsulfonyl-substituted compounds 5a–5c were obtained starting from 4-(methylsulfonyl)phenylacetic acid by conversion to its acid chloride with SOCl2 and subsequent reaction under Friedel–Crafts conditions with ethoxybenzene, anisole, and fluorobenzene, respectively (for detailed results of X-ray crystal structure analysis of the intermediate 5c see the Experimental section and the ESI‡).36 The sulfamoyl-substituted ethanone 5d was synthesized in two steps: 1-(4-fluorophenyl)-2-phenylethanone (4) was prepared in a yield of 51% by Friedel–Crafts reaction of phenylacetic acid chloride and fluorobenzene as described by Singh et al.37 followed by a chlorosulfonylation and ammonolysis at the para-position of the 2-phenyl ring. In detail this was performed by utilization of pure chlorosulfuric acid at temperatures about −78 °C rising slowly to room temperature and subsequent reaction of the crude product in ethyl acetate with aqueous ammonia solution what gave the desired product 5d in 34% yield.38 Compounds 5a–5d were then converted into the appropriate 3-sulfonylphenyl-2-phenyl-1H-indoles 3i–3l by Fischer indole synthesis using phenylhydrazine and BF3·Et2O as Lewis acid. However, the low yields about 20% prompted us to test the synthesis of 3j exemplarily by the McMurry pathway. For that, 2-amino-4′-(methylsulfonyl)benzophenone was allowed to react with 4-methoxybenzoyl chloride to form N-[2-(4-(methylsulfonyl)benzoyl)phenyl]-4-methoxybenzamide which was subsequently and without isolation of the intermediate reacted under McMurry conditions in a two step/one-pot procedure (Scheme 5) as described by us39 for the syntheses of 2-carbaboranyl-substituted indoles. This approach gave 3j in slightly better 32% yield over two steps. Crystals suitable for X-ray structure analysis could be obtained from a solution of 3j in ethyl acetate–petroleum ether 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 by slow evaporation at room temperature which unambiguously revealed the molecular structure of 3j (Fig. 2, for detailed results see the Experimental section and the ESI‡). Interestingly, in this compound the molecular geometry differs in comparison to 3a. The plane of the phenyl ring in 2-position is substantially twisted out of the plane of the indole-core (dihedral angle: 52.89°), likely due to the electron-rich methoxy-substituent in para-position. Consistently although not as stabilized as in compound 3a, the methylsulfonyl-substituted phenyl ring is again less twisted out of the plane of the indole core (dihedral angle: 39.62°) compared to the neighboring phenyl ring.
:50 by slow evaporation at room temperature which unambiguously revealed the molecular structure of 3j (Fig. 2, for detailed results see the Experimental section and the ESI‡). Interestingly, in this compound the molecular geometry differs in comparison to 3a. The plane of the phenyl ring in 2-position is substantially twisted out of the plane of the indole-core (dihedral angle: 52.89°), likely due to the electron-rich methoxy-substituent in para-position. Consistently although not as stabilized as in compound 3a, the methylsulfonyl-substituted phenyl ring is again less twisted out of the plane of the indole core (dihedral angle: 39.62°) compared to the neighboring phenyl ring.
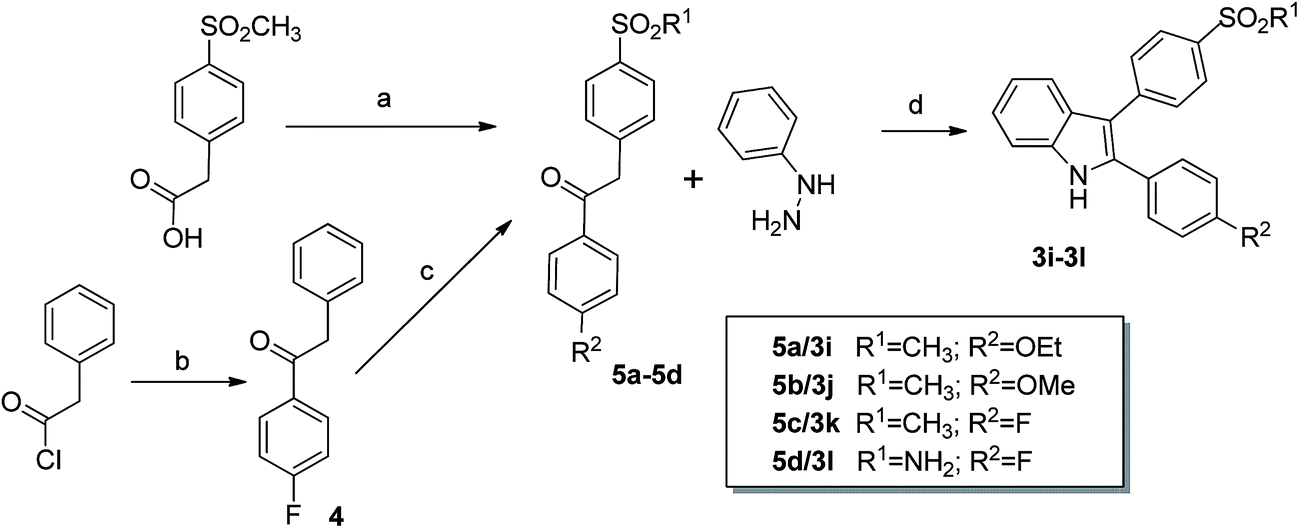 | ||
| Scheme 4 Synthesis of 3-sulfonyl-substituted indoles 3i–3l by Fischer indole synthesis. Reagents and conditions: (a) for 5a–5c: (i) SOCl2, (ii) R2C6H5, AlCl3, 15–20 °C; (b) AlCl3, fluorobenzene, DCM, rt; (c) for 5d: (i) ClSO3H, (ii) NH3(aq.), ethyl acetate, rt; (d) BF3·Et2O, CH3COOH, 130 °C. | ||
 | ||
| Scheme 5 Synthesis of compound 3j by McMurry cyclization. Reagents and conditions: (i) NEt3, THF, rt, (ii) TiCl4, Zn, THF, 65 °C. | ||
 | ||
| Fig. 2 Molecular Structure of compound 3j in the crystal (ORTEP plot: displacement thermal ellipsoids are drawn at 50% probability level). | ||
As shown before, demethylation during the BCl3-mediated Friedel–Crafts acylation hampered the synthesis of a corresponding 5-methoxy-substituted indole 3m via the McMurry-cyclization pathway. Thus, we decided to utilize another type of ring closure reaction, the Bischler–Möhlau cyclization for the synthesis of the methoxy-substituted derivative 3m (Scheme 6). The Bischler–Möhlau cyclisation has been used for the reaction of various methoxy-substituted anilines with 2-bromo-ketone derivatives.40,41 In our case, 1-(4-fluoro-phenyl)-2-sulfonylphenyl ethanone (5c) was brominated to give the precursor 5e in 52% yield.42 Then 5e was allowed to react with p-anisidine at 170 °C in ethanol under pressure to form 3m in 37% yield. The regioselectivity of the Bischler–Möhlau reaction is hardly predictable, so the corresponding two isomers with opposite substituent position could be formed (Scheme 6). Actually, during the workup a set of by-products was observed. However, we isolated one main product for its identification NOESY experiments were performed. Finally by the help of this technique the molecular structure of the 2-[4-(methylsulfonyl)phenyl]-3-(4-fluorophenyl)-5-methoxy-1H-indole (3m) was confirmed (Fig. 3, NOESY spectra are included in the ESI‡).
 | ||
| Scheme 6 Synthesis of 3m by Bischler–Möhlau cyclization. Reagents and conditions: (a) Br2, benzoyl peroxide (cat.), CCl4/CHCl3, 78 °C; (b) EtOH, 170 °C. | ||
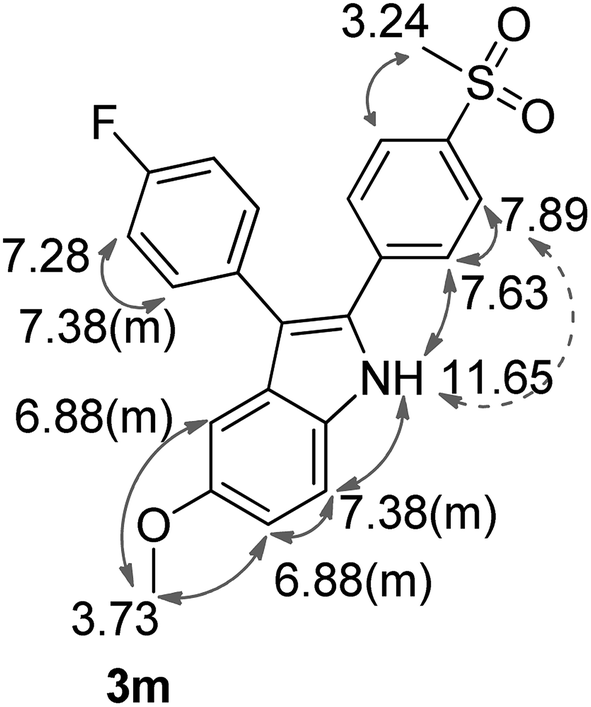 | ||
| Fig. 3 The chemical shifts of 3m in 1H-NMR (DMSO-d6, 400 MHz). The arrows (grey) indicate NOESY cross peaks. | ||
Optical properties
Optical properties have been previously reported for 2,3-diphenyl-1H-indoles and some derivatives were found to be highly fluorescent with emission maxima in the range of 410–475 nm.43–47 We recently presented the visualization of cyclooxygenase-2 by confocal laser induced cryofluorescence microscopy in human melanoma cells using the fluorescent indole 3g.17 With this in mind and because one of the COX assays used by us is based on the detection of a fluorescent dye with an excitation wavelength of 530–540 nm, we initially evaluated the optical properties of the compounds 3a–3m. For that purpose, the UV-vis absorbance and fluorescence excitation as well as emission spectra were recorded (Table 1). Fig. 4 shows the normalized fluorescence excitation and emission spectra of compounds 3j and 3m as examples for the indole based COX-2 inhibitors. In summary, all indoles are characterized by an excitation maxima at λ > 300 nm, a large stokes shift of 106–160 nm and a fluorescence emission maxima in the range of 444–492 nm. In comparison to 3g which was already used to visualize COX-2 in vitro, all tested indoles showed very similar fluorescence excitation and emission spectra so that they might be used for similar applications. Of note, 2-(4-sulfonylphenyl)-substituted indoles like 3m have an excitation maximum between 338–354 nm whereas 3-(4-sulfonylphenyl)-substituted indoles like 3j showed two bands with nearly similar intensity at 300–304 nm and 329–334 nm. As observed by X-ray crystallography for 3a and 3j, this also indicates that the interaction of the phenyl rings with the indole core differs substantially in dependence of the position of the sulfonyl moiety. Finally, no fluorescence signal is observed by excitation at 530 nm allowing the determination of COX-inhibitory activity with the fluorescence-based COX assay.
|
Absorption λabs [nm] (ε [104 M−1 cm−1]) | Excitation maximum λexc [nm] | Emission maximum λem [nm] | |||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| 3a | F | SO2CH3 | H | 236 (2.24), 249 (2.03), 336 (1.61) | 343 | 454 |
| 3b | F | SO2NH2 | H | 253 (2.08), 328 (1.70) | 338 | 450 |
| 3c | F | SO2CH3 | CH3 | 239 (5.62), 339 (2.03) | 345 | 475 |
| 3d | F | SO2NH2 | CH3 | 238 (4.06), 333 (2.10) | 339 | 451 |
| 3e | F | SO2NH2 | OH | 236 (4.33), 341 (2.05) | 345 | 481 |
| 3f | OCH3 | SO2CH3 | H | 237 (2.87), 256 (2.54), 332 (1.60) | 354 | 492 |
| 3g | OCH3 | SO2NH2 | H | 237 (3.71), 325 (1.61) | 345 | 482 |
| 3h | OCH3 | SO2NHAc | H | 256 (2.98), 323 (1.80) | 339 | 452 |
| 3i | SO2CH3 | OCH2CH3 | H | 238 (1.97), 300 (1.73), 340sh (0.93) | 303 | 451 |
| 3j | SO2CH3 | OCH3 | H | 238 (3.18), 300 (2.35), 344 (1.23) | 344 | 450 |
| 3k | SO2CH3 | F | H | 238 (4.47), 296 (2.17), 337 (1.27) | 300 | 444 |
| 3l | SO2NH2 | F | H | 237 (3.85), 296 (2.06), 329sh (1.36) | 303 | 443 |
| 3m | F | SO2CH3 | OCH3 | 237 (4.52), 344 (2.54) | 349 | 484 |

 | ||
| Fig. 4 Fluorescence excitation and emission spectra of compound 3j and 3m. Emission spectra were acquired with an excitation wavelength of 344 nm and 349 nm for 3j and 3m, respectively. | ||
Biological evaluation
|
Fluorescence-based COX-assay % inhibition | Redox-activity | EIA-based COX-assay % inhibition | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COX-1 | COX-2 | COX-1 | COX-2 | ||||||||||
| R1 | R2 | R3 | 10 μM | 1 μM | 1 μM | 0.1 μM | RDIENE | RHAVA | 10 μM | 1 μM | 1 μM | 0.1 μM | |
| a n.i. no inhibition. n.d. not determined. | |||||||||||||
| 3a | F | SO2CH3 | H | 38 | 28 | 60 | 28 | 1.21 | 1.25 | n.i. | 10 | 92 | 60 |
| 3b | F | SO2NH2 | H | 60 | 15 | 55 | 20 | 1.32 | 1.36 | 51 | n.i. | 93 | 82 |
| 3c | F | SO2CH3 | CH3 | 19 | 32 | 20 | 6 | 0.98 | 0.98 | n.d. | n.d. | ||
| 3d | F | SO2NH2 | CH3 | 45 | 20 | 30 | 10 | 0.98 | 0.99 | n.d. | n.d. | ||
| 3e | F | SO2NH2 | OH | 40 | 12 | 54 | 30 | 1.48 | 1.53 | 50 | 100 | 100 | 100 |
| 3f | OCH3 | SO2CH3 | H | 72 | 42 | 49 | 40 | 1.73 | 1.84 | 41 | n.i. | 90 | n.i. |
| 3g | OCH3 | SO2NH2 | H | 90 | 85 | 69 | 32 | 2.85 | 1.98 | 98 | 70 | 98 | 63 |
| 3h | OCH3 | SO2NHAc | H | 51 | 39 | 39 | 12 | 1.55 | 1.32 | 86 | 80 | 80 | n.d. |
| 3i | SO2CH3 | OCH2CH3 | H | 30 | 23 | 65 | 12 | 1.02 | 1.17 | n.d. | n.d. | ||
| 3j | SO2CH3 | OCH3 | H | 31 | 20 | 81 | 28 | 1.54 | 1.29 | n.i. | n.i. | 61 | 32 |
| 3k | SO2CH3 | F | H | 20 | 10 | 30 | 5 | 1.19 | 1.09 | n.d. | n.d. | ||
| 3l | SO2NH2 | F | H | 25 | 20 | 50 | 20 | 1.28 | 1.18 | n.i. | n.i. | 96 | 72 |
| 3m | F | SO2CH3 | OCH3 | 6 | 8 | 40 | 42 | 1.78 | 1.71 | 37 | 50 | 100 | 100 |
| Celecoxib | 30 | 28 | 88 | 80 | 0.97 | 0.96 | n.d. | n.d. | 100 | 65 | |||
| Melatonin | 53 | 39 | 32 | 19 | 1.71 | 1.52 | 30 | 17 | 20 | 15 | |||
| Quercetin | 3.36 | 2.88 | |||||||||||

Generally we found that all synthesized 2,3-diaryl-substituted indoles are inhibitors of COX-2 in the low micromolar or sub-micromolar level. This is in accordance with previous literature findings from other groups describing 3a, 3b, 3f, 3g and 3k as selective COX-2-inhibitors.9,10,48 The results from the assay used by us, showed that in 1 μM concentration 3a, 3b, 3e, 3g, 3i, 3j did inhibit more than 50% of COX-2 activity and four derivatives (3e, 3f, 3g, 3m) inhibited even better, between 30–40% in the 0.1 μM level. With view on regioisomers it is difficult to predict any significant impact from the position of the methylsulfonyl or the aminosulfonyl group to COX-2 affinity. For the regioisomers 3a/3k a higher activity is observed with the methylsulfone on the 2-phenyl ring, but for the pair 3b/3l values in the same range were found. Furthermore, there is no clear tendency if the methylsulfone substituent is superior to the sulfonamide because by direct comparison of two derivatives having the identical core but different sulfonyl substituents similar activities to COX-2 were found as demonstrated by the pairs 3a/3b, 3c/3d and 3f/3g. Compounds with a fluorine-substituent generally show a lower inhibition of COX-2 because in all investigated compounds its replacement by a methoxy group lead to increased inhibition as has been demonstrated for the cases 3a/3f, 3b/3g, and 3k/3j. Acetylation of the sulfonamide resulted likewise in loss of activity as visible in 3g versus 3h, a similar reduction is observed by replacement of a methoxy by an ethoxy group (3i versus 3j). Notable is the impact of the substituent in 5-position of the indole system (R3) on the COX-2 inhibitory activity. By direct comparison of a hydrogen (3a), a methyl- (3c) and a methoxy-group (3m) the latter gives the best inhibition to COX-2 whereas the methyl substituent lead to decreased affinity. Even a hydroxyl group at this position is well tolerated (3e) and results in 40% inhibition at 0.1 μM level.
In summary in the series of thirteen 2,3-diaryl substituted indoles the COX-2 inhibitory activity of the known compounds was confirmed and, compared to 3a, three even more potent derivatives, 3e, 3g and 3m could be identified.
Furthermore, we determined the inhibition profile of all compounds to COX-1 and found that most of the derivatives lacked COX-1 inhibitory activity as expected. However the methoxy-substituted compounds 3f–3g attracted our attention because 3f–3g appeared to be moderate inhibitors of COX-1, and especially 3g was even a more potent COX-1 than COX-2 inhibitor. The last finding was in harsh contrast to the literature, and we suspected false positive results since the fluorescence-based COX-assay is influenced by antioxidants as noted by the manufacturer. Particularly, the indole heterocycle is a well-characterized antioxidant pharmacophore49,50 and it had to be considered that possibly antioxidative properties of 3a–3m interfered with the assay and caused these deviations. This hypothesis was supported by our first findings in parallel in vitro experiments that indole 3g was able to inhibit not only the formation of PGE2 but also the formation of 8-iso-PGF2α generated via non-enzymatic oxidation after the exposure of endothelial cells in both monolayer and organo-type aortic ring models to ionizing radiation.51,52 Thus, we decided to evaluate the antioxidant capacity of the indole based COX-2 inhibitors 3a–3m.
As in the fluorescence-based assay, no clear tendency for the regioisomers was found since the regioisomers 3a/3k as well as 3f/3j showed similar COX-2/COX-1 selectivity although 3f seems slightly more potent than 3j. A similar statement can be made for the position of the aminosulfonyl and methylsulfonyl substituents because 3a/3b were both highly selective inhibitors of COX-2 and within the pair 3f/3g only 3f was a selective COX-2 inhibitor. In contrast to the fluorescence-based assay, also no clear tendency was observed for the comparison of fluorine with methoxy substituted compounds since in both groups very selective as well as unselective inhibitors were found.
Conclusions
The objective of this work was to develop and characterize in detail a series of 2,3-diaryl-substituted indoles selectively inhibiting cyclooxygenase-2, and preferably bearing a fluorine or methoxy-substituent, in order to gain a pool of versatile compounds promising for 18F- or 11C-PET radiotracer development. The position of the fluoro and methoxy group – on the 2-aryl respectively on the 3-aryl substituent – and of the appropriate methylsulfonyl or aminosulfonyl moiety should be altered with view on the resulting affinity towards COX-1 and COX-2.For that purpose thirteen derivatives of 2,3-diaryl-substituted indoles have been synthesized by applying different ring closure reactions, as Fischer-indole synthesis, McMurry cyclization or Bischler–Möhlau cyclization. Among them the McMurry cyclization proofed to be an excellent approach due to its regioselectivity, modularity and the good yields that were obtained. The spectroscopic and fluorescent properties of all 2,3-diaryl-substituted indoles revealed that all the compounds are fluorescent with emission maxima acceptable for biological investigations in vitro and partly in vivo.
To determine the COX inhibition activity two separate in vitro assays, one fluorescence-based and one enzyme immunoassay-based, were used. This exceptional approach was essential because it is known that each individual assay can have its limitations regarding the chemical and physical properties of the subjected compounds. In our case we found that the indoles do not only show fluorescence, some of them possess a considerable redox activity which is known to interact with the fluorescence-based assay, and as a consequence false results may be occur. Indeed, for 3g that showed the highest redox activity (RDIENE = 2.85, RHAVA = 1.98) of the tested indoles and appeared in the fluorescence-based assay as a more potent COX-1 than COX-2 inhibitor, the immunoassay-based determination revealed that this compound was slightly more affine towards COX-2.
Summarizing the in vitro behavior of all 2,3-diaryl-substituted indoles it can be stated that all compounds are inhibiting COX-2, but with a slight gradation. Some of them turned out to be unselective COX inhibitors in one (3e, 3f) respectively both (3g, 3h) of the assays and are hence unsuitable candidates for radiotracer development. Surprisingly the introduction of a methyl-substituent to the 5-position of the indole lead to a remarkable loss of activity, hence the derivatives 3c and 3d have to be excluded as well. Since a high selectivity for COX-2 is an important criterion for potential radiotracers, 3a, 3b, 3j, 3l and 3m emerged as most promising candidates in the following order of increasing inhibitory activity based on 0.1 μM concentrations: 3b ≤ 3l < 3a ≤ 3j < 3m.
The radiolabeled COX-2 inhibitor [18F]3a corresponding to the non-radioactive compound 3a has been recently evaluated by ourselves and showed no substantial tumor accumulation, due to fast elimination and insufficient affinity to COX-2.30 The COX-2 inhibitors 3b and 3l both have a fluoro-substituent making them to suitable candidates for radiotracer development with fluorine-18. However, they possess an aminosulfonyl group that is known to interact in vivo with carbonic anhydrases in the erythrocytes, resulting in a slow blood clearance. This was demonstrated for 125I-radiolabeled celecoxib derivatives56 and should be kept in mind for the development of a 18F-radiolabeled analogue of 3b and 3l. The derivative 3j does not contain a fluorine substituent. Hence, radiolabeling with carbon-11 could be a possible approach as demonstrated by Tanaka et al.57 but the low metabolic stability of the methoxy group in vivo is always limiting the radiotracer application.
Finally, 3m turned out as the most promising candidate for radiotracer development since this compound was potent and COX-2 selective in both assays showing furthermore in the EIA-based method a complete inhibition of COX-2 in the 0.1 μM level. The radiolabeling of 3m with fluorine-18 can be performed by using the McMurry cyclization30 as has been demonstrated for [18F]3a because the Bischler–Möhlau reaction as labeling strategy seems to be ineffective for the introduction of the radioisotope.
Experimental protocols
All commercial reagents and solvents were used without further purification unless otherwise specified. Flash chromatography was conducted using MERCK Silica Gel (mesh size 40–63 μm) and RP-18 silica gel (Lichroprep RP-18, 25–40 μm). Thin-layer chromatography (TLC) was performed on Merck silica gel F-254 aluminum plates and Alugram RP-18W/UV254. Visualization was carried out using UV (254 nm/366 nm). Analytical HPLC analysis were carried out with a Luna C18 column (250 × 4.6 mm, 5 μm, Phenomenex) using an isocratic eluent (acetonitrile/water + 0.1% TFA 70/30) by a gradient pump L2500 (Merck, Hitachi) with a flow rate of 1 mL min−1. The products were monitored by an UV detector L4500 (Merck, Hitachi) at 254 nm. All final compounds displayed ≥95% purity as determined by analytical HPLC. Mass spectra were obtained on a Quattro/LC mass spectrometer (MICROMASS) by electrospray ionization. Melting points were determined on a Galen III (Cambridge Instruments) melting points apparatus (Leica, Vienna, Austria) and are uncorrected. Nuclear magnetic resonance spectra were recorded on a Varian Inova-400. NMR spectra were referenced to the residual solvent shifts for 1H and 13C, and to CFCl3 for 19F spectra as internal standard. For some compounds, isotope effects on 13C NMR chemical shifts58 were observed in acetone-d6 which were caused by hydrogen/deuterium exchange while measurement. The signals with deuterium isotope shifts are indicated and the range from minimal to maximal value is given.Spectroscopic and fluorescent properties were determined at the “Synergy 4 Hybrid Multi-Mode Microplate Reader” from BioTek Company. The samples were dissolved in an appropriate amount of DMSO to give a 10 mM stock solution. To 20 μL of this stock solution was added 80 μL DMSO, 100 μL TWEEN 20 and 9800 μL PBS to yield a 20 μM test solution for the measurement of the extinction coefficient. Using BioCell 1 cm Quartz Vessels from BioTec Company, the extinction coefficients were determined from the absorption spectra. All extinction coefficients are given in ε/dm3 mol−1 cm−1 at the specified wavelength in nm. The fluorescence excitation and emission spectra were determined using a 100 μM solution of the test compound in a solution of 1% DMSO and 1% TWEEN 20 in PBS. In both experiments, baseline correction was done using a solution of 1% DMSO and 1% TWEEN 20 in PBS.
Syntheses
:10 → 50:50). Compound 1c was obtained as a yellow-brown solid (854 mg, 23%). mp 150–153 °C; Rf 0.51 (petroleum ether–ethyl acetate 50:50); 1H NMR (400 MHz; DMSO-d6) δ ppm: 6.50 (2H, br s, NH2), 6.68 (1H, d, 4J = 2.6 Hz, Hphenyl-6), 6.74 (1H, d, 3J = 8.8 Hz, Hphenyl-3), 6.86 (1H, dd, 3J = 8.8 Hz, 4J = 2.7 Hz, Hphenyl-4), 7.34 (2H, t, 3J = 8.8 Hz, HF-phenyl-3′/5′), 7.64 (2H, dd, 3J = 8.8 Hz, 4J = 5.7 Hz, HF-phenyl-2′/6′), 8.65 (1H, s, OH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 115.2 (d, 2J = 22 Hz), 116.5, 117.2, 118.2, 124.0, 131.2 (d, 3J = 9 Hz), 136.5 (d, 4J = 3 Hz), 145.3, 146.0, 163.5 (d, 1J = 249 Hz), 196.1; 19F NMR (376 MHz; DMSO-d6) δ ppm: −109.7; m/z ESI-MS (ES+) 232 [M + H]+ (100%).:70); 1H NMR (400 MHz; DMSO-d6) δ ppm: 3.83 (3H, s, OCH3), 6.51 (1H, t, 3J = 7.4 Hz, Harom), 6.79–6.86 (3H, m, NH2/Harom), 7.05 (2H, d, 3J 8.6 Hz, Hanisole), 7.22–7.32 (2H, m, Harom), 7.58 (2H, d, 3J = 8.6 Hz, Hanisole); 13C NMR (101 MHz; DMSO-d6) δ ppm: 55.4 (CH3O), 113.5, 114.1, 116.8, 117.2, 131.3, 131.9, 133.3, 133.6, 151.3, 161.8, 196.6 (CO).General procedure A for the synthesis of N-(2-benzoylphenyl)benzamides (2a–2g)
Under nitrogen atmosphere, to a solution of the appropriate 2-aminobenzophenone (0.87 mmol) and NEt3 (138 μL) in THF (1.8 mL) was added the appropriate 4-sulfonylbenzoyl chloride (0.87 mmol) in THF (1.0 mL). The mixture was stirred at room temperature for 2 h, filtered and the solid was washed with tetrahydrofuran. The filtrate was concentrated to dryness under reduced pressure and further purified as described below.:50). Starting from 1a (258 mg, 1.20 mmol) and 4-sulfamoylbenzoyl chloride (262 mg, 1.20 mmol), 2b was obtained as pale yellow solid (300 mg, 62%). mp 202–203 °C (lit.,11 204–206 °C); Rf 0.36 (petroleum ether–ethyl acetate 50/50); 1H NMR (400 MHz; CD3CN) δ ppm: 5.78 (2H, br s, SO2NH2), 7.20–7.30 (3H, m, 3J = 8.9 Hz, 3J = 7.9 Hz, Hphenyl), 7.63 (1H, dd, 3J = 7.9 Hz, 4J = 1.5 Hz, Hphenyl), 7.70 (1H, t, 3J = 7.9 Hz, 4J = 1.7 Hz, Hphenyl), 7.80 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6), 8.00 (2H, d, 3J = 8.6 Hz, Hphenyl), 8.05 (2H, d, 3J = 8.8 Hz, Hphenyl), 8.50 (1H, dd, 3J = 8.3 Hz, 4J = 0.9 Hz, Hphenyl), 11.15 (1H, br s, NH); 1H NMR (400 MHz; DMSO-d6) δ ppm: 7.30 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.37 (1H, t, 3J = 7.8 Hz, 3J = 6.8 Hz, 4J = 1.8 Hz, Hphenyl), 7.47–7.54 (3H, m, Hphenyl/SO2NH2), 7.63–7.71 (2H, m, Hphenyl), 7.76 (2H, dd, 3J 8.8 Hz, 4J = 5.6 Hz, HF-phenyl-2/6), 7.81 (2H, d, 3J = 8.7 Hz, Hphenyl), 7.88 (2H, d, 3J = 8.6 Hz, Hphenyl), 10.75 (1H, br s, NH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 115.3 (d, 2J = 22 Hz), 124.5, 125.0, 125.6, 128.0, 130.1, 131.4, 132.0, 132.4 (d, 3J = 9 Hz), 133.8 (d, 4J = 3 Hz), 136.1, 136.9, 146.7, 164.4, 164.6 (d, 1J = 251 Hz), 193.8; 19F NMR (376 MHz; DMSO-d6) δ ppm: −107.2; ESI-MS (ES+) m/z 421 [M + Na]+ (100%), 399 [M + H]+ (69), 462 [M + Na + CH3CN]+ (69).:30:5 → 47.5:47.5:5). Starting from 1b (315 mg, 1.37 mmol) and 4-(methylsulfonyl)benzoyl chloride (300 mg, 1.37 mmol), 2c was obtained as pale yellow crystalline solid (540 mg, 96%). mp 195–197 °C; Rf 0.40 (petroleum ether–ethyl acetate 50:50); 1H NMR (400 MHz; acetone-d6) δ ppm: 2.36 (3H, s, CH3), 3.19 (3H, s, SO2CH3), 7.31 (2H, t, 3J = 8.8 Hz, HF-phenyl-3/5), 7.48 (1H, d, 4J = 1.7 Hz, Hphenyl), 7.54 (1H, dd, 3J = 8.4 Hz, 4J = 2.1 Hz, Hphenyl), 7.87 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6), 8.12 (2H, d, 3J = 8.4 Hz, Hphenyl), 8.16 (2H, d, 3J = 8.5 Hz, Hphenyl), 8.44 (1H, dd, 3J = 8.4 Hz, 4J = 3.8 Hz, Hphenyl), 11.29 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 20.8, 44.1, 116.2 (d, 2J = 22 Hz), 123.0*, 126.7*, 128.7, 129.0, 133.6 (d, 3J = 9 Hz), 133.8*, 134.1*, 135.2*, 135.8 (d, 4J = 3 Hz)*, 137.9*, 140.3*, 145.1*, 164.6*, 166.1 (d, 1J = 252 Hz), 198.1*, * deuterium isotope shifts were observed in the range of 7 to 112 ppb; 19F NMR (376 MHz; CD3CN) δ ppm: −108.5; ESI-MS (ES−) m/z 410 ([M − H]−, 100%). Crystals suitable for X-ray crystallography were obtained by slow evaporation from ethyl acetate at room temperature. Detailed results of the single-crystal X-ray structure determination are given below and in the ESI.‡:50). Starting from 1b (204 mg, 0.89 mmol) and 4-sulfamoylbenzoyl chloride (194 mg, 0.89 mmol), 2d was obtained as colorless crystalline solid (252 mg, 69%). mp 230–231 °C; Rf 0.40 (petroleum ether–ethyl acetate 50:50); 1H NMR (400 MHz; CD3CN) δ ppm: 2.35 (3H, s, CH3), 5.77 (2H, br s, SO2NH2), 7.24 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.43 (1H, d, 4J = 1.6 Hz, Hphenyl), 7.51 (1H, dd, 3J = 8.4 Hz, 4J = 1.7 Hz, Hphenyl), 7.80 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6), 7.96–8.04 (4H, m, 3J = 8.7 Hz, Hphenyl), 8.32 (1H, d, 3J = 8.4 Hz, Hphenyl), 10.91 (1H, br s, NH); 13C NMR (101 MHz; CD3CN) δ ppm: 21.4, 116.9 (d, 2J = 22 Hz), 123.8, 127.7, 128.1, 129.5, 134.3 (d, 3J = 9 Hz), 134.6, 135.2, 135.9, 136.4 (d, 4J = 3 Hz), 138.2, 139.8, 147.7, 165.6, 166.9 (d, 1J = 237 Hz), 199.1; 19F NMR (376 MHz; CD3CN) δ ppm: −108.6; ESI-MS (ES−) m/z 411 [M − H]− (100%).:50 → 0:100). Starting from 1c (250 mg, 1.08 mmol) and 4-sulfamoylbenzoyl chloride (224 mg, 1.02 mmol), 2e was obtained as yellow solid (275 mg, 65%). mp 248–250 °C; Rf 0.56 (ethyl acetate); 1H NMR (400 MHz; acetone-d6) δ ppm: 6.75 (2H, br s, SO2NH2), 7.07 (1H, 4J = 2.9 Hz, Hphenyl), 7.18 (1H, dd, 3J = 8.9 Hz, 4J = 2.9 Hz, Hphenyl), 7.30 (2H, t, 3J = 8.8 Hz, HF-phenyl-3/5), 7.87 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6), 7.99–8.05 (4H, m), 8.25 (1H, d, 3J = 8.9 Hz, Hphenyl), 8.68 (1H, s, OH), 10.86 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 116.1 (d, 2J = 22 Hz), 119.2, 121.1, 125.3*, 127.3*, 128.6, 129.1, 132.0*, 133.5 (d, 3J = 9 Hz), 135.6 (d, 4J = 3 Hz), 138.9*, 147.8*, 154.2, 164.6, 166.1 (d, 1J = 252 Hz), 197.3, * deuterium isotope shifts were observed in the range of 11 to 101 ppb; 19F NMR (376 MHz; acetone-d6) δ ppm: −108.5; ESI-MS (ES+) m/z 437 [M + Na]+ (100%), 478 [M + Na + CH3CN]+, (72).:50); 1H NMR (400 MHz; DMSO-d6) δ ppm: 3.26 (3H, s, SO2CH3), 3.81 (3H, s, OCH3), 7.01 (2H, d, 3J = 8.8 Hz, Hanisole), 7.36 (1H, t, 3J = 7.4 Hz, Harom), 7.48 (1H, d, 3J = 7.6 Hz, 4J = 1.2 Hz, Harom), 7.64 (1H, t, 3J = 8.0 Hz, 4J = 1.3 Hz, Harom), 7.69 (2H, d, 3J = 8.8 Hz, Hanisole), 7.73 (1H, d, 3J = 7.9 Hz, Harom), 7.90 (2H, d, 3J = 8.4 Hz, Harom), 8.01 (2H, d, 3J = 8.4 Hz, Harom), 10.77 (1H, s, NH); ESI-MS (ES+) m/z: 432 [M + Na]+ (100%).:70). Starting from 1d (600 mg, 2.64 mmol) and 4-sulfamoylbenzoyl chloride (640 mg, 2.92 mmol), 2g was obtained as pale yellow solid (853 mg, 78%). mp 162–166 °C (lit.,11 173–174 °C); 1H NMR (400 MHz; DMSO-d6) δ ppm: 3.81 (3H, s, OCH3), 7.01 (2H, d, 3J = 8.8 Hz, Hanisole), 7.35 (1H, t, 3J = 7.6 Hz, 4J = 1.0 Hz, Harom), 7.48 (1H, d, 3J = 7.7 Hz, 4J = 1.5 Hz, Harom), 7.51 (2H, s, NH2), 7.64 (1H, t, 3J = 7.8 Hz, 4J = 1.5 Hz, Harom), 7.69 (2H, d, 3J = 8.8 Hz, Hanisole), 7.74 (1H, d, 3J = 7.6 Hz, Harom), 7.82 (2H, d, 3J = 8.5 Hz, Harom), 7.88 (2H, d, 3J = 8.4 Hz, Harom), 10.71 (1H, s, NH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 55.6 (OCH3), 113.6, 124.6, 124.9, 125.7, 128.0, 129.8, 130.2, 131.7, 131.7, 132.0, 136.2, 137.2, 146.6, 162.9, 164.3 (CO), 194.1 (CO); ESI-MS (ES+) m/z: 433 [M + Na]+ (100%).General procedure B for the synthesis of 2-(4-sulfonylphenyl)-3-phenyl-1H-indoles (3a–3g)
Under nitrogen atmosphere, TiCl4 (146.2 μL, 253 mg, 1.3 mmol) was added to a suspension of the appropriate benzamide (0.62 mmol) and Zn (170 mg, 2.5 mmol) in THF (5.5 mL). The resulting mixture was heated to 65 °C for 1.5 h. The solvent was removed and the mixture was purified as described below.400, 20300, 16100); fluorescence: λexc = 343, λem = 454 nm. Crystals suitable for X-ray crystallography were obtained by slow evaporation from ethyl acetate at room temperature. Detailed results of the single-crystal X-ray structure determination are given below and in the ESI.‡:50). Starting from 2b (220 mg, 0.54 mmol), 3b was obtained as colorless solid (168 mg, 85%). mp 238–239 °C (lit.,11 228–230 °C); Rf 0.26 (petroleum ether–ethyl acetate 50:50); UV/vis: λmax/nm 253, 328 (ε/dm3 mol−1 cm−1 20800, 17000); fluorescence: λexc = 338, λem = 450 nm; 1H NMR (400 MHz; CD3CN) δ ppm: 5.68 (2H, br s, SO2NH2), 7.13 (1H, t, 3J = 8.0 Hz, 3J = 7.1 Hz, Hindol), 7.18 (2H, t, 3J = 9.0 Hz, HF-phenyl-3/5), 7.26 (1H, t, 3J = 8.3 Hz, 3J = 7.1 Hz, 4J 1.1 Hz, Hindol), 7.39 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6), 7.49–7.54 (2H, m, Hindol), 7.59 (2H, d, 3J = 8.7 Hz, Hphenyl), 7.81 (2H, d, 3J = 8.7 Hz, Hphenyl), 9.78 (1H, br s, NH); 13C NMR (101 MHz; CD3CN) δ ppm: 113.1, 116.4, 117.1 (d, 2J = 22 Hz), 120.7, 122.1, 124.8, 127.9, 130.0, 130.1, 132.7 (d, 4J = 3 Hz), 133.5 (d, 3J = 8 Hz), 134.1, 138.0, 138.1, 143.5, 163.4 (d, 1J = 243 Hz); 19F NMR (376 MHz; CD3CN) δ ppm: −118.0; ESI-MS (ES−) m/z 365 [M − H]− (100%).:10). Starting from 2c (400 mg, 0.97 mmol), 3c was obtained as beige solid (221 mg, 60%). mp 239–242 °C; Rf 0.72 (chloroform–methanol 90:10); UV/vis: λmax/nm 239, 339 (ε/dm3 mol−1 cm−1 56200, 20300); fluorescence: λexc = 345, λem = 476 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 2.40 (3H, s, CH3), 3.13 (3H, s, SO2CH3), 7.08 (1H, dd, 3J = 8.4 Hz, 4J = 1.4 Hz, Hindol), 7.21 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.32 (1H, d, 4J = 0.6 Hz, Hindol), 7.38–7.45 (3H, m, 3J = 8.4 Hz, 3J = 8.8 Hz, 4J = 5.5 Hz, 2HF-phenyl/1Hindol), 7.71 (2H, d, 3J = 8.6 Hz, Hphenyl), 7.89 (2H, d, 3J = 8.6 Hz, Hphenyl), 10.72 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 21.6, 44.2, 112.2*, 115.8*, 116.5 (d, 2J = 21 Hz), 119.6, 125.8, 128.4, 129.3*, 129.8, 130.3, 132.3 (d, 4J = 3 Hz), 132.8 (d, 3J = 8 Hz), 133.2*, 136.2*, 138.8*, 140.6, 162.6 (d, 1J = 246 Hz), * deuterium isotope shifts were observed in the range of 36 to 143 ppb; 19F NMR (376 MHz; acetone-d6) δ ppm: −117.9; ESI-MS (ES−) m/z 378 [M − H]− (100%).:10; (2) petroleum ether–ethyl acetate 50:50) and preparative HPLC (RP-18, CH3CN–H2O (with 0.1% TFA) 50:50 → 70:30). Starting from 2d (220 mg, 0.54 mmol), 3d was obtained as colorless solid (44 mg, 21%). mp 242–244 °C; Rf 0.38 (petroleum ether–ethyl acetate 50:50); UV/vis: λmax/nm 238, 333 (ε/dm3 mol−1 cm−1 40600, 21000); fluorescence: λexc = 339, λem = 451 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 2.39 (3H, s, CH3), 6.60 (2H, br s, SO2NH2), 7.07 (1H, dd, 3J = 8.3 Hz, 4J = 1.4 Hz, Hindol), 7.21 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.33 (1H, d, 4J = 0.6 Hz, Hindol), 7.37–7.45 (3H, m, 3J = 8.9 Hz, 3J = 8.3 Hz, 4J = 5.5 Hz, 2HF-phenyl/1Hindol), 7.63 (2H, d, 3J = 8.6 Hz, Hphenyl), 7.84 (2H, d, 3J = 8.6 Hz, Hphenyl), 10.66 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 21.6, 112.1*, 115.2*, 116.4 (d, 2J = 21 Hz), 119.5, 125.6, 127.2, 129.1, 129.8*, 130.1, 132.5 (d, 4J = 3 Hz), 132.8 (d, 3J = 8 Hz), 133.6, 136.1*, 137.2*, 143.6*, 162.6 (d, 1J = 244 Hz), * deuterium isotope shifts were observed in the range of 33 to 144 ppb; 19F NMR (376 MHz; acetone-d6) δ ppm: −118.1; ESI-MS (MS−) m/z 379 [M − H]− (100%).:10). Starting from 2e (240 mg, 0.58 mmol), 3e was obtained as colorless solid (119 mg, 54%). mp 244–247 °C; Rf 0.19 (petroleum ether–ethyl acetate 50:50); UV/vis: λmax/nm 236, 341 (ε/dm3 mol−1 cm−1 43300, 20500); fluorescence: λexc = 345, λem = 481 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 6.60 (2H, br s, SO2NH2), 6.83 (1H, dd, 3J = 8.7 Hz, 4J = 2.3 Hz, Hindole-6), 6.95 (1H, d, 4J = 2.3 Hz, Hindole-4), 7.20 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.34 (1H, d, 3J = 8.6 Hz, Hindole-7), 7.40 (2H, dd, 3J = 8.8 Hz, 4J = 5.5 Hz, HF-phenyl-2/6), 7.61 (2H, d, 3J = 8.7 Hz, Hphenyl), 7.78 (1H, br s, OH); 7.83 (2H, d, 3J = 8.7 Hz, Hphenyl), 10.54 (1H, br s, NH); 13C NMR (101 MHz; CD3CN) δ ppm: 103.6*, 113.0*, 114.3*, 114.9*, 116.3 (d, 2J = 21 Hz), 127.2, 129.0, 130.3*, 132.4*, 132.6, 132.7 (d, 3J = 8 Hz), 134.1*, 137.3 (d, 4J = 4 Hz)*, 143.5*, 152.8*, 162.5 (d, 1J = 244 Hz), *deuterium isotope shifts were observed in the range of 12 to 140 ppb; 19F NMR (376 MHz; acetone-d6) δ ppm: 118.2; ESI-MS (MS+) m/z 383 [M + H]+ (100%).:70). Starting from 2f (360 mg, 0.88 mmol), 3f was obtained as a pale yellow solid (182 mg, 55%). mp 222–226 °C (lit.,11 218.5–220.5 °C); Rf 0.47 (petroleum ether–ethyl acetate 30:70); UV/vis: λmax/nm 237, 256, 332 (ε/dm3 mol−1 cm−1 28700, 25400, 16000); fluorescence: λexc = 354, λem = 492 nm; 1H NMR (400 MHz; DMSO-d6) δ ppm: 3.25 (3H, s, SO2CH3), 3.80 (3H, s, OCH3), 7.02 (2H, d, 3J = 8.8 Hz, Hanisole), 7.06 (1H, t, 3J = 8.0 Hz, 3J = 7.0 Hz, 4J = 0.9 Hz, Harom), 7.21 (1H, t, 3J = 8.1 Hz, 3J = 7.1 Hz, 4J = 1.1 Hz, Harom), 7.28 (2H, d, 3J = 8.7 Hz, Hanisole), 7.45 (1H, d, 3J = 8.0 Hz, Harom), 7.48 (1H, d, 3J = 8.1 Hz, Harom), 7.69 (2H, d, 3J = 8.7 Hz, Harom), 7.90 (2H, d, 3J = 8.6 Hz, Harom), 11.70 (1H, s, NH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 43.4, 55.0, 111.7, 114.4, 115.3, 119.1, 119.9, 122.8, 126.6, 127.1, 128.2*, 130.9, 131.5, 136.4, 137.5, 139.0, 158.0, *two carbon species with equivalent chemical shift; ESI-MS (ES+) m/z 400 [M + Na]+ (100%).:80). Starting from 2g (300 mg, 0.73 mmol), 3g was obtained as pale yellow solid (184 mg, 48%). mp 282–285 °C (lit.,11 280–282 °C), Rf 0.51 (petroleum ether–ethyl acetate 20:80); UV/vis: λmax/nm 237, 325 (ε/dm3 mol−1 cm−1 37100, 16100); fluorescence: λexc = 345, λem = 482 nm; 1H NMR (400 MHz; DMSO-d6) δ ppm: 3.80 (3H, s, OCH3), 6.79–7.08 (3H, m, 3J = 7.7 Hz, Hanisole/Harom.), 7.19 (1H, t, 3J = 7.9 Hz, Harom), 7.27 (2H, d, 3J = 7.9 Hz, Hanisole), 7.39 (2H, s, NH2), 7.43–7.49 (2H, m, Harom), 7.61 (2H, d, 3J = 8.2 Hz, Harom), 7.79 (2H, d, 3J 8.0, Harom), 11.64 (1H, s, NH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 55.1, 111.6, 114.4, 114.6, 119.0, 119.9, 122.6, 125.9, 126.8, 128.1*, 130.9, 132.0, 135.9, 136.3, 142.4, 158.0, *two carbon species with equivalent chemical shift; ESI-MS (ES+) m/z 401 [M + Na]+ (80%).:80). 3h was obtained as pale green powder (176 mg, 70%). mp 96–100 °C; Rf 0.32 (petroleum ether–ethyl acetate 20:80); UV/vis: λmax/nm 256, 323 (ε/dm3 mol−1 cm−1 29800, 18000); fluorescence: λexc = 339, λem = 452 nm; 1H NMR (400 MHz; DMSO-d6) δ ppm: 1.94 (3H, s, COCH3), 3.80 (3H, s, OCH3), 7.02 (2H, d, 3J = 8.8 Hz, Hanisole), 7.06 (1H, t, 3J = 8.0 Hz, 3J = 7.1 Hz, 4J = 0.8 Hz, Harom), 7.20 (1H, t, 3J = 8.2 Hz, 3J = 7.1 Hz, 4J = 1.1 Hz, Harom), 7.27 (2H, d, 3J = 8.7 Hz, Hanisole), 7.42–7.48 (2H, m, 3J = 8.2 Hz, Harom), 7.66 (2H, d, 3J = 8.6 Hz, Harom), 7.86 (2H, d, 3J = 8.6 Hz, Harom), 11.68 (1H, s, NH), 12.13 (1H, s, NH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 23.3, 55.1, 111.7, 114.5, 115.3, 119.2, 120.0, 122.9, 126.6, 127.8, 128.1, 128.2, 130.9, 131.6, 136.5, 137.4, 137.6, 158.1, 168.9; ESI-MS (ES+) m/z 443 [M + Na]+ (100%).:15) as a colorless solid (2.47 g, 51%). mp 80–82 °C (lit.,38 83 °C); Rf 0.36 (petroleum ether–ethyl acetate 85:15); 1H NMR (400 MHz; CD3CN) δ ppm: 4.32 (2H, s, CH2), 7.18–7.36 (7H, m, Hphenyl), 8.08 (2H, dd, 3J = 9.0 Hz, 4J = 5.5 Hz, HF-phenyl-2/6); 13C NMR (101 MHz; CD3CN) δ ppm: 45.9, 116.6 (d, 2J = 22 Hz), 127.7, 129.4, 130.7, 132.2 (d, 3J = 10 Hz), 134.4 (d, 4J = 3 Hz), 136.1, 166.6 (d, 1J = 252 Hz), 197.3; 19F NMR (376 MHz; CD3CN) δ ppm: −108.0; ESI-MS (ES+) m/z 215 [M + H]+ (100%).General procedure C for the synthesis of 2-[4-(methylsulfonyl)phenyl]-1-phenyl-ethanones 5a and 5b
The reaction was carried out under Schlenk-conditions. A suspension of 4-(methylsulfonyl)phenylacetic acid (1.714 g, 8 mmol) in dichloromethane (1.6 mL) and 4 drops of DMF was heated to 30 °C. Freshly distilled thionyl chloride (1.00 g, 0.60 mL, 8.48 mmol) was added and the mixture was stirred for 1.5 h at constant temperature. The solution was cooled to 15 °C and freshly sublimated AlCl3 (2.02 g, 15.15 mmol) was added in portions. After 15 min the appropriate benzene (9.66 mmol) was dropped to the mixture and it was stirred for 2 h. Then ethanol (2.8 mL) was added dropwise at 10 °C, followed by dichloromethane (20.6 mL) and water (10.2 mL) over a period of 20 min. The mixture was heated to 30 °C and the organic phase was separated, washed with 5 M HCl (6 mL) and saturated Na2CO3 solution (6 mL), and concentrated by distillation to a volume of 5 mL. Further purification was carried out as described below.:1 to obtain a further amount of 5a. Starting from 4-(methylsulfonyl)phenylacetic acid (428 mg, 2 mmol) and 4-ethoxybenzene (0.304 mL, 295 mg, 2.41 mmol), 5a was obtained as colorless solid (192 mg, 30%). mp 188–190 °C from ethanol (lit.,60 156–159 °C); Rf 0.41 (petroleum ether–ethyl acetate 50:50); 1H NMR (400 MHz; CDCl3) δ ppm: 1.45 (3H, t, 3J = 7.0 Hz, OCH2CH3), 3.04 (3H, s, SO2CH3), 4.11 (2H, q, 3J = 7.0 Hz, OCH2CH3), 4.34 (2H, s, CH2), 6.94 (2H, d, 3J = 8.9 Hz, Hphenyl), 7.46 (2H, d, 3J = 8.4 Hz, Hphenyl), 7.90 (2H, d, 3J = 8.4 Hz, Hphenyl), 7.98 (2H, d, 3J = 8.9 Hz, Hphenyl); 13C NMR (101 MHz; CDCl3) δ ppm: 14.8, 44.7, 44.9, 64.0, 114.6, 127.8, 129.2, 130.8, 131.0, 139.2, 141.5, 163.5, 194.9; ESI-MS (MS+) m/z 341 [M + Na]+ (100%), 319 [M + H]+ (53), 659 (24).:2) to yield 402 mg of compound 5b. Starting from 4-(methylsulfonyl)phenylacetic acid (1.714 g, 8 mmol) and 4-methoxybenzene (1.055 mL, 1.044 g, 9.66 mmol), 5b was obtained as colorless solid (1.24 g, 51%). mp 172–174 °C; Rf 0.34 (DCM–acetone 98:2); 1H NMR (400 MHz; CDCl3) δ ppm: 3.04 (3H, s, SO2CH3), 3.88 (3H, s, OCH3), 4.34 (2H, s, CH2), 6.96 (2H, d, 3J = 8.9 Hz, Hphenyl), 7.46 (2H, d, 3J = 8.4 Hz, Hphenyl), 7.90 (2H, d, 3J = 8.4 Hz, Hphenyl), 7.99 (2H, d, 3J = 8.9 Hz, Hphenyl); 13C NMR (101 MHz; CDCl3) δ ppm: 44.7, 44.9, 55.7, 114.2, 127.8, 129.4, 130.8, 131.0, 139.2, 141.5, 164.1, 194.9; ESI-MS (MS+) m/z 631 [2M + Na]+ (73%), 368 [M + Na + CH3CN]+ (100), 327 (56), 305 [M + H]+ (72).:50); 1H NMR (400 MHz; acetone-d6) δ ppm: 3.12 (3H, s, CH3), 4.59 (2H, s, CH2), 7.31 (2H, t, 3J = 8.8 Hz, HF-phenyl-3/5), 7.59 (2H, d, 3J = 8.3 Hz, Hphenyl), 7.91 (2H, d, 3J = 8.3 Hz, Hphenyl), 8.20 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6); 13C NMR (101 MHz; acetone-d6) δ ppm: 44.4, 45.3, 116.5 (d, 2J = 22 Hz), 128.1, 131.7, 132.2 (d, 3J = 9 Hz), 134.3 (d, 4J = 3 Hz), 140.8, 142.3, 166.6 (d, 1J = 252 Hz), 195.7; 19F NMR (376 MHz; acetone-d6) δ ppm: −107.7; ESI-MS (MS+) m/z 315 [M + Na]+ (100%). Single crystals were obtained as follows: 5c was crystallized from a mixture of acetone and acetonitrile saturated at the boiling heat which was slowly cooled down to room temperature overnight. This formed a small amount of tiny crystals. Then, the solution was allowed to evaporate slowly what generated crystals suitable for X-ray crystallography. Detailed results of the single-crystal X-ray structure determination are given below and in the ESI.‡:50); 1H NMR (400 MHz; DMSO-d6) δ ppm: 4.53 (2H, s, CH2), 7.32 (2H, br s, SO2NH2), 7.38 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.44 (2H, d, 3J = 8.3 Hz, Hphenyl), 7.78 (2H, d, 3J = 8.3 Hz, Hphenyl), 8.14 (2H, dd, 3J = 8.9 Hz, 4J = 5.5 Hz, HF-phenyl-2/6); 13C NMR (101 MHz; DMSO-d6) δ ppm: 44.3, 115.8 (d, 3J = 22 Hz), 125.6, 130.4, 131.3 (d, 3J = 10 Hz), 133.0 (d, 3J = 3 Hz), 139.2, 142.4, 165.1 (d, 3J = 252 Hz), 195.7; 19F NMR (376 MHz; DMSO-d6) δ ppm: −106.1; ESI-MS (MS+) m/z 316 [M + Na]+ (100%).:5). In this way, 5e was obtained as a yellow solid (1.009 g, 52%). mp 134–141 °C (lit.,47 140–141 °C); Rf 0.44 (ethyl acetate–petroleum ether 5:5); 1H NMR (400 MHz; acetone-d6) δ ppm: 3.15 (3H, s, SO2CH3), 7.03 (1H, s, CHBr), 7.33 (2H, t, 3J = 8.8 Hz, HF-phenyl-3/5), 7.91 (2H, d, 3J = 8.5 Hz, HSO2-phenyl), 7.99 (2H, d, 3J = 8.6 Hz, HSO2-phenyl), 8.25 (2H, dd, 3J = 9.0 Hz, 4J = 5.4 Hz, HF-phenyl-2/6); 13C NMR (101 MHz; acetone-d6) δ ppm: 44.2, 48.9, 116.9 (d, 2J = 22 Hz), 128.5, 131.4, 131.6 (d, 4J = 3 Hz), 133.1 (d, 3J = 10 Hz), 142.7, 142.8, 166.9 (d, 1J = 254 Hz), 190.3; 19F NMR (376 MHz; acetone-d6) δ ppm: −106.1.General procedure D for the synthesis of 3-(4-sulfonylphenyl)-2-phenyl-1H-indoles (3i–3l)
A mixture of phenylhydrazine (66 μL, 73 mg, 0.68 mmol) and the appropriate 1,2-diphenyl ethanone (0.68 mmol) was heated to 130 °C for 30 min. After cooling to room temperature, acetic acid (4.13 mL) and BF3·Et2O (36 μL, 40 mg, 0.29 mmol) were added and the mixture was heated to reflux for 1 h. Then, acetic acid was removed by distillation under reduced pressure. The resulting solid was suspended in water (3 mL), filtered, washed with a small amount of water and dried. The mixture was further purified as described below.:40). Starting from 5a (216 mg, 0.68 mmol), 3i was obtained as pale yellow solid (28 mg, 10%). mp 212–214 °C; Rf 0.56 (petroleum ether–ethyl acetate 50:50); UV/vis: λmax/nm 238, 300, 340 (ε/dm3 mol−1 cm−1 19700, 17300, 9300); fluorescence: λexc = 303, λem = 450 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 1.38 (3H, t, 3J = 7.0 Hz, CH2CH3), 3.15 (3H, s, SO2CH3), 4.08 (2H, q, 3J = 7.0 Hz, CH2CH3), 6.94 (2H, d, 3J = 8.8 Hz, Hphenyl), 7.12 (1H, t, 3J = 8.0 Hz, 3J = 7.1 Hz, 4J = 1.0 Hz, Hindol), 7.20 (1H, t, 3J = 8.1 Hz, 3J = 7.1 Hz, 4J = 1.1 Hz, Hindol), 7.42 (2H, d, 3J = 8.9 Hz, Hphenyl), 7.50 (1H, d, 3J = 8.1 Hz, Hindol), 7.63–7.69 (3H, m, 3J = 8.5 Hz, 2Hphenyl/1Hindol), 7.94 (2H, d, 3J = 8.5 Hz, Hphenyl), 10.71 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 15.1, 44.5, 64.1, 112.3*, 112.3*, 115.6, 119.2, 121.2, 123.1, 125.3*, 128.4, 128.9*, 130.8, 131.2, 136.9*, 137.4*, 139.2, 142.7, 160.1, *deuterium isotope shifts were observed in the range of 37 to 146 ppb; ESI-MS (APcI−) m/z 390 [M − H]− (100%).:40). Starting from 5b (206 mg, 0.68 mmol), 3j was obtained as pale yellow solid (60 mg, 23%). mp 241–243 °C; Rf 0.40 (petroleum ether–ethyl acetate 50:50); UV/vis: λmax/nm 238, 300, 344 (ε/dm3 mol−1 cm−1 31800, 23500, 12300); fluorescence: λexc = 344, λem = 450 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 3.15 (3H, s, SO2CH3), 3.83 (3H, s, OCH3), 6.96 (2H, d, 3J = 8.7 Hz, Hphenyl), 7.12 (1H, t, 3J = 7.5 Hz, Hindol), 7.20 (1H, t, 3J = 7.6 Hz, 4J = 0.9 Hz, Hindol), 7.43 (2H, d, 3J = 8.7 Hz, Hphenyl), 7.50 (1H, d, 3J = 8.1 Hz, Hindol), 7.59–7.69 (3H, m, 3J = 8.3 Hz, 2Hphenyl/1Hindole), 7.94 (2H, d, 3J = 8.3 Hz, Hphenyl), 10.71 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 44.5, 55.6, 112.3*, 112.3*, 115.1, 119.2, 121.2, 123.1, 125.5*, 128.4, 128.9*, 130.8, 131.2, 136.8*, 137.4*, 139.2, 142.6, 160.7, *deuterium isotope shifts were observed in the range of 38 to 145 ppb; m/z (ESI+) 378 [M + H]+ (100%). 5d was also synthesized in a two-step/one-pot procedure including aminolysis and McMurry reaction and purified by column chromatography ((1) petroleum ether–ethyl acetate 50:50 → 0:100, (2) petroleum ether–ethyl acetate 50:50) as previously described.39 Starting from 2-amino-4′-(methylsulfonyl)benzophenone (500 mg, 1.82 mmol) and p-methoxybenzoyl chloride, 5d was obtained as colorless solid having the same spectroscopic properties as described above (218 mg, 32%). From this batch, crystals suitable for X-ray crystallography were obtained from a solution of 3j in ethyl acetate–petroleum ether 50:50 by slow evaporation at room temperature. Detailed results of the single-crystal X-ray structure determination are given below and in the ESI.‡:50) and preparative HPLC (RP-18, CH3CN–H2O (with 0.1% TFA) 50:50 → 70:30). Starting from 5c (300 mg, 1.03 mmol), 3k was obtained as pale yellow solid (73 mg, 19%). mp 234–235 °C; Rf 0.57 (petroleum ether–ethyl acetate 50:50); UV/vis: λmax/nm 238, 296, 337 (ε/dm3 mol−1 cm−1 44700, 21700, 12700); fluorescence: λexc = 300, λem = 444 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 3.15 (3H, s, SO2CH3), 7.09–7.20 (3H, m, 3J = 8.9 Hz, 3J = 7.5 Hz, 4J = 1, 2Hphenyl/1Hindol), 7.23 (1H, t, 3J = 7.6 Hz, 4J = 1.0 Hz, Hindol), 7.46–7.58 (3H, m, 3J = 8.9 Hz, 3J = 7.9 Hz, 4J = 5.5 Hz, 2Hphenyl/1Hindol), 7.63–7.68 (3H, m, 3J = 8.4 Hz, 3J = 7.8 Hz, 2Hphenyl/1Hindol), 7.95 (2H, d, 3J = 8.4 Hz, Hphenyl), 10.83 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 44.4, 112.5, 113.3, 116.5 (d, 2J = 22 Hz), 119.5, 121.4, 123.5, 128.5, 128.7, 129.7 (d, 4J = 3 Hz), 131.7 (d, 3J = 8 Hz), 131.2, 135.7, 137.5, 139.5, 142.1, 163.4 (d, 1J = 246 Hz); 19F NMR (376 MHz; acetone-d6) δ ppm: −115.3; ESI-MS (ES−) m/z 364 [M − H]− (100%).:30) and preparative HPLC (RP-18, CH3CN–H2O with 0.1% TFA, 50:50 → 70:30). Starting from 5d (200 mg, 0.68 mmol), 3l was obtained as colorless solid (70 mg, 28%). mp 162–163 °C; Rf 0.23 (petroleum ether–ethyl acetate 70:30); UV/vis: λmax/nm 237, 296, 329 (ε/dm3 mol−1 cm−1 38500, 20600, 13600); fluorescence: λexc = 303, λem = 443 nm; 1H NMR (400 MHz; acetone-d6) δ ppm: 6.59 (2H, br s, SO2NH2), 7.10–7.20 (3H, m, 3J = 8.9 Hz, 3J = 7.5 Hz, 2Hphenyl/1Hindol), 7.22 (1H, dd, 3J = 7.6 Hz, 3J = 8.0 Hz, Hindol), 7.49–7.59 (5H, m, 3J = 8.9 Hz, 3J = 8.5 Hz, 3J = 8.2 Hz, 4J = 5.5 Hz, 4Hphenyl/1Hindol), 7.64 (1H, d, 3J = 8.0 Hz, Hindol), 7.91 (2H, d, 3J = 8.6 Hz, Hphenyl), 10.78 (1H, br s, NH); 13C NMR (101 MHz; acetone-d6) δ ppm: 112.4, 113.5, 116.5 (d, 2J = 22 Hz), 119.6, 121.3, 123.5, 127.3, 128.9, 129.8 (d, 4J = 3 Hz), 130.9, 131.6 (d, 3J = 8 Hz), 135.3, 137.5, 140.4, 142.5, 163.3 (d, 1J = 246 Hz); 19F NMR (376 MHz; acetone-d6) δ ppm: −115.5; ESI-MS (ES−) m/z: 365 [M − H]− (100%).:5); UV/vis: λmax/nm 237, 344 (ε/dm3 mol−1 cm−1 45200, 25400); fluorescence: λexc = 349, λem = 484 nm; 1H NMR (400 MHz; DMSO-d6) δ ppm: 3.24 (3H, s, SO2CH3), 3.73 (3H, s, OCH3), 6.82–6.94 (2H, m, Hindole), 7.28 (2H, t, 3J = 8.9 Hz, HF-phenyl-3/5), 7.33–7.44 (3H, m, HF-phenyl-2/6/Hindole), 7.63 (2H, d, 3J = 8.4 Hz, HSO2-phenyl), 7.89 (2H, d, 3J = 8.5 Hz, HSO2-phenyl), 11.65 (1H, s, NH); 13C NMR (101 MHz; DMSO-d6) δ ppm: 43.4, 55.3, 99.8, 112.6, 113.6, 114.1, 115.9 (d, 2J = 21 Hz), 127.2, 128.2, 128.3, 131.1 (d, 4J = 3 Hz), 131.7 (d, 3J = 8 Hz), 131.7, 132.6, 137.3, 139.1, 154.2, 161.0 (d, 1J = 243 Hz); 19F NMR (376 MHz; DMSO-d6) δ ppm: −116.4; ESI-MS (ES−) m/z 394 [M − H]− (100%).X-ray crystallography
The crystallographic data were collected with a Bruker-Nonius Apex-X8 and Bruker-Nonius APEX-II CCD diffractometer, respectively, with Mo-Kα radiation (λ = 0.71073 Å). The structures were solved using SHELXS-97 and refined against F2 on all data by full-matrix least squares with SHELXL-97.61 All non-hydrogen atoms were refined anisotropically; all hydrogen atoms bonded to carbon atoms were placed on geometrically calculated positions and refined using a riding model.‡LDL assay
In order to determine redox activity–structure relationships of COX-2 inhibitors low-density lipoprotein (LDL) copper-/iron-peroxidation models were used. Therefore, native albumin-free LDL (density 1.006–1.063 g mL−1) were isolated from the blood plasma of healthy, normolipidemic, normoglycemic male volunteers by sequential very fast ultracentrifugation (VFU) and prepared for oxidation experiments as previously described by us in detail.62 Blood plasma and LDL were all processed in subdued light to prevent the photooxidation of LDL. All buffers and solutions were made oxygen-free by degassing and purging with argon. LDL apoB-100 was measured by immunoelectrophoresis using ‘ready-to-use’ agarose gels (Sebia, Issy-les-Moulineaux, France). Immediately before oxidation of LDL, EDTA, and salt from the density gradient were removed using a size exclusion column (Econo-Pac 10DG, Bio-Rad) and phosphate-buffered saline (PBS, 10 mM sodium phosphate, 150 mM sodium chloride, pH 7.4) as the eluent.62For lipid oxidation, to 200 μL-aliquots of native LDL (125 μg apoB-100/mL, equal to 0.25 μM LDL) in 96 well plates (UV-Star plates, UV transparent to 200 nm, Greiner, Germany) were added 25 μL of an aqueous solution of CuSO4 (16 μM) and 25 μL of the solution of the compound to be tested. All compounds (novel COX-2 inhibitors, celecoxib, quercetin, and melatonin) were tested at 1 μM concentration. The same preparation without CuSO4 was used as control. The oxidative process was monitored using a Biotek Instruments Synergy 4 thermostatic UV/vis micro plate reader by following the formation of conjugated dienes at 234 nm every 5 min for 4 hours at 30 °C. This approach results in a curve exhibiting a lag phase, during which the absorbance does not increase significantly, a propagation phase, during which the absorbance increases rapidly, and a degradation phase, characterized by a slow fall in the absorbance.63,64 There is a positive (negative) correlation between lag phase duration and the concentration of antioxidants (prooxidants) contained in the LDL sample.63
For protein oxidation, aliquots of native LDL (125 μg apoB-100/mL, equal to 0.25 μM LDL) were subjected to a well characterized iron-catalyzed oxidation system containing 10 μM bovine hemin chloride and 100 μM H2O2 at 37 °C for 40 hours in the dark.65 The oxidative process was monitored by mass spectrometric determination of formation of γ-glutamyl semialdehyde, a specific product of protein oxidation, which by reduction forms 5-hydroxy-2-aminovaleric acid (HAVA) as described elsewhere in detail.65,66
In this study the results are expressed as ratios between the duration of lag phase in the presence and in the absence of the compound (RDIENE) and between the apoB-100 HAVA content in the absence and in the presence of the compound (RHAVA). Thus, in both approaches R values (RDIENE or RHAVA) higher than 1 indicate antioxidant activity, R values of about 1 mean that the compound has no effect and R values lower than 1 suggests prooxidant activity.
COX inhibitory assay
The COX inhibition activity against ovine COX-1 and human COX-2 presented in Table 2 was determined at the given concentrations using the fluorescence based COX assay “COX Fluorescent Inhibitor Screening Assay Kit” (catalog number 700100; Cayman Chemical, Ann Arbor, MI, USA) and the EIA based COX assay “COX inhibitor Screening Assay Kit” (catalog number 560131; Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer's instructions.Acknowledgements
The authors thank the Helmholtz Association for funding a part of this work through Helmholtz-Portfolio Topic “Technologie und Medizin – Multimodale Bildgebung zur Aufklärung des In vivo-Verhaltens von polymeren Biomaterialien”. This work is also part of the research initiative “Radiation-Induced Vascular Dysfunction (RIVAD)”. The excellent technical assistance of Peggy Wecke, Mareike Barth, Catharina Heinig, and Sebastian Meister is greatly acknowledged. Franz-Jacob Pietzsch is graduate student member of the Integrated Research Training Group “Matrixengineering” (within the Transregional Collaborative Research Centre 67 “Functional biomaterials for controlling healing processes in bone and skin – from material science to clinical application” funded by German Research Foundation) at Medical Faculty and University Hospital Carl Gustav Carus, Technische Universität, Dresden.References
- L. J. Marnett, Annu. Rev. Pharmacol. Toxicol., 2009, 49, 265–290 CrossRef CAS PubMed.
- P. Singh and A. Mittal, Mini-Rev. Med. Chem., 2008, 8, 73–90 CrossRef CAS.
- N. Chandna, J. K. Kapoor, J. Grover, K. Bairwa, V. Goyal and S. M. Jachak, New J. Chem., 2014, 38, 3662–3672 RSC.
- W. C. Black, C. Bayly, M. Belley, C. Chan, S. Charleson, D. Denis, J. Y. Gauthier, R. Gordon, D. Guay, S. Kargman, C. K. Lau, Y. Leblanc, J. Mancini, M. Ouellet, D. Percival, P. Roy, K. Skorey, P. Tagari, P. Vickers, E. Wong, L. Xu and P. Prasit, Bioorg. Med. Chem. Lett., 1996, 6, 725–730 CrossRef CAS.
- S. Olgen, E. Akaho and D. Nebioglu, Eur. J. Med. Chem., 2001, 36, 747–770 CrossRef CAS.
- J. A. Campbell, C. A. Broka, L. Gong, K. A. M. Walker and J. Wang, Tetrahedron Lett., 2004, 45, 4073–4075 CrossRef CAS PubMed.
- J. A. Campbell, V. Bordunov, C. A. Broka, M. F. Browner, J. M. Kress, T. Mirzadegan, C. Ramesha, B. F. Sanpablo, R. Stabler, P. Takahara, A. Villasenor, K. A. M. Walker, J. Wang, M. Welch and P. Weller, Bioorg. Med. Chem. Lett., 2004, 14, 4741–4745 CrossRef CAS PubMed.
- J. A. Campbell, V. Bordunov, C. A. Broka, J. Dankwardt, R. T. Hendricks, J. M. Kress, K. A. M. Walker and J. Wang, Tetrahedron Lett., 2004, 45, 3793–3796 CrossRef CAS PubMed.
- W. Hu, Z. Guo, F. Chu, A. Bai, X. Yi, G. Cheng and J. Li, Bioorg. Med. Chem., 2003, 11, 1153–1160 CrossRef CAS.
- W. Hu, Z. Guo, X. Yi, C. Guo, F. Chu and G. Cheng, Bioorg. Med. Chem., 2003, 11, 5539–5544 CrossRef CAS PubMed.
- Z. Guo, G. Cheng and F. Chu, US Pat., 20040058977 A1, 2004.
- M. S. Estevão, L. C. R. Carvalho, M. Freitas, A. Gomes, A. Viegas, J. Manso, S. Erhardt, E. Fernandes, E. J. Cabrita and M. M. B. Marques, Eur. J. Med. Chem., 2012, 54, 823–833 CrossRef PubMed.
- J. Kaur, A. Bhardwaj, Z. Huang and E. E. Knaus, Bioorg. Med. Chem. Lett., 2012, 22, 2154–2159 CrossRef CAS PubMed.
- B. B. Aggarwal and P. Gehlot, Curr. Opin. Pharmacol., 2009, 9, 351–369 CrossRef CAS PubMed.
- Z. Liao, K. A. Mason and L. Milas, Drugs, 2007, 67, 821–845 CrossRef CAS PubMed.
- M. R. Becker, M. D. Siegelin, R. Rompel, A. H. Enk and T. Gaiser, Melanoma Res., 2009, 19, 8–16 CrossRef CAS PubMed.
- C. Tondera, M. Laube, C. Wimmer, T. Kniess, B. Mosch, K. Großmann and J. Pietzsch, Biochem. Biophys. Res. Commun., 2013, 430, 301–306 CrossRef CAS PubMed.
- L. T. Soumaoro, H. Uetake, T. Higuchi, Y. Takagi, M. Enomoto and K. Sugihara, Clin. Cancer Res., 2004, 10, 8465–8471 CrossRef CAS PubMed.
- A. M. Minisini, G. Pascoletti, D. Intersimone, E. Poletto, P. Driol, R. Spizzo, C. A. Scott, F. Puglisi, G. Fasola and C. Di Loreto, Melanoma Res., 2013, 23, 96–101 CrossRef CAS PubMed.
- S. Meyer, T. J. Fuchs, A. K. Bosserhoff, F. Hofstadter, A. Pauer, V. Roth, J. M. Buhmann, I. Moll, N. Anagnostou, J. M. Brandner, K. Ikenberg, H. Moch, M. Landthaler, T. Vogt and P. J. Wild, PLoS One, 2012, 7, e38222 CAS.
- P. Zhan, Q. Qian and L. K. Yu, J. Thorac. Dis., 2013, 5, 40–47 Search PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- E. F. de Vries, Curr. Pharm. Des., 2006, 12, 3847–3856 CrossRef CAS.
- M. Laube, T. Kniess and J. Pietzsch, Molecules, 2013, 18, 6311–6355 CrossRef CAS PubMed.
- O. Tietz, A. Marshall, M. Wuest, M. Wang and F. Wuest, Curr. Med. Chem., 2013, 20, 4350–4369 CrossRef CAS.
- J. Prabhakaran, M. D. Underwood, R. V. Parsey, V. Arango, V. J. Majo, N. R. Simpson, R. Van Heertum, J. J. Mann and J. S. Kumar, Bioorg. Med. Chem., 2007, 15, 1802–1807 CrossRef CAS PubMed.
- E. F. de Vries, J. Doorduin, R. A. Dierckx and A. van Waarde, Nucl. Med. Biol., 2008, 35, 35–42 CrossRef CAS PubMed.
- M. J. Uddin, B. C. Crews, K. Ghebreselasie, I. Huda, P. J. Kingsley, M. S. Ansari, M. N. Tantawy, J. J. Reese and L. J. Marnett, Cancer Prev. Res., 2011, 4, 1536–1545 CrossRef CAS PubMed.
- D. D. Nolting, M. Nickels, M. N. Tantawy, J. Y. H. Yu, J. Xie, T. E. Peterson, B. A. Crews, L. Marnett, J. C. Gore and W. Pham, Front. Oncol., 2013, 2, 207, DOI:10.3389/fonc.2012.00207.
- T. Kniess, M. Laube, R. Bergmann, F. Sehn, F. Graf, J. Steinbach, F. Wuest and J. Pietzsch, Bioorg. Med. Chem., 2012, 20, 3410–3421 CrossRef CAS PubMed.
- M. Mor, G. Spadoni, G. Diamantini, A. Bedini, G. Tarzia, C. Silva, F. Vacondio, M. Rivara, P. Plazzi, D. Franceschinit, M. Zussot and P. Giusti, Adv. Exp. Med. Biol., 2003, 527, 567–575 CrossRef CAS.
- S. Suzen, S. S. Cihaner and T. Coban, Chem. Biol. Drug Des., 2012, 79, 76–83 CAS.
- D. P. Kudav, S. P. Samant and B. D. Hosangadi, Synth. Commun., 1987, 17, 1185–1187 CrossRef CAS.
- A. Trejo, H. Arzeno, M. Browner, S. Chanda, S. Cheng, D. D. Comer, S. A. Dalrymple, P. Dunten, J. Lafargue, B. Lovejoy, J. Freire-Moar, J. Lim, J. Mcintosh, J. Miller, E. Papp, D. Reuter, R. Roberts, F. Sanpablo, J. Saunders, K. Song, A. Villasenor, S. D. Warren, M. Welch, P. Weller, P. E. Whiteley, L. Zeng and D. M. Goldstein, J. Med. Chem., 2003, 46, 4702–4713 CrossRef CAS PubMed.
- C. J. Dinsmore and J. M. Bergman, PCT Int. Appl., WO2005030129 A2, 2005.
- N. Mathews, R. A. Ward and A. J. Whitehead, US-Pat., 6803463 B2, 2004.
- S. K. Singh, V. Saibaba, V. Ravikumar, S. V. Rudrawar, P. Daga, C. S. Rao, V. Akhila, P. Hegde and Y. K. Rao, Bioorg. Med. Chem., 2004, 12, 1881–1893 CrossRef CAS PubMed.
- J. J. Talley, S. Bertenshaw, D. J. Rogier Jr, M. Graneto, D. L. Brown, B. Devadas, H. Lu and J. A. Sikorski, PCT Int. Appl., WO9636617 A1, 1996.
- M. Laube, W. Neumann, M. Scholz, P. Lönnecke, B. Crews, L. J. Marnett, J. Pietzsch, T. Kniess and E. Hey-Hawkins, ChemMedChem, 2013, 8, 329–335 CrossRef CAS PubMed.
- Y. Vara, E. Aldaba, A. Arrieta, J. L. Pizarro, M. I. Arriortua and F. P. Cossio, Org. Biomol. Chem., 2008, 6, 1763–1772 CAS.
- V. Sridharan, S. Perumal, C. Avendaño and J. C. Menéndez, Synlett, 2006, 91–95 CAS.
- C. Farrerons Gallemi, I.-J. Miquel Bono, A. M. Fernandez Serrat, C. Monserrat Vidal, C. Lagunas Arnal, F. Gimenez Guasch and A. Fernandez Garcia, PCT Int. Appl., WO 2000008024 A1, 2000.
- M. Miyasaka, A. Fukushima, T. Satoh, K. Hirano and M. Miura, Chem.–Eur. J., 2009, 15, 3674–3677 CrossRef CAS PubMed.
- S. D. Koulocheri and S. A. Haroutounian, Eur. J. Org. Chem., 2001, 2001, 1723–1729 CrossRef.
- D. L. Horrocks, J. Chem. Phys., 1968, 49, 2913–2917 CrossRef CAS PubMed.
- I. B. Berlman, Spectrochim. Acta, Part A, 1971, 27, 473–489 CrossRef CAS.
- D. G. Kaiser, B. J. Bowman and A. A. Forist, Anal. Chem., 1966, 38, 977–980 CrossRef CAS.
- P. Prasit, D. Guay, Z. Wang, S. Leger and M. Therien, PCT Int. Appl., WO 9606840 A1, 1996.
- M. W. Tabor, E. Coats, M. Sainsbury and H. G. Shertzer, Adv. Exp. Med. Biol., 1991, 283, 833–836 CrossRef CAS.
- E. A. Lissi, M. Faure, N. Montoya and L. A. Videla, Free Radical Res. Commun., 1991, 15, 211–222 CrossRef CAS.
- J. Pietzsch, M. Laube, F. J. Pietzsch, R. Bergmann and T. Kniess, in Coronary Artery Disease: 2011 Update, ed. B. S. Lewis, M. Y. Flugelman and D. A. Halon, Medimond S.r.l – Monduzzi Editore International, Bologna, Italy, 2011, pp. 107–110 Search PubMed.
- S. Ullm, F. Sehn, M. Laube, C. Tondera, N. Bechmann, B. Mosch, T. Kniess and J. Pietzsch, in Proceedings of the 6th European Congress of Pharmacology, ed. A. Zarzuelo and R. Jimenez, Medimond S.r.l – Monduzzi Editore International, Pianoro (Bologna), Italy, 2013, pp. 87–90 Search PubMed.
- S. Suzen, P. Bozkaya, T. Coban and D. Nebiogu, J. Enzyme Inhib. Med. Chem., 2006, 21, 405–411 CrossRef CAS PubMed.
- A. Gozzo, D. Lesieur, P. Duriez, J. C. Fruchart and E. Teissier, Free Radical Res. Commun., 1999, 26, 1538–1543 CrossRef CAS.
- M. F. Walter, R. F. Jacob, C. A. Day, R. Dahlborg, Y. Weng and R. P. Mason, Atherosclerosis, 2004, 177, 235–243 CrossRef CAS PubMed.
- Y. Kuge, Y. Katada, S. Shimonaka, T. Temma, H. Kimura, Y. Kiyono, C. Yokota, K. Minematsu, K. Seki, N. Tamaki, K. Ohkura and H. Saji, Nucl. Med. Biol., 2006, 33, 21–27 CrossRef CAS PubMed.
- M. Tanaka, Y. Fujisaki, K. Kawamura, K. Ishiwata, Q. GeLeTu, F. Yamamoto, T. Mukai and M. Maeda, Biol. Pharm. Bull., 2006, 29, 2087–2094 CAS.
- M. S. Morales-Ríos, J. Espiñeira and P. Joseph-Nathan, Magn. Reson. Chem., 1987, 25, 377–395 CrossRef PubMed.
- K. Kobayashi, S. Fujita, S. Fukamachi and H. Konishi, Synthesis, 2009, 3378–3382 CrossRef CAS PubMed.
- M. C. Wilkinson, Org. Lett., 2011, 13, 2232–2235 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELXS/L-97, Programs for the Solutions and Refinements of Crystal Structures, University of Göttingen, Göttingen, 1997 Search PubMed.
- J. Pietzsch, R. Bergmann, K. Rode, C. Hultsch, B. Pawelke, F. Wuest and J. van den Hoff, Nucl. Med. Biol., 2004, 31, 1043–1050 CrossRef CAS PubMed.
- H. Esterbauer, G. Striegl, H. Puhl and M. Rotheneder, Free Radical Res. Commun., 1989, 6, 67–75 CrossRef CAS.
- S. Kopprasch, W. Leonhardt, J. Pietzsch and H. Kühne, Atherosclerosis, 1998, 136, 315–324 CrossRef CAS.
- J. Pietzsch, Biochem. Biophys. Res. Commun., 2000, 270, 852–857 CrossRef CAS PubMed.
- J. Pietzsch, F. J. Pietzsch and S. Kopprasch, Trends Chromatogr., 2009, 5, 15–20 CAS.
Footnotes |
| † This work is dedicated to the 75th birthday of Prof. Dr Bernd Johannsen. |
| ‡ Electronic supplementary information (ESI) available. CCDC 963601, 963602, 963609 and 963572. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra05650g |
| This journal is © The Royal Society of Chemistry 2014 |