
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExciton and charge transfer processes within singlet fission micelles†
Daniel Malinowski a,
Guiying Hebd,
Bernardo Salcido-Santacruzcd,
Kanad Majumderad,
Junho Kwona,
Matthew Y. Sfeir*bcd and
Luis M. Campos*a
a,
Guiying Hebd,
Bernardo Salcido-Santacruzcd,
Kanad Majumderad,
Junho Kwona,
Matthew Y. Sfeir*bcd and
Luis M. Campos*a
aDepartment of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: lcampos@columbia.edu
bDepartment of Physics, Graduate Center, City University of New York, New York, NY 10016, USA. E-mail: msfeir@gc.cuny.edu
cDepartment of Chemistry, Graduate Center, City University of New York, New York, NY 10016, USA
dPhotonics Initiative, Advanced Science Research Center, City University of New York, New York, NY 10031, USA
First published on 6th May 2025
Abstract
Multiexciton (ME) mechanisms hold great promise for enhancing energy conversion efficiency in optoelectronic and photochemical systems. In singlet fission (SF), the generation of two triplet excitons from a single photon provides a route to circumvent thermal energy losses and organic systems offer opportunities to modulate ME dynamics. However, the practical implementation of SF-based materials is hindered by poor triplet exciton mobility, interfacial recombination losses, and complex dynamics at heterogeneous interfaces. While studies of interfacial SF dynamics have demonstrated the potential for efficient charge and exciton transfer, experimental conditions and design of interfaces vary widely. To address this, we explore polymer-based self-assembled architectures as a tunable platform for studying mesoscale SF interfacial dynamics of (multi)exciton transfer, as well as electron and hole transfer. Specifically, we design amphiphilic block copolymers (BCPs) incorporating pendent tetracene moieties that self-assemble into micellar nanoparticles, placing the tetracenes in the amorphous core. These micelles provide a controlled environment to systematically introduce “dopants” to investigate interfacial dynamics. Importantly, the use of solvents within the micelle core can be also applied to impart polymer chain mobility.
Introduction
Multiexciton (ME) mechanisms have garnered significant interest due to their potential to amplify photonic, optoelectronic, and photochemical processes. In organic molecules, singlet fission (SF) involves the generation of two triplet excitons by absorption of a single photon through a spin-allowed, energy conserved process to form a bound triplet pair [TT] multiexciton that is delocalized across two or more chromophores. These molecular ME states can be generated with unity yield and can rapidly dephase into a set of long-lived individual triplet excitons, precluding the loss of excess energy as heat. As a result, SF-based materials provide vast opportunities to be implemented in efficient energy harvesting devices and photochemistry.1–3 However, the advantages of molecular multiexcitons are offset by several key challenges that hinder fundamental studies of interfacial charge and exciton transfer processes that are critical to these emerging concepts. Many of these challenges stem from the notoriously poor triplet exciton mobility in these materials, which limits the relevant active area for ME generation to only the molecules in direct vicinity of a heterointerface.4 Furthermore, the small relevant active area coupled with large, delocalized excited state wavefunctions5 facilitate parasitic losses due to singlet charge transfer and charge carrier annihilation at the interface.6–8 The sensitivity of SF materials to their topology at heterointerfaces can further complicate studies of interfacial dynamics (e.g. small molecules, multichromophore-containing polymers, and molecular crystals; Fig. 1a). Overall, there is a pressing need for strategies that facilitate systematic studies of how the microenvironment impacts SF interfacial dynamics, especially between different classes of materials and amorphous materials. | ||
| Fig. 1 (a) Conventional architectures comprising two types of singlet fission chromophores. (b) Examples of solid-state and solution phase designs to investigate multiexciton generation and exciton/charge transfer mechanisms at heterointerfaces (bilayers and collisions). (c) Macromolecular approach to access mesoscale structures (e.g. micelles) that can be readily doped with solvents to induce chain mobility of the hydrophobic core, as well as exciton/charge donors and acceptors. | ||
It has been shown that large variations in interfacial SF dynamics can be observed due to changes in the nature and scale of the assembled system. Several chromophores, most notably anthradithiophene and TIPS-anthracene, appear to undergo SF in the solid-state but not in molecular assemblies.9–13 Additional complexity is found when studying assemblies of two dissimilar SF chromophores, since excited state charge-transfer processes (e.g., pentacene–perylenediimide and pentacene–perfluoropentacene) can dominate over singlet fission when favored by the energetics and architecture of the mixed system.14–18 Furthermore, the nature of the interfacial interactions varies greatly as one moves from bulk solids to small molecules, resulting in vastly different singlet fission mechanisms, transport properties, and decay dynamics. For example, it has been observed that tetracene crystals doped with pentacene can exhibit both hetero- and conventional singlet fission processes, depending on the relative fractions of the two materials.6,19–22 In contrast, the unique and well defined interfacial interactions in diblock copolymers consisting of tetracene and pentacene do not show major contributions from heterochromophore singlet fission (hSF). In fact, these dynamics are largely independent of their relative fractions, with interfacial processes dominated by triplet exciton transfer.23 Yet in the limit of a small molecule heterodimer, extremely efficient intramolecular hSF is observed in a series of oligoacenes, with more controlled arrangements showing directed energy flow of triplet pairs to low energy sites.24–26 In all cases, the decay and transport processes of the different excited states (singlet, triplet, and triplet pair) must be considered when designing energy harvesting interfaces.
Studies of donor–acceptor interfaces beyond SF heterochromophores have also faced similar challenges stemming from the difficulty of engineering efficient interfaces that address the problem of poor triplet transport.27 One promising approach involves thin SF active layers combined with NIR semiconductors, such as quantum dots and modified silicon–acene interfaces (Fig. 1b).28–34 While these studies have suggested that interfacial triplet exciton transfer can occur, net losses are still observed under practical device conditions and quantification of key loss pathways is difficult. Another study of collision interactions between intramolecular singlet fission (iSF) chromophores has suggested that direct triplet pair transfer is both favorable and desirable for avoiding parasitic recombination processes.7 Mechanistic studies with small molecules have also revealed that charge transfer, rather than exciton transfer, is dominant when using appropriate electron acceptors (e.g., TCNQ and chloranil).35–37 In the realm of conjugated polymers, all-organic singlet fission devices have been fabricated, albeit their power conversion efficiencies were low.38 While these studies have yielded valuable insights, they also suffer from limited modularity. Therefore, developing a platform to systematically vary interfacial interactions using SF molecules with variable acceptors can lead to a deeper fundamental understanding of multiexciton transfer.
In order to efficiently screen donor–acceptor interactions for charge/exciton transfer with SF chromophores, we hypothesized that polymer assemblies can provide the means to investigate interfacial dynamics at the mesoscale (i.e. between the length scale of individual molecules and bulk crystals). Therefore, we designed a series of amphiphilic diblock copolymers (BCPs) to take advantage of modular architectures that can be assembled into nanostructures, driven by hydrophobic/hydrophilic interactions in solution.39 The polymers contained pendant tetracene moieties that serve as the hydrophobic core, alongside pendent triethylene glycol (TEG) chains to make up the hydrophilic outer shell of the micelles in water (Fig. 1c). These materials can self-assemble to effectively create nanoparticles with an amorphous tetracene core, which can additionally be swollen with non-polar solvents to impart mobility to the SF chromophores. The micelles can be further modified with other non-polar “dopants” that can serve as donors or acceptors when interfaced within the SF tetracene core. Three types of dopants were used in this study to understand their impact on SF dynamics of tetracene-based chromophores. Using a soluble pentacene derivative, (multi)exciton migration was observed, from tetracene to the lower energy pentacenes. Importantly, we also found that both electron and hole transfer processes were predominant when using tetracyanoquinodimethane (TCNQ as electron acceptor) and a triarylamine (TAA, as electron donor), respectively. In all cases, charge transfer only took place when the core of the micelles was doped, but not through collisional interactions between the unassembled polymers and the acceptors in solution. Employing the micelle approach thus represents a platform for the investigation of a diverse range of SF-driven photochemistry at heterointerfaces.
Results and discussion
A series of amphiphilic BCPs were synthesized by conventional methods (see the ESI,† for details). Briefly, the hydrophilic block was obtained by the polymerization of a triethylene glycol functionalized norbornene (TEGNo) monomer to an average of 40 repeat units (TEGPNo40). The tetracene-based norbornene monomer (TeNo) was subsequently added, targeting various degrees of polymerization, n, relative to the TEG-block to obtain the desired BCPs, TEGPNo40-b-TePNo[n], where n = 5, 10, 15, 25. The relative block ratios were confirmed by 1H-NMR and size exclusion chromatography (SEC, see ESI†).Steady-state absorption spectroscopy of TEGPNo40-b-TePNo[n] in an organic solvent showed similar features to a TePNo homopolymer we have previously reported (Fig. S12†).23 The spectra of these BCPs also display the typical tetracene vibronic structure, with a peak absorption at 542 nm and no shifting or broadening relative to the TeNo monomer. These characteristics suggest negligible chromophore aggregation in THF solution even with the addition of the hydrophilic block. The excited state dynamics of the tetracene block in the amphiphilic BCPs are characterized by transient absorption (TA) spectroscopy. We observe a SF process that extends over multiple timescales, consistent with variations in chromophore alignment along the polymer backbone. In TEGPNo40-b-TePNo[n], two time constants are sufficient to reproduce the ensemble averaged data and understand its evolution with polymer length. For example, in a comparison between the shortest and longest blocks of TEGPNo40-b-TePNo[n] (n = 5 & 25, respectively) in dilute solution, global analysis indicates that the rate constant for SF is slower when n = 5, τSF ∼ 45 ps and 1240 ps, compared to the longest BCP with n = 25, τSF ∼ 28 ps and 720 ps. Interestingly, this process differs slightly from the homopolymer TePNo20 previously reported, for which the SF dynamics could be characterized by a single time constant with τSF ∼ 330 ps.23 Still, we have observed similar length dependent differences in other tetracene-based BCPs and have attributed this effect to disruptions in chromophore interactions in diblock architectures, which were found to be less prevalent when increasing the size of the tetracene block.23 More details on the triplet pair dynamics in TEGPNo40-b-TePNo[n] can be found in the ESI.†
While these trends in dilute solution are consistent with previous reports of SF macromolecules,23,40 less is known about the multiexciton dynamics of self-assembled micellar nanostructures of amphiphilic BCPs where the core is based on SF chromophores. Therefore, we prepared a variety of pristine micelles, labeled TeM[n] (where n is the number of tetracene repeat units in the originating polymer) to differentiate from the unassembled BCP, by flash precipitation of TEGPNo40-b-TePNo[n] from THF to water (ESI†).41 Dynamic light scattering (DLS) measurements of the resulting micelles show a relatively monodisperse distribution of particle sizes, with z-average diameters ranging from 71 nm for TeM[5], to 135 nm for TeM[25] (Fig. 2b). Transmission electron microscopy imaging of TeM[25] micelles confirmed the expected spherical morphology (Fig. S17†). Importantly, the UV-vis absorption spectra of the micelles were clearly different than that of the BCPs in toluene. While the tetracene vibronic structure is maintained, the peak absorption displays a 4 nm bathochromic shift as well as line broadening. Both these observations are consistent with the formation of an amorphous aggregated phase (Fig. 2a) characteristic of type-I nanoparticles. Similar results have been previously reported for precipitated pentacene nanoparticles in the absence of solubilizing additives.42
 | ||
| Fig. 2 (a) Representative absorption spectra of a TeNo monomer, TePNo polymer, and TeM micelle show minimal differences. (b) Representative dynamic light scattering measurements of pristine TeM[n] micelles in water at 25 °C. (c) Representative TA spectra of pristine TeM[25] micelles and selected associated transient spectra. The relevant excited state dynamics are extracted from the ground state bleach (GSB) near 500 nm, the stimulated emission (SE) near 600 nm, and the broad excited state absorption (ESA) feature spanning 675–775 nm. (d) The shift of the GSB as a function of time is associated with exciton transport to lower energy sites in the polymer, where stronger interchromophore ordering occurs. Larger shifts are observed with longer TePNo blocks in pristine samples and in toluene-swollen micelles of all sizes. (e) The singlet fission dynamics are extracted by subtracting the dynamics at the singlet–triplet isosbestic point near 610 nm from the decay of the broad ESA signal from 700–750. Rate constants can be similarly determined from fitting the decay of SE signal. We note that the singlet decay of sTeM[5] and sTeM[25] are virtually indistinguishable from pristine TeM[25]. | ||
Transient absorption (TA) spectroscopy confirms that all TeM[n] micelles undergo SF upon excitation. The relevant exciton dynamics are readily determined from the analysis of three key spectral regions from which we are able to extract information about chromophore–chromophore interactions, exciton migration, and triplet pair formation. For example, a ground state bleach (GSB) feature corresponding to the vibrationally excited singlet state, S1, is prominent near 505 nm and appears as a sharp dip in a strong relatively featureless singlet excited state absorption (Fig. 2a). We find that the GSB minimum red-shifts over time, consistent with the picture of a heterogeneous material for which significant exciton migration occurs.43 In the micelles, the magnitude of the shift is found to increase with increasing tetracene block length (Fig. 2b, compare solid lines for n = 5 and 25). Additional TA data for the TeM[n] (n = 5, 10, 15) micelles are provided in the ESI.† As this heterogeneity results in dynamic peak shifts and multiple rate constants, standard global analysis techniques are not appropriate for obtaining deconvoluted spectra and accurate rate constants. Instead, we quantify this effect by comparing the position of the peak minima for TeM[n] (n = 5, 10, 15, 25) at select times ranging from 200 fs to 20 ps. Over this window, the n = 5 micelle shows the least spectral evolution (∼1 nm), the n = 10 (∼3 nm) and n = 15 (∼4 nm) micelles show intermediate shifts, and the n = 25 micelle exhibits the largest shift, ∼5 nm (Fig. S3†). We ascribe this behavior to exciton migration to lower energy sites with stronger interchromophore coupling within the micelle and deduce that increasing the size of TePNo block increases the level of interchromophore ordering.
Additionally, we can correlate these spectral shifts to the singlet fission dynamics of the micelles. The decay kinetics associated with the singlet state can be readily obtained from inspection of the stimulated emission (SE) feature near 600 nm as well as the strong singlet excited state absorption (ESA) spanning the NIR from 675–800 nm. As the triplet exciton also exhibits an overlapping broad featureless absorption in the NIR, we isolate the singlet dynamics by taking the difference between the extracted kinetics of the SE or ESA and the kinetics associated with the singlet–triplet isosbestic point near 610 nm (Fig. 2a). The latter provides information on the excited state population dynamics alone, independent of the (multi)exciton spin characteristics. A version of this analysis based on the ESA is shown in Fig. 2c and allows us to readily observe the decay of singlet excitons. Similar to the polymers, we apply a multiexponential fit to the extracted kinetics to account for variations in the chromophore alignment. We find that the time constants of the primary singlet decay components are significantly faster in TeM[25] compared to TeM[5] (∼2 and 13 ps versus 5 and 51 ps, respectively). As singlets evolve rapidly into a species with the well-characterized triplet exciton spectra, we conclude that the TeM[n] micelles indeed undergo SF with rates that are 40–60× faster than that of the associated unassembled free polymers in solution (Table S2†). Overall, we find the larger spectral shifts of the GSB are correlated to an enhanced SF rate, consistent with stronger interchromophore coupling in larger tetracene blocks. These data are reminiscent of dendritic systems for which the emergence of locally ordered hot spots was correlated to an increase in the system size.44 It is worth emphasizing that the polymer backbone and the greasy solubilizing alkyl groups on the chromophores did not play a major role in the exciton dynamics of SF. Instead, such architectural motifs played an essential role in the formation of micelles and providing a new modality to investigate SF in mesoscale systems.
The dependence of the singlet fission dynamics on the interchromophore coupling strength in pristine micelles suggests that this process can be manipulated by modifying its local microenvironment within their core. To explore this in more detail, we sought to introduce non-polar solvents in the core, as well as electron-rich/-deficient small molecule additives to alter the excited state dynamics of the TeM[n] micelles. First, we hypothesized that swelling the core using toluene can induce mobility of the tetracene chains, akin to the known characteristics of solvent-annealed BCPs.45 Swollen micelles, labeled sTeM[n], were prepared by addition of a small volume of the toluene to the bulk aqueous phase, then flash-precipitating TEGPNo40-b-TePNo[n]. DLS measurement confirmed the increase in particle diameters by approximately 50 nm via this procedure as compared to the unswollen micelles (Table S7†). The effect of toluene on the interchromophore coupling in swollen micelles was determined using TA spectroscopy and the same analysis procedure discussed above.
Qualitatively, we found that the dynamics of the swollen micelles are similar to unswollen, pristine micelles featuring a rapid singlet fission process that is followed by exciton migration to local sites with well-ordered chromophores. However, in contrast to pristine micelles (TeM[n]) we observed that both the singlet decay kinetics and the shift of the GSB feature are independent for all values of n in swollen micelles of sTeM[n]. In fact, the exciton dynamics of the sTeM[n] nanoparticles are nearly indistinguishable from pristine TeM[25] (Fig. 2b and c), with GSB shifts of ∼5 nm and picosecond singlet decay processes. Taken together, we conclude that swelling with toluene facilitates the formation of sites with well-ordered chromophores for all block lengths and that this enhanced ordering in the micelle core results in faster rates of SF. Such characteristics provide insights into the influence of ordering within mesoscale structures from a single family of macromolecular SF chromophores, lending tunability of the exciton dynamics within the microenvironment of the micelles. In turn, nanoscale assemblies provide advantages to understand the vast differences in multiexciton dynamics relative to bulk crystals and isolated small molecules.
The micellar structures developed for this study also provide opportunities to encapsulate hydrophobic guests within the core, protected by the hydrophilic corona. This level of control thus enables the facile inclusion of additives that provide opportunities to investigate the behavior of SF chromophores at heterointerfaces, akin to systems shown in Fig. 1a and b. For example, co-assemblies of tetracene and pentacene represent a canonical system for studying exciton migration in SF dynamics, and it has been found that the nature of their excited state interactions are dictated by their relative concentrations varied in bulk solids and small molecules.22,23 In a similar fashion, hydrophobic pentacene (Pen) additives can be added to the tetracene core, varying the concentration without altering the chemical structure of the amphiphilic BCP micelles. Therefore, micelles were prepared by flash precipitation from polymer solutions doped with 5% or 10% Pen (relative to tetracene concentration). The linear absorption spectra of TeM[n]/Pen micelles suggest that significant electronic interactions occur between the tetracene and pentacene chromophores (Fig. S13†). In THF solution, the spectra of TEGPNo40-b-TePNo[n] with co-dissolved TIPS-Pen molecules are identical to the sum of the individual components, with no observed peak shifts or line broadening. In doped TeM[n]/Pen micelles, however, the characteristic absorption peaks of both components are red-shifted and broadened, confirming successful incorporation of pentacene into the micelle core, where significant through-space electronic coupling occurs.
Due to the lower triplet energy of pentacene as compared to tetracene, we hypothesized that the excitons generated from photoexcitation of the micelle assembly would funnel to the low energy Pen sites. To identify key interfacial energy transfer processes, TA spectroscopy on TeM[n]/Pen micelles (n = 5 & 25) was performed using selective excitation wavelengths corresponding to either the tetracene (λex = 480 nm) or pentacene (λex = 600 nm) components (Fig. 3a and d). For tetracene-centered excitation at 480 nm, where minimal pentacene absorption is present, we clearly identify a fast-decaying tetracene singlet with a lifetime τSF < 10 ps, similar to what is observed in the TeM[n] micelles. This suggests the triplet pair formation in the micelle is minimally disturbed by the presence of the pentacene additive. However, new dynamics are observed on intermediate time scales, during which both tetracene- and pentacene-localized triplets are found to co-exist for hundreds of ps (Fig. 3a and b). Furthermore, a long-lived pentacene triplet signal is observed that persists for approximately 30 μs (Fig. 3b and c), consistent with its natural lifetime in dilute solution. The decay of the pentacene triplet is independent of both TePNo block size and pentacene doping percentage (Fig. 3c), suggesting that it results from an immobile, localized triplet exciton. These dynamics are consistent with rapid triplet exciton transfer from high energy tetracene sites to low energy pentacene sites, resulting in localized triplets.22,23,26
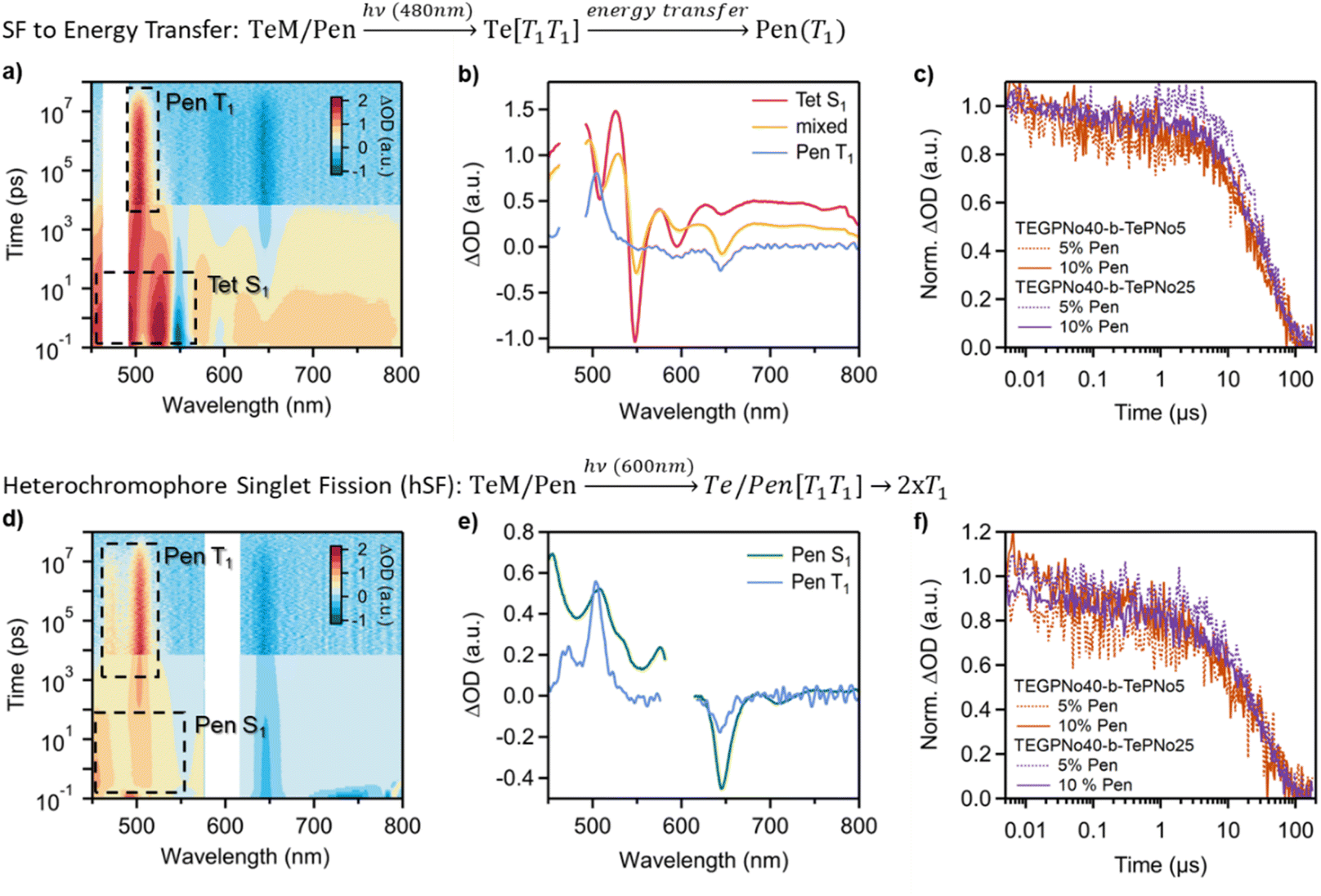 | ||
| Fig. 3 Excitation wavelength-dependent comparison of TA data of TEGPNo40-b-TePNo25, co-assembled with 10 mol% pentacene. (a) Transient absorption data for tetracene-centered excitation at 480 nm. (b) Analysis of the dynamics indicates that SF occurs in the micelle followed by energy transfer to pentacene. (c) All triplets are rapidly funneled to pentacene where they decay with the characteristic behavior of an individual, isolated triplet exciton. Here a kinetic cut of the data at 503 nm is used to illustrate the pentacene triplet decay dynamics. (d) Transient absorption data for pentacene-centered excitations at 600 nm and (e) the resulting analysis indicating that heterochromophore singlet fission (hSF) between pentacene and tetracene produces (f) long-lived isolated pentacene triplets. | ||
In contrast, the dynamics of the system for pentacene excitation at 600 nm show a different triplet formation mechanism involving interfacial hSF. Upon photoexcitation, we observed a pentacene-localized singlet that evolves into a long-lived pentacene triplet signal on timescales of 100–200 ps (Fig. 3d). This lifetime implicates SF as the primary mechanism for triplet formation, since inter-system crossing is weak in TIPS-pentacene and occurs on timescales exceeding 10 ns.46 Importantly, the decay of the pentacene triplet proceeds in a monoexponential fashion without contributions from geminate triplet–triplet annihilation. This behavior is similar to what is observed for 480 nm excitation, for which triplet exciton transfer to pentacene occurs, and suggests that triplet excitons are relatively isolated. We can thus rule out pentacene–pentacene SF as a substantial contributor to the observed dynamics. From these data, we infer a model for the excited state dynamics that includes hSF between the pentacene additives and the tetracene from the BCP. The absence of readily identifiable excited tetracene signatures in our data is attributed to the much lower triplet absorption cross-sections of TIPS-tetracene relative to TIPS-pentacene47 along with the fast migration of photogenerated triplets to lower energy pentacene sites.
Similar to what was observed in pristine micelles, the swollen core promotes inter-chromophore coupling between the polymeric tetracene and the Pen additives. For example, we observe a concentration dependence for pentacene singlet decay in pristine TeM[n]/Pen micelles, with the rate of singlet decay attributed to pentacene becoming faster by approximately 20% as the pentacene concentration is increased from 5% to 10%. However, in Pen-doped swollen micelles sTeM[n]/Pen the singlet decay is enhanced relative to pristine, un-swollen micelles and the signal remains relatively constant over the range of concentrations measured. Additionally, the triplet decays independently of micelle composition with a lifetime of ∼30 μs (Fig. 3f and Table S4†) as observed in all studied assemblies. We attribute these changes in hSF dynamics to a higher probability of favorable chromophore alignment when either the pentacene concentration is increased or when a good solvent is present within the core. It is notable that swelling promotes strong interchromophore interactions along the backbone and imparts mobility to small molecule additives, allowing stronger intermolecular interactions.
The examples above allow us to benchmark how the multiexciton dynamics vary within micellar architectures. Thus, we conjecture that the modularity to include dopants within the core leads to an unconventional framework to probe intermolecular donor–acceptor interactions with SF chromophores, which are generally challenging to tune in bulk crystalline/amorphous solids and in solution.29,35,38 Therefore, we introduced both a strong donor or strong acceptor as dopants within the core of the micelle. Analogous to the procedure introducing Pen to the core of the micelle, a one-to-one ratio of dopant relative to tetracene was incorporated by flash precipitation. In one case triarylamine 4-[bis(4-methoxyphenyl)amino]phenol (TAA) was used as an electron donor, and in another 7,7,8,8-tetracyanoquinodimethane (TCNQ) served as the electron acceptor. Although the onset of absorption of TAA is in the UV and overlaps significantly with tetracene, we can still confirm that tetracene vibronic bands in the visible are comparable to those obtained from pristine micelles, suggesting TAA does not interfere with self-assembly (Fig. S14†). The TCNQ absorption near 400 nm is more readily discernible in mixed solutions and assemblies. Similar to TAA, it has a negligible effect on the spectral region corresponding to tetracene absorption. Interestingly, the strength of the TCNQ absorption peak in TeM[n]/TCNQ assemblies is suppressed compared to the mixed solution (Fig. S15†), suggesting that electronic interaction between the TCNQ and tetracene chromophores does occur in the core.
While most efforts aimed at extracting charge from triplet excitons from SF have been mainly driven by using electron acceptors (hole transfer to the SF chromophore), we posited that electron donors (e.g., TAA) can also be used to “inject” charge to the SF chromophores. To test the hypothesis, we first carried out TA spectroscopy on unassembled BCP mixed with excess TAA in solution and observed that its singlet fission dynamics were identical to a solution of the BCP alone. However, in 1 molar equivalent TAA-doped n = 25 micelles (TeM[25]/TAA), marked changes in the transient spectra were clearly evident compared to solution and the TeM[25] micelle (Fig. 4a and b), suggesting a modified excited state process. As in pristine micelles, singlets are identified by their strong stimulated emission signal near 600 nm and are impulsively generated upon excitation. However, the singlet exciton of tetracene in the TAA co-assembly is more rapidly quenched (Fig. 4d). Furthermore, following the decay of the singlet, a new ESA feature is observed with a maximum near 750 nm, which is absent in the pristine micelle (Fig. 4b and e). By comparison to spectroelectrochemistry measurements of small molecule tetracene-TAA dimers (dashed blue line in Fig. 4e), we identify this species as the oxidized TAA˙+ (Fig. 4e). This confirms the presence of TAA within the core of the micelle and its efficient charge transfer interactions with the tetracene chromophores in the micelle. Electron transfer with TAA occurs on timescales < 100 ps in competition with SF and produces a transient charge transfer (CT) state. The cation signature from TAA˙+ decays with a lifetime of ∼10 ns, attributed to back electron transfer (BET). The small remaining population of tetracene free triplets decays with a lifetime of ∼30 μs (Fig. 4b). When the proportion of doped TAA was reduced to 0.1 molar equivalents, no identifiable TAA cation was observed.
 | ||
| Fig. 4 (a) TA data for pristine TeM[25] micelles showing triplet pair formation from the singlet, (b) micelles co-assembled with TAA electron donors that show the emergence of a TAA cation signal, and (c) micelles co-assembled with TCNQ electron acceptors that show the emergence of a TCNQ anion signal. (d) Kinetic traces corresponding the stimulated emission signal near 600 nm show more rapid singlet quenching in co-assemblies with electron donors or acceptors. (e) The transient spectra at key delay times allow us to identify the TAA cation by comparison to spectroelectrochemistry measurements (blue dotted) line. The singlet exciton is populated at early times (0.5 ps) and is identified by its strong stimulated emission signal (black). The characteristic ESA signal associated with TAA+ (blue) emerges following the decay of the singlet (>100 ps). (f) A similar analysis is performed to identify TCNQ− formation. Since the TCNQ− lifetime is relatively short, we take the difference between intermediate delay times (∼150 ps) and long delay times (∼4000 ps) to isolate its contribution and compare it to spectroelectrochemistry measurements (inset). | ||
In order to investigate SF core interactions with electron acceptors, we doped micelles with 1 molar equivalent of TCNQ. Akin to the donor dopant micelle system above, similar dynamics are observed in TeM[25]/TCNQ co-assemblies (Fig. 4c). The photoexcited singlet decays more rapidly than in pristine micelles, suggesting a new exciton quenching mechanism (Fig. 4d). At intermediate delay times (>100 ps) we observe a new NIR absorption signal that is absent in the pristine micelle solution (Fig. 4f). This feature is identified as TCNQ˙− that is formed due to electron transfer from the photoexcited tetracene to the TCNQ electron acceptor. This feature is less prominent than in the TAA-doped micelles, consistent with the expectation that the instantaneous population of TCNQ˙− is expected to be small due to its short lifetime, as compared to its formation. This is due to the well-known instability of the TCNQ ion relative to other species.48,49 To better isolate the anion spectra in the TA data, we use an analysis procedure in which we subtract the long delay time transient spectra for which we expect only triplets to be present (>4000 ps) from an intermediate time spectra that is a mix of tetracene triplets and charge separated species (∼150 ps). We can readily see a contribution that is a good match to the TCNQ˙− spectra obtained by spectroelectrochemistry (Fig. 4f, inset). We emphasize again that unlike in studies where charge transfer occurs sequentially after SF,35 in these doped micelles where significant interfacial interactions are present between the SF chromophore and the dopant, charge transfer is competitive with SF. These data highlight the flexibility of the micelle architecture in engineering a variety of excited state behaviors, showing that the same micelle can exhibit efficient singlet fission, electron transfer, or hole transfer depending upon the microenvironment of the core.
Experimental
Transient absorption spectroscopy
Synthetic methods
Conclusions
In summary, we have designed a modular platform to study donor–acceptor interactions with tetracene-based singlet fission chromophores in mesoscale environments by using amphiphilic diblock copolymers that can self-assemble into nanoscale micelles. We unveil a design to impart chain mobility that renders order and favorably affects triplet pair generation and various interactions within the core. This work illustrates that SF yield is not a sufficient figure of merit for designing SF-based devices. In different media, interfacial interactions, including charge and energy transfer, can occur on ultrafast timescales and compete with triplet pair interactions. Here, we illustrate this effect with nanoparticles that can encapsulate various small molecules and interact with the tetracene excitons. While the intrinsic SF yields of the polymers and micelles are high (>180%), doped micelles exhibit complex exciton transport, as well as hole and electron transfer mechanisms. These interactions depend intimately on the nature of the medium, as hole transfer was only observed when a triaryl amine was co-assembled in the core, but no interactions were found when the BCPs were homogeneously mixed with the donor in non-polar solvents. Given the sustained interest in singlet fission for light harvesting technologies and photochemistry, this platform offers a convenient means to screen various additives for energy extraction. The constituent TeNo monomer can also be facilely adapted to incorporate different SF chromophores in place of tetracene. Block copolymers also have potential to undergo self-assembly in the solid state, and varying compositions may lead to various morphologies. We envision such future studies will allow us to explore singlet fission dynamics in different mesoscale morphologies.54Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. M. synthesized and characterized the polymers and micelles. J. K. synthesized the monomer TEGNo. G. H. and K. M. performed the transient absorption spectroscopy measurements. D. M., G. H., and K. M. processed the TA data. B. S. S. collected spectroelectrochemistry reference data. M. Y. S. and L. M. C. supervised the project. D. M., M. Y. S., and L. M. C. wrote the manuscript with contributions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0022036, in addition to the National Science Foundation under grants DMR-2004683 and DMR-2004678. J. K. thanks the Seokcho Scholarship. The authors acknowledge the use of facilities and instrumentation supported by NSF through Columbia University, Columbia Nano Initiative, and the Materials Research Science and Engineering Center DMR-2011738.References
- M. C. Hanna and A. J. Nozik, Solar Conversion Efficiency of Photovoltaic and Photoelectrolysis Cells with Carrier Multiplication Absorbers, J. Appl. Phys., 2006, 100(7), 074510, DOI:10.1063/1.2356795.
- M. J. Y. Tayebjee, D. R. McCamey and T. W. Schmidt, Beyond Shockley–Queisser: Molecular Approaches to High-Efficiency Photovoltaics, J. Phys. Chem. Lett., 2015, 6(12), 2367–2378, DOI:10.1021/acs.jpclett.5b00716.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, Singlet Fission: Progress and Prospects in Solar Cells, Adv. Mater., 2017, 29(20), 1601652, DOI:10.1002/adma.201601652.
- T. Zhu and L. Huang, Exciton Transport in Singlet Fission Materials: A New Hare and Tortoise Story, J. Phys. Chem. Lett., 2018, 9(22), 6502–6510, DOI:10.1021/acs.jpclett.8b02181.
- X. Wang, T. Garcia, S. Monaco, B. Schatschneider and N. Marom, Effect of Crystal Packing on the Excitonic Properties of Rubrene Polymorphs, CrystEngComm, 2016, 18(38), 7353–7362, 10.1039/C6CE00873A.
- J. D. McNeill, D. Y. Kim, Z. Yu, D. B. O'Conno and P. F. Barbara, Near Field Spectroscopic Investigation of Fluorescence Quenching by Charge Carriers in Pentacene-Doped Tetracene, J. Phys. Chem. B, 2004, 108(31), 11368–11374, DOI:10.1021/jp049823r.
- G. He, K. R. Parenti, L. M. Campos and M. Y. Sfeir, Direct Exciton Harvesting from a Bound Triplet Pair, Adv. Mater., 2022, 34(40), 2203974, DOI:10.1002/adma.202203974.
- M. Hasan, S. Saggar, A. Shukla, F. Bencheikh, J. Sobus, S. K. M. McGregor, C. Adachi, S.-C. Lo and E. B. Namdas, Probing Polaron-Induced Exciton Quenching in TADF Based Organic Light-Emitting Diodes, Nat. Commun., 2022, 13(1), 254, DOI:10.1038/s41467-021-27739-x.
- K. Bhattacharyya and A. Datta, Polymorphism Controlled Singlet Fission in TIPS-Anthracene: Role of Stacking Orientation, J. Phys. Chem. C, 2017, 121(3), 1412–1420, DOI:10.1021/acs.jpcc.6b10075.
- D. M. De Clercq, M. I. Collins, N. P. Sloane, J. Feng, D. R. McCamey, M. J. Y. Tayebjee, M. P. Nielsen and T. W. Schmidt, Singlet Fission in TIPS-Anthracene Thin Films, Chem. Sci., 2024, 15(17), 6402–6409, 10.1039/D3SC06774B.
- S. Sanders, E. Kumarasamy, K. J. Fallon, M. Sfeir and L. Campos, Singlet Fission in a Hexacene Dimer: Energetics Dictate Dynamics, Chem. Sci., 2020, 11, 1079–1084, 10.1039/C9SC05066C.
- G. He, K. R. Parenti, P. J. Budden, J. Niklas, T. Macdonald, E. Kumarasamy, X. Chen, X. Yin, D. R. McCamey, O. G. Poluektov, L. M. Campos and M. Y. Sfeir, Unraveling Triplet Formation Mechanisms in Acenothiophene Chromophores, J. Am. Chem. Soc., 2023, 145(40), 22058–22068, DOI:10.1021/jacs.3c07082.
- G. Mayonado, K. T. Vogt, J. D. B. Van Schenck, L. Zhu, G. Fregoso, J. Anthony, O. Ostroverkhova and M. W. Graham, High-Symmetry Anthradithiophene Molecular Packing Motifs Promote Thermally Activated Singlet Fission, J. Phys. Chem. C, 2022, 126(9), 4433–4445, DOI:10.1021/acs.jpcc.1c10977.
- F. Unger, D. Lepple, M. Asbach, L. Craciunescu, C. Zeiser, A. F. Kandolf, Z. Fišer, J. Hagara, J. Hagenlocher, S. Hiller, S. Haug, M. Deutsch, P. Grüninger, J. Novák, H. F. Bettinger, K. Broch, B. Engels and F. Schreiber, Optical Absorption Properties in Pentacene/Tetracene Solid Solutions, J. Phys. Chem. A, 2024, 128(4), 747–760, DOI:10.1021/acs.jpca.3c06737.
- C. M. Mauck, K. E. Brown, N. E. Horwitz and M. R. Wasielewski, Fast Triplet Formation via Singlet Exciton Fission in a Covalent Perylenediimide-β-Apocarotene Dyad Aggregate, J. Phys. Chem. A, 2015, 119(22), 5587–5596, DOI:10.1021/acs.jpca.5b01048.
- A.-K. Hansmann, R. C. Döring, A. Rinn, S. M. Giesen, M. Fey, T. Breuer, R. Berger, G. Witte and S. Chatterjee, Charge Transfer Excitation and Asymmetric Energy Transfer at the Interface of Pentacene–Perfluoropentacene Heterostacks, ACS Appl. Mater. Interfaces, 2021, 13(4), 5284–5292, DOI:10.1021/acsami.0c16172.
- W. Kim, N. A. Panjwani, K. C. Krishnapriya, K. Majumder, J. Dasgupta, R. Bittl, S. Patil and A. J. Musser, Heterogeneous Singlet Fission in a Covalently Linked Pentacene Dimer, Cell Rep. Phys. Sci., 2024, 5(7), 102045, DOI:10.1016/j.xcrp.2024.102045.
- L. Wang, Y. Wu, J. Chen, L. Wang, Y. Liu, Z. Yu, J. Yao and H. Fu, Absence of Intramolecular Singlet Fission in Pentacene–Perylenediimide Heterodimers: The Role of Charge Transfer State, J. Phys. Chem. Lett., 2017, 8(22), 5609–5615, DOI:10.1021/acs.jpclett.7b02597.
- A. J. Campillo, S. L. Shapiro and C. E. Swenberg, Picosecond Measurements of Exciton Migration in Tetracene Crystals Doped with Pentacene, Chem. Phys. Lett., 1977, 52(1), 11–15, DOI:10.1016/0009-2614(77)85109-9.
- N. E. Geacintov, J. Burgos, M. Pope and C. Strom, Heterofission of Pentacene Excited Singlets in Pentacene-Doped Tetracene Crystals, Chem. Phys. Lett., 1971, 11(4), 504–508, DOI:10.1016/0009-2614(71)80395-0.
- J. Burgos, M. Pope, Ch. E. Swenberg and R. R. Alfano, Heterofission in Pentacene-doped Tetracene Single Crystals, Phys. Status Solidi B, 1977, 83(1), 249–256, DOI:10.1002/pssb.2220830127.
- C. Zeiser, L. Moretti, D. Lepple, G. Cerullo, M. Maiuri and K. Broch, Singlet Heterofission in Tetracene–Pentacene Thin-Film Blends, Angew. Chem., Int. Ed., 2020, 59(45), 19966–19973, DOI:10.1002/anie.202007412.
- G. He, L. M. Yablon, K. R. Parenti, K. J. Fallon, L. M. Campos and M. Y. Sfeir, Quantifying Exciton Transport in Singlet Fission Diblock Copolymers, J. Am. Chem. Soc., 2022, 144(7), 3269–3278, DOI:10.1021/jacs.1c13456.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, K. Appavoo, M. L. Steigerwald, L. M. Campos and M. Y. Sfeir, Exciton Correlations in Intramolecular Singlet Fission, J. Am. Chem. Soc., 2016, 138(23), 7289–7297, DOI:10.1021/jacs.6b00657.
- A. B. Pun, A. Asadpoordarvish, E. Kumarasamy, M. J. Y. Tayebjee, D. Niesner, D. R. McCamey, S. N. Sanders, L. M. Campos and M. Y. Sfeir, Ultra-Fast Intramolecular Singlet Fission to Persistent Multiexcitons by Molecular Design, Nat. Chem., 2019, 11(9), 821–828, DOI:10.1038/s41557-019-0297-7.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, Intramolecular Singlet Fission in Oligoacene Heterodimers, Angew. Chem., Int. Ed., 2016, 55(10), 3373–3377, DOI:10.1002/anie.201510632.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. V. Voorhis and M. A. Baldo, External Quantum Efficiency Above 100% in a Singlet-Exciton-Fission–Based Organic Photovoltaic Cell, Science, 2013, 340(6130), 334–337, DOI:10.1126/science.1232994.
- J. R. Allardice, A. Thampi, S. Dowland, J. Xiao, V. Gray, Z. Zhang, P. Budden, A. J. Petty, N. J. L. K. Davis, N. C. Greenham, J. E. Anthony and A. Rao, Engineering Molecular Ligand Shells on Quantum Dots for Quantitative Harvesting of Triplet Excitons Generated by Singlet Fission, J. Am. Chem. Soc., 2019, 141(32), 12907–12915, DOI:10.1021/jacs.9b06584.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Sensitization of Silicon by Singlet Exciton Fission in Tetracene, Nature, 2019, 571(7763), 90–94, DOI:10.1038/s41586-019-1339-4.
- V. Gray, D. T. W. Toolan, S. Dowland, J. R. Allardice, M. P. Weir, Z. Zhang, J. Xiao, A. Klimash, J. F. Winkel, E. K. Holland, G. M. Fregoso, J. E. Anthony, H. Bronstein, R. Friend, A. J. Ryan, R. A. L. Jones, N. C. Greenham and A. Rao, Ligand-Directed Self-Assembly of Organic-Semiconductor/Quantum-Dot Blend Films Enables Efficient Triplet Exciton-Photon Conversion, J. Am. Chem. Soc., 2024, 146(11), 7763–7770, DOI:10.1021/jacs.4c00125.
- E. Sundin, R. Ringström, F. Johansson, B. Küçüköz, A. Ekebergh, V. Gray, B. Albinsson, J. Mårtensson and M. Abrahamsson, Singlet Fission and Electron Injection from the Triplet Excited State in Diphenylisobenzofuran–Semiconductor Assemblies: Effects of Solvent Polarity and Driving Force, J. Phys. Chem. C, 2020, 124(38), 20794–20805, DOI:10.1021/acs.jpcc.0c06626.
- N. A. Pace, D. H. Arias, D. B. Granger, S. Christensen, J. E. Anthony and J. C. Johnson, Dynamics of Singlet Fission and Electron Injection in Self-Assembled Acene Monolayers on Titanium Dioxide, Chem. Sci., 2018, 9(11), 3004–3013, 10.1039/C7SC04688J.
- A. Kunzmann, M. Gruber, R. Casillas, J. Zirzlmeier, M. Stanzel, W. Peukert, R. R. Tykwinski and D. M. Guldi, Singlet Fission for Photovoltaics with 130 % Injection Efficiency, Angew. Chem., Int. Ed., 2018, 57(33), 10742–10747, DOI:10.1002/anie.201801041.
- A. Kunzmann, M. Gruber, R. Casillas, R. R. Tykwinski, R. D. Costa and D. M. Guldi, Tuning Pentacene Based Dye-Sensitized Solar Cells, Nanoscale, 2018, 10(18), 8515–8525, 10.1039/C8NR01502C.
- C. Hetzer, B. S. Basel, S. M. Kopp, F. Hampel, F. J. White, T. Clark, D. M. Guldi and R. R. Tykwinski, Chromophore Multiplication To Enable Exciton Delocalization and Triplet Diffusion Following Singlet Fission in Tetrameric Pentacene, Angew. Chem., Int. Ed., 2019, 58(43), 15263–15267, DOI:10.1002/anie.201907221.
- S. Nakamura, H. Sakai, H. Nagashima, Y. Kobori, N. V. Tkachenko and T. Hasobe, Quantitative Sequential Photoenergy Conversion Process from Singlet Fission to Intermolecular Two-Electron Transfers Utilizing Tetracene Dimer, ACS Energy Lett., 2019, 4(1), 26–31, DOI:10.1021/acsenergylett.8b01964.
- H. Liu, X. Wang, L. Ma, W. Wang, S. Liu, J. Zhou, P. Su, Z. Liu, Z. Li, X. Lin, Y. Chen and X. Li, Effects of the Separation Distance between Two Triplet States Produced from Intramolecular Singlet Fission on the Two-Electron-Transfer Process, J. Am. Chem. Soc., 2022, 144(34), 15509–15518, DOI:10.1021/jacs.2c03550.
- A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, D. N. Congreve and L. M. Campos, Triplet Harvesting from Intramolecular Singlet Fission in Polytetracene, Adv. Mater., 2017, 29(41), 1701416, DOI:10.1002/adma.201701416.
- J. Xia, E. Busby, S. N. Sanders, C. Tung, A. Cacciuto, M. Y. Sfeir and L. M. Campos, Influence of Nanostructure on the Exciton Dynamics of Multichromophore Donor–Acceptor Block Copolymers, ACS Nano, 2017, 11(5), 4593–4598, DOI:10.1021/acsnano.7b00056.
- L. M. Yablon, S. N. Sanders, H. Li, K. R. Parenti, E. Kumarasamy, K. J. Fallon, M. J. A. Hore, A. Cacciuto, M. Y. Sfeir and L. M. Campos, Persistent Multiexcitons from Polymers with Pendent Pentacenes, J. Am. Chem. Soc., 2019, 141(24), 9564–9569, DOI:10.1021/jacs.9b02241.
- A. J. Tilley, R. D. Pensack, E. L. Kynaston, G. D. Scholes and D. S. Seferos, Singlet Fission in Core–Shell Micelles of End-Functionalized Polymers, Chem. Mater., 2018, 30(13), 4409–4421, DOI:10.1021/acs.chemmater.8b01814.
- R. D. Pensack, C. Grieco, G. E. Purdum, S. M. Mazza, A. J. Tilley, E. E. Ostroumov, D. S. Seferos, Y.-L. Loo, J. B. Asbury, J. E. Anthony and G. D. Scholes, Solution-Processable, Crystalline Material for Quantitative Singlet Fission, Mater. Horiz., 2017, 4(5), 915–923, 10.1039/C7MH00303J.
- D. B. Sulas, A. E. London, L. Huang, L. Xu, Z. Wu, T. N. Ng, B. M. Wong, C. W. Schlenker, J. D. Azoulay and M. Y. Sfeir, Preferential Charge Generation at Aggregate Sites in Narrow Band Gap Infrared Photoresponsive Polymer Semiconductors, Adv. Opt. Mater., 2018, 6(7), 1701138, DOI:10.1002/adom.201701138.
- G. He, E. M. Churchill, K. R. Parenti, J. Zhang, P. Narayanan, F. Namata, M. Malkoch, D. N. Congreve, A. Cacciuto, M. Y. Sfeir and L. M. Campos, Promoting Multiexciton Interactions in Singlet Fission and Triplet Fusion Upconversion Dendrimers, Nat. Commun., 2023, 14(1), 6080, DOI:10.1038/s41467-023-41818-1.
- L. Navarro, A. F. Thünemann and D. Klinger, Solvent Annealing of Striped Ellipsoidal Block Copolymer Particles: Reversible Control over Lamellae Asymmetry, Aspect Ratio, and Particle Surface, ACS Macro Lett., 2022, 11(3), 329–335, DOI:10.1021/acsmacrolett.1c00665.
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Singlet Exciton Fission in Solution, Nat. Chem., 2013, 5(12), 1019–1024, DOI:10.1038/nchem.1801.
- H. Angliker, E. Rommel and J. Wirz, Electronic Spectra of Hexacene in Solution (Ground State. Triplet State. Dication and Dianion), Chem. Phys. Lett., 1982, 87(2), 208–212, DOI:10.1016/0009-2614(82)83589-6.
- L. Ma, P. Hu, C. Kloc, H. Sun, M. E. Michel-Beyerle and G. G. Gurzadyan, Ultrafast Spectroscopic Characterization of 7,7,8,8-Tetracyanoquinodimethane (TCNQ) and Its Radical Anion (TCNQ−), Chem. Phys. Lett., 2014, 609, 11–14, DOI:10.1016/j.cplett.2014.06.029.
- L. Ma, P. Hu, H. Jiang, C. Kloc, H. Sun, C. Soci, A. A. Voityuk, M. E. Michel-Beyerle and G. G. Gurzadyan, Single Photon Triggered Dianion Formation in TCNQ and F4TCNQ Crystals, Sci. Rep., 2016, 6(1), 28510, DOI:10.1038/srep28510.
- E. Kumarasamy, S. N. Sanders, A. B. Pun, S. A. Vaselabadi, J. Z. Low, M. Y. Sfeir, M. L. Steigerwald, G. E. Stein and L. M. Campos, Properties of Poly- and Oligopentacenes Synthesized from Modular Building Blocks, Macromolecules, 2016, 49(4), 1279–1285, DOI:10.1021/acs.macromol.5b02711.
- M. S. Sanford, J. A. Love and R. H. Grubbs, A Versatile Precursor for the Synthesis of New Ruthenium Olefin Metathesis Catalysts, Organometallics, 2001, 20(25), 5314–5318, DOI:10.1021/om010599r.
- Y. Kim, J. E. Whitten and T. M. Swager, High Ionization Potential Conjugated Polymers, J. Am. Chem. Soc., 2005, 127(34), 12122–12130, DOI:10.1021/ja052828x.
- C. I. Müller and C. Lambert, Electrochemical and Optical Characterization of Triarylamine Functionalized Gold Nanoparticles, Langmuir, 2011, 27(8), 5029–5039, DOI:10.1021/la1051244.
- E. A. Murphy, S. J. Skala, D. Kottage, P. A. Kohl, Y. Li, C. Zhang, C. J. Hawker and C. M. Bates, Accelerated Discovery and Mapping of Block Copolymer Phase Diagrams, Phys. Rev. Mater., 2024, 8(1), 015602, DOI:10.1103/PhysRevMaterials.8.015602.
Footnote |
| † Electronic supplementary information (ESI) available: Details of transient absorption spectroscopy, materials synthesis, and characterization. See DOI: https://doi.org/10.1039/d5sc01479d |
| This journal is © The Royal Society of Chemistry 2025 |