
Anion doping promotes electrocatalyst reconfiguration for efficient C–C bond cleavage of 4-methylcyclohexanol†
Yuhang
Wang
a,
Ying
Chen
a,
Lihao
Liu
a,
Linhan
Ren
a,
Jieyu
Wang
a,
Kai
Li
a,
Jiahui
He
a,
Suiqing
Li
a,
Jinfu
Cai
a,
Chuang
Qi
a,
Pan
Hu
a,
Yongyong
Cao
 *b,
Xing
Zhong
*a and
Jianguo
Wang
*a
*b,
Xing
Zhong
*a and
Jianguo
Wang
*a
aInstitute of Industrial Catalysis, State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310032, P.R. China. E-mail: zhongx@zjut.edu.cn; jgw@zjut.edu.cn
bCollege of Biological, Jiaxing University, Jiaxing, Zhejiang 314001, P. R. China. E-mail: cyy@zjxu.edu.cn
First published on 20th January 2025
Abstract
Electrochemical cleavage of the C–C bond is primarily employed to transform organic molecules derived from biomass into valuable short-chain chemicals. However, achieving high reaction activation for the cleavage of C–C bonds under mild conditions continues to present a significant challenge. We prepared NiX/GF (X represents doped anionic elements, S, Se, P) and obtained NiX-R/GF through reconfiguration by electrochemical activation using cyclic voltammetry. Among the prepared materials, Ni2P-R/GF exhibited favorable electrochemical activity and achieved a high yield (88.1%) of 3-methyladipic acid through the oxidation of 4-methylcyclohexanol. Comprehensive ex/in situ electrochemical experiments demonstrate that incorporating P results in a reduced reconfiguration potential and a heterogeneous interface between NiOOH and Ni2P. Density functional theory (DFT) calculations demonstrate that the enhanced performance exhibited by the reconfigured electrocatalyst is attributed to its unique geometric and electronic structural characteristics. The strategy of anion doping to enhance the electrochemical oxidation performance of anodes presents a sustainable approach for breaking C–C bonds.
Green foundation1. The present study proposes an eco-friendly electrocatalytic synthesis of 3-methyladipic acid, which is a promising alternative to conventional oxidation processes that generate polluting emissions.2. The Ni2P-R/GF electrocatalyst reported herein demonstrated remarkable efficacy in the electrooxidation of 4-methylcyclohexanol, achieving 98.0% conversion and 89.9% selectivity. The method demonstrated an efficient electrooxidation capability, which facilitated the C–C bond cleavage process to produce the desired carboxylic acid products while minimizing the emission of hazardous substances. Furthermore, the utilization of non-homogeneous catalysts facilitates the recycling of catalysts, thereby aligning with the principles of green chemistry. 3. Future research endeavours could concentrate on exploring methodologies to augment the scale of continuous flow 3-methyladipic acid production, further reducing costs and energy consumption, thereby advancing the potential future industrial applications of this chemical. |
Introduction
The catalytic cleavage of C–C bonds facilitates the efficient synthesis of novel compounds with desirable properties. However, achieving high reaction activation for the cleavage of C–C bonds under mild conditions remains a significant challenge.1 As an illustrative example, the commonly employed preparation methods for 3-methyladipic acid (3-MAA) are as follows: the use of 4-methylcyclohexanol (4-MA) as a substrate allows for the realization of C–C bond breakage through the application of nitric acid oxidation, trifluoroacetic acid oxidation, or the use of ruthenium trichloride and osmium tetrachloride as a non-homogeneous catalyst. The cleavage reaction of C–C bonds requires using reagents and catalysts that are potentially hazardous or expensive, leading to a substantial increase in the production costs of the acid (Scheme 1). Electrochemical synthesis is an efficient, environmentally friendly, and controllable method for organic synthesis.2,3 A substantial body of evidence from numerous studies has demonstrated that transition metal catalysts, due to their rich electronic structures, enhance the covalent bonding between the transition metal and oxygen. Consequently, they have been widely employed in organic electrosynthesis.4–6 More recently the electrochemical cleavage of C–C bonds has been reported, primarily for the conversion of biomass-derived organic molecules, such as cyclohexanol, into high-value short-chain chemicals.7–9 Nevertheless, the coexistence of purely chemical and electrochemical steps in the oxidation of cyclohexanol, combined with the slow reaction kinetics associated with the multi-electronic process from secondary alcohol to carboxylic acids, presents a significant limitation to the practical application of this approach.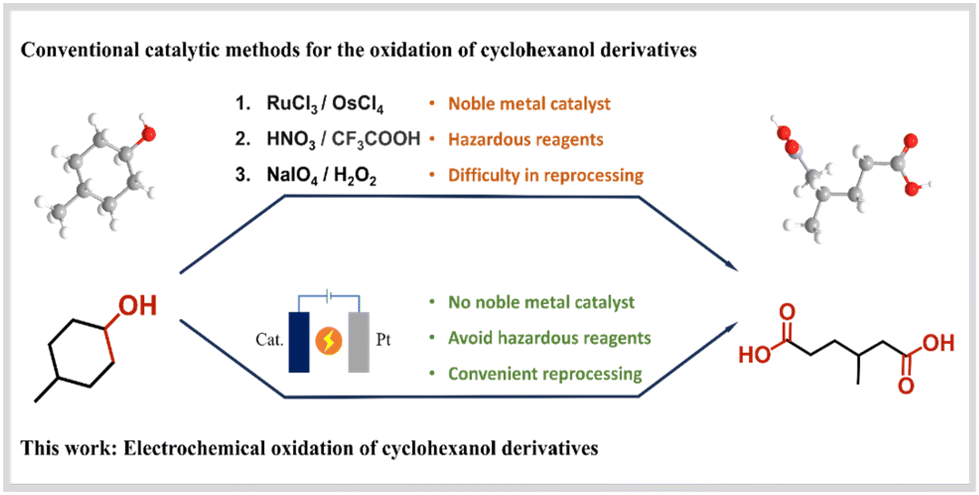 | ||
| Scheme 1 Schematic comparison of 3-methyladipic acid production pathways. | ||
Therefore, it is crucial to develop highly active, low-cost catalysts and to employ in situ electrochemical methods to elucidate the reaction mechanism of intermediates on the electrode surface and in solution. Nickel-based electrocatalysts are extensively utilized in oxygen evolution and nucleophilic oxidation reactions due to their low price and high performance.10,11 Various modification design methods for nickel-based electrocatalysts have been widely reported. Both cation and anion doping have emerged as effective strategies for the rational design of these electrocatalysts.12,13 Ion doping can result in the generation of oxygen vacancies, the occurrence of phase transitions, the exposure of active sites, and the modulation of the electronic structure of metal centers.14–16 In these reports, ion-regulated Ni-based electrocatalysts served as the initial electrocatalysts, which subsequently underwent an irreversible reconfiguration process to form nickel oxyhydroxide (NiOOH), which is generally considered to be the active catalytic species.17,18 Despite their similar chemical compositions, Ni-based electrocatalytically reconstituted NiOOH, which is derived from ionic modulation, typically demonstrates better catalytic activity when compared to directly synthesized NiOOH.19,20 In recent years, electrocatalytic oxidation of 4-MA has been reported (Table S1†); however, the effects of doped anions and recombinant electrocatalysts on C–C bond cleavage remain to be explored. Consequently, it is essential to investigate the mechanism underlying the conversion of 4-MA over reconstituted anion-doped Ni-based electrocatalysts.
This study initially investigates the effects of modifying nickel-based catalysts with various anions (P, S, Se) on their properties related to the electrooxidation of 4-MA. Ni-based electrocatalysts were synthesized on graphite felts (GF) using a one-step hydrothermal method, with the anions doped into the Ni-based catalysts (Ni(OH)2, Ni2P, NiS2, NiSe) through calcination in a tube furnace. Subsequently, the reconfigured nickel-based electrocatalysts (Ni(OH)2-R, Ni2P-R, NiS2-R, NiSe-R) were produced by in situ anodic electrochemical reconfiguration. This process results in the formation of an NiOOH layer on the surface of the electrocatalyst, subsequently enhancing the electrocatalytic activity. Electrochemical experiments demonstrated that Ni2P-R exhibits high reactivity (3-MAA yields of 88.1%, at 1.55 V vs. RHE) among various samples featuring the same active center, Ni3+. In situ Raman spectroscopy demonstrated that anionic doping optimized the local ligand environment, making it possible to make the catalyst more active through the electrochemical reconfiguration method, thereby improving its oxidation characteristics. In situ electrochemical infrared spectroscopy was employed to elucidate the changes in the surface groups of the catalyst during the reaction process, offering insight into the underlying reaction mechanism. Furthermore, experimental results showed that the reconfigured Ni2P-R catalysts feature a distinct heterojunction interface. Subsequent density functional theory (DFT) calculations revealed that the unique geometric and electronic properties of the heterojunction influence the adsorption and desorption behavior, thereby enhancing catalytic reactivity. These insights can inform the development of surface modulation techniques to enhance the activity of electrocatalysts employed in 4-MA oxidation, extending beyond the application of nickel-based electrocatalysts.
Experimental
Pre-treatment of graphite felt
The graphite felt (GF) was sliced into pieces measuring approximately 3 cm × 4 cm, and then these GF pieces were placed in a muffle furnace and heated to 500 °C for 30 min.Preparation of Ni(OH)2/GF
To prepare Ni(OH)2/GF, Ni(NO3)2·6H2O (2 mmol), CO(NH2)2 (1 mmol), and NH4F (1 mmol) were dissolved in 80 mL of DI water. After the addition of the treated GF, the mixture was homogeneously dispersed by ultrasound and transferred to a PTFE liner. Ni(OH)2/GF was obtained by hydrothermally heating the dispersion at 120 °C for 12 hours.Preparation of NiX/GF (X = P, S, or Se)
To synthesize Ni2P/GF, Ni(OH)2/GF was put next to sodium hypophosphite monohydrate (NaH2PO2, 600 mg) under an Ar atmosphere in the middle of a tube furnace. After flushing with Ar for ∼30 min, the sample was heated to 300 °C with a heating rate of 5 °C min−1 for 1 h and then programmed to cool to ambient temperature. The other catalysts were synthesized in a similar way to that of the above procedure, except that the anionic precursors for the tube furnace heating were S and Se powders. As a comparison, the anion-undoped Ni(OH)2-H/GF was obtained by direct heating in a tube furnace without any anion source.Synthesis of NiX-R/GF
NiX-R/GF was prepared by electrochemical activation of the NiX/GF samples via application of cyclic voltammetry (CV) in 1 M KOH medium at 100 mV s−1 from 1.0–1.6 V vs. RHE for 200 cycles without iR compensation. NiX/GF was the working electrode, while a Pt plate acted as the counter electrode, and Hg/HgO was utilized as the reference electrode to establish a three-electrode system.Structural characterizations
The morphology and microstructure of these electrocatalysts were investigated by scanning electron microscopy (SEM, Hitachi FE-SEM S-4700) and scanning transmission electron microscopy (TEM, JEM-ARM300F). Powder X-ray diffraction (XRD) was conducted using an X-ray diffractometer with graphite monochromatized Cu Kα irradiation (λ = 1.54 Å). Based on the XRD results, the corresponding cell parameters of each sample were calculated using Jade software. X-ray photoelectron spectroscopy (XPS) data were recorded with an X-ray photoelectron spectrometer (Thermo Scientific ESCALAB 250Xi). The Brunauer–Emmett–Teller (BET) specific surface areas and pore distributions were carried out on an ASAP2460 analyzer. In situ Raman spectra were characterized utilizing the confocal Raman imaging microscope (Renishaw InVia, 532 nm laser) under different potentials (1.20–1.70 V vs. RHE) employing an Ivium-n-Stat electrochemical workstation in a three-electrode system. Inductively coupled plasma optical emission spectrometry (ICP-OES) data were obtained using the Agilent720ES.Results and discussion
Morphology analysis of electrocatalysts
The Ni(OH)2/GF electrocatalyst precursor was synthesized on GF using a one-step hydrothermal method. The subsequent calcination of the NiX/GF electrocatalyst in a tube furnace utilized S, Se powder, and Na2HPO2 particles as anion sources (Fig. 1a). Scanning electron microscopy (SEM) images revealed that the Ni(OH)2/GF precursor exhibited a dense nanosheet structure, in contrast to the smooth surface of the GF (Fig. S1 and S2†). The morphology of NiSe/GF closely resembled that of Ni(OH)2/GF. In contrast, NiS2/GF displayed multiple holes on the nanosheet surfaces, while Ni2P/GF exhibited fine whisker-like structures at the nanosheet edges (Fig. 1b–d). These structural variations may enhance the accessibility of the electrocatalytic sites. Furthermore, energy-dispersive X-ray spectroscopy (EDS) mapping confirmed that the predominant elements Ni, X, and O (X = S, Se, P) were uniformly distributed across the nanosheets, indicating the successful loading of NiX (X = P, S, Se) onto the GF (Fig. 1e–g). | ||
| Fig. 1 (a) Schematic illustration of the synthesis route for the Ni2P/NiS2/NiSe. All the experimental procedures are kept the same except for the anion source. SEM images of Ni2P/GF (b), NiS2/GF (c) and NiSe/GF (d). Elemental mapping of Ni2P/GF (e), NiS2/GF (f) and NiSe/GF (g). | ||
The structures of the electrocatalysts were further characterized using transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). TEM images revealed that NiX/GF exhibited a lamellar structure, consistent with the SEM results (Fig. S3†). However, the TEM images of different anion-doped electrocatalysts revealed significant differences. Ni2P/GF displayed a sheet-like structure with Ni2P distributed across the surface. TEM images of NiS2/GF revealed nanoparticles aggregated into nanosheets, while those of NiSe/GF displayed a greater number of nanoparticles and nanowires at the edges of the nanosheets (Fig. S4†). HRTEM revealed lattice fringes with a spacing of 0.221 nm on the Ni2P/GF surface,21–23 corresponding to the (111) lattice plane of Ni2P (Fig. 2a). Similarly, in NiS2/GF, lattice fringes with spacings of 0.232 nm and 0.254 nm were observed, which were associated with the (121) and (−201) lattice planes of NiS2, respectively (Fig. 2b).24 In the case of NiSe/GF, distinct lattice fringes were only observed on the nanoparticles at the edges of the nanosheets, exhibiting a pitch of 0.272 nm, which corresponds to the (101) crystal plane of NiSe (Fig. 2c).25 These observations indicate the successful synthesis of Ni2P, NiS2, and NiSe on GF.
 | ||
| Fig. 2 (a–c) HRTEM images of Ni2P/GF (a), NiS2/GF (b), and NiSe/GF (c). (d) XRD patterns for Ni2P, NiS2, NiSe, Ni(OH)2, and Ni(OH)2-H. (e) N2 adsorption–desorption isotherms of the Ni2P, NiS2, NiSe, and Ni(OH)2 catalysts. (f) Cdl calculations of Ni2P/GF, NiS2/GF, NiSe/GF, and Ni(OH)2/GF. High-resolution XPS spectra of Ni 2p levels for Ni2P (g), NiS2 (h), and NiSe (i). | ||
The composition of the electrocatalysts was analyzed using X-ray diffraction (XRD). The pronounced diffraction peaks of the precursor Ni(OH)2 at 12.1°, 23.8°, 33.7°, 35.2°, and 59.6° correspond to the standard pattern of α-Ni(OH)2 (JCPDS No. 00-022-0444), representing the (001), (002), (110), (111), and (300) lattice planes of the electrocatalyst, respectively. Ni(OH)2-H was obtained by calcining the precursor Ni(OH)2 in a tube furnace without an anion source. The original diffraction peak at 12.1° shifts to a higher angle, the diffraction peak at 23.8° disappears, and the diffraction peaks at 33.7° and 59.6° exhibit increased intensity (Fig. 2d). These changes indicate a reduction in crystal plane spacing and an increase in crystallinity during the calcination process. Meanwhile, the characteristic peaks of Ni(OH)2-H were identified in the Ni2P, NiS2, and NiSe samples. For example, in the XRD pattern of Ni2P, the diffraction peaks at 40.7°, 44.6°, and 47.4° correspond to the (111), (201), and (210) crystal planes of Ni2P (JCPDS No. 01-074-1385), indicating the presence of diffraction peaks of both substances in the Ni2P electrocatalysts. Similarly, for NiS2 and NiSe, the diffraction peaks align with the standard patterns of Ni(OH)2-H, NiS2, and NiSe, respectively (Fig. 2d and Fig. S5†). These results suggest that incomplete phase transitions may occur during the synthesis of NiX/GF (where X = P, S, and Se), leading to the formation of an NiX–Ni(OH)2 hybrid phase. However, no lattice fringes related to Ni(OH)2 were observed in HRTEM. This absence was attributed to XRD primarily detecting the bulk phase of the catalyst, resulting in limited exposure of surface Ni(OH)2 crystallites, or it may be due to a decrease in Ni(OH)2 crystallinity after annealing, which manifests as an amorphous layer on the catalyst surface.
To evaluate the specific surface area and pore structure of the electrocatalysts, Brunauer–Emmett–Teller (BET) testing and pore size distribution analysis using the Barrett–Joyner–Halenda (BJH) method were performed. The results indicate that Ni(OH)2, NiSe, NiS2, and Ni2P exhibit type IV isotherms featuring prominent H3-type hysteresis loops and no discernible saturated adsorption platforms, signifying irregular pore structures (Fig. 2e). Pore size distribution ranging analysis reveals that the pore sizes of Ni(OH)2, NiSe, NiS2, and Ni2P are predominantly mesoporous, with distribution of between 2 and 50 nm (Fig. S6†). Furthermore, the electrochemical surface area (ECSA) was evaluated based on the calculated electrochemical double layer capacitance (Cdl) in the non-faradaic regions using cyclic voltammetry (Fig. S7†). As illustrated in Fig. 2f, the Cdl of Ni(OH)2 is 10.9 mF cm−2, considerably higher than that of NiSe (6.1 mF cm−2), NiS2 (3.8 mF cm−2), and Ni2P (3.5 mF cm−2). This suggests that the active electrochemical region is diminished as a result of anionic doping in the Ni(OH)2 material, which aligns with the BET results. The variations in the specific surface area, pore size, and active surface area of anion-doped catalysts may lead to differences in reactivity.
X-ray photoelectron spectroscopy (XPS) was utilized to determine the chemical composition of the surfaces of Ni2P, NiS2, NiSe, and Ni(OH)2 samples and to evaluate the influence of anion doping on the electronic structure of these materials. The analysis of the samples indicated the presence of the elements Ni, P, S, and Se (Fig. S8†). Additionally, the high-resolution Ni 2p spectra (Fig. 2g–i) demonstrated that anion-doped nickel catalysts, as determined through deconvolution, commonly exhibit six primary peaks along with two satellite peaks (labeled as “Sat.”).26 In the case of Ni2P, the peaks with binding energies (BE) of 856.9 and 874.6 eV correspond to Ni 2p3/2 and Ni 2p1/2 of Ni2+; the peaks at 859.1 and 876.7 eV are attributed to Ni3+ resulting from oxidation on the Ni2P surface; while the two peaks at 853.1 and 870.7 eV are associated with Niδ+ (δ close to 0).27–29 The binding energies of the corresponding peaks derived from the deconvolution of NiS2, NiSe, and Ni(OH)2 in the high-resolution spectra of Ni 2p are documented in Table S2.† A comparison of the peak positions indicates that the peaks corresponding to Ni2+ and Ni3+ shift towards higher binding energies following P elemental doping. Notably, the Niδ+ peaks appearing after the introduction of P indicate an enhancement of the electronic coupling of Ni–P. Furthermore, in comparison to S and Se doping, P doping resulted in more significant shifts in the binding energy of Ni2+ and stronger Niδ+ peaks. This suggests that P doping has a greater influence on the electronic structure of Ni, thereby promoting electron transfer and enhancing the proton affinity of the electrocatalysts. Additionally, the anions were characterized using XPS (Fig. S9†). In the P 2p spectrum, the characteristic peaks at 129.2 eV and 130.5 eV correspond to the 2p3/2 and 2p1/2 orbitals of phosphorus, respectively. Furthermore, the peaks at 133.8 eV correspond to phosphorus oxides.30 In the S 2p spectrum, the characteristic peaks at 162.6 eV and 163.8 eV represent the 2p1/2 and 2p3/2 orbitals of sulfur, which align well with the values for metal-coordinated S22− dimers.31,32 In the Se 3d spectrum, the Se 3d3/2 and Se 3d5/2 peaks at 54.2 eV and 55.2 eV suggest the presence of Se2− ions, while the peak near 59.0 eV corresponds to SeOx species.33 The binding energies of P 2p3/2, S 2p3/2, and Se 3d5/2 are shifted by 0.7 eV, 0.2 eV, and 0.4 eV, respectively, compared to the standard values in the XPS handbook, which also indicates the presence of Ni–P, Ni–S, and Ni–Se interactions, and the binding energies of P have larger shifts in agreement with the above findings. The shift of the binding energy to a higher level indicates a reduction in electron density near the Ni atom. This change enhances the electron affinity of the Ni atom, making it more favorable for the rapid adsorption of substrate intermediate radicals.
Morphology analysis of reconfigured electrocatalysts
The surface reconfiguration of NiX/GF was accomplished through cyclic voltammetry (CV) over 200 cycles in a 1.0 M KOH electrolyte, utilizing a sweep rate of 100 mV s−1, across a potential range of 1.0–1.6 V vs. RHE. The reconfigured electrocatalysts were designated as NiX-R/GF. Fig. S10† illustrates the variation of CV curves from the 5th to the 200th cycle. The area of the Ni oxidation peaks (Ni2+ to Ni3+) and the oxygen evolution reaction (OER) currents increased with the number of cycles. Furthermore, peak cathodic current (Ipc) and peak anodic current (Ipa) were extracted at different cycles and at a voltage of 1.5 V vs. RHE to illustrate their evolution (Fig. 3a and Fig. S11†). All NiX/GF samples exhibited a similar trend, wherein the oxidation and reduction currents gradually increased before reaching equilibrium. Initially, the current decreased and then increased, likely due to the oxidation of P and S monomers on the surface. Notably, Ni2P achieved a stable current density exhibiting a higher anodic current peak and a lower peak cathodic current with fewer CV cycles compared to NiSe, NiS2, and Ni(OH)2. This suggests that Ni2P undergoes the reconstruction process more rapidly and possesses enhanced electrochemical activity.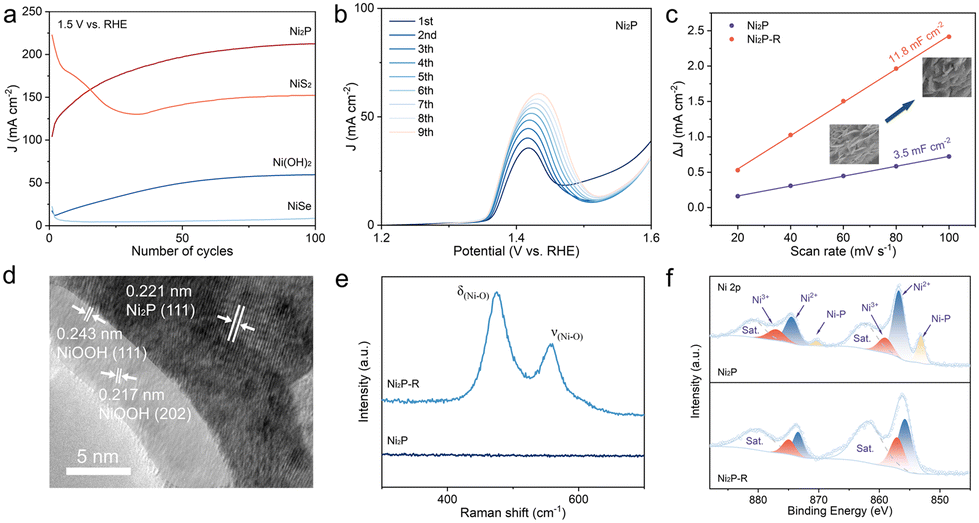 | ||
| Fig. 3 (a) Current variation curves during electrochemical activation at a voltage of 1.5 V vs. RHE. (b) LSV curves of Ni2P in 1 M KOH. (c) Cdl calculations of Ni2P and Ni2P-R. (d) HRTEM image of Ni2P-R. (e) Raman spectrogram and (f) high-resolution XPS spectra of Ni 2p levels for Ni2P and Ni2P-R. | ||
To achieve a more accurate understanding of the changes in electrochemical properties influenced by anions during the reconfiguration process, multiple linear scanning voltametric curves of Ni2P, NiS2, NiSe and Ni(OH)2 were recorded in 1 M KOH solution at a scan rate of 5 mV s−1 (Fig. 3b and Fig. S12†). The oxidation peaks of the anion-modified catalysts exhibited a forward shift and a gradual increase compared to those of Ni(OH)2, indicating a reduction in the potential required for generating active species. Moreover, the electrochemical surface area (ECSA) was assessed by comparing the Cdl within the non-faradaic zone, calculated through CV (Fig. S13†). As illustrated in Fig. 3c, the Cdl of Ni2P-R/GF was 11.8 mF cm−2, whereas that of Ni2P/GF before activation was merely 3.5 mF cm−2, representing a 3.4-fold enhancement. This suggests that the electrochemical activation process resulted in a significantly larger active surface area for the activated electrocatalyst. The inset in Fig. 3c presents SEM images of Ni2P before and after activation, demonstrating that the surface of the nanosheets displays a more refined lamellar structure. A similar structure was also observed on the surface of NiS2-R (Fig. S14†). This structure may facilitate the formation of additional active sites on the electrocatalysts, thereby enhancing mass transfer between the electrocatalysts and the reactant molecules.
In the high-resolution transmission electron microscopy (HRTEM) of Ni2P-R, a less crystalline NiOOH layer was observed at the edges of the nanosheets, displaying lattice fringes of 0.243 nm and 0.217 nm corresponding to the (111) and (202) crystalline planes of NiOOH, respectively (JCPDS No. 97-016-5961) (Fig. 3d).34 In addition, HRTEM reveals the presence of a distinct NiOOH/Ni2P heterogeneous interface, which could potentially influence the electrochemical performance of the reconfigured catalysts.35,36 The Raman spectra of Ni2P and Ni2P-R, before and after activation, exhibits two signal peaks at 471.5 cm−1 and 552.4 cm−1 in the activated Ni2P-R, corresponding to the bending vibration (δNi–O) and the stretching vibration (νNi–O) of Ni3+–O in NiOOH, respectively (Fig. 3e).37–39 In the high-resolution Ni 2p spectra, the peak corresponding to Ni–P was no longer present in the activated Ni2P-R, indicating the precipitation of anions on the surface. Furthermore, the proportion of the Ni3+ peak area increased significantly from SNi3+/SNi2+ = 0.28 to 0.60 (Fig. 3f). These findings suggest that anion-doped electrocatalysts generate a substantial quantity of Ni3+ on the surface following electrochemical activation, which is believed to enhance electrochemical activity by serving as an active site for oxidation reactions. In addition, in the high-resolution O 1s spectra of Ni2P-R (Fig. S15a and b†), there are four fitted peaks at binding energies of 529.4, 530.6, 532.0 eV, and 535.2 eV corresponding to the peaks of lattice oxygen, metal hydroxide, adsorbed oxygen, and adsorbed water, respectively, and the extra peaks of lattice oxygen indicate the formation of NiOOH compared to that of unreconstructed Ni2P.40 In addition, in P 2p of Ni2P-R, the Ni–P peak disappeared and the P–O bond still existed (Fig. S15c†), which indicated that Ni2P on the catalyst surface was transformed into NiOOH during electrochemical reconstruction, and that phosphorus oxide species existed on the surface although part of elemental P precipitated out. A small amount of POx doping is beneficial to optimize the coordination environment near the Ni site to enhance the oxidation capacity.19 Inductively coupled plasma optical emission spectrometry (ICP-OES) measurements were carried out on the electrolyte after CV electrolysis of Ni2P (Table S3†). The results showed that the P content in the electrolyte was 29.6198 mg L−1 and the Ni content was 0.1851 mg L−1, which indicated that the catalyst reconstruction process was accompanied by the leaching of P, while Ni was hardly lost.
Electrochemical and reactive properties
LSV tests were conducted on reconstituted NiX-R in 1.0 M KOH solution at a sweep rate of 5 mV s−1. The results demonstrated that Ni2P-R/GF exhibited a higher current density than other reconstituted electrocatalysts, regardless of the presence or absence of the substrate 4-MA. Additionally, the oxidation potential of Ni2P-R/GF was significantly advanced, indicating enhanced reactivity and reduced energy requirements in the electrochemical oxidation of 4-MA. This advancement suggests that Ni2P-R/GF promotes the oxidation reaction at a lower voltage, thereby decreasing energy consumption, which is a notable advantage for Ni2P-R in the electrochemical oxidation of 4-MA (Fig. 4a, b and Fig. S16†). Upon the addition of 4-MA, two distinct oxidation peaks were observed in the LSV curves of Ni2P-R at voltages of 1.45 V and 1.55 V vs. RHE. It is hypothesized that these peaks correspond to the oxidation of Ni2+ to Ni3+ and the subsequent oxidation of the substrate by the Ni3+ active site, respectively. As illustrated in Fig. 4c, the Nyquist plots in 1.0 M KOH solution indicate that the plots for Ni(OH)2-R at 1.50 V vs. RHE are close to being approximately linear. This signifies that the electrode processes on the surface of Ni(OH)2-R are predominantly governed by diffusion, resulting in sluggish reaction kinetics. In contrast, the Nyquist plots for Ni2P-R display a semicircular shape, indicating that the rate-limiting step in the electrode process is a charge transfer step. This suggests that reactants can rapidly reach the electrode surface, indicating that Ni2P-R exhibits superior electrochemical reactivity compared to Ni(OH)2-R. Furthermore, a comparison of the Nyquist plots before and after the addition of 4-MA demonstrates that the substrate addition leads to a reduction in the radius of the semicircle, indicating that the substrate undergoes rapid charge transfer on the electrocatalyst's surface. This observation suggests that the oxidation reaction of 4-MA is more electrochemically favorable than the OER. A comparison of the Nyquist plots for NiSe-R, NiS2-R, and Ni2P-R in 1 M KOH with 4-MA at 1.50 V vs. RHE indicates that Ni2P-R exhibits a smaller semicircular radius (Fig. S17†), signifying lower charge transfer resistance at the Ni2P-R surface in the presence of the substrate. This finding is favorable for the reaction. To further understand the role of anionic modification, the Tafel slopes on Ni(OH)2-R and anion-modified Ni-based catalysts were measured for analyzing the kinetics of the OER and the 4-MA oxidation reaction. Notably, the current density within the Tafel region during LSV testing increases with potential due to the oxidation of Ni2+ to Ni3+. To minimize the interference of Ni3+ oxidation on the Tafel slope measurement of 4-MA, the catalyst was first oxidized at 1.8 V vs RHE for 10 min and then LSV tested in reverse scan mode (Fig. S18a and b†), thereby deriving the Tafel slope.41 In the absence of substrate, the Tafel slope of Ni2P-R (41.1 mV dec−1) was lower than that of the other catalysts, indicating that P modification enhances the kinetics of the OER (Fig. S18c†). After the introduction of substrate, the Tafel slope on Ni2P-R (32.3 mV dec−1) was lower than that on Ni(OH)2-R (82.8 mV dec−1), suggesting that the effect of P-modification on the kinetics of the 4-MA oxidation reaction was more pronounced than that on the OER (Fig. S18d†). These findings imply that while P modification promotes the OER to some extent, it significantly accelerates the kinetics of the 4-MA oxidation reaction. | ||
| Fig. 4 (a) LSV curves of Ni2P-R/GF, NiS2-R/GF, NiSe-R/GF, and Ni(OH)2-R/GF in 1 M KOH. (b) LSV curves of Ni2P-R/GF in 1 M KOH with or without 4-MA. (c) Nyquist plots of Ni(OH)2-R and Ni2P-R in 1 M KOH and Ni2P-R in 1 M KOH with 4-MA. (d) HPLC yields of 3-MAA with different oxidation potentials for 2 h. (e) Recycling experiments of Ni2P-R in 1 M KOH with 10 mM 4-MA solution at 1.55 V vs. RHE for 2h. (f) Relative concentration of the reaction intermediates and products during the reaction at 1.55 V vs. RHE using Ni2P-R/GF electrodes. | ||
The oxidation reaction of 4-MA was conducted using the prepared electrocatalysts, and the yield of 3-MAA was quantified using high-performance liquid chromatography with a refractive index detector (HPLC-RID) (Fig. S19†). The reaction conditions were evaluated with Ni2P-R/GF as the electrocatalyst, applying voltages ranging from 1.40 to 1.70 V vs. RHE in increments of 0.05 V, for two hours at each potential, with a deviation of the results of less than 5%. The results showed that the yield of 3-MAA increased with increasing voltage. However, the faradaic efficiency (FE) exhibited a distinct volcano trend, initially rising and then declining. The Ni2P-R/GF achieved higher liquid-phase yield and faradaic efficiency at 1.55 V, indicating that this voltage is the optimal reaction voltage (Fig. 4d). Additionally, the effects of reaction temperature, electrolyte composition, and electrolyte concentration were investigated (Fig. S20† and Table S4†). The optimal conditions were established as 1.55 V vs. RHE, a reaction temperature of 40 °C, and 1 M KOH as the electrolyte. Under these conditions, a comparison of various electrocatalysts revealed that the reconstituted catalysts yielded higher 3-MAA production quantities, as illustrated in Fig. S21.† Among these, Ni2P-R achieved a higher yield of 58.9%, surpassing the performance of the other electrocatalysts. The Ni2P-R demonstrated superior reactivity compared to Ni(OH)2-R, NiS2-R, and NiSe-R. Additionally, the stability of Ni2P-R was evaluated through cycling experiments. The yield decreased after every three cycles; however, its reaction performance was partially restored through reactivation via cyclic voltammetry. This indicates that the reconstituted catalyst possesses high stability, and its activity can be revived using a straightforward activation method (Fig. 4e). Due to the difficulty in detecting 4-MA and 4-methylcyclohexanone (4-MK) via HPLC, nuclear magnetic resonance (NMR) spectroscopy was employed to monitor the trends of substrate, products, and by-products using deuterated-dimethyl sulfoxide (DMSO) as an internal standard (Fig. S21 and S22†). The accumulation of 4-MK intermediates during the reaction was evident, alongside the production of 3-MGA and 3-MAA. At a total charge of 300 C, 4-MA was nearly completely converted (98.0%), while 3-MAA achieved a higher yield (88.1%) and selectivity (89.9%). Consequently, the reconstructed electrocatalysts doped with phosphorus demonstrate exceptional electrochemical oxidation performance for 4-MA. However, the source of their activity remains uncharacterized, necessitating an exploration of its origin through a series of in situ electrochemical techniques.
In situ experiments to reveal reaction mechanisms
To provide a more detailed illustration of the reconfiguration phenomena occurring on the Ni2P/GF surface, in situ Raman analysis was employed. Fig. 5a illustrates the in situ electrochemical Raman spectra of Ni(OH)2 in 0.5 M KOH recorded between 1.20 V and 1.70 V vs. RHE. Before reaching 1.40 V, no discernible Raman peaks were observed within the range of 200–1200 cm−1. Upon reaching 1.40 V, two new peaks emerged at 471.5 cm−1 and 552.4 cm−1, exhibiting increased sharpness. The peaks observed at 1.45 V are attributed to bending vibrations (δNi–O) and stretching vibrations (νNi–O). As the potential was increased to 1.65 V, the intensity of these peaks diminished, and at 1.70 V, they almost disappeared. This attenuation can be attributed to the OER, which generates bubbles on the electrocatalyst surface, thereby weakening the Raman signal. The electrochemical Raman experiments conducted on Ni2P yielded comparable results to those observed for Ni(OH)2 under identical conditions. This suggests that Ni2P undergoes reconstruction at lower potentials than Ni(OH)2, indicating that doping with phosphorus facilitates the reconstruction process. The OER at 1.60 V results in the formation of bubbles that interfere with the Raman signal, as evidenced by the disappearance of the carbon peaks at 1348 cm−1 and 1598 cm−1.42 Cyclic voltammetry activation in the 1.00–1.60 V vs. RHE potential range, in conjunction with in situ Raman testing (Fig. S24†), demonstrates that the characteristic peaks corresponding to Ni3+–O appear after fewer cycles and stabilize at around 100 cycles. This trend is consistent with the CV curves, indicating that this electrochemical activation process effectively generates Ni3+ active sites on the electrocatalyst surface. Two potential reaction mechanisms have been identified for the nucleophilic oxidation of transition metal-based electrocatalysts. The initial mechanism proposes direct oxidation facilitated by adsorbed hydroxyl groups.43,44 In contrast, the second mechanism involves an indirect oxidation process, in which the nucleophilic reagent undergoes a spontaneous reaction with the transition metal (oxygen) hydroxide.45–47 To demonstrate the spontaneous reactivity of Ni3+ toward the substrate, in situ electrochemical Raman experiments were conducted in 0.5 M KOH solution supplemented with 4-MA. The circuit was activated at 1.50 V for 200 seconds to excite Ni3+ on the catalyst surface. Subsequently, the circuit was disengaged, and the Raman spectrum of the material was recorded at 100-second intervals. The characteristic peaks at 471.5 cm−1 and 552.4 cm−1 were observed to diminish over time, essentially disappearing after approximately 600 seconds. Conversely, the same experiment conducted in a solution-free substrate demonstrated that the diffraction peaks remained unaltered after 600 seconds (Fig. S25†). This outcome suggests that Ni3+ exhibits spontaneous reactivity towards 4-MA.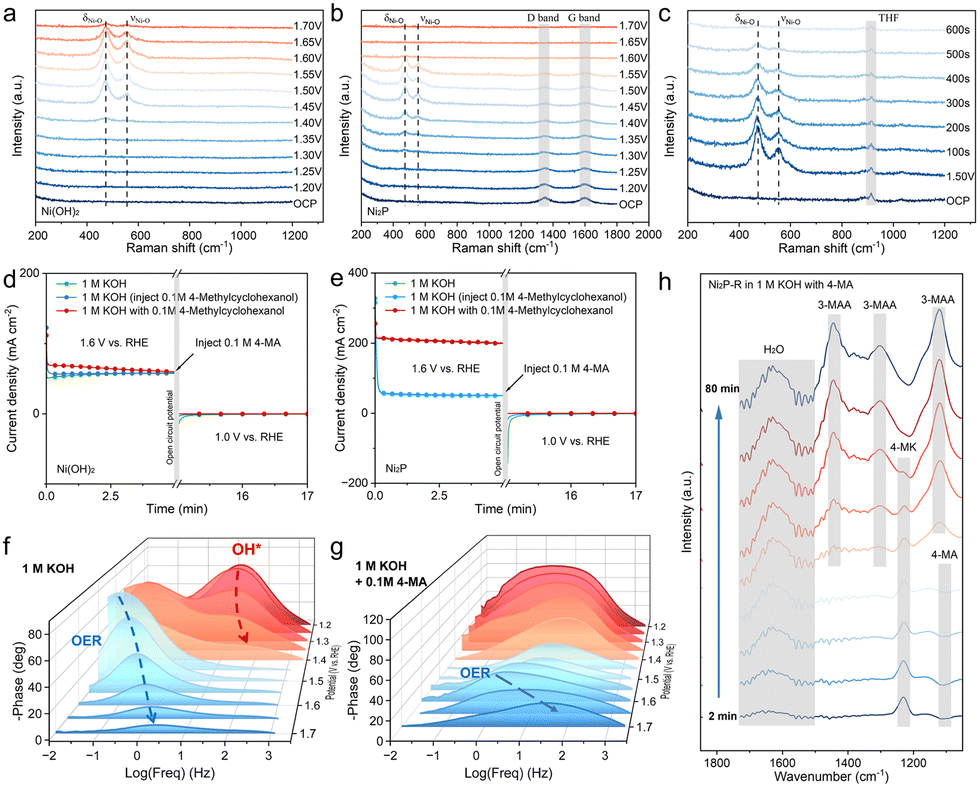 | ||
| Fig. 5 In situ Raman spectra for (a) Ni(OH)2 and (b) Ni2P in 0.5 M KOH. (c) In situ Raman spectra for Ni2P in 0.5 M KOH with 0.1 M 4-MA. Multi-potential step experiments for (d) Ni(OH)2 and (e) Ni2P, consisting of three stages: (1) 1.6 V vs. RHE to enrich NiOOH; (2) OCP to detect the possible reaction between NiOOH and 4-MA; and (3) 1.0 V vs. RHE to observe the reduction of NiOOH. (f) Bode plots of Ni2P for the OER and (g) Ni2P-R for 4-MA oxidation at different potentials. (h) In situ FT-IR spectra of 4-MA oxidation on Ni2P-R at 1.5 V vs. RHE. | ||
The distinctive peak observed at 914 cm−1 in Fig. 5c is attributed to tetrahydrofuran (THF).48 The addition of THF was performed to enhance the aqueous solubility of 4-MA, thereby facilitating its contact with the catalyst surface. However, in situ Raman experiments on Ni2P in 0.5 M KOH with 0.1 M substrate solution demonstrated that the peak of NiOOH does not disappear and that its appearance is delayed during the reaction (Fig. S26†). This phenomenon may be attributed to the low solubility of the substrate in the solution. Only a limited quantity of substrate is adsorbed on the catalyst surface, and the regeneration rate of NiOOH is considerably faster than the consumption rate. However, a delay in the decrease of the NiOOH peak potential was observed, indicating that the addition of the substrate may have inhibited the OER. Consequently, the oxidation of 4-MA occurring on the NiOOH surface may be more favorable compared to the OER.
The reaction mechanism of NiOOH-electrocatalyzed 4-MA was further investigated using the multiple potential step (STEP) method (Fig. 5d and e). Initially, an oxidation potential of 1.6 V vs. RHE, which exceeds the potential for the oxidation of Ni2+ to Ni3+, was applied to the prepared electrocatalysts (Ni(OH)2-R and Ni2P-R) in 100 mL of 1 M KOH solution, resulting in the formation of NiOOH on the catalyst surface. Subsequently, the applied potential was converted to an open circuit potential (OCP) and 0.1 M substrate was added to the solution. Finally, the voltage was set to 1.0 V vs. RHE, causing NiOOH to be electrochemically reduced to Ni(OH)2. During this period, the potential reaction between the active substance and the substrate was investigated. As illustrated in Fig. 5a, the reduction current response at 1.0 V with the addition of the substrate exhibited a diminished response compared to the reduction current response without the substrate during the OCP phase, indicating that the substrate was partially consumed. Previous studies have suggested that this phenomenon may be attributed to the trapping of protons (HNu) and electrons (eNu−) from the substrate by NiOOH. The following process occurs:49,50
| NiOOH + HNu + eNu− = Ni(OH)2 |
A series of analogous experiments were conducted on Ni2P-R, yielding similar conclusions to those observed in the Ni(OH)2-R experiments (Fig. 5e). Furthermore, the STEP method performed with the substrate in solution resulted in a notable enhancement in current at 1.60 V for the Ni2P-R catalyst compared to Ni(OH)2-R. Additionally, the reduction current disappeared at 1.0 V after OCP, indicating heightened reactivity toward 4-MA.
To investigate the catalytic kinetics and surface behavior of the catalysts in OER and the oxidation of 4-MA, in situ electrochemical impedance spectroscopy (EIS) tests were conducted on Ni2P both in the presence and absence of substrate in 1.0 M KOH. The response in the high-frequency region was associated with the surface electrooxidation of the electrocatalyst, while the response in the low-frequency region was attributed to a non-uniform charge distribution, possibly caused by the presence of surface-adsorbed oxidizing substances. Bode plots at voltages ranging from 1.20 to 1.70 V vs. RHE, without substrate addition, exhibit a distinct response peak in the high-frequency region between 1.20 and 1.40 V, which diminishes with increasing voltage. This response may correspond to the adsorption and oxidation of OH* on the electrocatalyst surface, facilitating the generation of Ni3+ After the voltage reaches 1.45 V, an electrochemical response peak appears in the low-frequency region and shifts towards the high-frequency region as the voltage increases, accompanied by a decrease in the phase angle. The response occurring beyond 1.45 V is regarded as the onset of the OER.10 Nyquist plots reveal a straight line, suggesting diffusion control, observed up to 1.40 V. A semicircle indicative of charge transfer control appears at 1.40 V, accompanied by a slanted straight line that indicates diffusion control. Beyond 1.50 V, the curve demonstrates an increase in impedance concomitant with rising voltage, alongside a reduction in the phase angle. The curve exhibits a semicircle with a decreasing radius as the voltage increases and oxygen precipitation becomes more prominent after 1.50 V. Collectively, the Bode and Nyquist plots indicate that Ni2P facilitates a possible OER beyond 1.45 V, with this response becoming increasingly favorable as the voltage rises (Fig. 5f and Fig. S27a†). Upon the addition of the reactive substrate, the response peak at 1.40 V decreases sharply compared to the OER process and shifts to a higher frequency region. This shift indicates that the introduction of the substrate accelerates the oxidation of additional adsorbed substrate molecules, thereby promoting faster interfacial charge transfer. Consequently, at this voltage, the catalyst exhibits a higher tendency to oxidize the substrate.51,52 The phase angle of the response peak in the high-frequency region reaches its minimum at 1.55 V, confirming the results of the oxidation reaction. Thus, 1.55 V is identified as the optimal reaction potential for substrate oxidation. Beyond 1.60 V, the phase angle of the response peak increases and shifts toward the low-frequency region, suggesting competition between the OER and substrate oxidation. This indicates that substrate oxidation can occur on the catalyst surface between 1.40 and 1.60 V. Analysis of the Nyquist plots reveals that the curve radius initially decreases before increasing, becoming larger at 1.60 V. This change suggests that at this voltage, the competition between the OER and substrate oxidation impacts surface charge transfer, resulting in increased impedance (Fig. 5g and Fig. S27b†).
To investigate changes in adsorbates on the electrocatalyst surface during the oxidation reaction and to study the reaction process, in situ attenuated total reflection infrared (ATR-IR) spectroscopy was carried out using Ni2P-R in 1.0 M KOH solution containing 0.1 M 4-MA (Fig. 5h). The electrode potential for oxidation of 4-MA was maintained at 1.55 V vs. RHE, and Fourier transform infrared (FT-IR) spectra were continuously recorded. The broad peak at 1640 cm−1 and the nearby successive small peaks are considered to be the bending vibration of H2O (Fig. S28†). The peaks near 1442 cm−1, 1302 cm−1, and 1120 cm−1 show similar intensity variations and are considered to be the characteristic peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, and C–OH of the carboxylic acids, which collectively represent 3-MAA.53 The peak at 1230 cm−1 is considered to be the characteristic peak of 4-MK,9 and the peak near 1100 cm−1 is considered to be the characteristic peak of C–OH of 4-MA. This observation shows that the characteristic peak of 4-MK appears first and disappears with time, accompanied by the enhancement of the characteristic peak of 3-MAA and the weakening of the characteristic peak of 4-MK. The electrochemical oxidation of 4-MA involves the formation of 4-MK by oxidation and the formation of a product of 3-MAA by further oxidation.
O, C–O, and C–OH of the carboxylic acids, which collectively represent 3-MAA.53 The peak at 1230 cm−1 is considered to be the characteristic peak of 4-MK,9 and the peak near 1100 cm−1 is considered to be the characteristic peak of C–OH of 4-MA. This observation shows that the characteristic peak of 4-MK appears first and disappears with time, accompanied by the enhancement of the characteristic peak of 3-MAA and the weakening of the characteristic peak of 4-MK. The electrochemical oxidation of 4-MA involves the formation of 4-MK by oxidation and the formation of a product of 3-MAA by further oxidation.
Mechanistic studies of electrocatalytic oxidation
To elucidate the exceptional catalytic performance of the NiOOH/Ni2P heterostructure for the electrooxidation of 4-MA, DFT calculations were performed to investigate the electronic structures of Ni2P, NiOOH, and NiOOH/Ni2P catalysts, as well as the intrinsic relationship between adsorption and desorption behavior (Fig. 6a). The total density of states (TDOS) analysis reveals a lower d-band center (εd) for NiOOH/Ni2P (−1.87 eV) compared to Ni2P (−1.70 eV) and NiOOH (−1.77 eV) as shown in Fig. 6b. The result of charge density difference (CDD) further highlights significant charge redistribution at the interface in NiOOH/Ni2P (Fig. 6c). These findings suggest that the unique structural properties of NiOOH/Ni2P effectively modulate the adsorption and desorption of organic molecules. Further analysis of the adsorption behavior of the reactants, intermediates, and products on the surfaces of the different catalysts (Fig. 6d–f) reveals that the NiOOH/Ni2P heterostructure exhibits stronger adsorption of reactants and intermediates. This is particularly true for the enolate isomers that are prone to bond-breaking reactions, while the heterostructure also enhances the desorption of products. Fig. 6g shows the adsorption of reactants at the stepped NiOOH/Ni2P interface. A charge transfer of 1.74 |e| is observed, effectively activating the hydroxyl group (OH) in the reactants (Fig. 6h). Therefore, the interface of NiOOH/Ni2P is more favorable for the accumulation and reaction of enolate intermediates, while facilitating product desorption. This enhancement in catalytic performance is attributed to the stepped interface structure and the optimal charge distribution at that site in the NiOOH/Ni2P heterostructure. In conclusion, Fig. 6i illustrates the potential reaction pathways of 4-MA at the NiOOH/Ni2P interface. | ||
| Fig. 6 (a) Schematic structure of Ni2P, NiOOH, and NiOOH/Ni2P catalysts for the DFT simulation. (b) TDOS analysis of Ni2P, NiOOH, and NiOOH/Ni2P catalysts. (c) CDD analysis of the NiOOH/Ni2P catalyst. (d–f) Adsorption and desorption energy values of the Ni2P, NiOOH, and NiOOH/Ni2P catalysts for reactants (d), intermediate (e), and product (f). CDD and Bader charge analysis (g) and ELF (h) plots of reactant adsorption. (i) Schematic diagram of the 4-MA reaction pathway. | ||
Conclusions
In summary, this study introduces a novel strategy for enhancing the electrochemical oxidation activity of 4-MA by leveraging CV activation of nickel-based catalysts doped with anionic elements (P, S, Se). Following the electrocatalyst performance tests, the Ni2P-R/GF catalyst, which demonstrated high activity, was selected for further investigation in the oxidation of 4-MA. This resulted in the achievement of high 4-MA conversion and 3-MAA selectivity. Experiments conducted ex/in situ demonstrate that the oxidation mechanism of 4-MA involves an indirect electrooxidation process facilitated by NiOOH active centers, which are generated through electrochemical reconstruction. The doping of P enhances the electrocatalyst's ability to generate additional active centers at lower potentials. Furthermore, DFT analysis revealed that the enrichment of reactants and intermediates at the NiOOH/Ni2P heterojunction interface of Ni2P-R/GF facilitated the cleavage of C–C bonds on the catalyst surface, while also promoting product desorption, thereby increasing the yield of 3-MAA. Therefore, Ni2P-R/GF is considered to be a promising catalyst for 4-MA oxidation. This study introduces an efficient approach for designing high-performance electrocatalysts for 4-MA oxidation reactions via reconstruction. This method has the potential to guide the development of electrocatalysts for various applications in the field of electrocatalytic biomass valorization.Author contributions
Wang YH: major experimental production, formal analysis, methodology, investigation, conceptualization, data curation, writing – original draft. Chen Y, Wang JY: experimental production, methodology, visualization, conceptualization. Liu LH, Li SQ: validation, formal analysis, data curation, writing – review & editing. Ren LH, He JH, Li K, Hu P, Qi C: visualization, writing – review & editing. Cao YY: theoretical calculations, supervision, funding acquisition, writing – review & editing. Zhong X: methodology, funding acquisition, resources, supervision, project administration, writing – review & editing. Wang JG: methodology, funding acquisition, resources, supervision, project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors acknowledge the financial support from the National Key R & D Program of China (2022YFA1504200), the Zhejiang Provincial Natural Science Foundation of China (No. LR22B060003), the National Natural Science Foundation of China (22322810, 22078293, 22141001, and 22008211), and the Fundamental Research Funds for the Provincial Universities of Zhejiang (RF-C2023004).References
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef PubMed.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- Z. Lu, S. Li, Y. Lu, S. Wang and Y. Zou, Acta Phys.-Chim. Sin., 2024, 40, 2306003 CrossRef.
- Y. Fang, C. Dai, X. Liu, Y. Wang, C. Ju, S. He, R. Shi, Y. Liu, J. Zhang, Y. Zhu and J. Wang, Nano Energy, 2024, 127, 109754 CrossRef.
- S. Guo, M. Ma, R. Ge, H. Algadi and Q. Shao, Adv. Compos. Hybrid Mater., 2023, 6, 158 CrossRef.
- Z. Li, X. Li, H. Zhou, Y. Xu, S.-M. Xu, Y. Ren, Y. Yan, J. Yang, K. Ji, L. Li, M. Xu, M. Shao, X. Kong, X. Sun and H. Duan, Nat. Commun., 2022, 13, 5009 CrossRef PubMed.
- F. Liu, X. Gao, Z. Guo, E. C. M. Tse and Y. Chen, J. Am. Chem. Soc., 2024, 146, 15275–15285 CrossRef PubMed.
- R. Wang, Y. Kang, J. Wu, T. Jiang, Y. Wang, L. Gu, Y. Li, X. Yang, Z. Liu and M. Gong, Angew. Chem., Int. Ed., 2022, 61, e202214977 CrossRef PubMed.
- W. Chen, J. Shi, Y. Wu, Y. Jiang, Y.-C. Huang, W. Zhou, J. Liu, C.-L. Dong, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316449 CrossRef PubMed.
- L. Dai, Z.-N. Chen, L. Li, P. Yin, Z. Liu and H. Zhang, Adv. Mater., 2020, 32, 1906915 CrossRef PubMed.
- S. Li, S. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, Y. Wang, X. Li, X. Zhong and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202306553 CrossRef.
- J. He, S. Li, C. Li, K. Li, Y. Xu, M. Wang, S. Zhao, J. Zhang, X. Zhong, X. Li, Z. Zhang and J. Wang, AIChE J., 2023, 69, e18153 CrossRef.
- C. Huang, Q. Zhou, D. Duan, L. Yu, W. Zhang, Z. Wang, J. Liu, B. Peng, P. An, J. Zhang, L. Li, J. Yu and Y. Yu, Energy Environ. Sci., 2022, 15, 4647–4658 RSC.
- D. Ma, Z. Zhao, Y. Wang, X. Yang, M. Yang, Y. Chen, J. Zhu, H. Mi and P. Zhang, Adv. Mater., 2024, 36, 2310336 CrossRef PubMed.
- X. Wei, K. Li, X. Zhang, Q. Tong, J. Ji, Y. Cai, B. Gao, W. Zou and L. Dong, Appl. Catal., B, 2022, 317, 121694 CrossRef.
- M. Cai, Q. Zhu, X. Wang, Z. Shao, L. Yao, H. Zeng, X. Wu, J. Chen, K. Huang and S. Feng, Adv. Mater., 2023, 35, 2209338 Search PubMed.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li, H. Peng and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10577–10582 CrossRef CAS.
- S. Li, R. Ma, J. Hu, Z. Li, L. Liu, X. Wang, Y. Lu, G. E. Sterbinsky, S. Liu, L. Zheng, J. Liu, D. Liu and J. Wang, Nat. Commun., 2022, 13, 2916 CrossRef CAS.
- H. Zhong, X. Wang, G. Sun, Y. Tang, S. Tan, Q. He, J. Zhang, T. Xiong, C. Diao, Z. Yu, S. Xi, W. S. V. Lee and J. Xue, Energy Environ. Sci., 2023, 16, 641–652 RSC.
- H. Liu, J. Gao, X. Xu, Q. Jia, L. Yang, S. Wang and D. Cao, Chem. Eng. J., 2022, 448, 137706 CrossRef CAS.
- L. Liu, Y. Hou, Y. Gao, N. Yang, J. Liu and X. Wang, Electrochim. Acta, 2019, 295, 340–346 CrossRef CAS.
- Z. Peng, S. Li, R. Li, K. Sun, J. Li, Y. Liu, Z. Liu, Y. Wang, J. Ge and K. Xiang, Appl. Catal., B, 2024, 342, 123450 CrossRef CAS.
- H. Liu, Q. He, H. Jiang, Y. Lin, Y. Zhang, M. Habib, S. Chen and L. Song, ACS Nano, 2017, 11, 11574–11583 CrossRef CAS.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- M. Zhang, T. Wang, H. Cao, S. Cui and P. Du, J. Energy Chem., 2020, 42, 71–76 CrossRef.
- C. Tang, R. Zhang, W. Lu, Z. Wang, D. Liu, S. Hao, G. Du, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2017, 56, 842–846 CrossRef CAS.
- D. Zeng, W. Xu, W.-J. Ong, J. Xu, H. Ren, Y. Chen, H. Zheng and D.-L. Peng, Appl. Catal., B, 2018, 221, 47–55 CrossRef CAS.
- W. Zhu, X. Fu, A. Wang, M. Ren, Z. Wei, C. Tang, X. Sun and J. Wang, Appl. Catal., B, 2022, 317, 121726 CrossRef CAS.
- X. Liu, Q. Yu, X. Qu, X. Wang, J. Chi and L. Wang, Adv. Mater., 2024, 36, 2307395 CrossRef CAS PubMed.
- Z. Guo and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 5898–5902 CrossRef CAS PubMed.
- D. Susac, L. Zhu, M. Teo, A. Sode, K. C. Wong, P. C. Wong, R. R. Parsons, D. Bizzotto, K. A. R. Mitchell and S. A. Campbell, J. Phys. Chem. C, 2007, 111, 18715–18723 CrossRef.
- Z. Chen, Y. Gao, F. Chen and H. Shi, Chem. Eng. J., 2021, 413, 127474 CrossRef.
- K. Li, L. Ren, S. Li, L. Liu, J. He, Y. Xu, M. Wang, S. Zhao, Y. Wang, Y. Chen, J. Wang, X. Zhong and J. Wang, AIChE J., 2024, e18531 CrossRef.
- J. Woo, J. Choi, J. Choi, M.-Y. Lee, E. Kim, S. Yun, S. Yoo, E. Lee, U. Lee, D. H. Won, J. H. Park, Y. J. Hwang, J. S. Yoo and D. K. Lee, Adv. Funct. Mater., 2024, 2413951 Search PubMed.
- R. Luo, Y. Li, L. Xing, N. Wang, R. Zhong, Z. Qian, C. Du, G. Yin, Y. Wang and L. Du, Appl. Catal., B, 2022, 311, 121357 CrossRef.
- M. Wang, J. Li, S. Li, L. Liu, J. He, K. Li, Y. Xu, S. Zhao, W. Zhou, C. Li, X. Zhong, Z. Zhang, Z. Yao and J. Wang, Chem. Eng. Sci., 2024, 285, 119589 Search PubMed.
- D. Zhou, S. Wang, Y. Jia, X. Xiong, H. Yang, S. Liu, J. Tang, J. Zhang, D. Liu, L. Zheng, Y. Kuang, X. Sun and B. Liu, Angew. Chem., Int. Ed., 2019, 58, 736–740 CrossRef PubMed.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S.-T. Lee and T. Ma, Nat. Commun., 2019, 10, 2149 CrossRef.
- C. Sun, P. Zhao, Y. Yang, Z. Li and W. Sheng, ACS Catal., 2022, 12, 11830–11837 CrossRef.
- X. Liu, Y.-Q. Zhu, J. Li, Y. Wang, Q. Shi, A.-Z. Li, K. Ji, X. Wang, X. Zhao, J. Zheng and H. Duan, Nat. Commun., 2024, 15, 7685 CrossRef PubMed.
- S. Zhao, S. Wang, C. Qiu, Z. Xu, L. Ren, Y. Wang, S. Li, J. He, K. Li, Y. Xu, M. Wang, X. Zhong and J. Wang, Chem. Eng. Sci., 2024, 286, 119653 CrossRef.
- Y. Wang, Y.-Q. Zhu, Z. Xie, S.-M. Xu, M. Xu, Z. Li, L. Ma, R. Ge, H. Zhou, Z. Li, X. Kong, L. Zheng, J. Zhou and H. Duan, ACS Catal., 2022, 12, 12432–12443 CrossRef.
- P. Zhou, X. Lv, S. Tao, J. Wu, H. Wang, X. Wei, T. Wang, B. Zhou, Y. Lu, T. Frauenheim, X. Fu, S. Wang and Y. Zou, Adv. Mater., 2022, 34, 2204089 Search PubMed.
- W. Chen, Y. Wang, B. Wu, J. Shi, Y. Li, L. Xu, C. Xie, W. Zhou, Y.-C. Huang, T. Wang, S. Du, M. Song, D. Wang, C. Chen, J. Zheng, J. Liu, C.-L. Dong, Y. Zou, J. Chen and S. Wang, Adv. Mater., 2022, 34, 2105320 Search PubMed.
- B. Zhou, Y. Li, Y. Zou, W. Chen, W. Zhou, M. Song, Y. Wu, Y. Lu, J. Liu, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 22908–22914 CrossRef CAS PubMed.
- Y.-Q. Zhu, H. Zhou, J. Dong, S.-M. Xu, M. Xu, L. Zheng, Q. Xu, L. Ma, Z. Li, M. Shao and H. Duan, Angew. Chem., Int. Ed., 2023, 62, e202219048 Search PubMed.
- D. Das, J. Solution Chem., 2008, 37, 947–955 Search PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K.-S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 Search PubMed.
- W. Chen, L. Xu, X. Zhu, Y.-C. Huang, W. Zhou, D. Wang, Y. Zhou, S. Du, Q. Li, C. Xie, L. Tao, C.-L. Dong, J. Liu, Y. Wang, R. Chen, H. Su, C. Chen, Y. Zou, Y. Li, Q. Liu and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 7297–7307 Search PubMed.
- K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou, L. Tao and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 20253–20258 Search PubMed.
- X. Lin, Z. Wang, S. Cao, Y. Hu, S. Liu, X. Chen, H. Chen, X. Zhang, S. Wei, H. Xu, Z. Cheng, Q. Hou, D. Sun and X. Lu, Nat. Commun., 2023, 14, 6714 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc05682e |
| This journal is © The Royal Society of Chemistry 2025 |