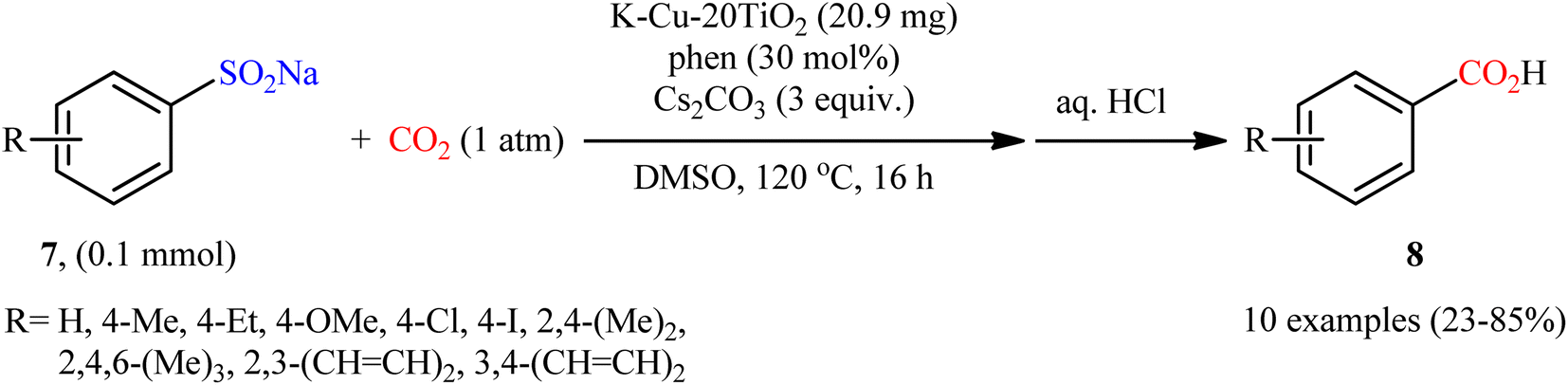DOI:
10.1039/D4RA02405B
(Review Article)
RSC Adv., 2024,
14, 15680-15690
Recent trends in incorporation of CO2 into organosulfur compounds via C–S bond cleavage
Received
29th March 2024
, Accepted 29th April 2024
First published on 15th May 2024
Abstract
Desulfurative functionalization of organosulfur compounds to form various carbon–carbon and carbon–heteroatom bonds has become established as a powerful tool in organic chemistry. In this context, desulfurative carboxylation of this class of compounds using carbon dioxide (CO2) as a sustainable and renewable source of carboxyl has recently been developed as an efficient option for the synthesis of carboxylic acid derivatives. The aim of this Focus Review is to summarize the major progress in this appealing research field with particular emphasis on the mechanistic features of the reactions. Literature has been surveyed until the end of February 2024, according to the data collected using SciFinder and Google Scholar engines.
1. Introduction
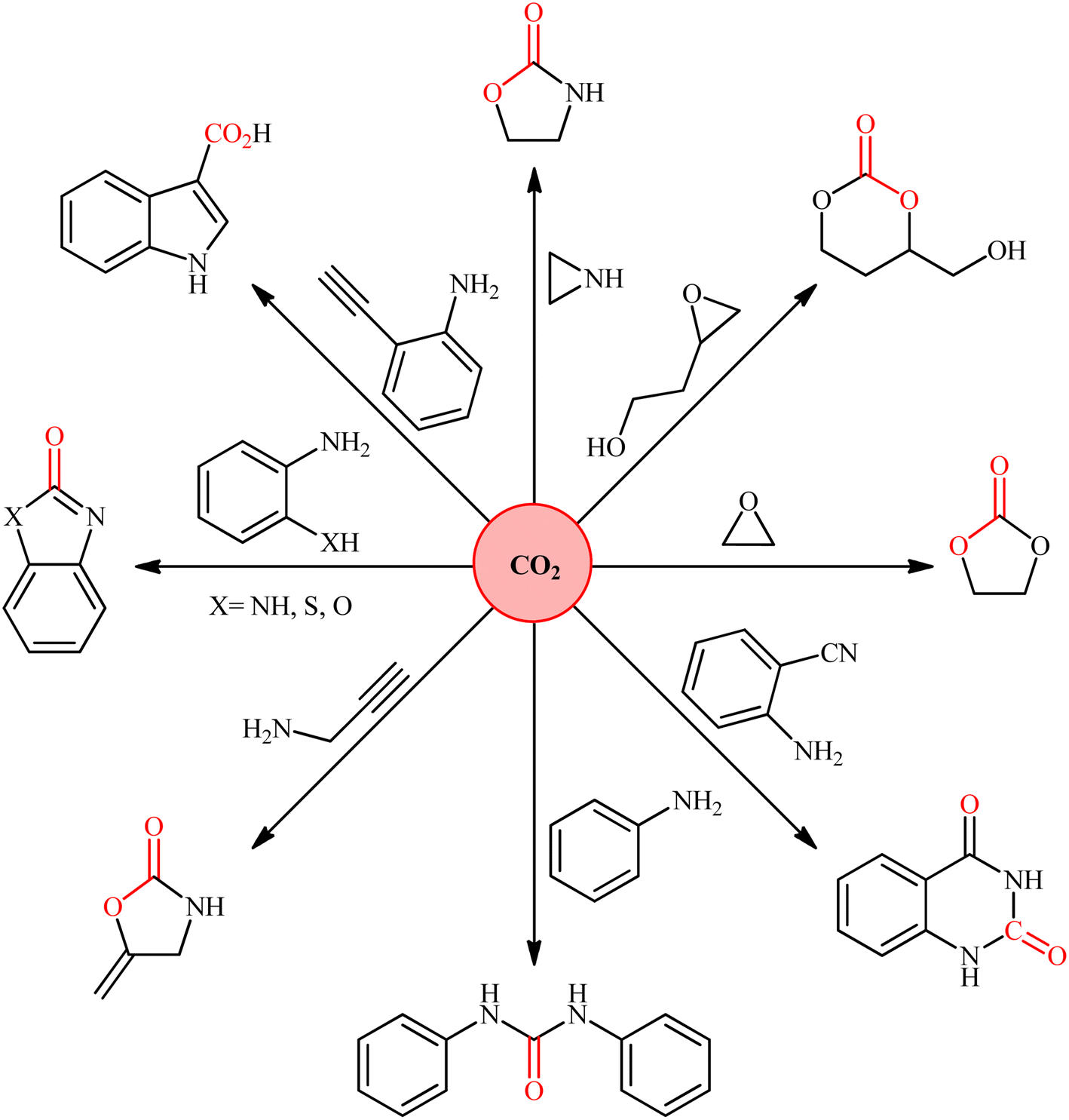
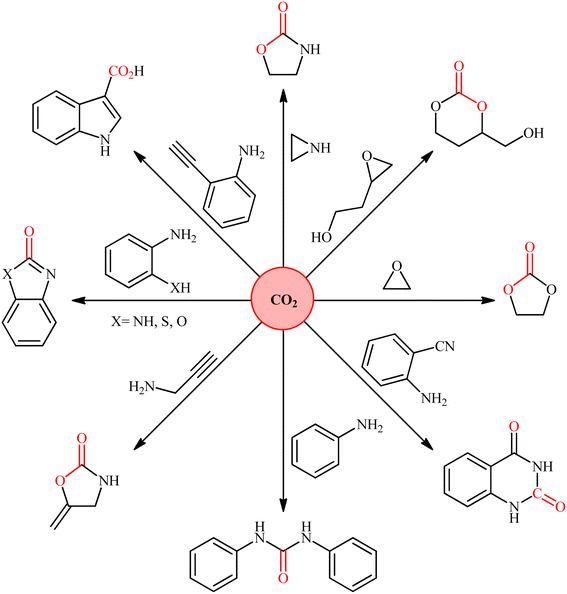
Nowadays, the incorporation of carbon dioxide (CO2) into organic substrates is of increasing interest for the production of value-added chemicals and fuels, because CO2 is both regarded as the chief anthropogenic greenhouse gas responsible for global climate change and a plentiful, inexpensive, and environmentally benign one-carbon (C1) feedstock (Fig. 1).1 However, due to its high thermodynamic stability and chemical inertness, the efficient utilization of CO2 as building block in the production of useful chemicals is a challenging issue.2 A strategy to overcome the low reactivity of this greenhouse gas is based on the reactions with high free energy substrates.3 One of the most promising approaches in this area is the conversion of CO2 into carboxylic acids, a vital class of organic compounds that play a pivotal role in numerous applications in the fields of pharmaceuticals, agriculture, food, fuel and other industries.4,5 Traditionally, strong nucleophilic reagents such as Grignard reagents were used in such carboxylation reactions.6 However, the main drawback with Grignard reagents is their poor compatibility with a variety of functional groups as well as their high air/moisture sensitivity.7 In order to bypass the need for these highly reactive and hard to control reagents, the catalytic carboxylation of less reactive nucleophiles such as organozinc,8 organotin9 organoboron10 have been developed. However, instability and moisture sensitivity of organozinc reagent, toxicity of organotin reagent, and laborious preparation and purification of organoborons limited the utility of these methods.11 Alternatively, many studies have been focused on the use of organohalides as pre-nucleophiles for the fixation of CO2.12 Nevertheless, they have also several drawbacks; such as: (i) the high cost of organic bromides and iodides, (ii) very low reactivity of organic chlorides, and (iii) environmental toxicity of some organic halides.13 The use of organosilicon compounds as the alternative or complementary nucleophilic reagents for the fixation of CO2 have been the subject of a number of recent studies.11 However, due to the poor polarization of C–Si bond, the majority of the existing protocols are relying on the use of fluoride ions for activation of this bond. In light of the above-mentioned facts, there is still further need for broadening the scope of nucleophiles within the realm of carboxylation reactions using CO2, which may extend the synthetic application of CO2 as a source of carboxyl and lead to the development of novel and effective methods for the synthesis of carboxylic acids.
 |
| | Fig. 1 Selected examples of conversion of CO2 to valuable chemicals. | |
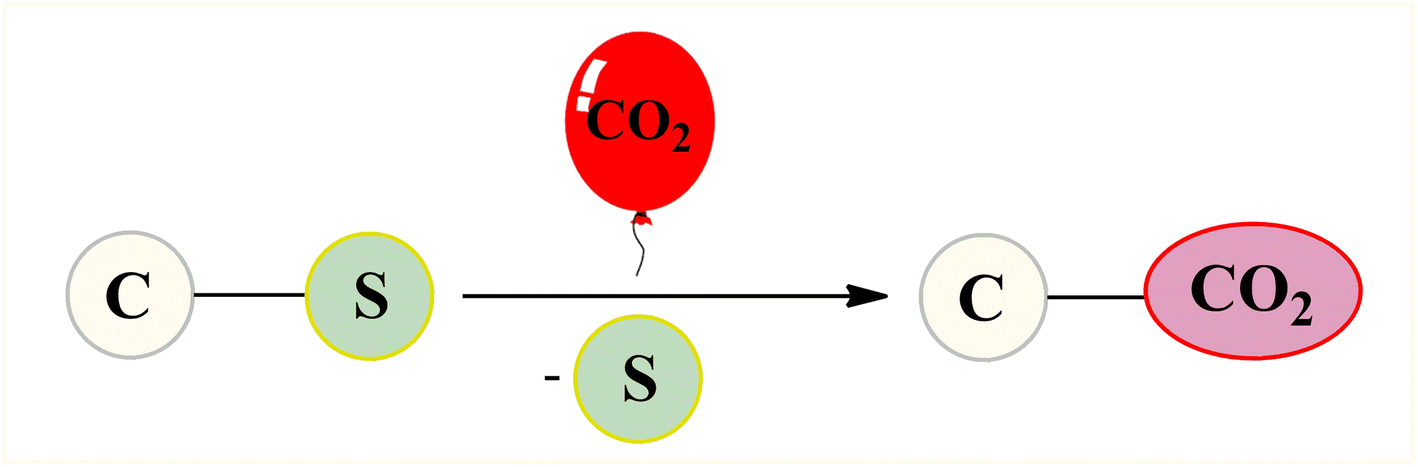
Given the versatility and ubiquity of organosulfur compounds as well as low dissociation energy of C–S bond, desulfurative functionalization of organosulfur compounds through C–S bond cleavage has recently attracted significant attention in organic synthesis and great progress has been achieved in this area.14 In this regard, synthesis of carboxylic acid derivatives through desulfurative carboxylation of various organosulfur compounds (i.e., sulfonium salts, sulfinates, and sulfones) using CO2 as the carboxyl source has been the subject of several papers in recent years. In 2020, Nogi and Yorimitsu highlighted this synthetic strategy in their interesting review paper entitled “catalytic carbonylation and carboxylation of organosulfur compounds via C–S bond cleavage”;15 albeit with only one example on the above-mentioned reactions. To the best of our knowledge, a comprehensive review has not appeared on this appealing research field in the literature as yet. In continuation of our ongoing works on CO2-fixation reactions,16 we summarize here the available literature in incorporation of CO2 into organosulfur compounds via C–S bond cleavage (Fig. 2), by hoping that it will stimulate researchers to develop more-efficient and improved methods for utilization of CO2 in the chemical synthesis.
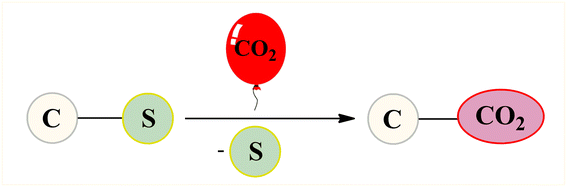 |
| | Fig. 2 Desulfurative carboxylation of organosulfur compounds with CO2. | |
Since sulfur is bigger in size than oxygen the carbon–sulfur (C–S) bond is weaker compared to the carbon–oxygen (C–O) bond. Therefore, the carbon–oxygen (C–O) bond has higher bond dissociation energy than the carbon–sulfur (C–S) bond.
2. Carboxylation of sulfonium salts
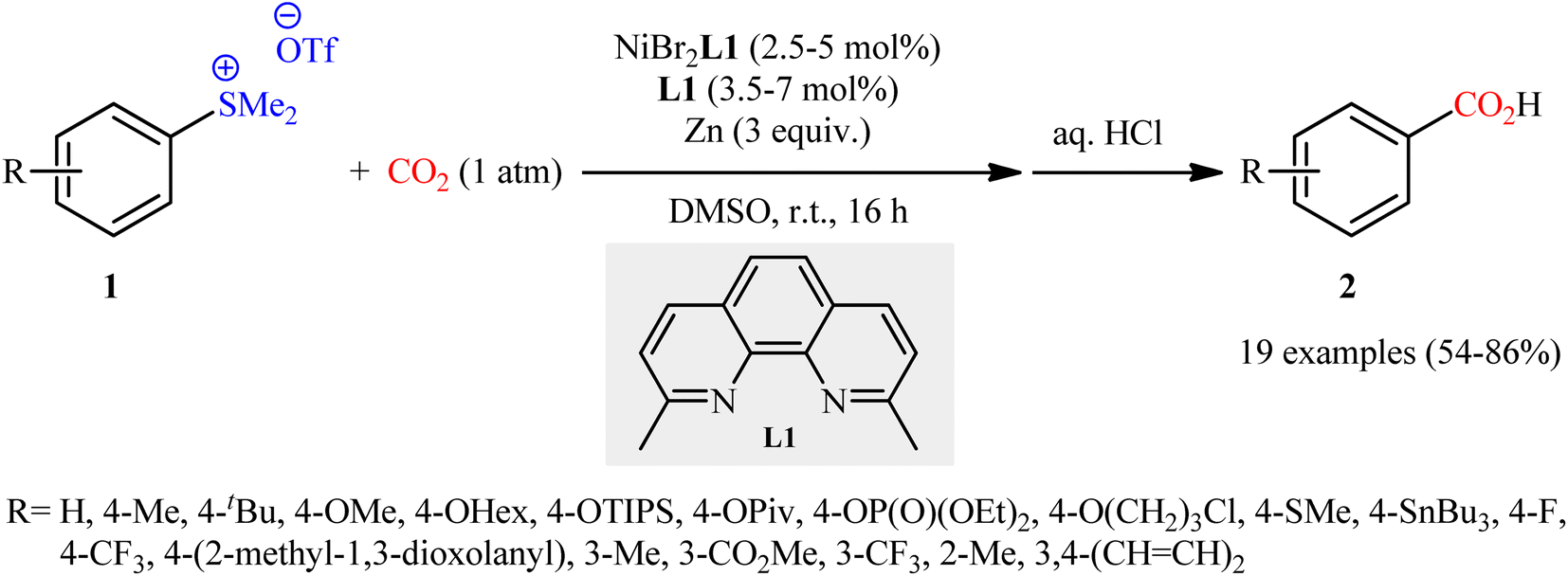
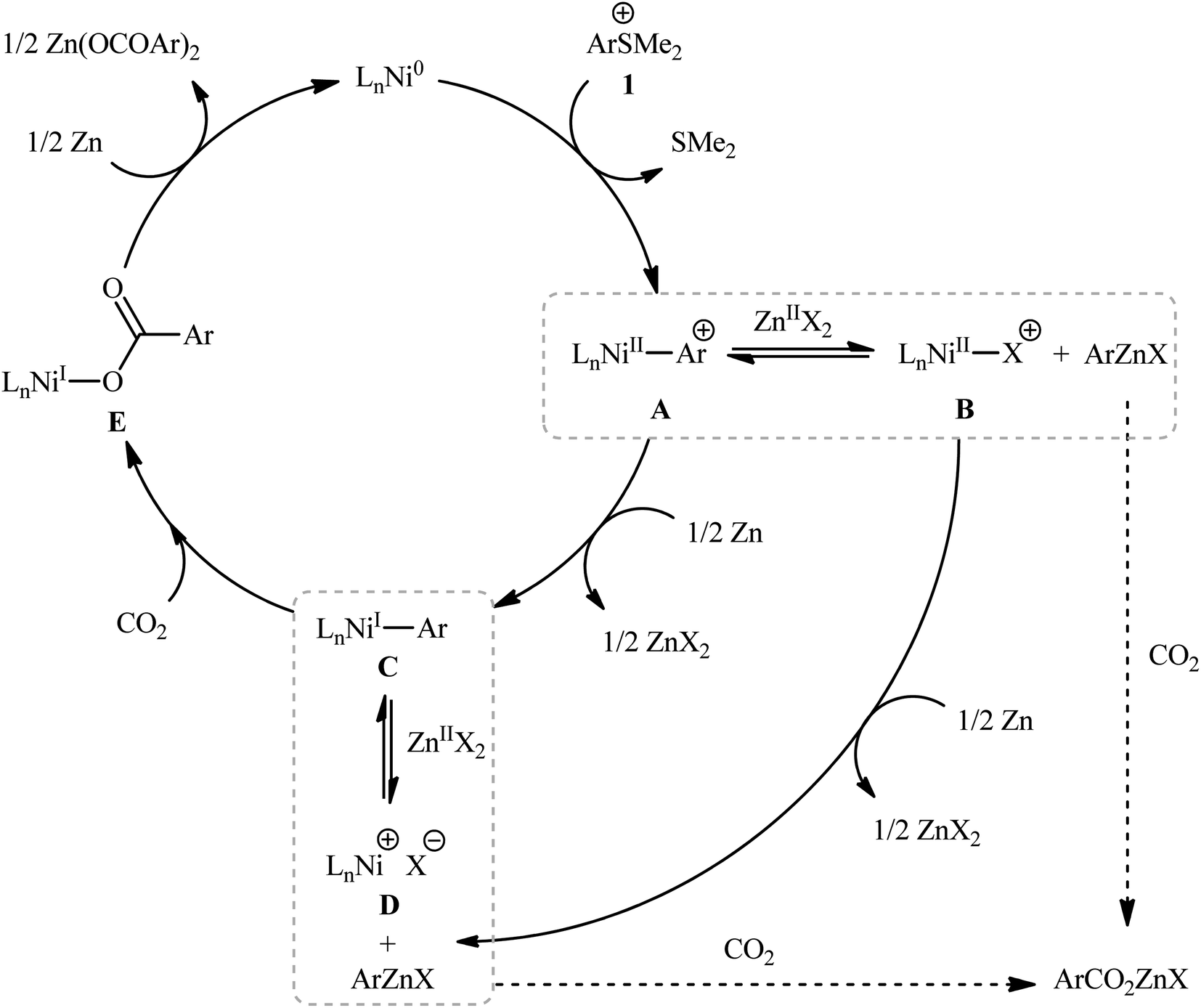
Sulfonium salts (SR3+) are tricoordinate S(IV) species that behave as excellent leaving groups due to their highly electron deficient sulfur atom.17 As evidenced in the literature, this class of versatile electrophilic reagents have been extensively used recently in construction of various carbon–carbon and carbon–heteroatom bonds through C–S bond cleavage.18 The first report on the usefulness of aryl sulfonium salts as aryl transfer reagents in carboxylation reactions with CO2 was published at the outset of 2020 by Yanagi et al.19 It was disclosed that treatment of aryl dimethylsulfonium triflate salts 1 with nickel–neocuproine complex and Zn powder under CO2 atmosphere furnished corresponding aromatic carboxylic acids 2 in moderate to high yields after hydrolysis (Scheme 1). It was found that the electronic nature of the substituents in the phenyl ring periphery of substrates had a major impact on the facility of this reaction. Thus, the presence of electron-donating groups (e.g., alkyl, alkoxy) tended to increase the reaction rate, and the electron-withdrawing groups (e.g., CF3, CO2Me) slowed down the process and facilitate undesired reductive demethylation by Zn. The results also indicated that the outcome of this desulfurative carboxylation was not dependent on the effects of the substituents; the toylsulfonium salts with methyl group at ortho, meta and para positions afford similar yields. The system was also amenable to the carboxylation of vinyl sulfonium salts as exemplified by the formation of cinnamic acid (36%) from the corresponding β-styrylsulfonium. However, trialkylsulfonium salts failed to produce any product under the optimized conditions. In this study, the authors also elegantly showed that in combination with the sulfonium moiety installation, this methodology provides an efficient protocol for the site-selective C–H carboxylation of arenes. While the detailed mechanistic picture remains unclear, the authors have suggested a plausible sequence as is depicted in Scheme 2, which includes Ni-to-Zn transmetalation steps. Based on the proposed mechanistic cycle, in this transformation Zn metal not only serves the role of terminal reductant, but also facilitates the activation of C–S bond by in situ generation of arylzinc species.
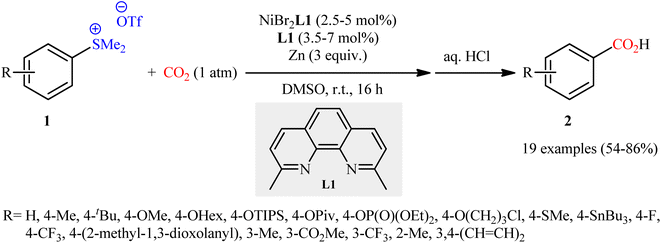 |
| | Scheme 1 Ni-catalyzed carboxylation of arylsulfonium salts 1 with CO2. | |
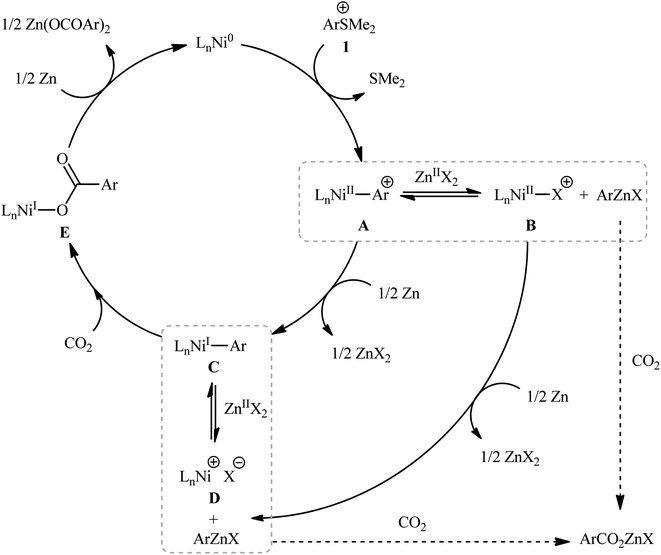 |
| | Scheme 2 Mechanistic proposal for the formation of carboxylic acids 2. | |
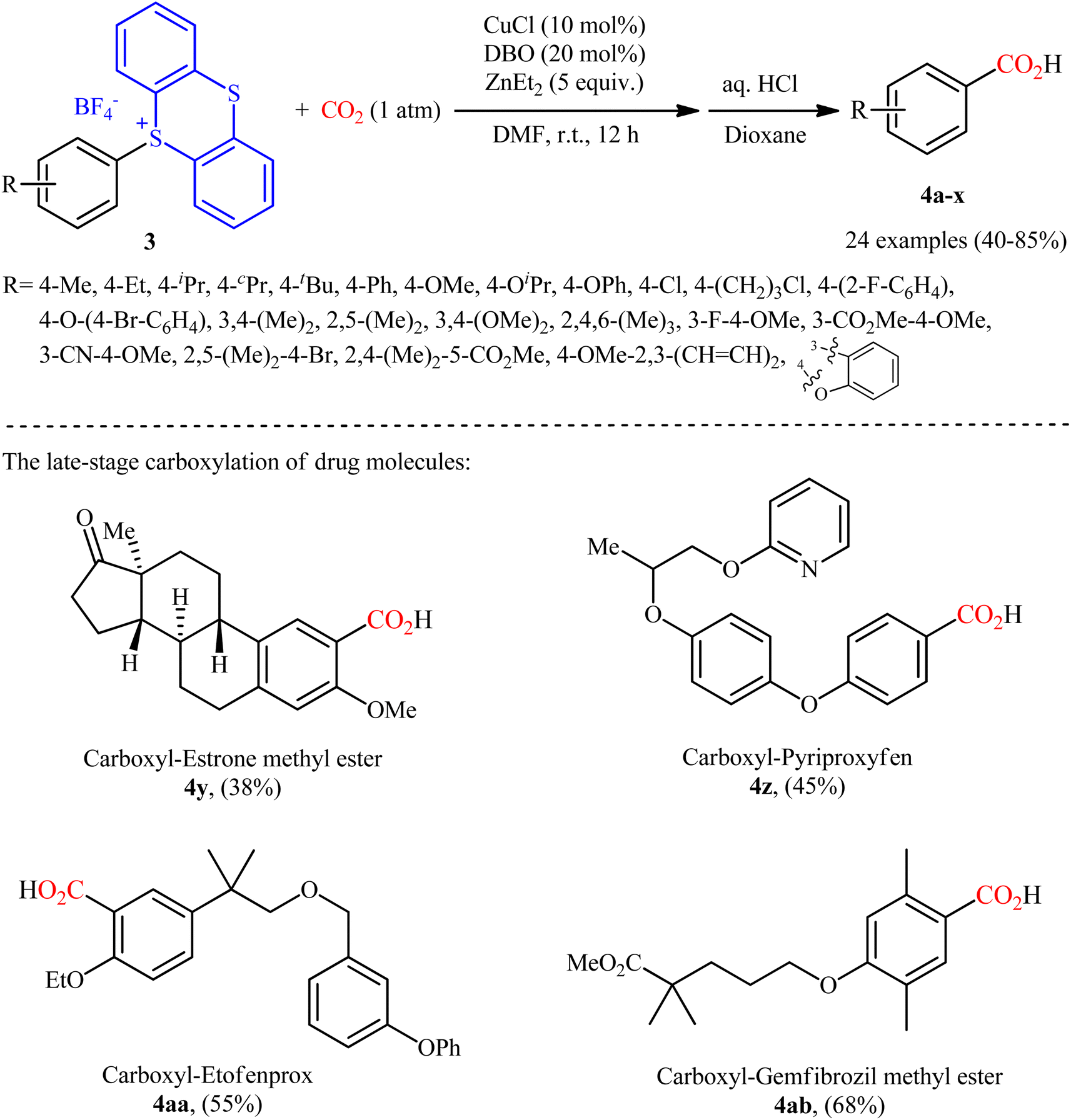
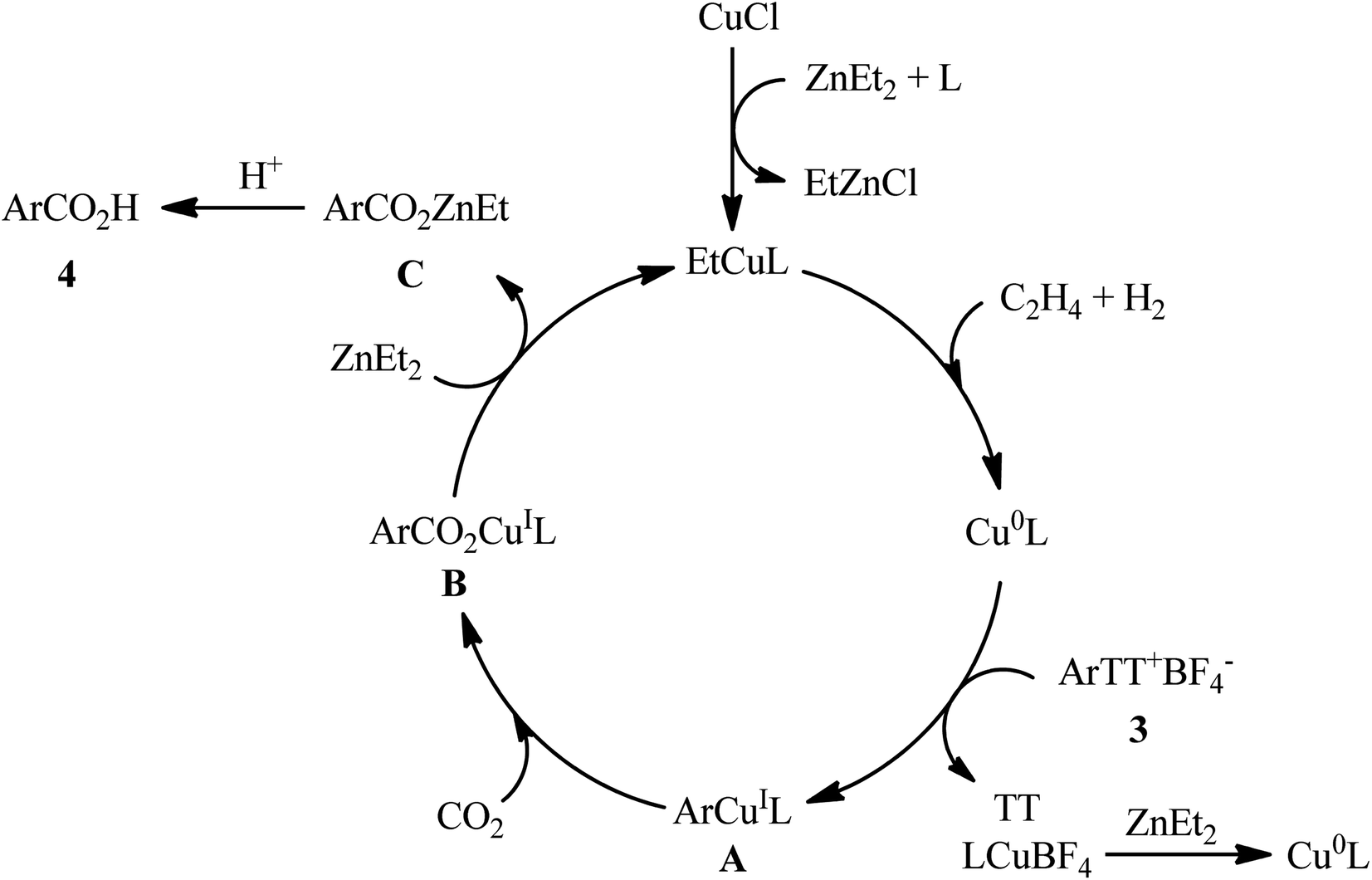
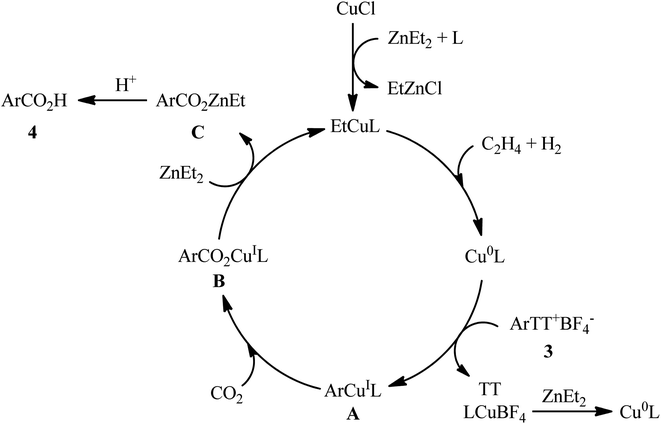
Thianthrenium salts (TT) are a class of sulfonium salts characterized by a positively charged sulfur atom carrying three carbon–sulfur bonds and a neutral sulfur atom.20 Due to their promising electrophilic features, this family of organosulfur compounds have recently attracted considerable attention from the synthetic community.20,21 In late 2022, Wang and co-workers studied the possibility of synthesizing benzoic acid derivatives through Cu-catalyzed desulfurative carboxylation of aryl thianthrenium salts with CO2.22 In order to determine the most appropriate catalytic system, the activities of various palladium and copper catalysts (e.g., CuI, CuBr, CuCl, Cu2O, CuOAc, Pd(TFA)2, PdCl2), ligands (e.g., PPh3, xantphos, neocuproine, 1,10-phen, DMEDA, TMEDA, DBO, BTMO), and reductants (e.g., Sm, Zn, ZnMe2, ZnEt2, AlEt3, AlEt2Cl) were carefully screened in the carboxylation of p-tolyl thianthrenium salt with atmospheric CO2 as a model reaction. The merge of low-cost CuCl and N,N′-dibenzyloxalamide (DBO) with ZnEt2 was proved to be most efficient system, which gave the highest yield of the expected carboxylic acid product in DMF at room temperature. Using the optimized conditions, a panel of 28 of aryl thianthrenium salts 3 smoothly underwent desulfurative carboxylation and gave the corresponding carboxylic acids 4 with yield ranging from 38% to 85% (Scheme 3). Intriguingly, this methodology was also effectively applied to the late-stage carboxylation of drug molecules, such as pyriproxyfen, estrone, etofenprox, and gemfibrozil. In order to further value, the applicability of the method, the authors investigated one-pot C–H thianthrenation/carboxylation of a small series of simple arenes, and the desired products were obtained in synthetically useful yields. Based on a series of control experiments and previous literature reports, the author suggested a mechanistic course for this CO2-fixation reaction, which is outlined in Scheme 4. Initially, the reaction of CuCl with ZnEt2 generates CuEt intermediate. Next, this intermediate release H2 and C2H4, and affords the catalytically active Cu0 species, which after a single electron transfer (SET) to aryl thianthrenium salts 3 renders aryl CuI intermediate A, CuBF4 and TT. Subsequently, the insertion of CO2 into the C–CuI bond of intermediate A leads to carboxylate CuI intermediate B. Finally, reaction of this intermediate B with ZnEt2 furnishes the zinc carboxylate C that after protonation affords the observed products 4.
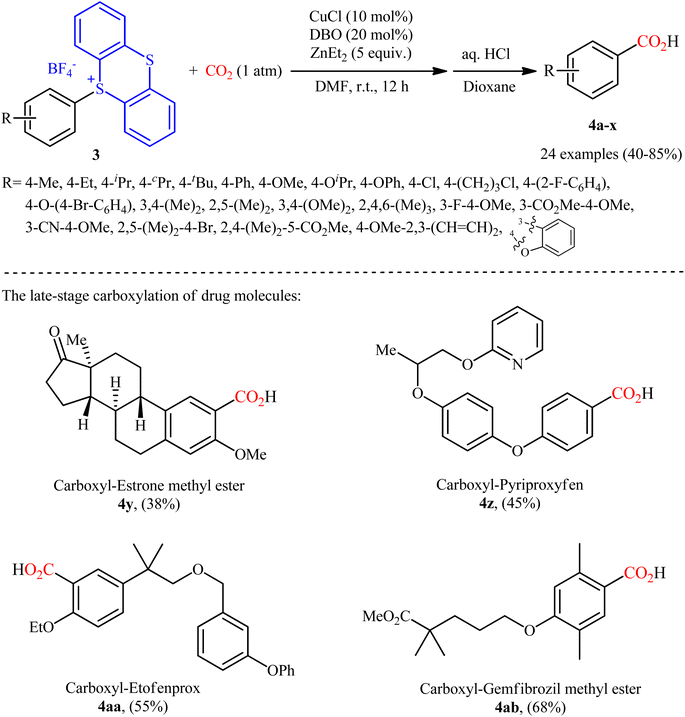 |
| | Scheme 3 Cu-catalyzed carboxylation of aryl thianthrenium salts 3 with CO2. | |
 |
| | Scheme 4 Proposed mechanism for the reaction in Scheme 3. | |
3. Carboxylation of sodium sulfinates
Sodium sulfinates (RSO2Na) are among the most important sulfonyl compounds in organic synthesis, chiefly because of their high stability, reactivity, easy accessibility, and user-friendly features.23 They have emerged as highly versatile building blocks for preparing a diverse array of value-added organosulfur compounds (e.g., sulfides, sulfones, sulfonamides, thiosulfonates), and for the construction of various carbon–carbon/heteroatom bonds via the extrusion of sulfur dioxide (SO2).24
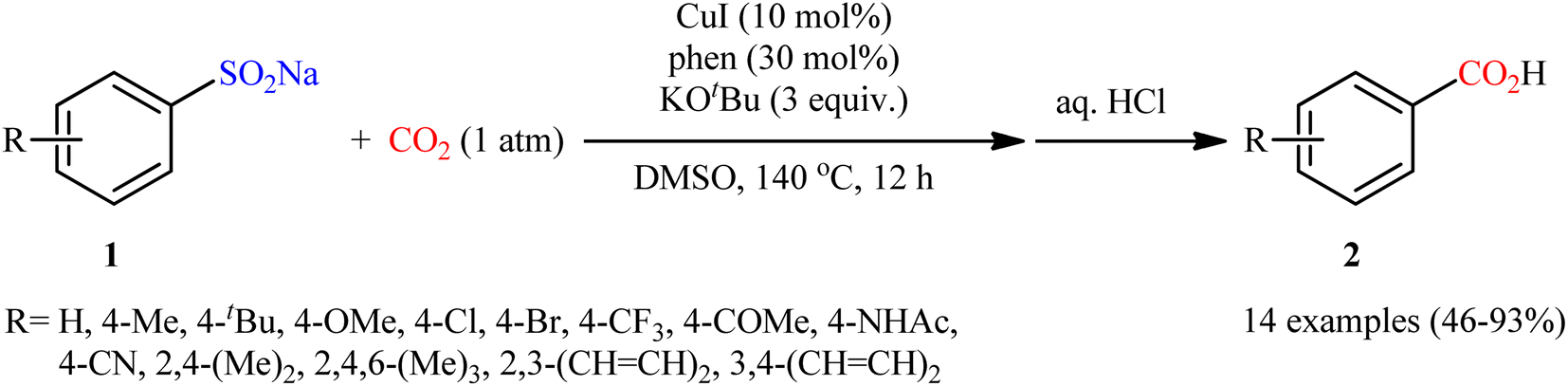
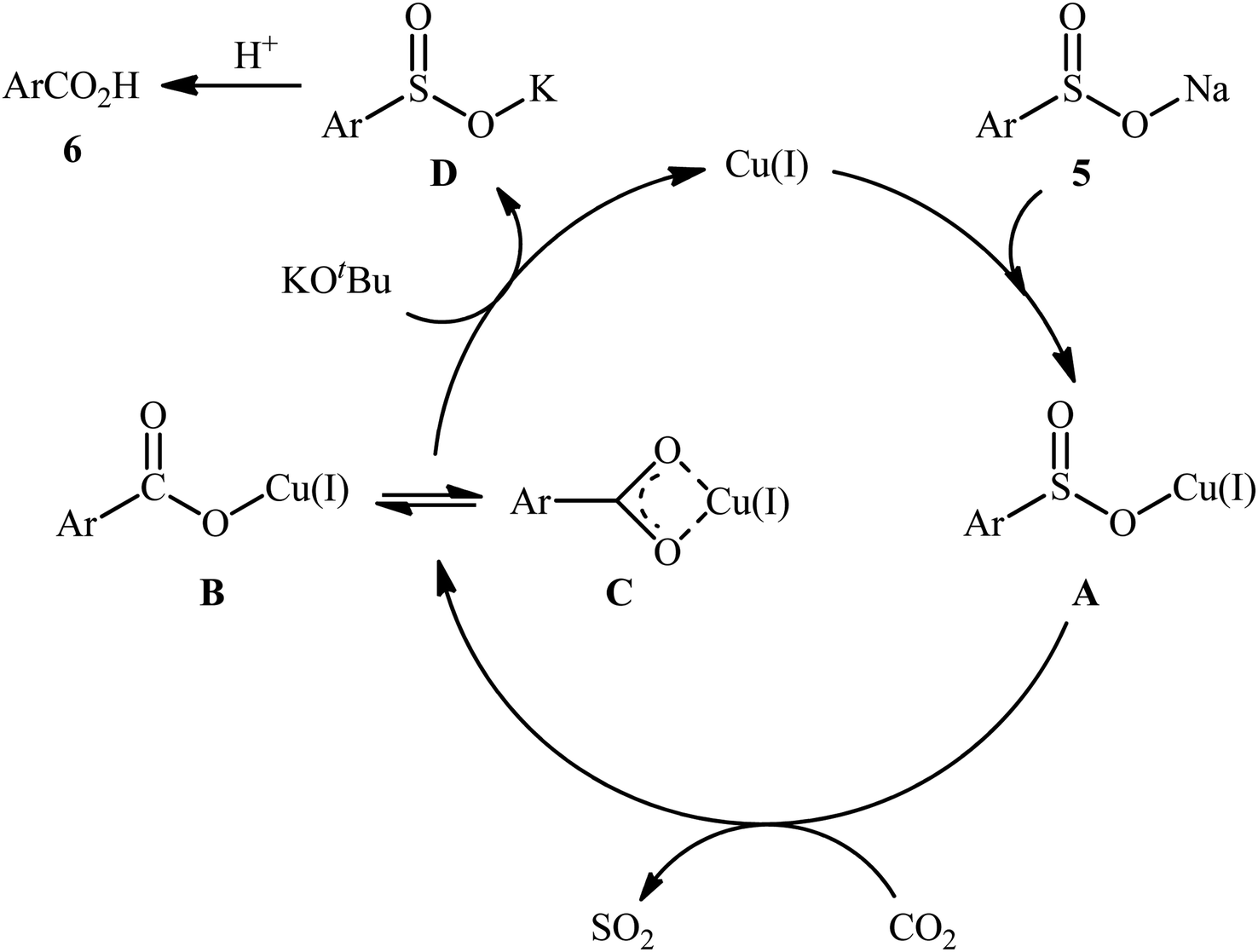
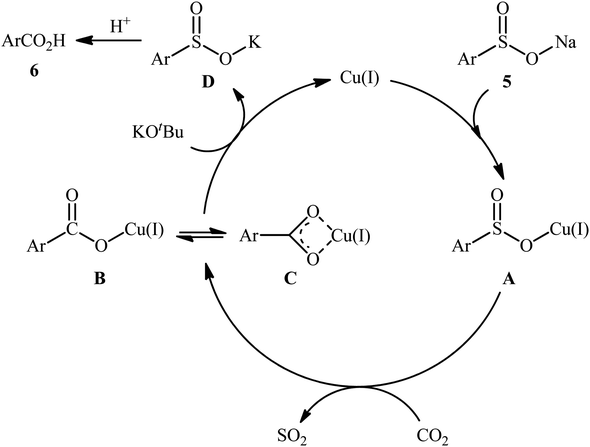
In 2015, Cheng and co-workers communicated the first example on carboxylation of sodium sulfinates with CO2 via C–S bond cleavage.25 It was disclosed that treatment of various sodium arylsulfinates 5 with atmospheric CO2 in the presence of CuI/phen/KOtBu combination as a catalytic system, resulted in the formation of the corresponding benzoic acid derivatives 6 in moderate to excellent yields; in addition, a tolerance for alkenyl sulfinates (i.e., sodium 2-phenylethenesulfinate) was also demonstrated. As shown in Scheme 5, a relatively wide panel of important functional groups (e.g., Cl, Br, CF3, OMe, COMe, NHCOMe, CN) at different positions of phenyl rings of sodium arylsulfinates were well tolerated by this protocol, thus indicating its broad applicability. Notably, the steric effect had a profound impact on the facility of this reaction. For example, sodium 2,4,6-trimethylarenesulfinate gave high yield of the corresponding carboxylic acid (82%), while the bulkier sodium 2,4,6-triisopropylarenesulfinate failed to produce any product under the identical conditions. According to mechanistic studies, this reaction starts with the generation of intermediate A via coordination of sodium sulfinate 5 to copper(I). Subsequently, desulfonylative carboxylation of intermediate A assisted by CO2 furnishes copper benzoate species B or C, followed by their metathesis by KOtBu to give the potassium carboxylate D with concomitant regeneration of the copper(I) catalyst. Finally, protonation of D by the acid affords the observed carboxylic acids 6 (Scheme 6).
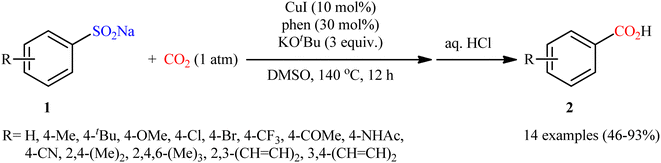 |
| | Scheme 5 Cheng's synthesis of carboxylic acids 6. | |
 |
| | Scheme 6 Proposed mechanism for the formation of carboxylic acids 6. | |
Encouraged by this work, the group of Peng reported the synthesis of a mesoporous ternary metal oxide (K–Cu–20TiO2) via a sol gel method using F68 (polyethylene–polypropylene glycol), Ti(OBu)4, Cu(NO3)2·3H2O and KNO3 in a solution composed of glacial acetic acid, hydrochloric acid and ethanol (AcHE).26 The catalytic activity of the synthesized composite was evaluated in the desulfonylative carboxylation of a series of sodium arylsulfinates 7 with atmospheric CO2 in the presence of phen as a ligand and Cs2CO3 as a base (Scheme 7). The obtained results from this study showed that functional groups including ether, chloro, and iodo were tolerated by the reaction conditions employed and the desired carboxylic acids 8 were formed in relatively poor to high yields. However, no examples were given with arylsulfinates bearing a coordinating moieties such as ester, amine, and nitrile moiety. Unfortunately, like Chang's work, a major drawback of this methodology is the requirement for an elevated reaction temperature (120 °C), which may limit its range of applications. Recycling tests established that the catalyst can be reused at least for five consecutive trials, with only negligible loss of the activity. The authors proposed a mechanism analogous to that of Cheng and co-workers for CuI-catalyzed desulfonylative carboxylation depicted in Scheme 6.
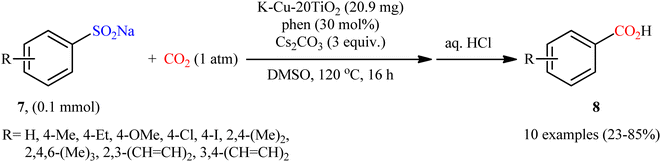 |
| | Scheme 7 Peng's synthesis of carboxylic acids 8. | |
4. Carboxylation of sulfones
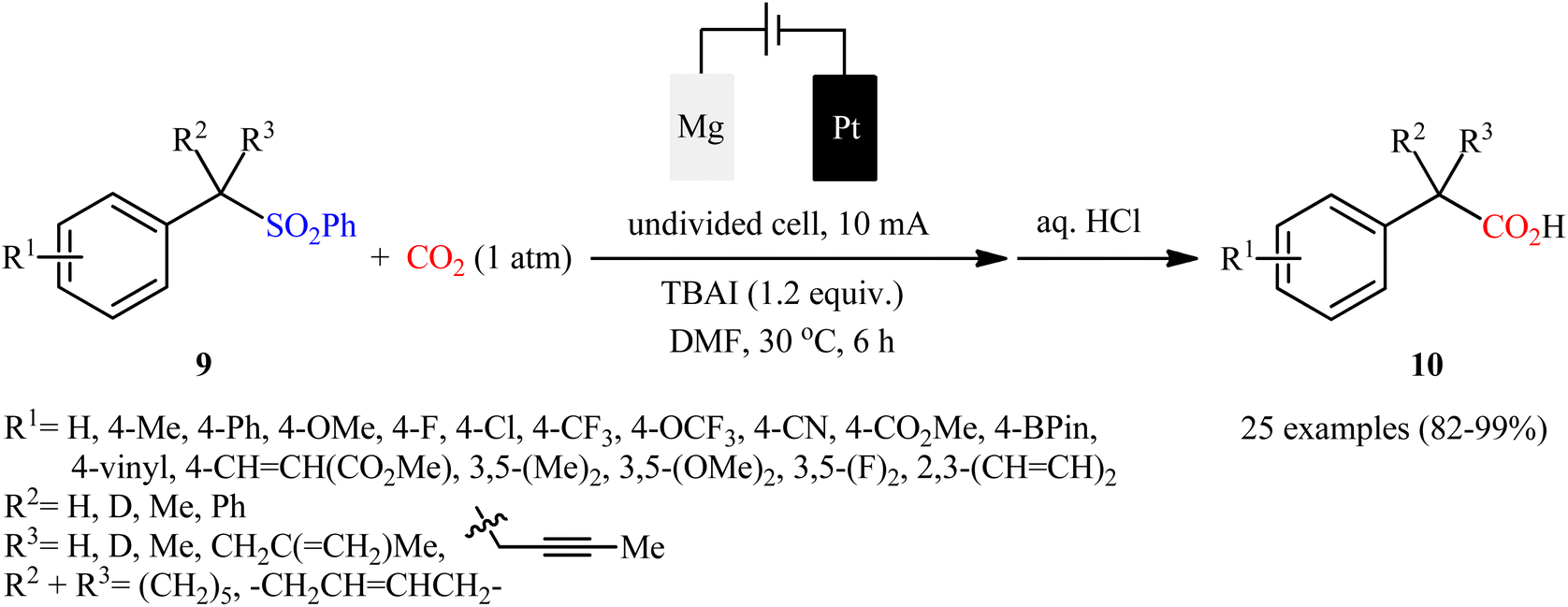
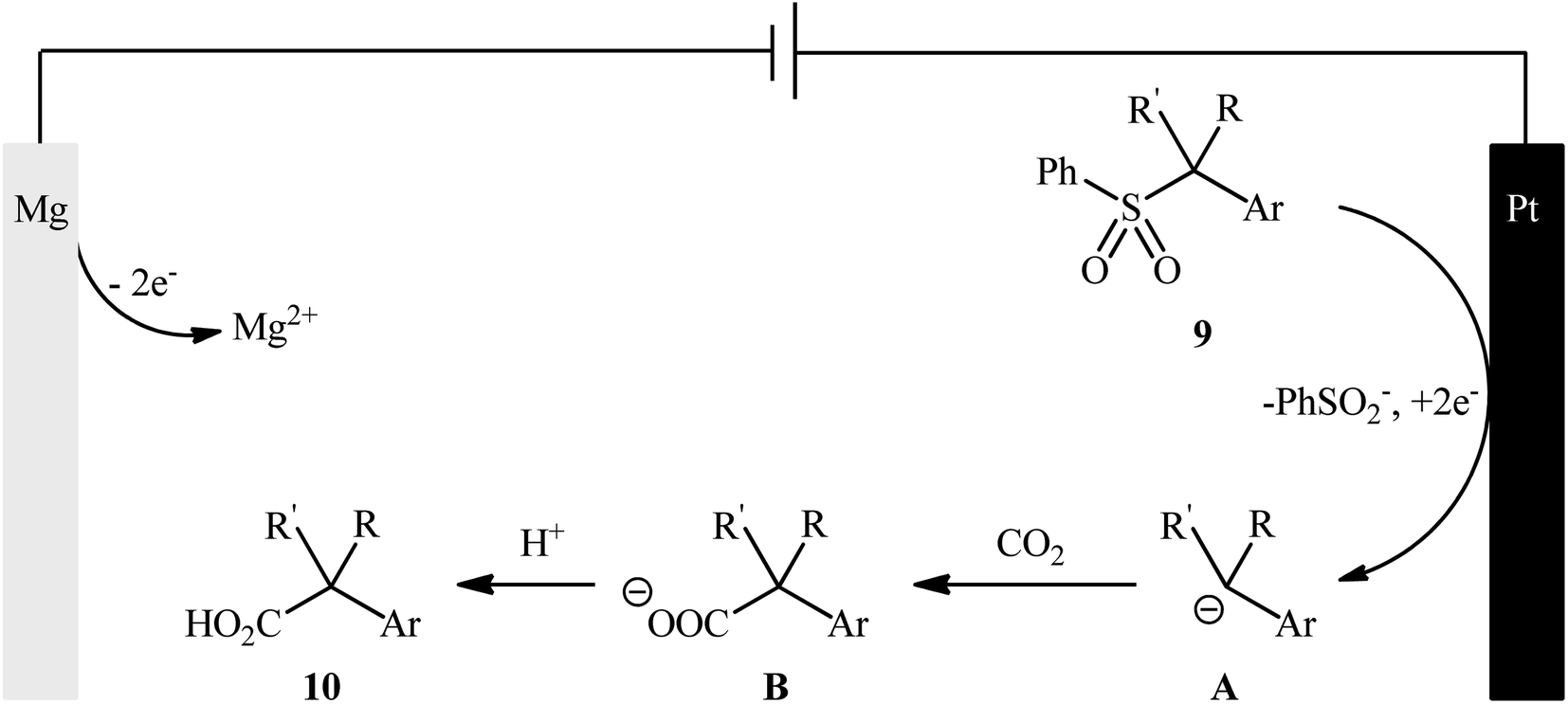
Sulfones are one of the latest substrates that were joined to the story of desulfurative carboxylation reactions with CO2. In 2021, Yu and co-workers developed a robust desulfonylative electrocarboxylation reaction of (benzylsulfonyl)benzenes 9 with CO2 for the synthesis of diverse benzylic carboxylic acids 10 (Scheme 8).27 In an undivided cell assembled with a magnesium anode and a platinum cathode, the best reaction conditions were achieved with tetrabutylammonium iodide (TBAI) as the electrolyte and DMF as the solvent, with a constant current of 10 mA at 30 °C. In this study, a relatively broad reaction scope was evaluated by testing different electron-donating and electron-withdrawing functional groups at the phenyl ring of benzyl moieties, as well as a series of functionalized secondary and tertiary sulfones. Moreover, a tolerance for 2-(thiophen-3-yl)acetic acid and 3-methylbut-3-enoic acid was also demonstrated. In addition to these results, the authors also successfully applied this electrocarboxylation protocol to the high yielding synthesis of nonsteroidal anti-inflammatory drug ibuprofen. Mechanistically, this transformation is believed to proceed through the formation of a benzylic carbanion A via cathodic reduction of sulfone 9, followed by its nucleophilic attack to CO2 to give the carboxylated product 10 (Scheme 9).
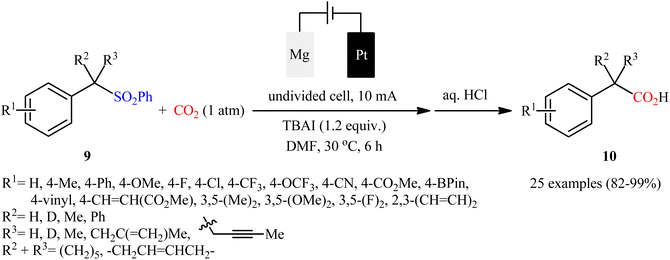 |
| | Scheme 8 Yu's synthesis of benzylic carboxylic acids 10. | |
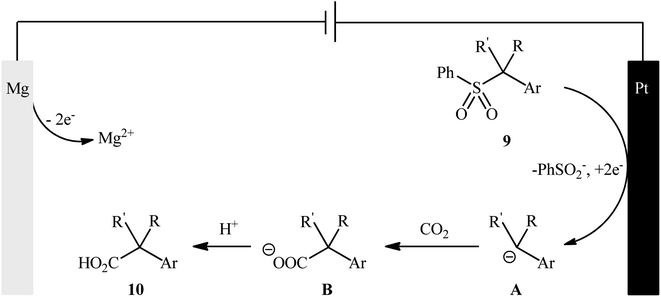 |
| | Scheme 9 A plausible mechanism for the formation of benzylic carboxylic acids 10. | |
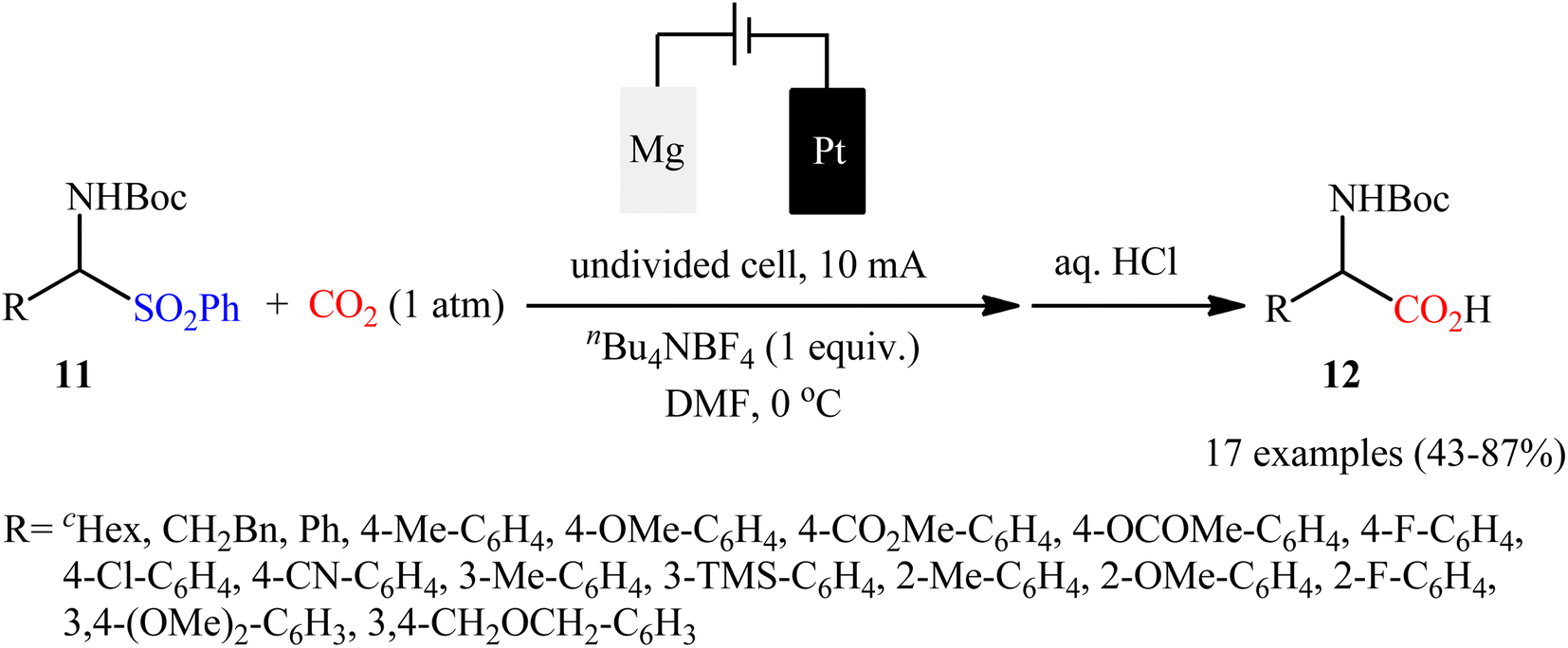
Concurrently, in a related investigation, Senboku and co-workers realized electrocarboxylation of N-Boc-α-aminosulfone derivatives 11 with CO2 under atmospheric pressure, to form the corresponding N-Boc-α-amino acids 12 by cleaving the C–S bond.28 An undivided cell equipped with a Pt plate cathode and an Mg rod anode under a current of 10 mA and nBu4NBF4 as the electrolyte in DMF were the best reaction conditions, and 17 examples were obtained in yields of 43 to 87% (Scheme 10). The results showed that N-Boc-α-aminosulfones having an aryl substituent at the α position were high yielding compared to N-Boc-α-aminosulfones bearing an alkyl substituent at the same position. Electron-rich alcohols gave higher yields than electron-deficient ones. Interestingly, the electronic and steric effects of the substituents on the aromatic units had a negligible influence on the outcome of the reaction; therefore, the presence of different electron-donating and with-drawing fictional groups in o-, m-, or p-positions on the phenyl ring were well tolerated by the present electrochemical carboxylation. According to the authors proposed mechanism, this carboxylation reaction most likely proceeds via a radical pathway.
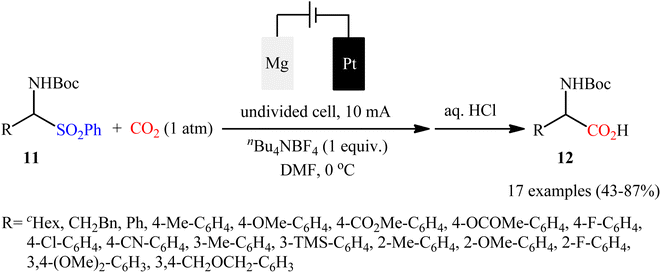 |
| | Scheme 10 Senboku's synthesis of N-Boc-α-amino acids 12. | |
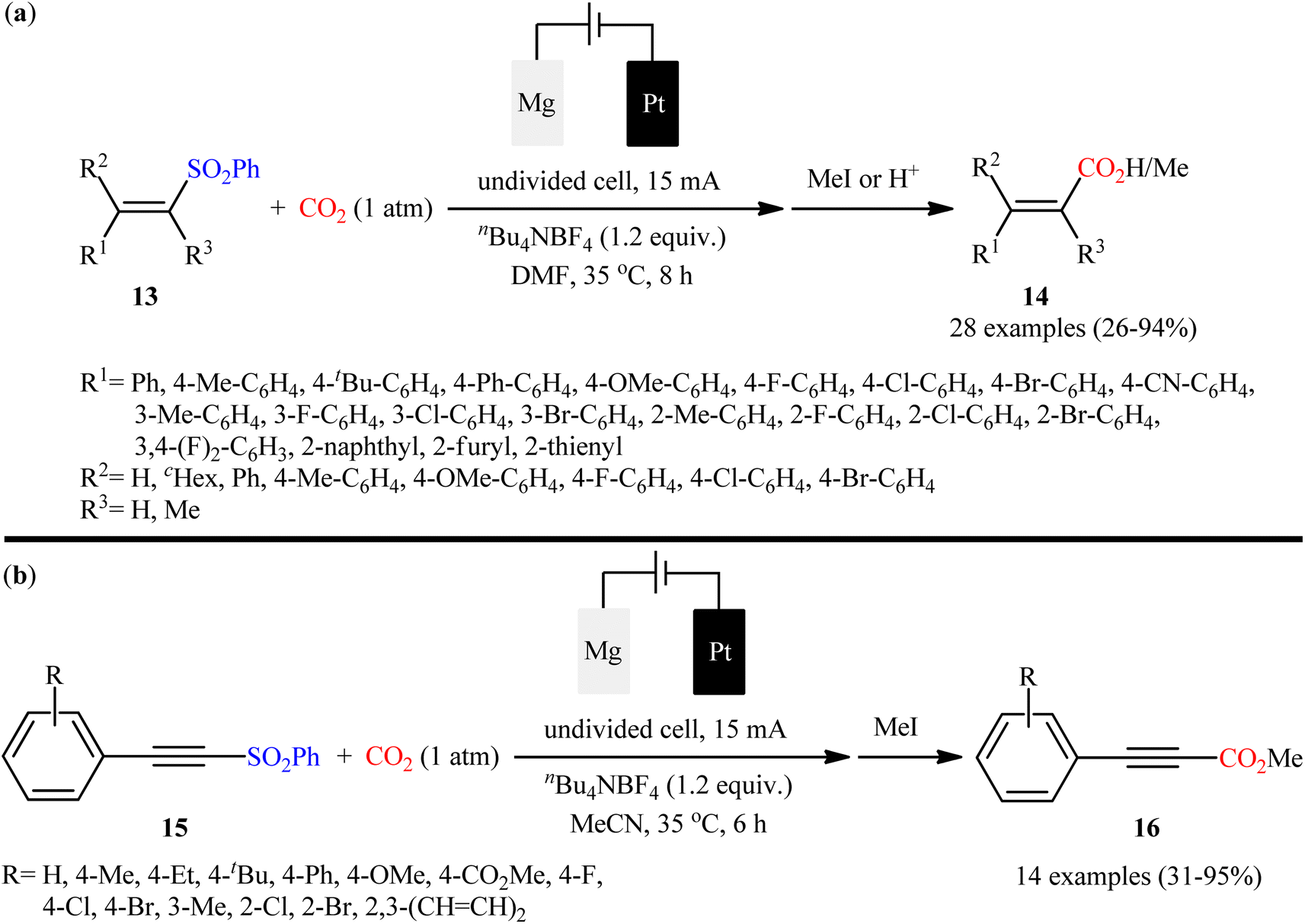
In their subsequent studies, very recently, Ye and colleagues unraveled the electrosynthesis of cinnamic acid derivatives 14 via desulfonylative carboxylation of respective α, β-unsaturated sulfones 13 with atmospheric CO2 using their undivided cell under constant current of 15 mA.29 As shown in Scheme 11a, an array of mono, di, tri, and tetrasubstituted vinyl sulfones were compatible with this protocol and afforded the target α,β-unsaturated carboxylic acids in poor to excellent yields with complete (E)-selectivity. The reaction, however, appears to be limited to vinyl sulfones containing (hetero)aryl groups in the alkene terminus. According to the authors, selective formation of more stable (E)-vinyl anion from the fragmentation of starting vinyl sulfone is responsible for the (E)-selectivity of products. Apart from vinyl sulfones, the protocol was also compatible for carboxylation of alkynyl sulfones 15 (Scheme 11b). In this case, MeCN was found to be better solvent than DMF for the reaction. Just like vinyl sulfones, the scope of alkynyl sulfones were limited to the aromatic substrates. Delightedly, this protocol was also compatible with aryl sulfones as evidenced by synthesis of benzoic acid, 1-naphthoic acid, and 2-thiophenecarboxylic acid in moderate to good yields (62–79%) from the corresponding aryl sulfones.
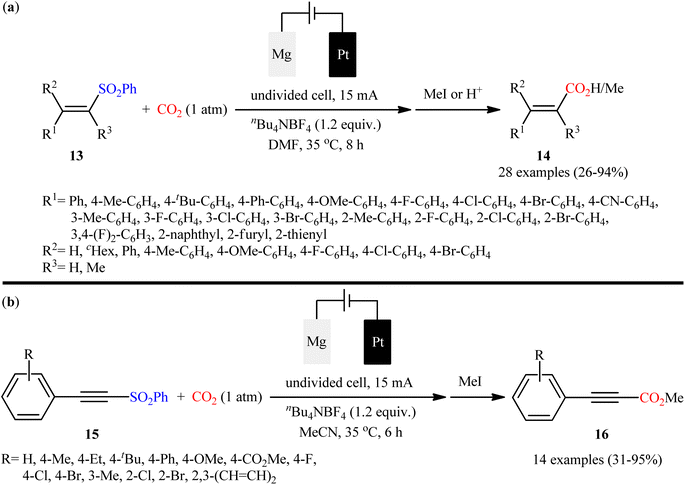 |
| | Scheme 11 Ye synthesis of (a) cinnamic acid derivatives 14; (b) methyl propiolates 16. | |
5. Carboxylation of sulfoxides
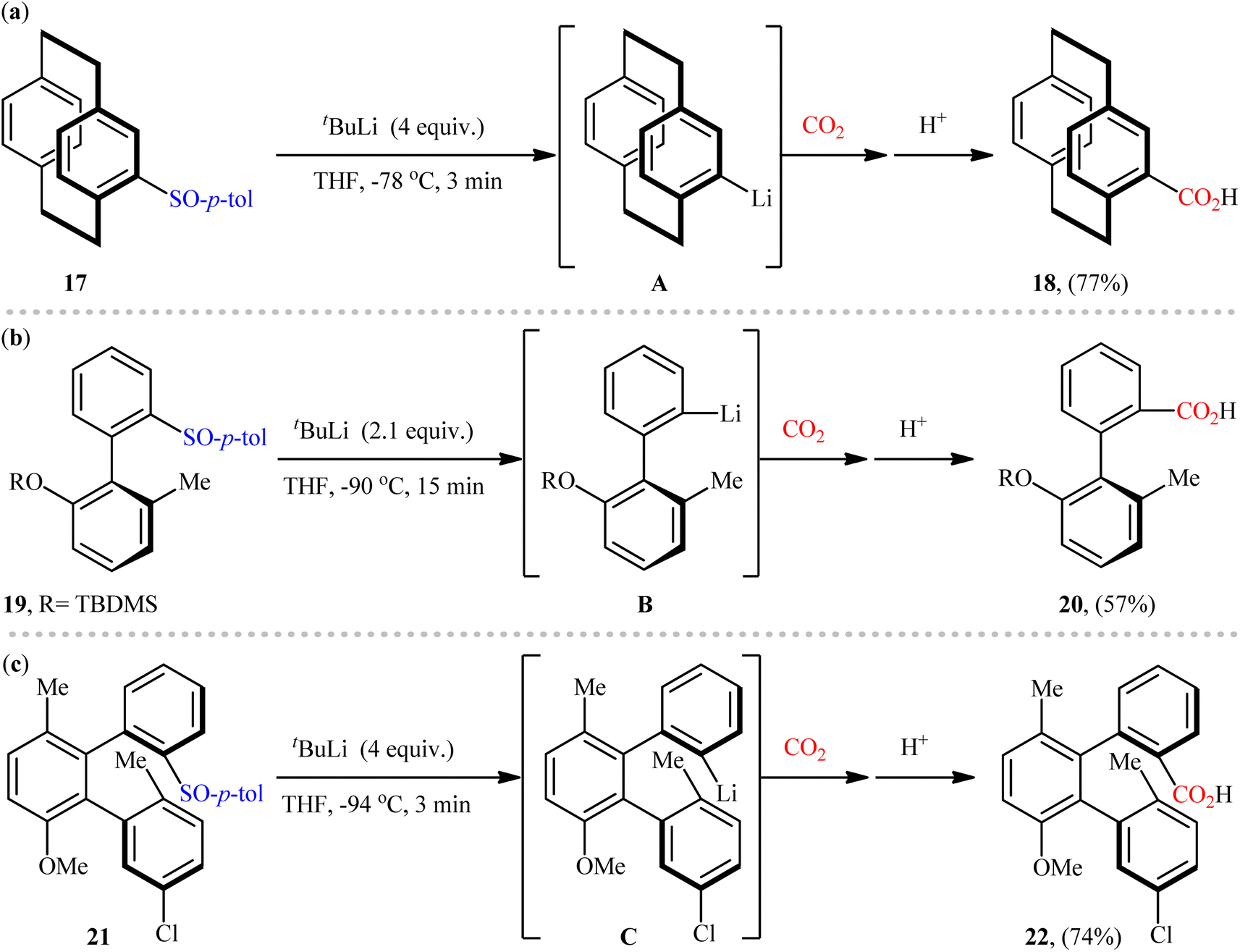
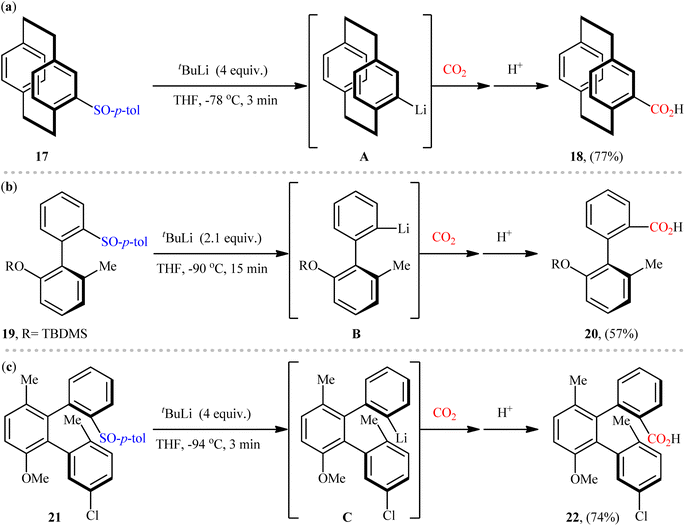
Despite the numerous reports that have been published on the use of aryl sulfoxides as aryl transfer reagents for the formation of various C(aryl)–C/X bonds,30 the reported examples for the synthesis of carboxylic acid derivatives via carboxylation of the corresponding sulfoxides with CO2 through C–S bond cleavage are scarce, and to the best of our knowledge there is only three single examples on this chemistry. One of the earliest reports of the applicability of this page of carboxylic acid synthesis, has been reported by the research group of Rowlands in 2005,31 when 4-tolylsulfinyl-substituted [2.2]paracyclophane 17 underwent sulfoxide–lithium exchange using tBuLi to form 4-lithio[2.2]paracyclophane A, which after electrophilic trapping using CO2 (dry ice) afforded the corresponding carboxylic acid 18 in a yield of 77% (Scheme 12a). Nearly a decade later, Delord, Colobert, and co-workers applied these principles for the synthesis of chiral carboxylic acid 20 from tolylsulfinyl-substituted axially chiral biaryl 19 in moderate yield with outstanding enatioselectivity (Scheme 12b).32,33 Shortly after, using the same scenario, this research group was able to synthesis of an enantiomerically pure carboxylated ortho-terphenyl 22 in high yield from the respective sulfinyl-substituted terphenyl scaffold 21 (Scheme 12c).34 Later, Castellano's research team applied this protocol as the key strategic step in the synthesis of a small library of 4-amidated [2.2]paracyclophanes.35
 |
| | Scheme 12 Available examples on the carboxylation of sulfoxides with CO2. | |
6. Carboxylation of thioethers
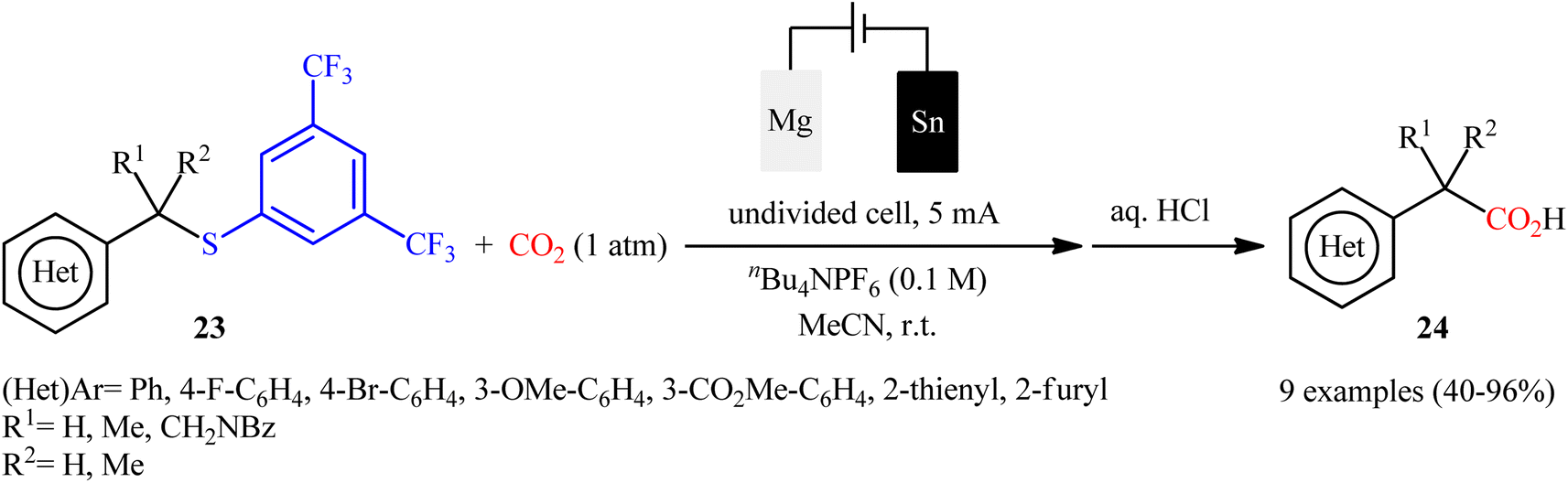
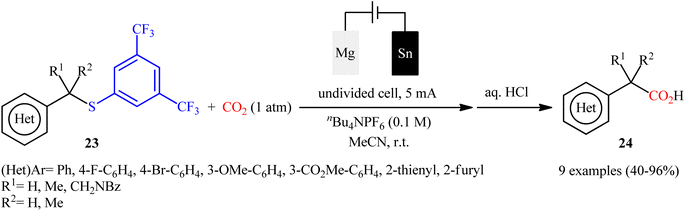
One of the latest organosulfur compounds to join the story of desulfurative carboxylation reactions using CO2 is thioethers. In 2023, Ahlquist–Lundberg and co-workers developed a catalyst-free electrochemical approach for selective desulfurative carboxylation of benzylic 3,5-bistrifluoromethylthiophenyl ethers 23 with CO2 under ambient conditions.36 The authors identified tin and magnesium as the optimal cathode and anode, respectively, and tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the supporting electrolyte. The established electrocarboxylation was conducted in anhydrous acetonitrile under ambient air, tolerated several important functional groups, and provided the target benzylic carboxylic acids 24 in moderate to excellent yields (Scheme 13); in addition, a tolerance for a propargylic 3,5-bistrifluoromethylthiophenyl ether was also demonstrated. Of note, in all provided examples, the reaction proceeded with complete selectivity for C(sp3)–S bond cleavage. After a series of mechanistic investigations, it was confirmed that this transformation most likely proceeds via a radical pathway.
 |
| | Scheme 13 Electroreductive desulfurative carboxylation of benzylic 3,5-bistrifluoromethylthiophenyl ethers 23 with CO2. | |
7. Conclusion
In this review, we have summarized the available literature on desulfurative carboxylation of organosulfur compounds employing CO2 as an ideal carboxyl source. Although several sulfur-containing compounds (e.g., sulfonium salts, sodium sulfinates, sulfones, sulfoxides) have been described for this page of carboxylic acid synthesis, it is still desirable to explore other alternative organosulfur compounds (e.g., thiols, disulfides, sulfinic acids, sulfonyl hydrazides) that may be improving the efficiency, atom-economy, and minimizing the energy and chemical waste. On the other hand, the number of reported examples on the existing protocols are too narrow and there is further need to study the scope and limitations of these reactions. Furthermore, the majority of reactions covered in this review were carried out under electrochemical conditions. Despite many advantages of electroorganic processes, there are challenges for its use on industrial scales. The need for supporting electrolytes is one of the main limitations of electro-organic transformations, which are sometimes very expensive or even the source of unacceptable pollutants. On the other hand, recovery of the supporting electrolyte can cause technical difficulties and increase costs. Another drawback of electrosynthesis is the long reaction time due to the sensitivity of substrates to higher current densities, which hampers broader acceptance of the method in organic chemical laboratories. Despite recent advances, this field of research is still in its infancy and further investigations and improvements are still needed to reach maturity. We hope that this review will inspire and motivate scientists to conduct future research on this interesting field.
Conflicts of interest
There are no confilicts to declare.
References
-
(a) A. Rafiee, K. R. Khalilpour, D. Milani and M. Panahi, J. Environ. Chem. Eng., 2018, 6, 5771–5794 CrossRef CAS;
(b) G. Yuan, C. Qi, W. Wu and H. Jiang, Curr. Opin. Green Sustainable Chem., 2017, 3, 22–27 CrossRef;
(c) R. Guil-López, N. Mota, J. Llorente, E. Millán, B. Pawelec, J. L. G. Fierro and R. M. Navarro, Materials, 2019, 12, 3902 CrossRef PubMed;
(d) G. Naik, N. Sarki, V. Goyal, A. Narani and K. Natte, Asian J. Org. Chem., 2022, 11, e202200270 CrossRef CAS;
(e) T. A. Qassem, H. Bahair, H. Tariq, F. Behmagham and F. Shahimi, Chem. Rev. Lett., 2023, 6, 235–244 CAS;
(f) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed.
-
(a) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC;
(b) L. Wang, S. Que, Z. Ding and E. Vessally, RSC Adv., 2020, 10, 9103–9115 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
-
(a) S. S. Bharate, Pharm. Res., 2021, 38, 1307–1326 CrossRef CAS PubMed;
(b) H. Cai, X. Gan, Z. Jin and G. Hao, J. Agric. Food Chem., 2023, 71, 9973–9993 CrossRef CAS PubMed;
(c) L. Zhang, X. Hu, K. Hu, C. Hu, Z. Zhang, Q. Liu, S. Hu, J. Xiang, Y. Wang and S. Zhang, J. Power Sources, 2018, 403, 137–156 CrossRef CAS;
(d) B. N. Bhadra, I. Ahmed, H. J. Lee and S. H. Jhung, Coord. Chem. Rev., 2022, 450, 214237 CrossRef CAS.
-
(a) J. Luo and I. Larrosa, ChemSusChem, 2017, 10, 3317–3332 CrossRef CAS PubMed;
(b) S. S. Yan, Q. Fu, L. L. Liao, G. Q. Sun, J. H. Ye, L. Gong, Y. Z. Bo-Xue and D. G. Yu, Coord. Chem. Rev., 2018, 374, 439–463 CrossRef CAS;
(c) C. K. Ran, L. L. Liao, T. Y. Gao, Y. Y. Gui and D. G. Yu, Curr. Opin. Green Sustainable Chem., 2021, 32, 100525 CrossRef CAS;
(d) N. Salehi and B. Azizi, J. Chem. Lett., 2021, 2, 2–8 Search PubMed;
(e) W. Huang, J. Lin, F. Deng and H. Zhong, Asian J. Org. Chem., 2022, 11, e202200220 CrossRef CAS;
(f) A. K. O. Aldulaimi, H. R. Saud, M. Ubaid, M. H. Sami, A. H. Adhab and F. Shahimi, Chem. Rev. Lett., 2024, 7, 148–158 CAS.
- A. Correa and R. Martin, Angew Chem. Int. Ed. Engl., 2009, 48, 6201–6204 CrossRef CAS PubMed.
- F. Behmagham, S. M. Saied, M. D. Azeez, R. R. Abbass, A. H. Adhab and E. Vessally, RSC Adv., 2023, 13, 32502–32517 RSC.
-
(a) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681–2683 CrossRef CAS PubMed;
(b) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826–7827 CrossRef CAS PubMed.
- M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057–5058 CrossRef CAS.
- W. Xu, D. Guo, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2021, 45, 101403 CrossRef CAS.
- W. Xu, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2021, 43, 101358 CrossRef CAS.
- M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739–6749 CrossRef PubMed.
-
(a) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828–33844 RSC;
(b) M. Abdoli and H. Saeidian, J. Sulfur Chem., 2015, 36, 556–582 CrossRef CAS.
-
(a) S. G. Modha, V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042–5055 RSC;
(b) S. Huang, M. Wang and X. Jiang, Chem. Soc. Rev., 2022, 51, 8351–8377 RSC;
(c) K. Yang, Q. Li, Z. Y. Li and X. Sun, Chem. Commun., 2023, 59, 5343–5364 RSC.
- K. Nogi and H. Yorimitsu, Chem.–Asian J., 2020, 15, 441–449 CrossRef CAS PubMed.
- Selected reviews:
(a) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS;
(b) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef CAS;
(c) S. Farshbaf, L. Z. Fekri, M. Nikpassand, R. Mohammadi and E. Vessally, J. CO2 Util., 2018, 25, 194–204 CrossRef CAS;
(d) S. Arshadi, A. Banaei, S. Ebrahimiasl, A. Monfared and E. Vessally, RSC Adv., 2019, 9, 19465–19482 RSC;
(e) M. Li, S. Abdolmohammadi, M. S. Hoseininezhad-Namin, F. Behmagham and E. Vessally, J. CO2 Util., 2020, 38, 220–231 CrossRef CAS.
-
(a) A. Y. B. Ángel, P. R. O. Campos and E. E. Alberto, Org. Biomol. Chem., 2023, 21, 223–236 RSC;
(b) R. Fan, C. Tan, Y. Liu, Y. Wei, X. Zhao, X. Liu, J. Tan and H. Yoshida, Chin. Chem. Lett., 2021, 32, 299–312 CrossRef CAS;
(c) M. Mondal, S. Chen and N. J. Kerrigan, Molecules, 2018, 23, 738 CrossRef PubMed.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
- T. Yanagi, R. J. Somerville, K. Nogi, R. Martin and H. Yorimitsu, ACS Catal., 2020, 10, 2117–2123 CrossRef CAS.
- H. Meng, M. S. Liu and W. Shu, Chem. Sci., 2022, 13, 13690–13707 RSC.
-
(a) X. Y. Chen, Y. Wu and P. Wang, Synthesis, 2022, 54, 3928–3940 CrossRef CAS;
(b) X. Wu, P. Gao and F. Chen, Eur. J. Org Chem., 2023, 26, 202300864 CrossRef.
- S. Tang, X. Zhao, L. Yang, B. Li and B. Wang, Angew. Chem., Int. Ed., 2022, 134, 202212975 CrossRef.
- X. Ye, X. Wu, S. R. Guo, D. Huang and X. Sun, Tetrahedron Lett., 2021, 81, 153368 CrossRef CAS.
-
(a) S. Liang, K. Hofman, M. Friedrich and G. Manolikakes, Eur. J. Org Chem., 2020, 4664–4676 CrossRef CAS;
(b) R. J. Reddy and A. H. Kumari, RSC Adv., 2021, 11, 9130–9221 RSC.
- S. Sun, J. T. Yu, Y. Jiang and J. Cheng, Adv. Synth. Catal., 2015, 357, 2022–2026 CrossRef CAS.
- Y. Chen, X. Dai, W. Zhang, T. Wu, L. Chen and Z. Peng, RSC Adv., 2022, 12, 772–776 RSC.
- J. S. Zhong, Z. X. Yang, C. L. Ding, Y. F. Huang, Y. Zhao, H. Yan and K. Y. Ye, J. Org. Chem., 2021, 86, 16162–16170 CrossRef CAS PubMed.
- H. Senboku, Y. Minemura, Y. Suzuki, H. Matsuno and M. Takakuwa, J. Org. Chem., 2021, 86, 16077–16083 CrossRef CAS PubMed.
- Z. X. Yang, L. Lai, J. Chen, H. Yan, K. Y. Ye and F. E. Chen, Chin. Chem. Lett., 2023, 34, 107956 CrossRef CAS.
-
(a) C. B. Rauhut, L. Melzig and P. Knochel, Org. Lett., 2008, 10, 3891–3894 CrossRef CAS PubMed;
(b) M. A. Fascione, R. Brabham and W. B. Turnbull, Chem.–Eur. J., 2016, 22, 3916–3928 CrossRef CAS PubMed;
(c) K. Yamamoto, S. Otsuka, K. Nogi and H. Yorimitsu, ACS Catal., 2017, 7, 7623–7628 CrossRef CAS;
(d) Q. Chen, S. Wu, S. Yan, C. Li, H. Abduhulam, Y. Shi, Y. Dang and C. Cao, ACS Catal., 2020, 10, 8168–8176 CrossRef CAS;
(e) W. X. Li, B. W. Yang, X. Ying, Z. W. Zhang, X. Q. Chu, X. Zhou, M. Ma and Z. L. Shen, J. Org. Chem., 2022, 87, 11899–11908 CrossRef CAS PubMed.
- P. B. Hitchcock, G. J. Rowlands and R. Parmar, Chem. Commun., 2005, 4219–4221 RSC.
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed. Engl., 2014, 126, 14091–14095 CrossRef.
- Q. Dherbassy, G. Schwertz, C. K. Hazra, T. Wesch, J. Wencel-Delord and F. Colobert, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1339–1351 CrossRef CAS.
- Q. Dherbassy, J. P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed. Engl., 2018, 130, 4758–4762 CrossRef.
- W. R. Henderson, D. E. Fagnani, J. Grolms, K. A. Abboud and R. K. Castellano, Helv. Chim. Acta, 2019, 102, 1900047 CrossRef.
- J. Kuzmin, J. Röckl, N. Schwarz, J. Djossou, G. Ahumada, M. Ahlquist and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202304272 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Shakir Mahmood Saeedd,
Ameer Hassan Idane,
Nahed Mahmood Ahmed Alsultanyf,
Sattar Arshadig,
Farnaz Behmagham*h and
Esmail Vessally
c,
Shakir Mahmood Saeedd,
Ameer Hassan Idane,
Nahed Mahmood Ahmed Alsultanyf,
Sattar Arshadig,
Farnaz Behmagham*h and
Esmail Vessally