
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lattice solvent- and substituent-dependent spin-crossover in isomeric iron(II) complexes†
Senthil Kumar
Kuppusamy
 *a,
Asato
Mizuno‡
b,
Lea
Kämmerer
c,
Soma
Salamon
c,
Benoît
Heinrich
d,
Corinne
Bailly
e,
Ivan
Šalitroš
*fg,
Heiko
Wende
*c and
Mario
Ruben
*abh
*a,
Asato
Mizuno‡
b,
Lea
Kämmerer
c,
Soma
Salamon
c,
Benoît
Heinrich
d,
Corinne
Bailly
e,
Ivan
Šalitroš
*fg,
Heiko
Wende
*c and
Mario
Ruben
*abh
aInstitute of Quantum Materials and Technologies (IQMT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: senthil.kuppusamy2@kit.edu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
cUniversity of Duisburg-Essen, Faculty of Physics and Center for Nanointegration Duisburg-Essen (CENIDE), Lotharstraße 1, 47057 Duisburg, Germany
dInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), CNRS-Université de Strasbourg, 23, rue du Loess, BP 43, 67034 Strasbourg Cedex 2, France
eService de Radiocristallographie, Fédération de Chimie Le Bel UAR2042 CNRS-Université de Strasbourg, 1 rue Blaise Pascal, BP 296/R8, 67008 Strasbourg cedex, France
fCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic
gDepartment of Inorganic Chemistry, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Bratislava SK-81237, Slovakia
hCentre Européen de Sciences Quantiques (CESQ), Institut de Science et d'Ingénierie, Supramoléculaires (ISIS), 8 allée Gaspard Monge, BP 70028, 67083 Strasbourg Cedex, France
First published on 22nd May 2024
Abstract
Spin-state switching in iron(II) complexes composed of ligands featuring moderate ligand-field strength—for example, 2,6-bi(1H-pyrazol-1-yl)pyridine (BPP)—is dependent on many factors. Herein, we show that spin-state switching in isomeric iron(II) complexes composed of BPP-based ligands—ethyl 2,6-bis(1H-pyrazol-1-yl)isonicotinate (BPP-COOEt, L1) and (2,6-di(1H-pyrazol-1-yl)pyridin-4-yl)methylacetate (BPP-CH2OCOMe, L2)—is dependent on the nature of the substituent at the BPP skeleton. Bi-stable spin-state switching—with a thermal hysteresis width (ΔT1/2) of 44 K and switching temperature (T1/2) = 298 K in the first cycle—is observed for complex 1·CH3CN composed of L1 and BF4− counter anions. Conversely, the solvent-free isomeric counterpart of 1·CH3CN—complex 2a, composed of L2 and BF4− counter anions—was trapped in the high-spin (HS) state. For one of the polymorphs of complex 2b·CH3CN—2b·CH3CN-Y, Y denotes yellow colour of the crystals—composed of L2 and ClO4− counter anions, a gradual and non-hysteretic SCO is observed with T1/2 = 234 K. Complexes 1·CH3CN and 2b·CH3CN-Y also underwent light-induced spin-state switching at 5 K due to the light-induced excited spin-state trapping (LIESST) effect. Structures of the low-spin (LS) and HS forms of complex 1·CH3CN revealed that spin-state switching goes hand-in-hand with pronounced distortion of the trans-N{pyridyl}-Fe–N{pyridyl} angle (ϕ), whereas such distortion is not observed for 2b·CH3CN-Y. This observation points that distortion is one of the factors making the spin-state switching of 1·CH3CN hysteretic in the solid state. The observation of bi-stable spin-state switching with T1/2 centred at room temperature for 1·CH3CN indicates that technologically relevant spin-state switching profiles based on mononuclear iron(II) complexes can be obtained.
Introduction
Molecular materials that show switching of a physical property—for example, magnetic1–12 or electric13–17—accompanied by hysteresis are candidates desirable for applications. Several classes of magnetic-molecular systems such as organic radicals,18–20 single-molecule magnets (SMMs),3,21–23 and spin-crossover (SCO) complexes24–31 are known to show bistable switching characteristics. However, to the best of our knowledge, SCO complexes are the only class of molecules that frequently show bistable switching near room-temperature (RT).4,12 For realistic applications, SCO complexes showing a sizable thermal hysteresis width (ΔT1/2)—around 70 K—with the switching temperature (T1/2) centred around RT are desirable.4 However, preparing an SCO complex that can undergo hysteretic switching around RT is not a trivial task because spin-state switching in the solid state is controlled by system-dependent factors, such as lattice solvent and disorder. Therefore, it is necessary to conduct a systematic search starting from a basic ligand skeleton featuring a moderate ligand field allowing spin-state switching of a coordinated metal ion, iron(II) in the context of this study. 2,6-Bis(1H-pyrazol-1-yl)pyridine (BPP)-based ligands (Fig. 1a) are a class of tridentate nitrogen-donor ligands that facilitate spin-state switching of the coordinated iron(II) centre, as first reported by Halcrow and co-workers in 2001.32 The propensity to modify the BPP skeleton with functional groups at the pyridine and pyrazole rings allows the systematic study of spin-state switching of the iron(II)–BPP complexes, useful for establishing structure–SCO property correlations.33 Recently, technologically relevant spin-state switching characteristics of iron(II) complexes (complexes a–d, Fig. 1b) composed of a BPP-based ligand—ethyl 2,6-bis(1H-pyrazol-1-yl)isonicotinate (BPP-COOEt, L1)—have been reported. Complex a underwent above room-temperature low-spin (LS)-to-high-spin (HS) switching and remained trapped in the HS-state after the loss of lattice acetone solvent.34 On the other hand, complex b, co-crystallized with lattice acetonitrile, underwent bi-stable spin-state switching with T1/2 = 233 K and ΔT1/2 = 100 K in the first cycle.9 However, in the subsequent cycles spin-state switching of complex b is rendered unstable due to self-grinding of the crystals. In a subsequent report, some of us have reported on the stable spin-state switching characteristics of complexes c and d and elucidated on the role of pronounced molecular distortion as a factor contributing to the occurrence of hysteretic spin-state switching.35 In this contribution, we have studied iron(II) complexes composed of BPP-COOEt (L1) and (2,6-di(1H-pyrazol-1-yl)pyridin-4-yl)methylacetate (BPP-CH2OCOMe, L2) ligands (Fig. 1a). Complex 1·CH3CN (Fig. 1b), composed of the BPP-COOEt ligand and BF4− counter anions, undergoes bi-stable spin-state switching (ΔT1/2 = 44 K) with T1/2 centred at RT (298 K) in the first cycle and the switching is not stable upon repeated cycling. To study the impact of substituents on the switching characteristics, we have prepared a new ligand L2, which features the same molecular formula as L1—the ligands L1 and L2 are structural isomers. Complex 2a (Fig. 1c) composed of L2 crystallized without the lattice solvent and showed no spin-state switching from the HS state upon cooling from 300 K. Conversely, for complex 2b·CH3CN-Y (Fig. 1c), a gradual SCO without thermal hysteresis is observed. Remarkably, complexes 1·CH3CN and 2b·CH3CN-Y also underwent light-induced LS-to-HS switching at 5 K following the light-induced excited spin-state trapping (LIESST) mechanism. The molecular structures of the LS and HS forms of 1·CH3CN obtained from single-crystal X-ray diffraction (SC-XRD) studies revealed a pronounced angular distortion of the trans-N{pyridyl}-Fe–N{pyridyl} angle (ϕ) in the HS-form relative to the LS-form. Additionally, the conformation of the ethyl substituent of 1·CH3CN is also switched upon spin-state switching. In contrast, no significant variation of ϕ was observed for the LS and HS forms of 2b·CH3CN-Y. These results indicate the contributory role of molecular distortion in spurring the bi-stable spin-state switching observed for 1·CH3CN. Overall, the contrasting magnetic characteristics of the complexes show a complex interplay between structural- and spin-isomerism, and a structural control over SCO is achievable following molecular engineering principles. Crucially, complex 1·CH3CN is one of the remarkable examples of a mononuclear iron(II) complex that undergoes temperature-induced bistable SCO with a T1/2 centred at RT. Importantly, our attribution that pronounced molecular distortion and a change in substituent conformation contribute to the opening of the thermal hysteresis loop could enable future designs of iron(II)-based SCO complexes with application-oriented spin-state switching characteristics.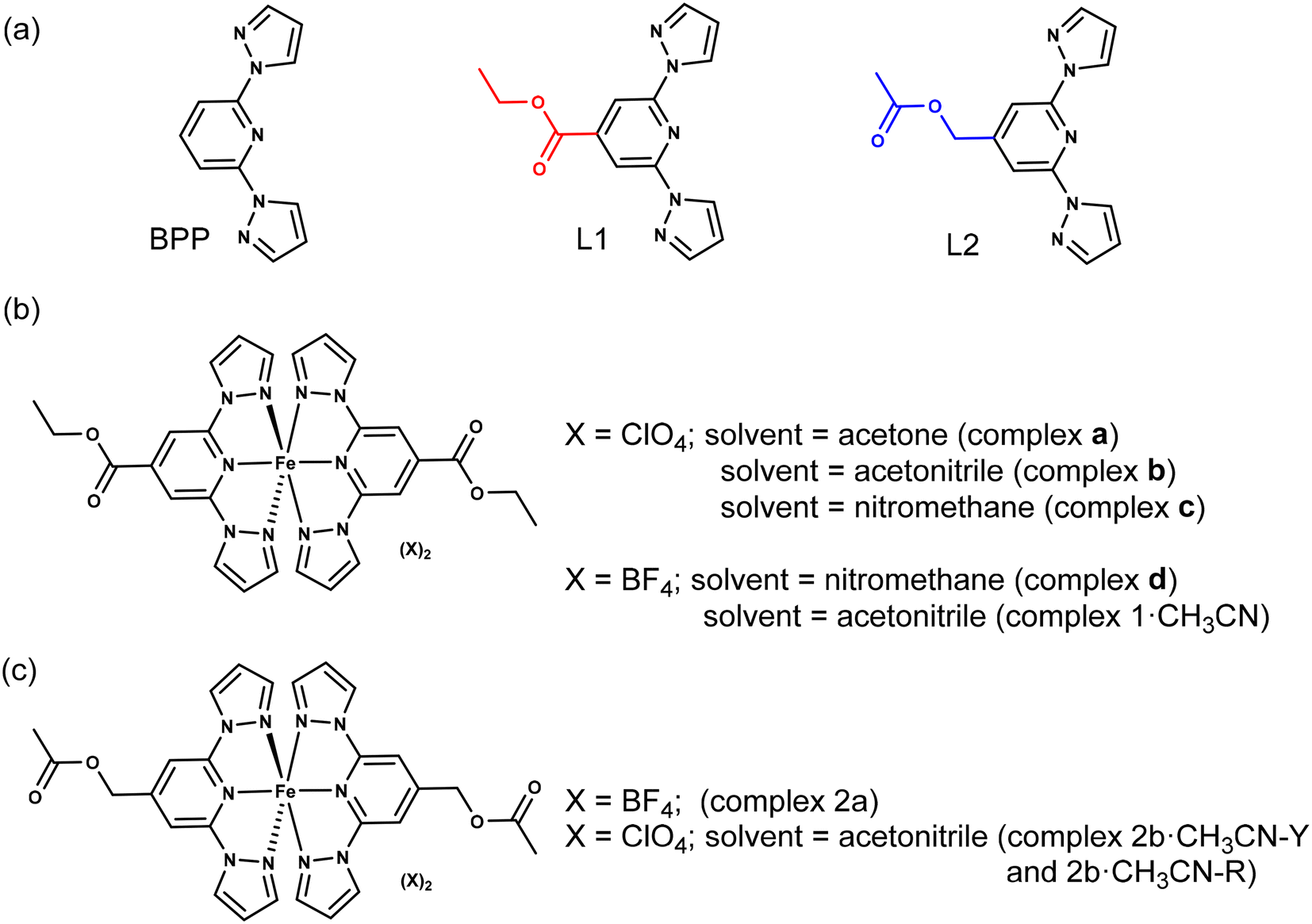 | ||
| Fig. 1 Molecular structures of ligands and iron(II) complexes discussed in this study. (a) Structures of parent BPP ligand and its functional variants L1 and L2. The ligands L1 and L2 are structural isomers; red and blue colours highlight functional group isomerism. The aim of this study is to elucidate how the nature of the substituent controls spin-state switching in iron(II) complexes composed of L1 and L2. (b) Structures of L1-based complexes a, b, c, d, and 1·CH3CN. (c) Structures of L2-based complexes 2a and 2b·CH3CN. For 2b·CH3CN, two different polymorphs—2b·CH3CN-Y and 2b·CH3CN-R—were obtained; Y and R denote yellow and red, respectively, colours of the crystals. Iron(II) complexes a, b, c, and d have been studied previously.9,34,35 | ||
Experimental
Materials and methods
Anhydrous solvents, [Fe(BF4)2·6H2O], and [Fe(ClO4)2·6H2O] were purchased from commercial sources and used as received. Glassware was dried in a vacuum oven at 423 K prior to the experiments. The complexation reactions were performed under an argon (Ar) atmosphere. Experimental protocols used to study the physical properties of the ligands and complexes (section S1), and synthesis procedures used to prepare the ligands and complexes (section S2) are described in the ESI.†Caution: perchlorate salts are potentially hazardous; care should be taken while handling them. We have encountered no difficulties during the preparation, characterization, and magnetic studies of perchlorate containing complexes—2b·CH3CN-Y and 2b·CH3CN-R—discussed in this study.
Results and discussion
Synthesis of the ligands and complexes
Ligand L1 was synthesized following a previously reported procedure.36 Ligand L2 was synthesized from (2,6-di(1H-pyrazol-1-yl)pyridin-4-yl)methanol, as detailed in section S2 of the ESI.† Treatment of [Fe(X)2·6H2O] (X = BF4 or ClO4) as a solid with the acetonitrile solution of the respective ligand resulted in the formation of complexes 1·CH3CN, 2a, or 2b·CH3CN (Scheme 1). Slow diffusion of diethyl ether into the acetonitrile solutions of the reaction mixtures kept inside a fridge at 4 °C over a period of 2–3 weeks yielded a crop of wine-red crystals of complex 1·CH3CN and pale-yellow crystals of 2a, suitable for single-crystal X-ray diffraction (SC-XRD) studies. In the case of 2b·CH3CN, crystallizing with lattice acetonitrile, two different polymorphs coloured yellow (2b·CH3CN-Y) and red (2b·CH3CN-R) were obtained. After harvesting from the mother liquor, the crystals of 1·CH3CN were washed with diethyl ether and dried under vacuum for 4 h. Elemental analysis of the dried crystals revealed that lattice acetonitrile solvent is retained after the drying process (section S2†). In the case of 2b·CH3CN-Y, drying under vacuum resulted in the loss of the lattice solvent. Therefore, the yellow crystals were left to dry inside a fume-hood for an hour after washing with diethyl ether. We have also noted slow release of the lattice solvent from the crystals of 2b·CH3CN-Y over the course of a few days while storing under ambient conditions. The crystals of 2b·CH3CN-R lost the lattice solvent rapidly after removing from the mother liquor with the consequent cracking and yellow-brown coloration of the crystals. A complete loss of the lattice solvent was inferred from the elemental analysis data of 2b·CH3CN-R. See section S2 and Fig. S1–S6 of the ESI† for the characterization data of the ligands and complexes.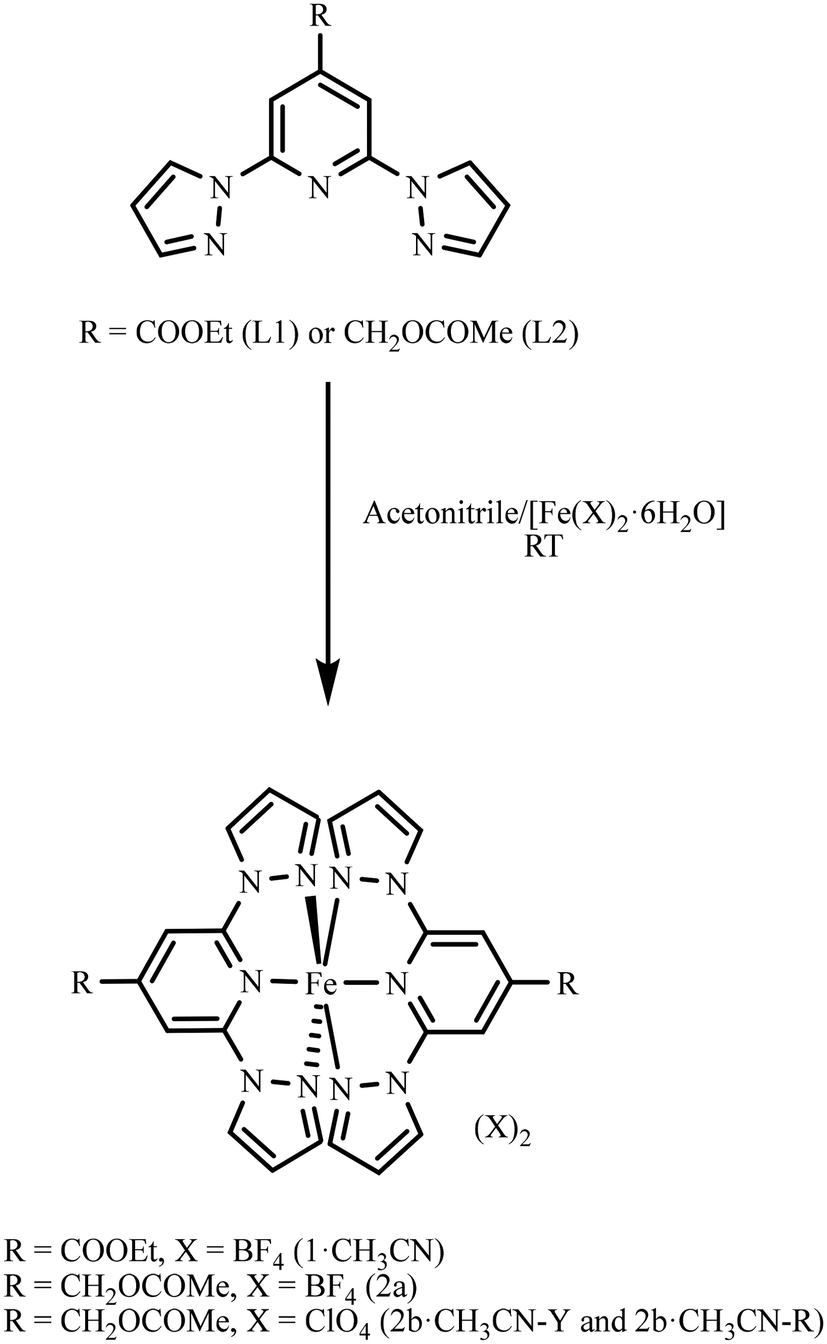 | ||
| Scheme 1 Preparation of complex 1·CH3CN from L1 and complexes 2a and 2b·CH3CN from L2. Two different polymorphs coloured yellow (2b·CH3CN-Y) and red (2b·CH3CN-R) were obtained for 2b·CH3CN. | ||
Electronic absorption spectra of ligands L1 and L2 and complexes 1·CH3CN, 2a, and 2b·CH3CN-Y
To get insights into the effect of substituents on the absorption spectroscopic characteristics of ligands and complexes, UV-Vis absorption spectroscopic studies of L1, L2, 1·CH3CN, 2a, and 2b·CH3CN-Y were performed in acetonitrile solvent. To make comparisons, spectroscopic studies of the parent BPP ligand and its complex—[Fe(BPP)2](BF4)2—were also performed. In general, two sets of transitions in the 250-to-275 nm and 275-to-400 nm regions were observed for the ligands and their corresponding complexes (Fig. 2 and Table 1). The first and second set of transitions are tentatively attributed to the π → π* and n → π* transitions of the ligand skeletons. The absorption maxima of BPP and L2 are comparable, elucidating the negligible electronic effect of the OCH2COCH3 substituent on the BPP skeleton. On the other hand, absorption maxima of L1 were bathochromically shifted relative to the parent BPP ligand, reflecting on the significant inductive effect of the COOEt substituent on the BPP skeleton. We tentatively attribute the bathochromic shifting observed for L1, relative to BPP, to the stabilization of the lowest unoccupied molecular orbital (LUMO) of the ligand imparted by the electron withdrawing COOEt substituent. Complexation of the ligands with iron(II) induced negligible changes in the absorption characteristics of the ligands, as inferred from the comparable nature of the absorption maxima of the ligands and their corresponding complexes (Fig. 2a and b). Remarkably, for 1·CH3CN, a shoulder with a maximum centred at 461 nm was observed (Fig. S7†), attributed to the singlet metal-to-ligand charge transfer (1MLCT) transition associated with the LS form of the complex. Such a transition was not observed for [Fe(BPP)2](BF4)2, complexes 2a, and 2b·CH3CN-Y, indicating the HS state of the complexes in the solution.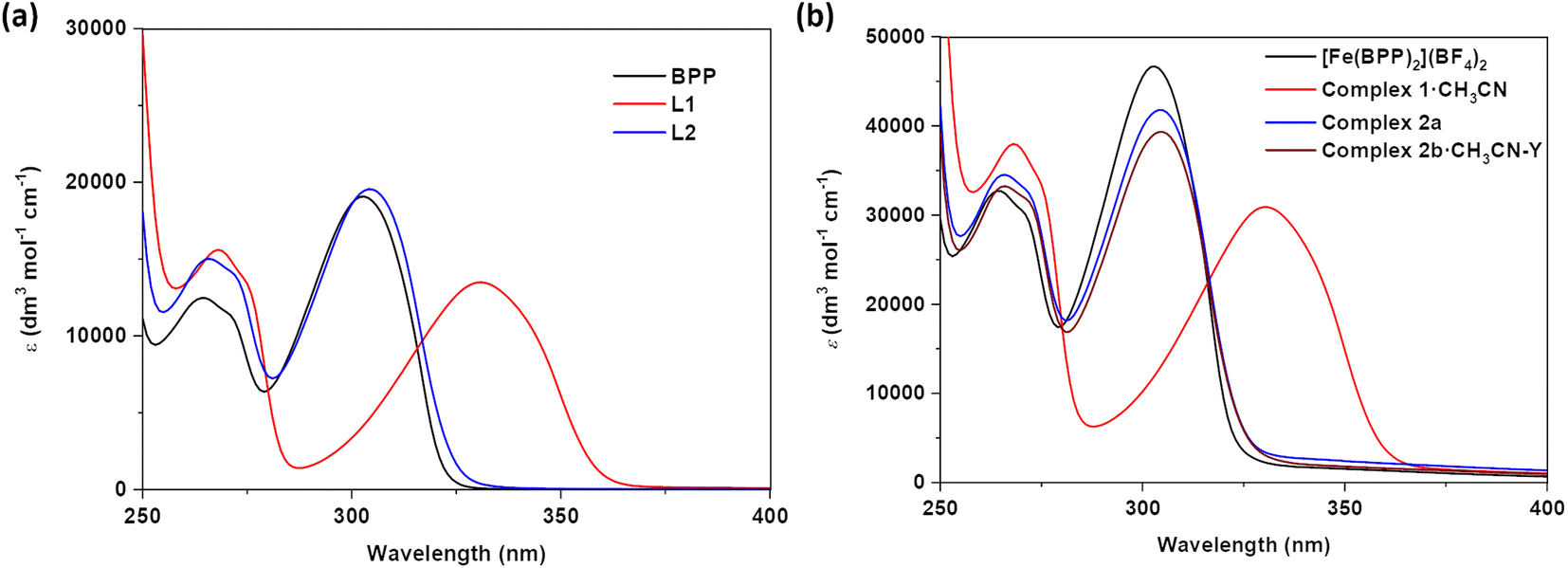 | ||
| Fig. 2 Electronic absorption spectroscopic studies. UV-Vis electronic absorption spectra of (a) ligands and (b) complexes measured in acetonitrile solution. Functionalization of the parent BPP skeleton with COOEt substituent caused bathochromic shifting of the absorption maxima of L2, whereas OCH2COCH3 substituent caused no such shift in L2. | ||
| Entry | λ max. (CH3CN)/nm (ε)/dm3 mol−1 cm−1 |
|---|---|
| BPP | 265 (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 567), 272 (11399), 303 (19136) 567), 272 (11399), 303 (19136) |
| L1 | 268 (15711), 275 (13559), 331 (13559) |
| L2 | 265 (15008), 272 (13884), 304 (19654) |
| [Fe(BPP)2](BF4)2 | 264 (32791), 271 (30011), 302 (46736) |
| 1·CH3CN | 268 (38147), 274 (33538), 330 (30875), 461 (708) |
| 2a | 265 (34708), 272 (32440), 304 (41893) |
| 2b·CH3CN-Y | 265 (33216), 272 (31577), 304 (39479) |
Structural studies of L1; low spin (LS) and high spin (HS) forms of complex 1·CH3CN; HS form of 2a; LS form of 2b·CH3CN-R; and LS and HS forms of 2b·CH3CN-Y
Slow evaporation of the CDCl3 solution of L1 in an NMR tube resulted in the formation of single crystals of L1 suitable for X-ray diffraction (XRD) studies (Table S1†). In the molecular structure shown in Fig. 3a, the coordinating β-nitrogens of the pyrazole rings are in the trans position with respect to the pyridyl nitrogen, in line with the previously reported structures of bpp-based ligands.37–39 The trans orientation of the nitrogen atoms indicates the need for the rotation of the pyrazole rings to coordinate with iron(II) centres.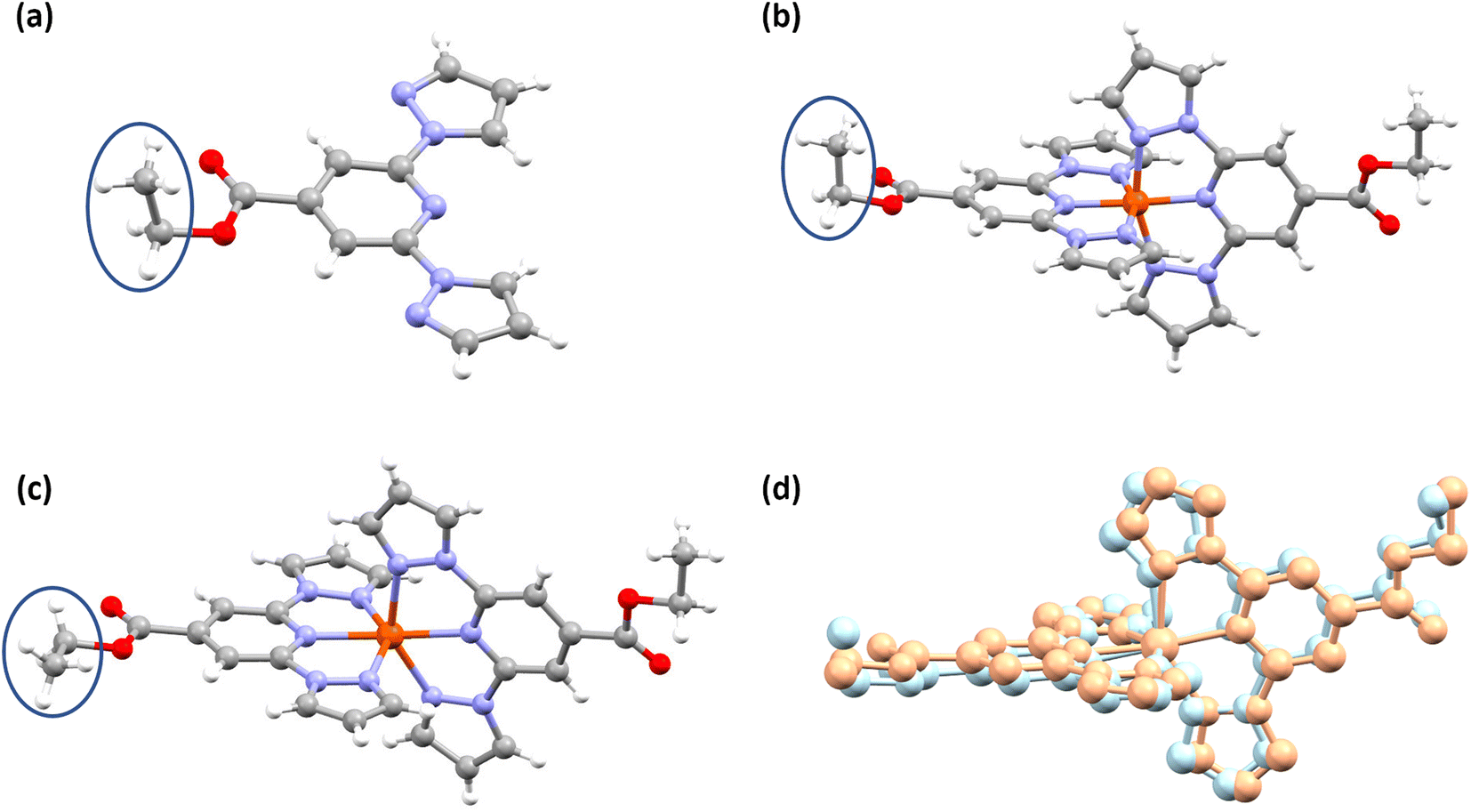 | ||
| Fig. 3 Structures of L1 and the corresponding complex—1·CH3CN—obtained from single-crystal X-ray diffraction (SC-XRD) studies. Molecular structures of (a) L1, (b) LS- and (c) HS-forms of complex 1·CH3CN, and (d) overlay of LS (light blue) and HS (light orange) structures of 1·CH3CN determined at 300 K and 345 K, respectively. The lattice acetonitrile molecules and BF4− anions in b–d are not shown for clarity. The most abundant parts of the disordered moieties are shown for b and c. Colour code: white, hydrogen; grey, carbon; blue, nitrogen; red, oxygen; orange, iron. The blue circles in a–c indicate the positions of one of the ethyl groups of the COOEt substituents in L1 and the corresponding complex in the LS and HS states. The LS-to-HS switching of 1·CH3CN involves pronounced angular distortion and conformational switching of the circled ethyl groups. Such structural variations associated with the LS and HS forms of 1·CH3CN cause bi-stable spin-state switching of the complex, as discussed below. | ||
Structure determination of wine-red crystals at 173 K (Fig. 3b and Table S2†) and 300 K (Fig. S8 and Table S1†) obtained from the reaction between L1 and iron(II) salt revealed that complex 1·CH3CN crystallized with one lattice acetonitrile solvent. The iron(II) centres in the crystals are in the LS state as inferred from the Fe–N bond lengths and angular parameters summarized in Tables 2 and S3.† We have also obtained the HS structure of 1·CH3CN at 345 K (Fig. 3c and Table S2†) from a LS 1·CH3CN crystal by heating it in the diffractometer at a rate of 0.5 K min−1.
| Parameter | 1·CH3CN (LS) | 1·CH3CN (HS) | 2a (HS) | 2b·CH3CN-Y (LS) | 2b·CH3CN-Y (HS) |
| T/K | 173 | 345 | 173 | 120 | 300 |
| rFe–N | 1.942 | 2.161 | 2.177 | 1.952 | 2.160 |
| N3–Fe1–N8 (ϕ) | 172.87 | 159.20 | 168.75 | 177.37 | 178.68 |
| N6–Fe1–N10 (ψ) | 160.53 | 145.15 | 146.01 | 159.7 | 147.63 |
| N1–Fe1–N5 (ψ) | 160.43 | 145.32 | 145.15 | 159.87 | 147.29 |
| α | 80.29 | 72.94 | 72.79 | 79.89 | 73.74 |
| Σ | 84.40 | 158.67 | 160.10 | 89.58 | 148.09 |
| θ | 86.70 | 80.73 | 84.67 | 85.86 | 85.00 |
| Θ | 315.18 | 637.22 | 540.14 | 294.20 | 474.74 |
A comparison between the structures of the LS and HS forms 1·CH3CN indicates that the spin-state switching is accompanied by conformational switching of one of the ethyl substituents (Fig. 3b and c) and pronounced angular distortion (Table 2 and Fig. 3d). Noteworthy are the trans-N{pyridyl}-Fe–N{pyridyl} angle (ϕ), N{pyrazolyl}-Fe–N{pyrazolyl} clamp angle (ψ), average of four cis-N{pyridyl}-Fe–N{pyrazolyl} angles (α), distortion index (Σ),40 angle between the planes of two ligands (θ), and trigonal distortion parameter (Θ), as shown in Table 2. Upon LS-to-HS switching, significant deviation from close to octahedral-to-trigonal prismatic structure has been observed for 1·CH3CN.
Structural investigation of complex 2a obtained at 173 K indicates that the complex is in the HS form. The complex cation (Fig. 4a and Table S4†) is distorted, as inferred from the angular parameters listed in Table 2. However, the distortion is less pronounced in HS-2a relative to HS-1·CH3CN. In the case of 2b·CH3CN-Y, the LS and HS forms of the complex (Fig. 4b and c and Table S4†) showed no appreciable deviation of ϕ value from the ideal value of 180° (Table 2). Other angular parameters associated with the LS and HS forms of 2b·CH3CN-Y are comparable with the respective ones obtained for the LS and HS forms of 1·CH3CN. A notable exception is the Σ value, which is relatively larger for the LS 2b·CH3CN-Y than the one obtained for the LS 1·CH3CN. Conversely, the Σ value of the HS 1·CH3CN is found to be greater than the value obtained for the HS 2b·CH3CN-Y. Overall, the variation of Σ upon spin-state switching is larger for 1·CH3CN relative to 2b·CH3CN-Y, indicating the fact that the former complex follows a more distorted pathway than the latter upon spin-state switching.
 | ||
| Fig. 4 Molecular structures of complexes composed of L2 obtained from single-crystal X-ray diffraction (SC-XRD) studies. (a) HS form of complex 2a, (b) LS form of complex 2b·CH3CN-Y, (c) HS form of complex 2b·CH3CN-Y, and (d) LS form of 2b·CH3CN-R. The lattice acetonitrile molecules, where appropriate, and counter anions are not shown for clarity. Colour code: white, hydrogen; grey, carbon; blue, nitrogen; red, oxygen; orange, iron. | ||
The red polymorph of 2b, 2b·CH3CN-R, also crystallized with lattice acetonitrile solvent. See Table S5† for the structural data. The polymorph is in the LS state at 173 K as inferred from the molecular structure (Fig. 4d) and angular parameters (Table S6†) obtained from SC-XRD studies. The average of Fe–N bond lengths and angular parameters of 2b·CH3CN-R are comparable with the values obtained for the LS 2b·CH3CN-Y, as listed in Table S6.†
For complex 1·CH3CN in the HS and LS states, the complex cations in the crystal lattice organize in a pseudo 1D-chain like manner along the c-axis (Fig. S9 and S11†). The pseudo 1D-chains stack (inter-sheet organization) along the crystallographic b-plane. Molecular organization in the lattices is governed by intermolecular interactions between the complex cation, counter anion, and acetonitrile solvent entities as depicted in Fig. S10 and S12.† Noteworthy are the short contacts between the lattice acetonitrile solvent and the complex cation (see Table S7† for details).
For complex 2a in the HS form, complex cations are ordered in an alternative manner along the b- and c-axes (Fig. S13†). Short contacts between F atoms of the counter cation and H atoms of the pyridine/pyrazole moieties along with an O–H contact, involving carbonyl oxygen of the –COOEt substituent and pyrazole-based hydrogen atom, between adjacent complex cations direct the molecular organization in the lattice (Fig. S14 and Table S8†). In the cases of the HS and LS forms of 2b·CH3CN-Y, a zig-zag molecular organization (Fig. S15 and S17†) in the crystal lattice is observed, when viewed along the crystallographic axis a. The lattice is held together by inter molecular contacts between the counter anion and complex cation (see Fig. S16 and S18 and Tables S9† for details). Similar molecular organization (Fig. S19†) and intermolecular contacts (Fig. S20 and Table S10†), as in 2b·CH3CN-Y, are observed in the crystal lattice of LS 2b·CH3CN-R.
For the estimation of short contacts, the criterion d(x…y) = ∑r(vdW)[x, y] − 0.2 Å was used, where d(x…y) is the distance between two atoms and ∑r(vdW)[x, y] is the sum of van der Waals radii of atoms x and y. Note, the short contact estimation following the above criterion is associated with significant pitfalls as discussed by G. P. Schiemenz.41 Considering the above point, the discussion presented in this study mainly intends to elucidate how intermolecular short contacts direct the molecular organization in the crystal lattices of complexes discussed in this study.
Spin-state switching characteristics of 1·CH3CN and 2b·CH3CN-Y and magnetic studies of HS 2a
Magnetic susceptibility measurements of gently ground crystals of 1·CH3CN, 2a, and 2b·CH3CN-Y were performed under 0.1 T applied magnetic field at a scan rate of 3 K min−1. Complex 1·CH3CN exhibited an abrupt SCO with 44 K and 52 K hysteresis loops for the first (T1/2↑ = 320 K and T1/2↓ = 276 K) and second (T1/2↑ = 320 K and T1/2↓ = 268 K) cycles, respectively, as depicted in Fig. 5a.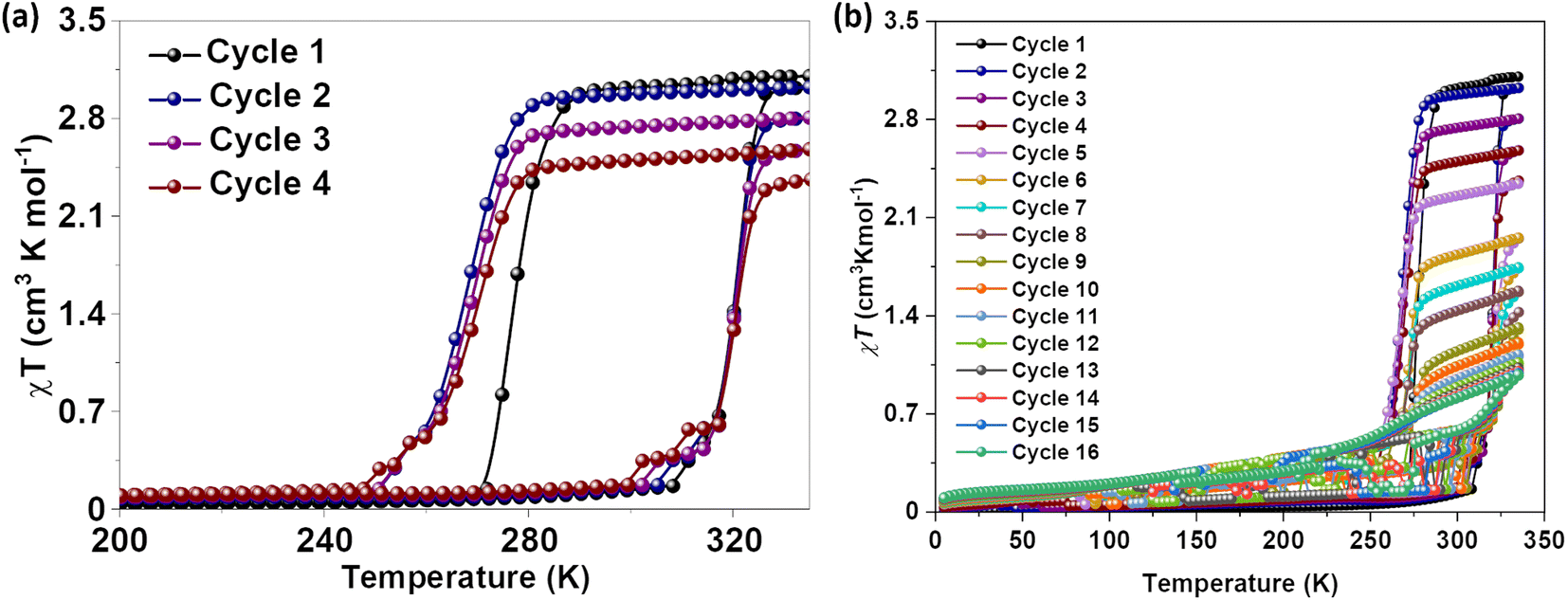 | ||
| Fig. 5 Temperature-induced spin-state switching characteristics of 1·CH3CN. (a) χT versus T plots of wine-red crystals of 1·CH3CN measured at a scan rate of 3 K min−1. The sample showed bi-stable—abrupt and hysteretic—spin-state switching with ΔT1/2 = 44 K and T1/2 = 298 K in the first cycle. (b) Repeated heat–cool cycling resulted in the decrease of χT values with a concomitant increase of thermal hysteresis width due to the stabilization of the HS state in the cooling mode. Spin-state switching in 1·CH3CN is accompanied by pronounced angular distortion, see Table 2, with concomitant self-grinding of the crystals, causing cycle-dependent evolution of the switching characteristics. | ||
The obtained χT products of 3.18 cm3 K mol−1 and 0.18 cm3 K mol−1 at 335 K and 200 K, respectively, for the first cycle (Table 3), indicate the presence of pure HS and predominantly LS states of the complex at those temperatures. The above point elucidates temperature-induced spin state switching of 1·CH3CN. Upon repeated scanning, a steady decrease of χT values was observed until the 11th scan (Fig. 5b). Starting from the 12th cycle, a relatively stabilized χT versus T plots were observed (Fig. 5b). This is attributed to the trapping of the sample in a mixed spin phase composed predominantly of the LS state.42 The cycling is also accompanied by a steady increase of the χT value in the 5 K-to-275 K region, as recently observed for an iron(II)-BPP system reported by Halcrow and co-workers.43 A fraction of the complex molecules in the mixed phase, obtained after the 11th cycle, undergoes SCO, as depicted in Fig. 5b.
| SQUID (sample 1) | DSC (sample 1) | ||||
|---|---|---|---|---|---|
| χT (HS)/cm3 K mol−1 | T 1/2/K | ΔT/K | T 1/2/K | ΔT/K | |
| Cycle 1 | 3.18 | 298 | 44 | 298 | 44 |
| Cycle 2 | 3.02 | 294 | 52 | 294 | 56 |
| Cycle 3 | 2.79 | 294 | 51 | 293 | 60 |
| Cycle 4 | 2.56 | 295 | 50 | 292 | 66 |
To check for the effect of in situ solvent removal on the spin-state switching characteristics of 1·CH3CN, the sample was annealed in the SQUID chamber at 400 K for thirty minutes. A cool–heat cycle performed after the annealing step revealed that the sample is trapped in the HS state, as depicted in Fig. S21.† The results obtained from continuous cycling depicted in Fig. 5 elucidate that spin-state switching induces self-grinding in the lattice, causing the spin-state switching to become unstable upon repeated cycling. On the other hand, the purposeful removal of the lattice solvent in the SQUID sample chamber traps the complex in the HS state, demonstrating the helping hand of the lattice solvent in mediating spin-state switching.
To check for the reproducible nature of the switching process and the effect of scan rate on the switching characteristics, we have prepared a second batch of 1·CH3CN crystals and probed their spin-state switching characteristics. When measured at a scan rate of 3 K min−1, the sample underwent temperature-induced bi-stable spin-state switching (Fig. S22†) with a thermal hysteresis width of 48 K and T1/2 = 298 K (T1/2↑ = 322 K and T1/2↓ = 274 K). Such results are comparable with the values obtained from the data shown in Fig. 5a (cycle 1). When a crop of fresh crystals obtained from the second batch was studied at a scan rate of 1 K min−1 (Fig. S23†), a thermal hysteresis width of 41 K and T1/2 = 304 K (T1/2↑ = 325 K and T1/2↓ = 284 K) were obtained. While the T1/2 obtained for the heating branch is comparable to the previous results, the T1/2 obtained for the cooling branch markedly differs from that of others. We attribute this difference to the nature of the sequence employed to collect the data. In previous cases, the data were collected from 340 K after keeping the sample at that temperature for thirty minutes in the SQUID chamber, see the Experimental section (S1†) for details. On the other hand, the first cooling branch at 1 K min−1 scan rate was obtained after the first heating branch (Fig. S23†). Such an experimental protocol could have caused a slightly different grinding of the crystals upon spin-state switching. Overall, the reproducible nature of bi-stable spin-state switching in complex 1·CH3CN is unambiguously elucidated by studying the switching properties of two different batches of the complex.
To check for the LIESST44 active nature of 1·CH3CN, we have prepared a third batch of crystals. Prior to photomagnetic studies, the sample was cooled to 5 K at a scan rate of 3 K under an applied field of 0.1 T. The subsequent irradiation of the complex using red laser light (λ = 637 nm, laser intensity was adjusted to 10 mW cm−2) caused an increase of the magnetic moment (Fig. 6a, grey solid circles). A gradual increase of the χT value was observed under laser irradiation. After three hours, χT = about 1 cm3 K mol−1 was obtained, indicating that about 28% of the iron(II) centres are switched to the HS state caused by the LIESST effect and no further increase in the χT value was observed after that. Temperature-dependent investigation of the photoexcited metastable HS fraction in the absence of the laser irradiation resulted in a gradual decrease of χT value in two steps, ultimately reaching values typical of the LS state of the compound at 100 K. The gradual nature of the LIESST curve precluded the determination of specific T(LIESST) temperatures. The heat–cool cycles of the sample performed after the LIESST study indicate that the cooperative spin-state switching is preserved in the sample (Fig. 6a). The T1/2 = 305 K and ΔT1/2 = 42 K (Table S11†) obtained for the batch in the first cycle (scan rate = 3 K min−1) are comparable to the values obtained for the second batch of the complex measured at a scan rate of 1 K min−1 (Fig. S23†). See the discussion in the previous paragraph. This implies a dependence of SCO parameters on the measurement protocol: experiments performed by cooling the HS sample from 340 K yielded T1/2 = 298 K, whereas T1/2 = 305 K was obtained when starting from the LS state.
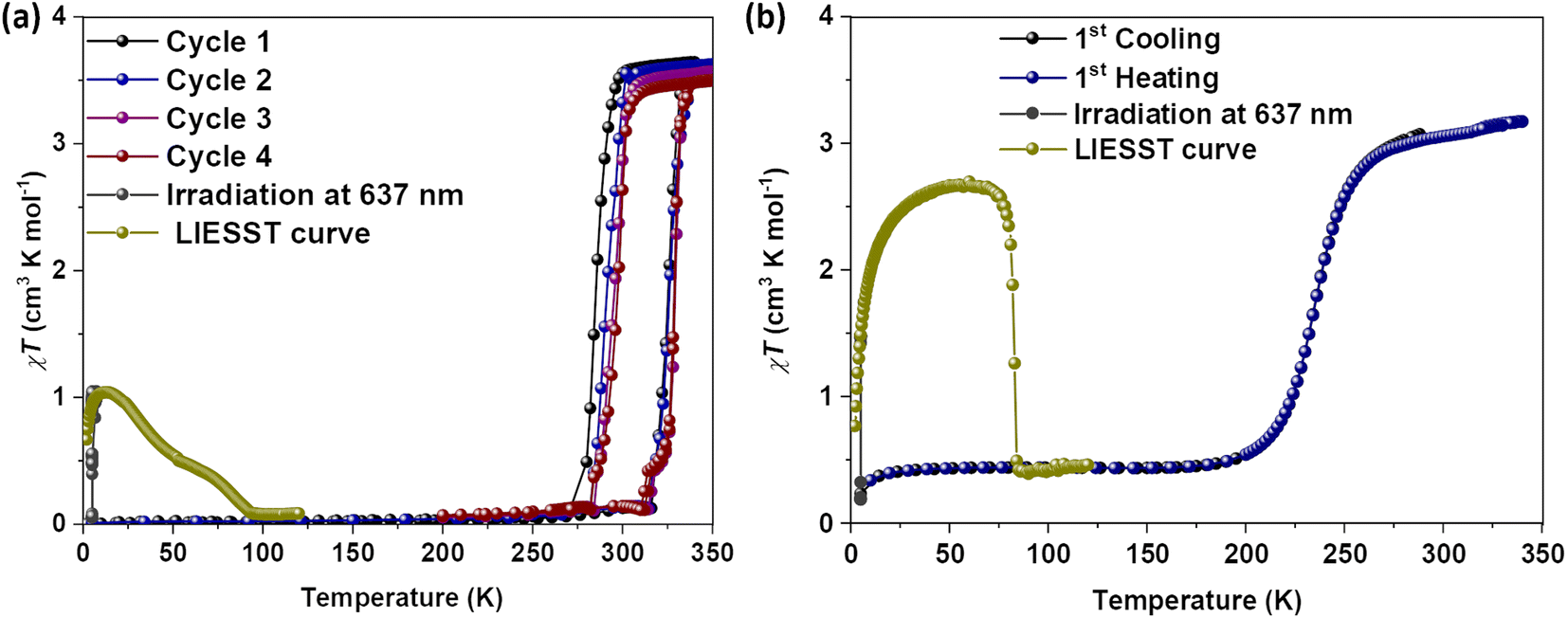 | ||
| Fig. 6 Temperature- and light-induced spin-state switching of 1·CH3CN and 2b·CH3CN-Y. (a) χT versus T plots of wine-red crystals of 1·CH3CN measured at a scan rate of 3 K min−1, showing temperature-dependent spin-state switching. Upon irradiation with 637 nm light at 5 K, the complex underwent light-induced LS-to-HS switching (grey filled circles) due to the LIESST effect. The green filled circles represent the temperature-dependent switching of the metastable HS state to the LS state. (b) χT versus T plot of complex 2b·CH3CN-Y—the complex shows a gradual temperature-induced spin-state switching (T1/2 = 234 K) with no thermal hysteresis. The complex also undergoes light-induced LS-to-HS switching (black filled circles), when irradiated with 637 nm light at 5 K. Temperature-induced conversion of the metastable HS state to the LS state starts around 50 K (green solid circles). A point noteworthy here is not only the thermal switching but also LIESST-mediated SCO is influenced by the nature of substituent, elucidating a connection between structural and spin isomerism. | ||
Unlike 1·CH3CN, complex 2a remained in the HS state upon cooling from 300 K to 5 K, as inferred from the χT versus T plot shown in Fig. S24.† This observation is consistent with the HS state of solvent-free complex 1, indicating the role of lattice solvent in mediating spin-state switching. At 300 K, the χT value of 3.98 cm3 K mol−1 was obtained. The value is larger than the spin only value (3 cm3 K mol−1) expected for an isolated HS iron(II) centre with S = 2 and L = 0. Upon cooling, the χT value remained almost constant until 50 K and then decreased to 0.95 cm3 K mol−1 at 1.8 K. As far as HS iron(II) systems are concerned, such a drop could be attributed to the depopulation of excited stark levels of the ground-state multiplet, presence of magnetic anisotropy, and intermolecular antiferromagnetic interactions. Considering the mononuclear nature of the complex and the shortest inter iron–iron distance of 8.69(4) Å in the crystal lattice, the contribution from intermolecular antiferromagnetic interactions is ruled out.
On the other hand, the steep decrease observed for 2a at low temperatures could be due to the presence of magnetic anisotropy. To clarify this point, we have performed isothermal field (M) versus magnetization (H) and alternating current (AC) magnetic susceptibility measurements. Isothermal M versus H data (Fig. S25a†) collected in the temperature range of 2 to 10 K until 7 T revealed that magnetization curves (M versus H/T, see Fig. S25b†) are non-superimposable, especially in the high-field regime. Such observation along with the magnetization values of 3.24μB (2 K) at 7 T indicates the possible presence of magnetic anisotropy in the complex. To probe the existence of slow relaxation of magnetization in the complex, AC measurements at zero and applied DC magnetic fields were performed at 1.8 K. Unfortunately, no out-of-phase susceptibility peaks (χ′′) were observed either at zero or applied DC fields (Fig. S26†), indicating that complex 2a is not a single-molecule magnet (SMM) at and above 1.8 K. This result is not surprising considering the fact that iron(II)-based SMMs are not frequently observed45 due to efficient quantum tunnelling of magnetization facilitating fast under barrier magnetization relaxation.
Magnetic measurements of compound 2b·CH3CN-Y revealed temperature-induced SCO behavior below room temperature (Fig. 6b). The complex is in the LS state in the 5-to-175 K temperature range. The χT value of about 0.40 cm3 K mol−1 in the temperature range is indicative of a remnant HS fraction. Heating the sample above 175 K resulted in a gradual LS-to-HS switching. The χT value of 3.17 cm3 K mol−1 was obtained at 340 K, in the range expected for a mononuclear iron(II) complex in the HS state. The cooling branch retraced the heating branch, and no thermal hysteresis was observed. For 2b·CH3CN-Y, T1/2 = 234 K was obtained, and the value is in the range reported for BPP-based iron(II) complexes. Irradiation of 2b·CH3CN-Y at 5 K with a red laser (λ = 637 nm) induced LS-to-HS switching as a consequence of the LIESST effect. After three hours of irradiation, χT = 1.5 cm3 K mol−1 was obtained; no further increase of the χT value was observed upon further irradiation. Heating of the metastable HS sample in the dark—that is, in the absence of laser irradiation—resulted in an increase of the χT value. At 50 K, χT = 2.7 cm3 K mol−1 was obtained, indicating that approximately 85% of the iron(II) centers underwent LS-to-HS switching. Such an increase of the χT value, until 50 K, while heating the sample in the absence of light irradiation is attributed to the population of the zero-field split high energy stark levels of the HS term corresponding to the photoexcited metastable HS 2b·CH3CN-Y. Heating the sample above 50 K led to an abrupt decrease of the HS fraction back to the LS state, indicating the decay of the metastable HS state. T(LIESST) = 83 K is calculated for the complex from the minimum in the ∂(χT)/∂T versus T curve ( data not shown). For the family of iron(II)-BPP complexes, the relation T(LIESST) = 150 − 0.3T1/2 was established. Using the relation T(LIESST) = 80 K is calculated for 2b·CH3CN-Y, comparable with the experimental value of 83 K. This elucidates that there exists a linear dependence between T(LIESST) and T1/2 for 2b·CH3CN-Y, as previously observed for iron(II) complexes composed of BPP-based and other ligand systems.46
57Fe Mössbauer spectroscopic studies of 1·CH3CN
In addition to the magnetometry measurements, 57Fe Mössbauer spectroscopy measurements were performed on 1·CH3CN to obtain the spin state composition at different temperatures. Fig. 7 shows the temperature-dependent 57Fe Mössbauer spectra of 1·CH3CN at 300 K and 335 K (heating) and 300 K and 200 K (cooling). The LS and HS contributions were fitted with two discrete doublets and the fit parameters obtained are given in Table 4. At 300 K, 100% LS state of the complex was inferred after fitting the data (Fig. 7 and Table 4). Upon heating to 335 K, the original LS doublet (300 K) lost intensity, and a second HS doublet with higher quadrupole and isomer shift forms. A composition of 74.6% HS and 25.4% LS was obtained based on the fitted resonant areas shown in Table 4. Upon cooling to 300 K, the spin-state equilibrium shifted towards the LS state and a LS:HS ratio of 67.3%:32.7% was obtained. Further cooling to 200 K resulted in a complete spin state switching and the complex was found to be in 100% LS state.
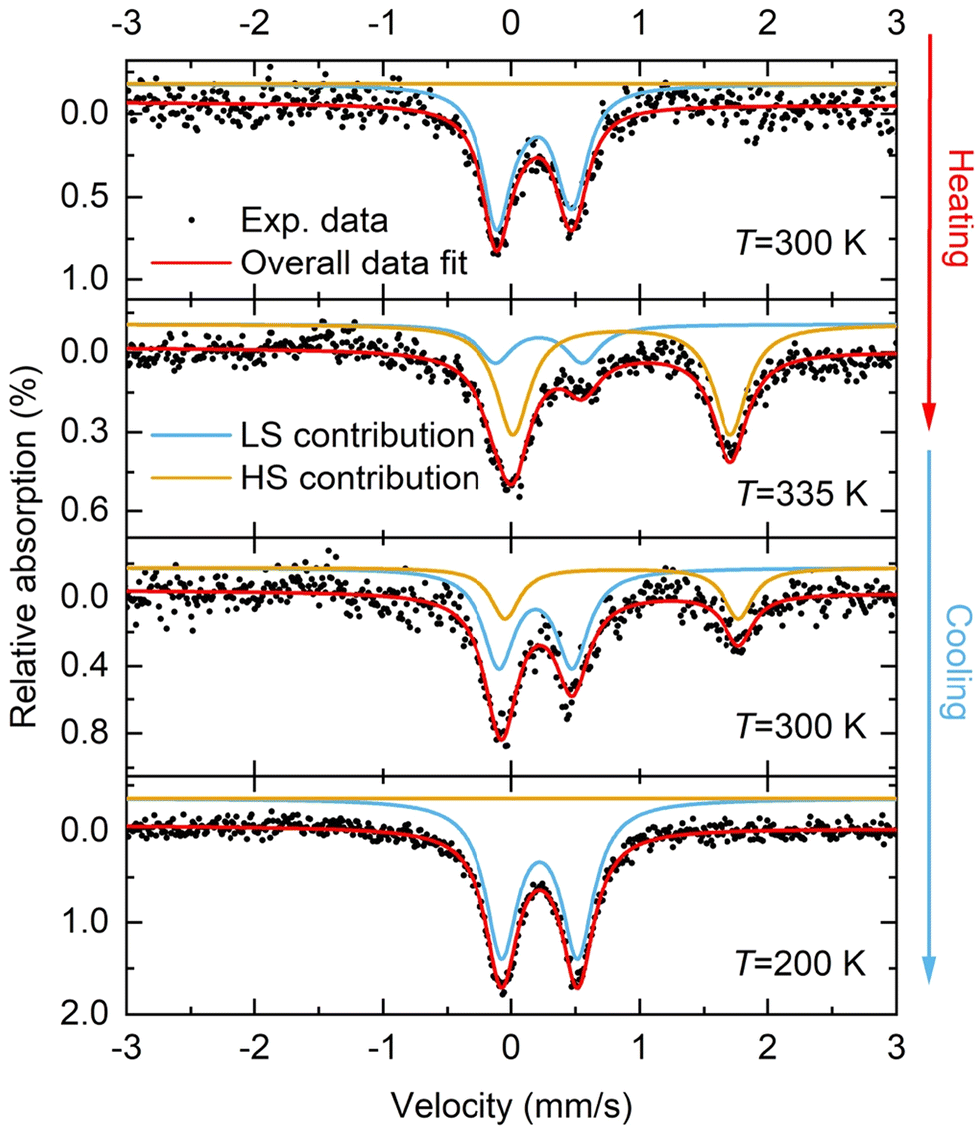 | ||
| Fig. 7 Temperature-dependent 57Fe Mössbauer spectroscopic studies of 1·CH3CN. The complex was heated from 300 to 335 K, leading to the observation of partial LS → HS switching: 74.6% HS and 25.4% LS. Cooling the sample back to 300 K resulted in the observation of a LS:HS mixture in a 67.3%:32.7% ratio, indicating the hysteretic nature of the spin-state switching process. Cooling down to 200 K resulted in a pure LS state, in agreement with the magnetometry data. Note: 335 K is the maximum achievable temperature limit with the instrument used to collect the 57Fe Mössbauer data. | ||
| Temperature [K] | LS doublet | HS doublet | ||||
|---|---|---|---|---|---|---|
| δ LS [mm s−1] | ΔEQ(LS) [mm s−1] | Resonant area [%] | δ HS [mm s−1] | ΔEQ(HS) [mm s−1] | Resonant area [%] | |
| 300 | 0.268 | 0.548 | 100 | — | — | 0 |
| 335 | 0.323 | 0.678 | 25.4 | 0.967 | 1.694 | 74.6 |
| 300 | 0.296 | 0.571 | 67.3 | 0.969 | 1.818 | 32.7 |
| 200 | 0.327 | 0.591 | 100 | — | — | 0 |
The results obtained from the 57Fe Mössbauer spectroscopic studies indicate the existence of hysteretic spin-state switching in 1·CH3CN, in agreement with the magnetic susceptibility measurements discussed above. However, a 100% HS state at 335 K (heating) and 300 K (cooling) was not observed in the 57Fe Mössbauer spectroscopic studies. In line with the thermal behavior of the 1·CH3CN powder (see below), we attribute this discrepancy to the partial loss of lattice solvent during the Mössbauer studies, where the sample is kept above room temperature for several hours.
Solution-state spin-state switching characteristic of 1·CH3CN
In addition to the magnetometry measurements performed on the crystalline form of 1·CH3CN, magnetometry studies of the complex in the solution phase were performed using a SQUID magnetometer. For the studies, a 7.42 mmol solution of the complex in acetonitrile was used.Fig. 8 shows the mass magnetization as a function of temperature for three measured cycles. The inset shows the range from 180 to 360 K to highlight the observed thermal hysteresis. The measurement shows a much smaller thermal hysteresis width than the one observed in the crystalline state with ΔT = 25 K (T↑ = 265 K, T↓ = 290 K) for the first cycle. In addition, the freezing and melting of the solvent acetonitrile were observed at Tfreeze = 203 K and Tmelt = 217 K, respectively. The thermal hysteresis remains almost the same for all the three cycles performed, indicating the absence of self-grinding in the solution phase. Such a result is contrary to the unstable nature of switching profiles obtained in the solid-state and elucidates the fact that stable hysteretic spin-state switching of the complex can be observed in the solution state.
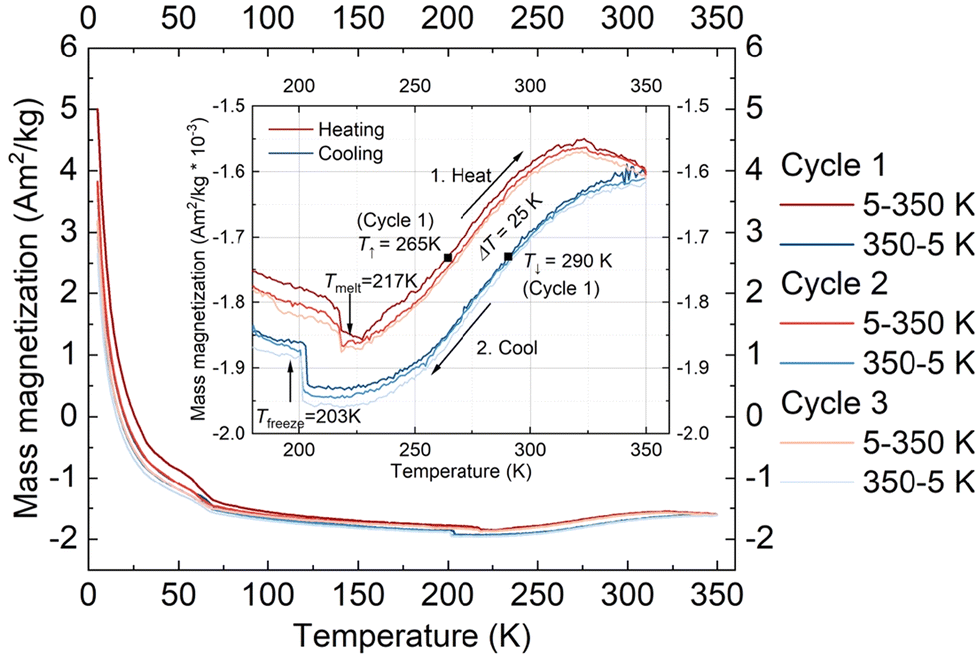 | ||
| Fig. 8 Spin-state switching characteristics of 1·CH3CN in acetonitrile solution. Mass magnetization of acetonitrile solution of complex 1·CH3CN as a function of temperature at an applied magnetic field of 0.1 T. For the three heating–cooling cycles presented, the heating branch is shown in red, and the cooling branch is shown in blue, both going to brighter colours with a higher cycle number. Highlights, like the freezing and melting of the solvent, as well as T↑ = 265 K, T↓ = 290 K, and ΔT = 25 K (all cycle 1) are shown. In contrast to the measurement of the powder form of 1·CH3CN, the top branch is the heating branch, and the lower branch is the cooling branch, as labelled in the inset: 1. heat and 2. cool. | ||
At present, we are not clear about factors causing hysteretic SCO of 1·CH3CN in the solution state. Probable ones are aggregation of the complex in the solution and strong intermolecular interactions between the complex entity and acetonitrile solvent molecules.
Differential scanning calorimetric (DSC) and small- and wide-angle X-ray scattering (SWAXS) studies of 1·CH3CN
To provide supplementary confirmation for the occurrence of temperature-induced spin-state switching in 1·CH3CN, differential scanning calorimetric (DSC) studies were performed, as shown in Fig. 9a. The T1/2 and ΔT1/2 values obtained from the DSC studies (Table 3) are comparable with the values obtained from the χT versus T plots shown in Fig. 5a, confirming the bi-stable spin-state switching characteristic of 1·CH3CN.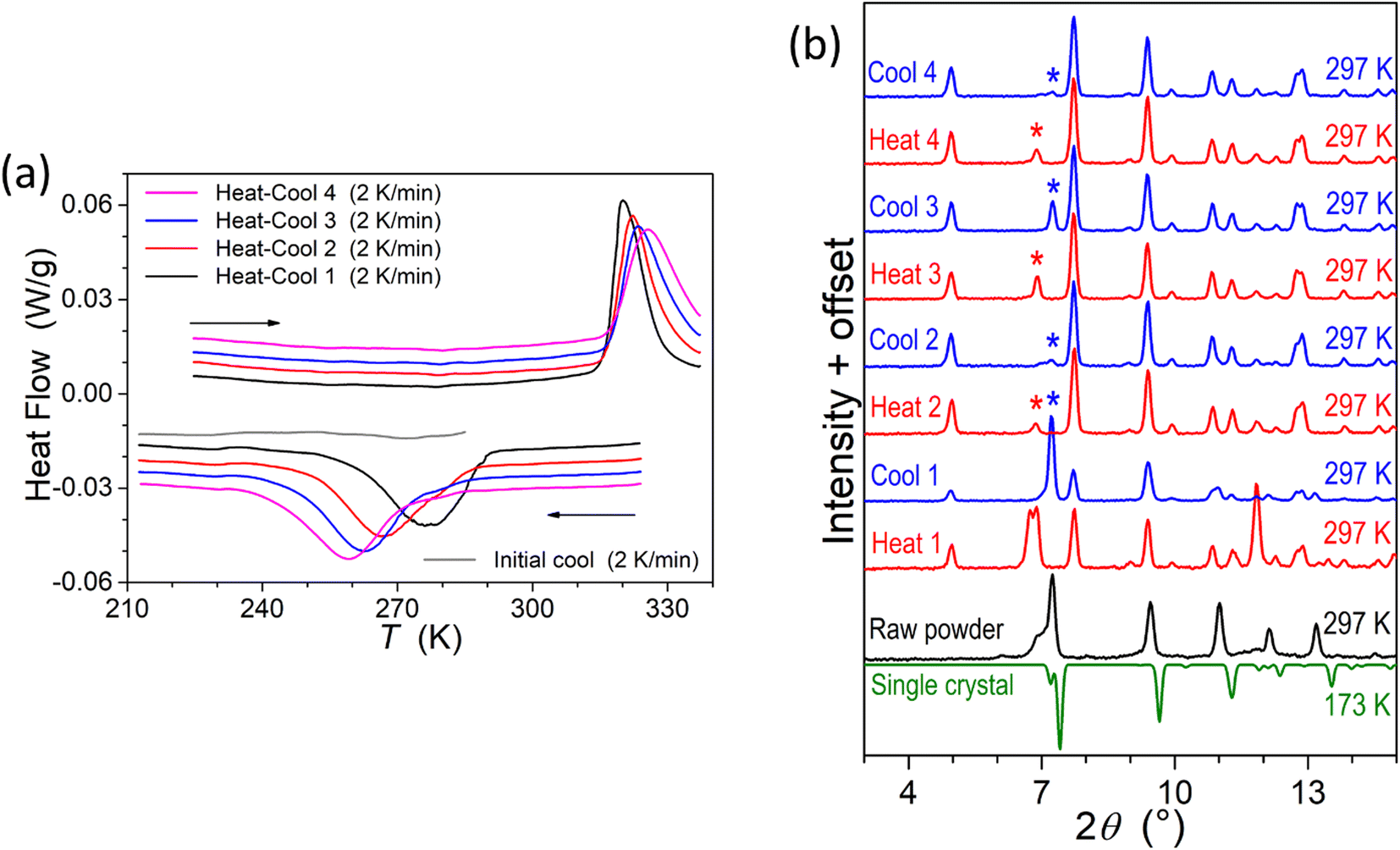 | ||
| Fig. 9 Differential scanning calorimetric (DSC) and small- and wide-angle X-ray scattering (SWAXS) studies of complex 1·CH3CN. (a) DSC profiles showing temperature-induced spin-state switching in the 210-to-330 K temperature range. (b) SWAXS patterns of 1·CH3CN showing transitions between crystalline phases in response to repeated heat–cool cycling. The patterns were collected at 297 K after heating and cooling the sample ex situ at 330 K and 77 K, respectively. The irreversible changes in the SWAXS patterns obtained after repeated heat–cool cycling indicates that there exists phase variations contributing to the irreversible alteration of switching parameters, including the hysteresis width, upon repeated heat–cool cycling, as shown in Fig. 5b. | ||
Enthalpy (ΔH) and entropy (ΔS) variations associated with the LS-to-HS and HS-to-LS switching branches of the first cycle have also been estimated from the DSC plots shown in Fig. 9a. The estimated values of ΔH = 13.2 kJ mol−1 (LS-to-HS); 14.4 kJ mol−1 (HS-to-LS) and ΔS = 41.25 J mol−1 K−1 (LS-to-HS); 52.17 J mol−1 K−1 (HS-to-LS) are comparable with the values reported for iron(II) SCO complexes.47,48 The differing values of thermodynamic parameters in the LS-to-HS and HS-to-LS switching branches are attributed to different vibrational modes associated with the LS and HS forms of 1·CH3CN.
We have also performed SWAXS studies of complex 1·CH3CN to get a glimpse of lattice variations during the spin-state switching process upon repeated cycling. As shown in Fig. 9b, the SWAXS pattern obtained from a ground sample of 1·CH3CN is comparable with the calculated pattern obtained from the single-crystal data of the LS crystal. This observation confirming the retainment of crystalline order in the ground sample is remarkable, considering the fact that 1·CH3CN crystals are ground prior to magnetic measurements. The differences between the SWAXS and calculated patterns obtained at 297 K and 173 K, respectively, are attributed to temperature- and grinding-induced variations of lattice parameters in relation with the displacement of the co-crystallized solvent. Indeed, grinding may strongly impact the structure49 and even cause loss of crystallinity and transformation to an amorphous state, as we recently observed for complex c (Fig. 1).35 Repeated heat–cool cycles of the ground 1·CH3CN caused an irreversible transformation of diffraction profiles, serving as the proof of the evolution of the crystalline phases during the repeated heat–cool cycling.
To shed light on the fate of lattice acetonitrile in 1·CH3CN during the repeated heat–cool cycling, thermogravimetric analyses (TGA) of various forms of the complex were performed. The crystalline 1·CH3CN underwent a 5% weight loss around 400 K, corresponding to the loss of one molecule of acetonitrile from the lattice (Fig. S27a†). The weight loss obtained from the TGA is comparable with the calculated value of 4.9% for a loss of one molecule of acetonitrile from the lattice. In contrast, no such weight loss is observed for a sample dried at 423 K (Fig. S27a†), revealing the complete removal of acetonitrile molecules from the lattice. TGA analysis of the sample obtained after SWAXS studies also showed a lack of pronounced weight loss (Fig. S27b†) observed for 1·CH3CN. Moreover, the TGA profiles of the deliberately desolvated sample at 423 K and the sample obtained after the repeated SWAXS measurements are comparable. From the above observations, we infer that repeated spin-state switching of 1·CH3CN is accompanied by a gradual loss of lattice acetonitrile molecules, forming a 1·xCH3CN (x < 1) lattice with variable solvent content. Note that the solvent removal process during the SWAXS measurements is accelerated due to the ex situ heating and cooling of the sample, which is in contrast to the magnetic measurements performed inside a SQUID sample chamber, where the sample is repeatedly scanned under a low-pressure He-atmosphere without exposure to the external conditions. This implies that the switching-induced lattice solvent release could progress differently between magnetometry and SWAXS conditions.
The first synthesis of BPP-COOEt ligand used to prepare 1·CH3CN discussed in this study was reported almost 20 years ago.36 However, the utility of the ligand to prepare iron(II)-based mononuclear complexes was not reported until 2018.34 The first reported iron(II) complex (a, Fig. 1) of the ligand, showed an irreversible LS-to-HS switching with the switching temperature centred around 330 K. Once heated above 350 K, complex a lost the lattice acetone solvent and the solvent-free version was trapped in the HS-state in the subsequent cooling. Our attempts to study similar complexes as acetonitrile (complex b; Fig. 1) and nitromethane (complexes c and d; Fig. 1) solvates led to the observation of hysteretic spin-state switching.9,35 Remarkably, complex c showed stable hysteretic spin-state switching characteristics with T1/2 = 288 K; ΔT1/2 = 62 K.35
In a different scenario, Sato and co-workers used BPP-COOEt to prepare a mononuclear cobalt(II) complex, which underwent switching of orbital angular momentum due to switching of coordination number from seven-to-six, and vice versa,50 demonstrating an altogether different switching mechanism contrast to the spin-multiplicity-based switching demonstrated in this study. The above discussion indicates that coordination complexes based on a simple ligand system such as BPP-COOEt can feature exotic magnetic properties.
In a recent study, we observed pronounced molecular distortion upon spin-state switching of complexes c and d (Fig. 1 and Tables S12 and S13†).35 Such distortion coupled with conformational variation of the Et group stabilizes the complexes either in a LS- or HS-state; creating an energy barrier, whereby thermal hysteresis loops are observed upon spin-state switching. Recently, Real and co-workers reported a mononuclear iron(II) complex with a wide 105 K thermal hysteresis loop and T1/2 = 308 K. They elucidated that molecular distortion and conformational switching cause hysteretic spin-state switching of the complex.4 The angular parameters collected in Table 3 reveal that the HS-form of complex 1·CH3CN features a distorted molecular geometry relative to its LS counterpart showing close to the ideal octahedral geometry. The obtained variations in ϕ and θ—Δϕ = 13.67° and Δθ = 5.97°—indicate that the complex needs to traverse a significant energy barrier imposed by the crystal lattice to switch from one spin-state to another; that is, HS-to-LS and vice versa. Moreover, conformation of one of the ethyl groups of 1·CH3CN varies upon spin-state switching (Fig. 3b and c, blue circles). In the solid-state, positions of the ethyl groups of L1 and LS form of 1·CH3CN are comparable as can be seen in Fig. 3a and b. To compare the structural variation coupled with spin-state switching in iron(II) complexes composed of BPP-COOEt ligands, we have compiled the difference between the angular parameters of the LS and HS forms of complexes c, d, and 1·CH3CN (Fig. 1) in Tables S12–S14.† All the complexes showed pronounced angular variation upon spin-state switching with HS forms of the complexes featuring large Θ values. Remarkably, the ΔΘ(LS–HS) values observed for 1·CH3CN (322.04°), c (302.07°), and d (318.22°) are the largest so far observed for BPP-based iron(II) complexes, surpassing the previous best of 215° reported by Halcrow and co-workers.51
The successful determination of the structures of LS and HS forms of 1·CH3CN enabled us to compare the angular parameters obtained for the complex with the previously reported iron(II) complexes composed of BPP-based ligands and BPP-COOEt-based complexes (Fig. 10a). The task is made easy by Halcrow and co-workers, who have recently reported a compilation of angular parameters of LS and HS forms of iron(II)-BPP complexes and structural distortion pathways.37 The authors have shown that a select band of HS-complexes (yellow squares in the pale grey region of Fig. 10b) that undergo strong angular distortion upon LS-to-HS switching are capable of showing abrupt and hysteretic spin-state switching.
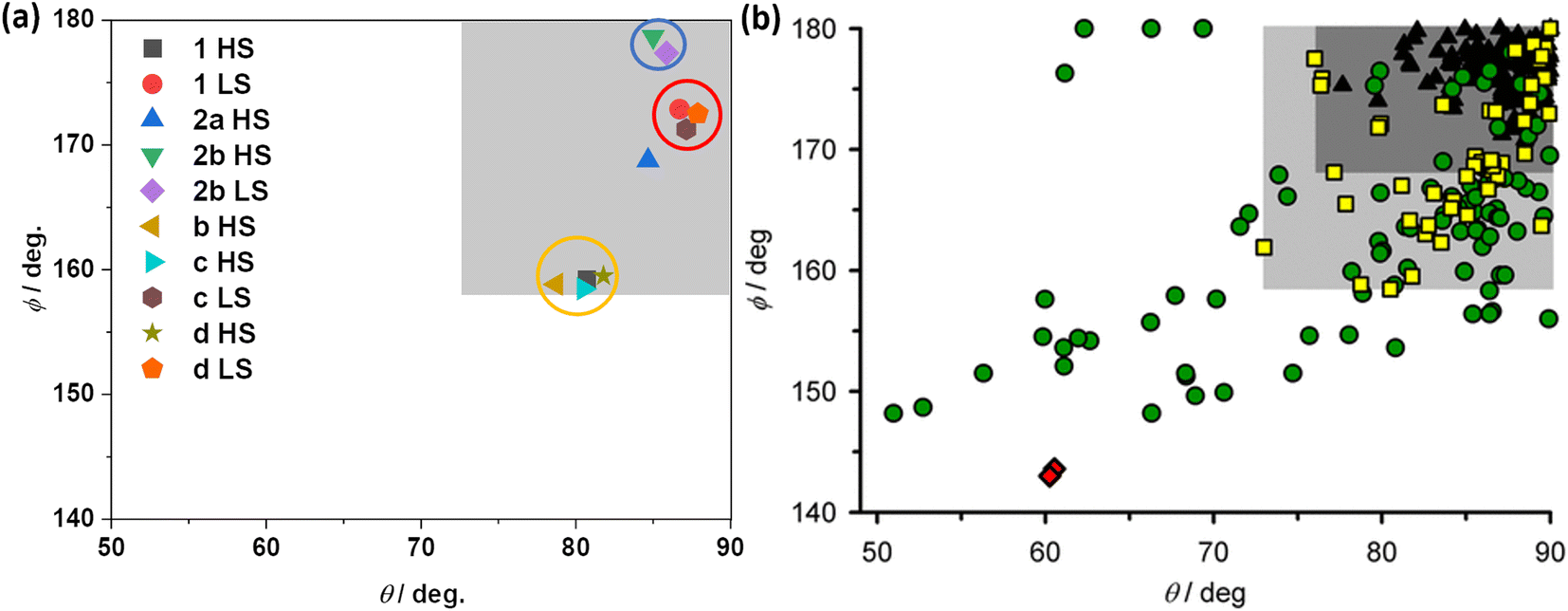 | ||
| Fig. 10 Angular parameters of HS and LS states of iron(II)-BPP complexes. (a) Angular parameters of HS and LS forms of complexes 1·CH3CN, b, c, d, and 2b·CH3CN-Y. For complexes b and 2a only HS structures are available, and the angular parameters obtained from the structures are shown. (b) A compilation of angular parameters reported for iron(II)-BPP complexes. (b) is reproduced from the study by Halcrow and co-workers.37 Copyright © 2024 The authors. Open access licensed under CC-BY 4.0. | ||
The angular parameters obtained for the LS form of 1·CH3CN and the related complexes c and d are clustered together in Fig. 10a at the top right corner, as highlighted with a red circle. Similarly, the HS forms of the complexes—1·CH3CN, b, c, and d—are also clustered in closer proximity, as highlighted with a yellow circle. Remarkably, the HS complexes fall in the pale grey region identified by Halcrow and co-workers (Fig. 10b), a position where an iron(II)-BPP complex is expected to show bi-stable SCO with sizable thermal hysteresis loop, albeit rarely. The above points elucidate that iron(II)-BPPCOOEt complexes belong to a not-frequently reported class of SCO systems featuring pronounced angular distortion and the consequent bi-stable spin-state switching.
In the case of HS complex 2a, the angular parameters reveal a less distorted coordination geometry relative to its isomeric HS counterparts—1·CH3CN, b, c, and d. As in Fig. 10a, the ϕ versus θ point is placed in the dark grey region of the angular landscape (Fig. 10b), where SCO is more probable relative to the structures in the pale grey region. However, complex 2a remains stable in the HS state upon cooling. Such an observation is attributed to the lack of co-crystallized lattice solvent molecules aiding spin-state switching via elastic intermolecular interactions. This attribution is supported by the SCO active nature of the 2b·CH3CN-Y, crystallizing with lattice acetonitrile. The angular parameters associated with the LS and HS forms of 2b·CH3CN-Y (Table S15†) is placed in the dark grey region (Fig. 10a), where SCO is probable. Crucially, the lack of significant variation of the angular parameters between the HS and LS forms of 2b·CH3CN-Y and the gradual SCO accompanied with no thermal hysteresis serve as an additional proof of the contributing role of distortion in spurring bi-stable SCO in 1·CH3CN.
Overall, complex 1·CH3CN reported in this study is one of the remarkable mononuclear iron(II) spin-crossover complexes showing hysteretic spin-state switching with T1/2 centred at RT. However, the SCO is lattice-solvent-dependent and significant variations in the lattice parameters, as inferred from the SWAXS studies, accompany the spin-state switching. Such aspects render the SCO unstable to thermal cycling, impeding the practical utility of the complex as a molecular switch or memory. On the other hand, the attribution that pronounced angular distortion and conformational variation of a functional group contribute to the opening of thermal hysteresis in 1·CH3CN adds to the extensive knowledge collected on SCO systems over almost a century. Remarkably, a direct proof of molecular distortion causing bi-stable SCO in 1·CH3CN can be obtained from the studies on 2b·CH3CN-Y. The HS form of the latter complex is significantly less distorted relative to the HS 1·CH3CN. Moreover, the gradual SCO of 2b·CH3CN-Y is accompanied with much smaller variations of the angular parameters relative to the ones obtained for 1·CH3CN.
As a perspective, the grand old phenomenon, SCO, continues to evolve keeping abreast with the contemporary developments in molecular magnetism and related topics.5,52–63 It is our opinion, agreeing with the remarks made by the editors of this special collection, that the topic is fascinating to study, one of the pillars of molecular magnetism, and offers opportunities to perform basic science research,64 allowing us chemists to elucidate how molecular structure variation imbues a physical property change, as exemplified in this script relating molecular distortion with thermal hysteresis.
Conclusions
In this study, we show how structural isomerism influences spin isomerism. Complexes 1·CH3CN and 2a featuring BF4− counter anions are a case. 1·CH3CN crystallizes with a lattice solvent and undergoes bi-stable spin state switching, whereas lattice solvent-free 2a is trapped in the HS state, elucidating the helping hand of the co-crystallized lattice solvent in facilitating spin-state switching. Pronounced angular distortion and conformation variation of the ethyl substituent accompany spin-state switching of 1·CH3CN, causing a sizable thermal hysteresis width. As far as the angular parameters are considered, the HS form of 1·CH3CN features one of the largest reported values of the distortion parameter (Θ) comparable to the previously reported isostructural solvates (b–c, Fig. 1b) of the complex. The difference (ΔΘ) in the values of Θ estimated from the HS and LS structures of the complex series is also the largest reported for the iron(II)-BPP family of complexes. The perchlorate anion containing complex of L2—2b·CH3CN—crystallizes as two polymorphs—2b·CH3CN-Y and 2b·CH3CN-R. The switching in polymorph 2b·CH3CN-Y, crystallizing with lattice acetonitrile, is gradual and no hysteresis is observed. Analysis of the structures of the LS and HS forms of 2b·CH3CN-Y reveals that the trans-N{pyridyl}-Fe–N{pyridyl} angles (ϕ) are 177.37° and 178.68°, respectively. This indicates that switching in 2b·CH3CN-Y is accompanied by a small variation of ϕ and supports our hypothesis that a strong variation of ϕ upon spin-state switching contributes to the opening of the thermal hysteresis loop. Overall, the picture-perfect centering of the T1/2 at 298 K and ΔT1/2 = 44 K observed for 1·CH3CN places the complex as one of the less frequently encountered mononuclear iron(II) complexes featuring bi-stable spin-state switching characteristics. The results are encouraging to pursue iron(II) complexes with a sizable thermal hysteresis width with T1/2 centered in the vicinity of RT, a fundamental scientific quest in the topic.Author contributions
S.K.K.: conceptualization, synthesis and characterization of the ligands and complexes, magnetic measurements, writing of the original and revised drafts, review and editing. A.M.: SC-XRD studies, review and editing. L.K.: 57Fe Mössbauer and solution-phase magnetic studies and writing of the corresponding parts, review and editing. S.S.: solution-phase magnetic studies and review and editing. B.H.: SWAXS, TGA, and DSC studies, writing of the corresponding parts, review and editing. C.B.: SC-XRD studies, review and editing. I.S.: magnetic measurements, LIESST measurements, writing of the corresponding part, review and editing. H.W.: review and editing and funding acquisition. M.R.: review and editing and funding acquisition. All the authors have read and agreed to the submission of the manuscript.Data availability
Crystallographic data for complexes 1·CH3CN and 2 have been deposited at the CCDC under 1560719 (1·CH3CN, LS, 173 K); 2330938 (1·CH3CN, LS, 300 K); 2330939 (1·CH3CN, HS, 345 K); 1913041 (2a, HS, 173 K); 1953349 (2b·CH3CN-R, LS, 173 K); 1953350 (2b·CH3CN-Y, LS, 120 K); 2340087 (2b·CH3CN-Y, HS, 300 K). The data can be obtained from the Cambridge Crystallographic Data Center.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank Lydia Karmazin, University of Strasbourg, for the structure determination of L1. Grant Agency Innovation FRC is acknowledged for the financial support for the project self-assembly of spin-crossover (SCO) complexes on graphene. A. M. acknowledges the Alexander von Humboldt (AvH) Foundation for a postdoctoral fellowship, a Grant-in-Aid for the JSPS Research Fellowship program, and the financial support by JSPS KAKENHI Grant Numbers JP22KJ3097 and JP23K13723. M. R. thanks the DFG Priority Program 1928 “COORNETS” for generous support. L. K. and H. W. acknowledge the funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 278162697 – CRC 1242, Project A05. S. S. and H. W. acknowledge the financial support by the German Research Foundation (DFG) via the CRC/TRR 247 (Project-ID 388390466, sub-project B02). The authors thank Ulrich von Hörsten for his technical assistance with the 57Fe Mössbauer spectroscopy measurements. I. S. acknowledges the Slovak (Slovak Research and Development Agency: APVV-19-0087, APVV-22-0172, DS-FR-22-0010 and Scientific Grant Agency of the Ministry of Education of Slovak Republic VEGA 1/0029/22) and Czech (Grant Agency of the Czech Republic No. 22-23760S) grant agencies for the financial support. The authors thank Karlsruhe Institute of Technology (KIT) for providing open access of the article through programme DEAL.References
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- M. Seredyuk, K. Znovjyak, F. J. Valverde-Muñoz, I. da Silva, M. C. Muñoz, Y. S. Moroz and J. A. Real, J. Am. Chem. Soc., 2022, 144, 14297–14309 CrossRef CAS PubMed.
- S.-G. Wu, L.-F. Wang, Z.-Y. Ruan, S.-N. Du, S. Gómez-Coca, Z.-P. Ni, E. Ruiz, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2022, 144, 14888–14896 CrossRef CAS PubMed.
- G. Gallé, D. Deldicque, J. Degert, Th. Forestier, J.-F. Létard and E. Freysz, Appl. Phys. Lett., 2010, 96, 041907 CrossRef.
- B. Weber, W. Bauer and J. Obel, Angew. Chem., Int. Ed., 2008, 47, 10098–10101 CrossRef CAS PubMed.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- K. Senthil Kumar, B. Heinrich, S. Vela, E. Moreno-Pineda, C. Bailly and M. Ruben, Dalton Trans., 2019, 48, 3825–3830 RSC.
- O. Kahn, J. Kröber and C. Jay, Adv. Mater., 1992, 4, 718–728 CrossRef CAS.
- M. A. Halcrow, Chem. Lett., 2014, 43, 1178–1188 CrossRef CAS.
- B. Schäfer, C. Rajnák, I. Šalitroš, O. Fuhr, D. Klar, C. Schmitz-Antoniak, E. Weschke, H. Wende and M. Ruben, Chem. Commun., 2013, 49, 10986 RSC.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed.
- A. S. Tayi, A. K. Shveyd, A. C.-H. Sue, J. M. Szarko, B. S. Rolczynski, D. Cao, T. J. Kennedy, A. A. Sarjeant, C. L. Stern, W. F. Paxton, W. Wu, S. K. Dey, A. C. Fahrenbach, J. R. Guest, H. Mohseni, L. X. Chen, K. L. Wang, J. F. Stoddart and S. I. Stupp, Nature, 2012, 488, 485–489 CrossRef CAS PubMed.
- H. Liu, Y. Ye, X. Zhang, T. Yang, W. Wen and S. Jiang, J. Mater. Chem. C, 2022, 10, 13676–13689 RSC.
- S. Mohapatra, S. Cherifi-Hertel, S. K. Kuppusamy, G. Schmerber, J. Arabski, B. Gobaut, W. Weber, M. Bowen, V. Da Costa and S. Boukari, J. Mater. Chem. C, 2022, 10, 8142–8167 RSC.
- S. Horiuchi, Y. Tokunaga, G. Giovannetti, S. Picozzi, H. Itoh, R. Shimano, R. Kumai and Y. Tokura, Nature, 2010, 463, 789–792 CrossRef CAS PubMed.
- W. Fujita and K. Awaga, Science, 1999, 286, 261–262 CrossRef CAS PubMed.
- A. Paul, A. Gupta and S. Konar, Cryst. Growth Des., 2021, 21, 5473–5489 CrossRef CAS.
- A. Mizuno, R. Matsuoka, T. Mibu and T. Kusamoto, Chem. Rev., 2024, 124, 1034–1121 CrossRef CAS PubMed.
- Z. Hu, Y. Wang, A. Ullah, G. M. Gutiérrez-Finol, A. Bedoya-Pinto, P. Gargiani, D. Shi, S. Yang, Z. Shi, A. Gaita-Ariño and E. Coronado, Chem, 2023, 9, 3613–3622 CAS.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- P. Gütlich, Eur. J. Inorg. Chem., 2013, 2013, 581–591 CrossRef.
- A. Dürrmann, G. Hörner and B. Weber, Cryst. Growth Des., 2023, 23, 1743–1754 CrossRef.
- M. Paez-Espejo, M. Sy and K. Boukheddaden, J. Am. Chem. Soc., 2018, 140, 11954–11964 CrossRef CAS PubMed.
- R. Torres-Cavanillas, M. Gavara-Edo and E. Coronado, Adv. Mater., 2024, 36, 2307718 CrossRef CAS PubMed.
- V. Rubio-Giménez, S. Tatay and C. Martí-Gastaldo, Chem. Soc. Rev., 2020, 49, 5601–5638 RSC.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119 RSC.
- J. M. Holland, C. A. Kilner, M. Thornton-Pett, M. A. Halcrow, J. A. McAllister and Z. Lu, Chem. Commun., 2001, 577–578 RSC.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS.
- V. García-López, M. Palacios-Corella, A. Abhervé, I. Pellicer-Carreño, C. Desplanches, M. Clemente-León and E. Coronado, Dalton Trans., 2018, 47, 16958–16968 RSC.
- N. Suryadevara, A. Mizuno, L. Spieker, S. Salamon, S. Sleziona, A. Maas, E. Pollmann, B. Heinrich, M. Schleberger, H. Wende, S. K. Kuppusamy and M. Ruben, Chem. – Eur. J., 2022, 28, e202103853 CrossRef CAS PubMed.
- T. Vermonden, D. Branowska, A. T. M. Marcelis and E. J. R. Sudhölter, Tetrahedron, 2003, 59, 5039–5045 CrossRef CAS.
- I. Capel Berdiell, E. Michaels, O. Q. Munro and M. A. Halcrow, Inorg. Chem., 2024, 63, 2732–2744 CrossRef CAS PubMed.
- S. K. Kuppusamy, A. Mizuno, A. García-Fuente, S. Van Der Poel, B. Heinrich, J. Ferrer, H. S. J. Van Der Zant and M. Ruben, ACS Omega, 2022, 7, 13654–13666 CrossRef CAS PubMed.
- I. Šalitroš, O. Fuhr, M. Gál, M. Valášek and M. Ruben, Chem. – Eur. J., 2017, 23, 10100–10109 CrossRef PubMed.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, J. Mater. Chem., 2002, 12, 2546–2551 RSC.
- G. P. Schiemenz, Z. Naturforsch., B: J. Chem. Sci., 2007, 62, 235–243 CrossRef CAS.
- Y. Miyazaki, T. Nakamoto, S. Ikeuchi, K. Saito, A. Inaba, M. Sorai, T. Tojo, T. Atake, G. S. Matouzenko, S. Zein and S. A. Borshch, J. Phys. Chem. B, 2007, 111, 12508–12517 CrossRef CAS PubMed.
- R. Kulmaczewski, L. J. Kershaw Cook, C. M. Pask, O. Cespedes and M. A. Halcrow, Cryst. Growth Des., 2022, 22, 1960–1971 CrossRef CAS PubMed.
- S. Decurtins, P. Gütlich, C. P. Köhler, H. Spiering and A. Hauser, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef CAS.
- A. J. Valentine, A. M. Geer, T. J. Blundell, W. Tovey, M. J. Cliffe, E. S. Davies, S. P. Argent, W. Lewis, J. McMaster, L. J. Taylor, D. Reta and D. L. Kays, Dalton Trans., 2022, 51, 18118–18126 RSC.
- J. Létard, P. Guionneau, O. Nguyen, J. S. Costa, S. Marcén, G. Chastanet, M. Marchivie and L. Goux-Capes, Chem. – Eur. J., 2005, 11, 4582–4589 CrossRef PubMed.
- S. K. Kulshreshtha and R. M. Iyer, Chem. Phys. Lett., 1984, 108, 501–504 CrossRef CAS.
- W. Nicolazzi and A. Bousseksou, C. R. Chim., 2018, 21, 1060–1074 CrossRef CAS.
- Y. Hirai, S. Van Baaren, T. Ohmura, T. Nakanishi, T. Takeda, Y. Kitagawa, Y. Hasegawa, R. Métivier and C. Allain, Adv. Opt. Mater., 2023, 11, 2203139 CrossRef CAS.
- S.-Q. Su, S.-Q. Wu, M. L. Baker, P. Bencok, N. Azuma, Y. Miyazaki, M. Nakano, S. Kang, Y. Shiota, K. Yoshizawa, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2020, 142, 11434–11441 CrossRef CAS PubMed.
- E. Michaels, I. Capel Berdiell, H. B. Vasili, C. M. Pask, M. J. Howard, O. Cespedes and M. A. Halcrow, Cryst. Growth Des., 2022, 22, 6809–6817 CrossRef CAS.
- S. P. Vallone, A. N. Tantillo, A. M. dos Santos, J. J. Molaison, R. Kulmaczewski, A. Chapoy, P. Ahmadi, M. A. Halcrow and K. G. Sandeman, Adv. Mater., 2019, 31, 1807334 CrossRef PubMed.
- L. Zhao, Y.-S. Meng, Q. Liu, O. Sato, Q. Shi, H. Oshio and T. Liu, Nat. Chem., 2021, 13, 698–704 CrossRef CAS PubMed.
- M. Gavara-Edo, R. Córdoba, F. J. Valverde-Muñoz, J. Herrero-Martín, J. A. Real and E. Coronado, Adv. Mater., 2022, 2202551 CrossRef CAS PubMed.
- B. Kumar, A. Paul, D. J. Mondal, P. Paliwal and S. Konar, Chem. Rec., 2022, 22, e202200135 CrossRef CAS PubMed.
- M. Oppermann, F. Zinna, J. Lacour and M. Chergui, Nat. Chem., 2022, 14, 739–745 CrossRef CAS PubMed.
- Y. Jiang, L. C. Liu, H. M. Müller-Werkmeister, C. Lu, D. Zhang, R. L. Field, A. Sarracini, G. Moriena, E. Collet and R. J. D. Miller, Angew. Chem., Int. Ed., 2017, 56, 7130–7134 CrossRef CAS PubMed.
- S.-Q. Su, S.-Q. Wu, Y.-B. Huang, W.-H. Xu, K.-G. Gao, A. Okazawa, H. Okajima, A. Sakamoto, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2022, 61, e202208771 CrossRef CAS PubMed.
- J.-P. Xue, Y. Hu, B. Zhao, Z.-K. Liu, J. Xie, Z.-S. Yao and J. Tao, Nat. Commun., 2022, 13, 3510 CrossRef CAS PubMed.
- R. Akiyoshi and S. Hayami, Chem. Commun., 2022, 58, 8309–8321 RSC.
- S. Johannsen, S. Ossinger, J. Grunwald, A. Herman, H. Wende, F. Tuczek, M. Gruber and R. Berndt, Angew. Chem., Int. Ed., 2022, 61, e202115892 CrossRef CAS PubMed.
- S. Johannsen, S. Schüddekopf, S. Ossinger, J. Grunwald, F. Tuczek, M. Gruber and R. Berndt, J. Phys. Chem. C, 2022, 126, 7238–7244 CrossRef CAS.
- D. Li, Y. Tong, K. Bairagi, M. Kelai, Y. J. Dappe, J. Lagoute, Y. Girard, S. Rousset, V. Repain, C. Barreteau, M. Brandbyge, A. Smogunov and A. Bellec, DOI:10.48550/ARXIV.2206.13767.
- S. Dehnen, Angew. Chem., Int. Ed., 2018, 57, 10386–10387 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1560719, 1913041, 1953349, 1953350, 1988543, 2330938, 2330939 and 2340087. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00429a |
| ‡ Current address: Department of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |