
Understanding the role of linker flexibility in soft porous coordination polymers†
Yamil J.
Colón
 *a and
Shuhei
Furukawa
b
*a and
Shuhei
Furukawa
b
aDepartment of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: ycolon@nd.edu
bInstitute for Integrated Cell-Materials Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 15th October 2019
Abstract
Soft porous coordination polymers are a new class of materials that have the potential to combine favorable features of metal–organic polyhedra (MOP) and frameworks (MOFs) with those of soft materials: permanent porosity and chemical specificity with processability and optical, electronic, and mechanical responses. Fundamental studies are needed to guide the future technological applications of these materials. We employ topologically-based algorithms to generate crystalline structures in the fcu topology and use classical molecular modeling to calculate their mechanical properties. The generated crystals used dimetal paddlewheel based MOP with cuboctahedral topology [M2(bdc)2]12 (M = Co, Cu, Rh, and Zn; bdc = 1,3-benzenedicarboxylate) as the nodes and 1,4-diazabicyclo[2.2.2]octane (DABCO), bipyridine, 1,4-bis(imidazol-1-ylmethyl)benzene (bix), 4,4′-bis(imidazol-1-ylmethyl)biphenyl (bibPh), and 1,1′-(1,12-dodecanediyl)bis[1H-imidazole] (bidod) as the linkers. Structures containing DABCO or bipyridine are found to have bulk moduli approximately an order of magnitude higher than those containing bix, bibPh, or bidod. The differences in mechanical properties are associated with the linker flexibility, as evidenced by free energy calculations on the radius of gyration of the linkers. We also calculate free energies as a function of radius of gyration of chosen nanoparticles.
 Yamil J. Colón | Yamil J. Colón is an Assistant Professor in the Department of Chemical and Biomolecular Engineering at the University of Notre Dame. He received his B.S. from the University of Notre Dame (2009) and completed his Ph.D. under the supervision of Prof. Randall Snurr at Northwestern University (2015). He then joined the University of Chicago and Argonne National Laboratory as a postdoctoral researcher with Professor Juan J. de Pablo. Dr Colón's research interests involve the computational design and discovery of materials for energy-related applications. |
Design, System, ApplicationHerein we combine crystal enumeration algorithms with classical molecular mechanics and enhanced sampling techniques to calculate the mechanical properties of hypothetical crystals and the free energy of conformation of linkers and nanoparticles. Part of the systems of interests are known to form crystals or amorphous, soft porous coordination polymers. Drastic differences in the bulk modulus values are observed for the systems. We identify the flexibility of the linkers used as the key design factor that will give rise to crystalline or amorphous soft porous coordination polymers. A distinct design feature of the chemistry of linkers that give rise to the soft porous coordination polymers is the presence of sp3 carbons connecting rigid aromatic rings. These findings will lead to the realization of new soft porous coordination polymers. Lastly, our results suggest that the crystalline state of the studied systems that are amorphous can be metastable. This can be potentially exploited to engineer and design molecular systems that can reversibly go from amorphous to crystalline and vice versa. Such a materials response can find use in many applications including energy storage, separations, catalysis, sensing, etc. |
Introduction
Metal–organic frameworks, or MOFs, are an exciting class of crystalline, porous materials lauded for their structural and functional tailorability. In many ways, what affords these materials their designability is the molecular building block (MBB) approach to their realization. The geometry and connectivity of the building blocks can be controlled through judicious choice or design and the resulting crystal structure can then be reliably predetermined by taking advantage of the coordination-driven self-assembly process of these materials. This type of approach has been successfully exploited in both experimental and computational settings for a multitude of applications including methane storage,1,2 carbon capture,3,4 hydrogen storage,5–9 catalysis,10–13 drug delivery,14–17 sensing,18–20etc.21–28Usually, the molecular building blocks that comprise MOFs are inorganic nodes and organic linkers. Recent synthetic efforts have focused on incorporating supermolecular building blocks (SBBs) as the nodes.29–31 SBBs are usually metal–organic polyhedra (MOP) or metal–organic cages (MOCs), which are discrete molecules combined from inorganic and organic moieties. Similar to MOFs, they provide design opportunities through judicious choice of building blocks. For instance, the size and connectivity of the cage can be controlled by the length and the angle of the organic linker. Due to the combination of inorganic and organic parts available to MOP, the palette of geometry and connectivity as nodes in crystalline structures is expanded when compared to simple inorganic nodes. This new complexity can then be exploited to achieve crystals with new and complex topological nets.32–36
In a great example of the MBB approach for MOF synthesis, Guillerm and co-workers demonstrated the use of an 18-connected MOP to form a MOF with the gea topology.37 Interestingly, the first gea MOF the authors reported utilized a nonanuclear carboxylate-based cluster as the node in the crystalline structure. From that they hypothesized and demonstrated that another node with the same geometry and connectivity, the MOP as SBB, would yield a MOF with the same topology. This strategy of top-down generation of MOFs, where building blocks of interest are mapped onto a known topology, has also been demonstrated computationally to generate and subsequently screen a number of crystalline structures, including those containing SBBs.38–40
In a similar fashion, researchers have exploited the use of a 24-connected MOP to form MOFs with the rht topology.31,41–45 MOFs with the rht topology have the property that they cannot catenate; two or more frameworks that occupy each other's pore space by interpenetration. As such, they have been looked at for various gas storage applications with great success.46–50 We stress that these are only a few examples of the use of MOP in MOFs. For further reading on SBBs, their chemistry, and topological approaches we refer the readers to excellent reviews by Guillerm et al.32 and Kim et al.33
Of particular interest to our research endeavour is the use of 12-connected, cuboctahedral MOP to generate MOFs with the fcu topology.51,52 Various examples in the literature include the use of MOP-1 analogues, a cage formed with 12 metal paddlewheels and 24 bent ditopic ligands, connected by DABCO or bipyridine in order to form MOFs. The metals in the paddlewheels, which are considered the connections points of the SBB, can be Cu, Co, or Zn.53–55
Ligands can also be considered the connections points, as is in the case of DUT-49.51 The SBB contains 6 Cu paddlewheels and 12 carbazole-based linkers as octahedral cages. Krause et al. reported DUT-49 exhibits negative adsorption behaviour: gas is desorbed despite increasing pressure.56,57 Through a combined experimental and computational approach, they demonstrated the negative adsorption behaviour is due to reversible pore volume reduction within the crystal, enabled by the deformation of the organic linker that connects the SBBs. Notably, the SBB did not show any reduction in pore volume.
Carné-Sánchez et al. have leveraged the permanent porosity displayed by the SBBs to design amorphous, soft materials with the SBBs: soft porous coordination polymers.58,59 By exploiting the processing conditions, the authors were able to form colloids, colloidal gels, and aerogels that contain permanent porosity, imbued by the SBB. The SBB used was a MOP-1 analogue that used the metals as the connection points and Rh as the metal in the paddlewheels. The SBBs are connected through ditopic imidazole linkers.
The DUT-49 and amorphous Rh-MOP system examples highlight the role that the connecting linker between the SBBs can play in crystal structural deformations or in the formation of soft and flexible amorphous systems. These examples suggest the mechanical properties of these systems can be tuned via the flexibility of the organic linker. Recent experimental efforts by Lv et al. strengthen this notion, as they find that linker rigidity can significantly improve the stability of Zr-based MOFs.60
The prospect of combining advantageous properties from MOFs and other porous materials with soft matter, makes soft porous coordination polymers attractive for a large number of applications. As an example, Liu and co-workers reported a novel membrane system comprised of hyper-cross-linked MOP.61 They demonstrated the membranes have high water permeabilities, as well as self-healing and antimicrobial capabilities. As these materials are poised to usher technological advances, fundamental studies are needed to understand their properties.
Herein, we employ a computational approach to explore the role of linker flexibility for soft porous materials. To do so, we use top-down generation algorithms to make crystalline structures with fcu nets using the linkers of interest and cuboctahedral MOP with varying metals on the paddlewheels (Co, Cu, Rh, and Zn) as SBBs and calculate the mechanical properties of the crystalline structures using classical molecular mechanics. Then, we use molecular dynamics simulations along with enhanced sampling techniques to calculate the free energy as a function of the radius of gyration of the various organic linkers used in the systems of interest: 1,4-diazabicyclo[2.2.2]octane (DABCO), bipyridine, 1,4-bis(imidazol-1-ylmethyl)benzene (bix), 4,4′-bis(imidazol-1-ylmethyl)biphenyl (bibPh), and 1,1′-(1,12-dodecanediyl)bis[1H-imidazole] (bidod). Lastly, to illustrate the linker effect on the systems, we calculate the free energy of the radius of gyration of nanoparticles of combinations of MOPs and linkers.
Methods
All-atom simulations were performed for the following building blocks and structures:• Linkers: DABCO, bipyridine, bix, bibPh, and bidod.
• Nodes: MOP-1 analogues as [M2(bdc)2]12 (bdc = 1,3-benzenedicarboxylate) were used while varying the metal on the paddlewheel. Metals of interest are: Co, Cu, Rh, and Zn. Metal atoms facing the inside of the cage were coordinated by a water molecule. Structure of the MOP was obtained using the crystallographic structure of Rh-MOP62 with coordinated water molecules in the inside of the cage. Rh atoms were substituted for Co, Cu, or Zn to generate the new cages.
• Crystal structures: all possible combinations of nodes and linkers were considered in crystalline structures with the fcu topology.
• Nanoparticles: discrete nanoparticles for the Rh-MOP with DABCO as the linker and Rh-MOP with bibPh as the linker.
MOP-1 analogues (cuboctahedron cages) with various metals (Co, Cu, Rh, and Zn used for the paddlewheels) and linkers were mapped onto an fcu net using the top-down crystal generator, ToBaCCo 3.0 (https://github.com/tobacco-mofs/tobacco_3.0).38–40,63 Linkers were energy minimized prior to the crystal generation. Nitrogen atoms at the ends of the linkers and the metals on the outside of the MOPs were considered as connection points. The resulting crystal structures, as output .cif files from the code, were used as input for Boyd and co-workers' software to generate LAMMPS64 input files with the corresponding universal force field (UFF) parameters.65 Structures were then energy minimized at least twice using LAMMPS, the first minimization held the unit cell dimensions constant while the second minimization allowed for the unit cell dimensions to change. Fig. 1 shows the various building blocks used in this work as well as a resulting structure from the top-down crystal generation process using the fcu topology.
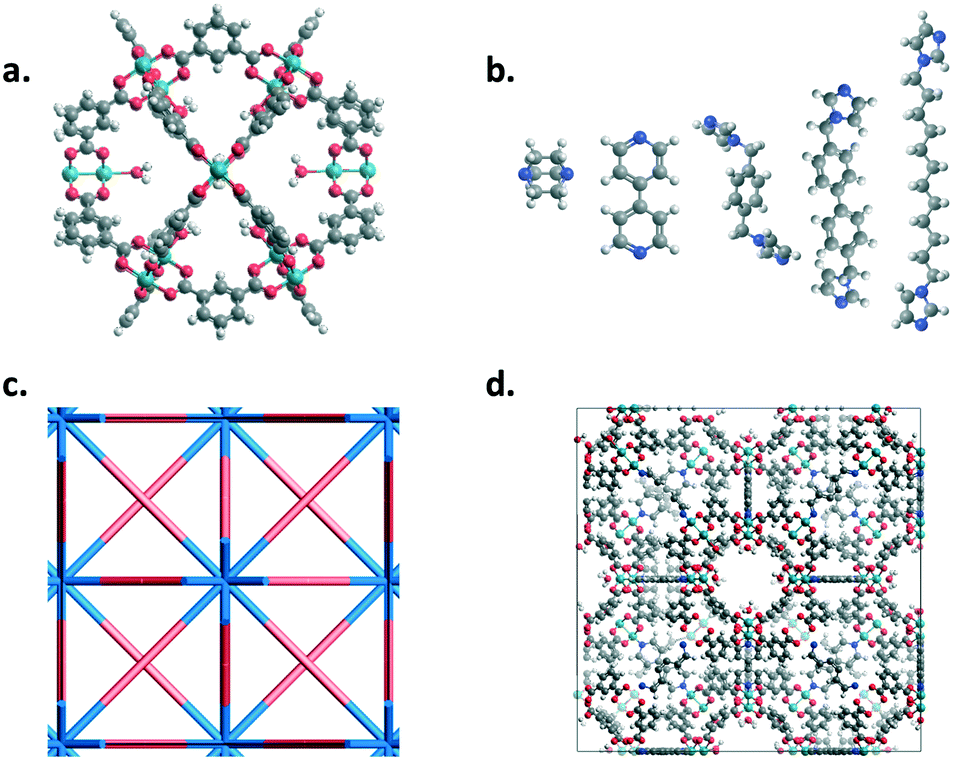 | ||
| Fig. 1 Panel a. shows the 12-connected MOP structure with coordinated water molecules on the inside of the cage. Connection points of the MOP are the metal atoms on the outside of the cage which can be either Co, Cu, Rh, or Zn. Panel b. shows the linkers of interest in this work. The connection points are the nitrogen atoms at the ends of the linkers. From left to right they are DABCO, bipyridine, bix, bibPh, and bidod. Panel c. shows the fcu topology where the 12-connected MOPs and linkers of interest were mapped. Panel d. shows an example of a resulting crystalline structure after the top-down generation using a MOP structure as the node and bipyridine as the linker. C = gray, H = white, O = red, N = blue, and Co, Cu, Zn, Rh = turquoise. | ||
Once the structures were minimized, mechanical properties of the crystalline structures were calculated at 0 K by isotropically expanding and contracting the unit cell of interest.65,66 Expansion and contractions were performed with increments of 0.05% with a maximum strain of 1% for the unit cell length. After each unit cell expansion or contraction, the energy was minimized. The resulting energy versus unit cell volume values were then fit using the Murnaghan equation of state67 to extract the bulk modulus of the structures:
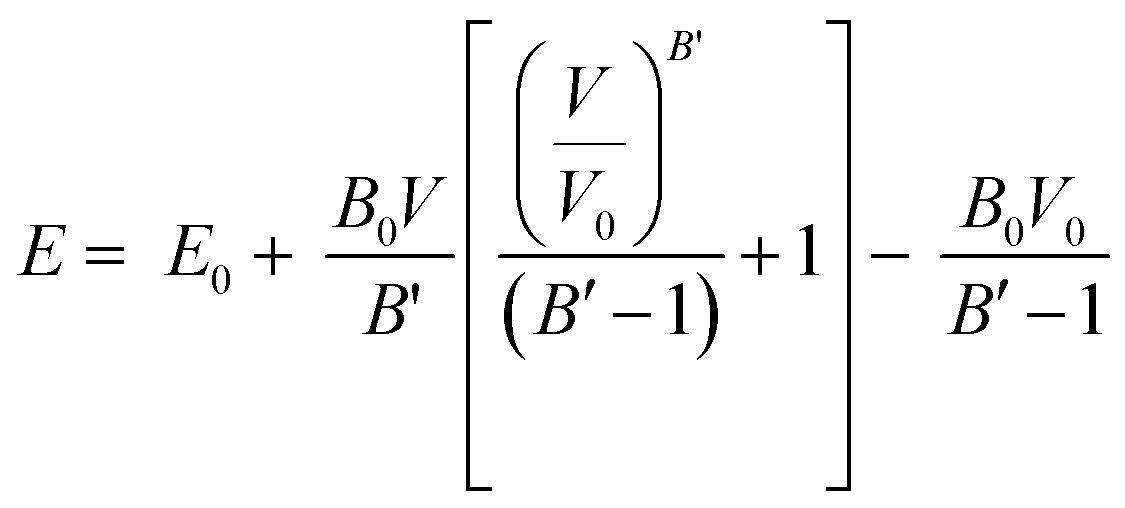 | (1) |
Nanoparticles of Rh-MOP with DABCO and Rh-MOP with bibPh were also considered. Fig. 2 shows how the nanoparticles were generated starting from the crystalline arrangement. The nanoparticles were formed by taking 8 unit cells of the generated crystalline structures and eliminating atoms pertaining to bonds that crossed periodic boundaries. More atoms were subsequently deleted to ensure proper chemical connectivity. As a result, discrete nanoparticles were formed consisting of 13 MOP cages and the associated linkers at each connection point of the MOP. The outermost MOP contain dangling linkers.
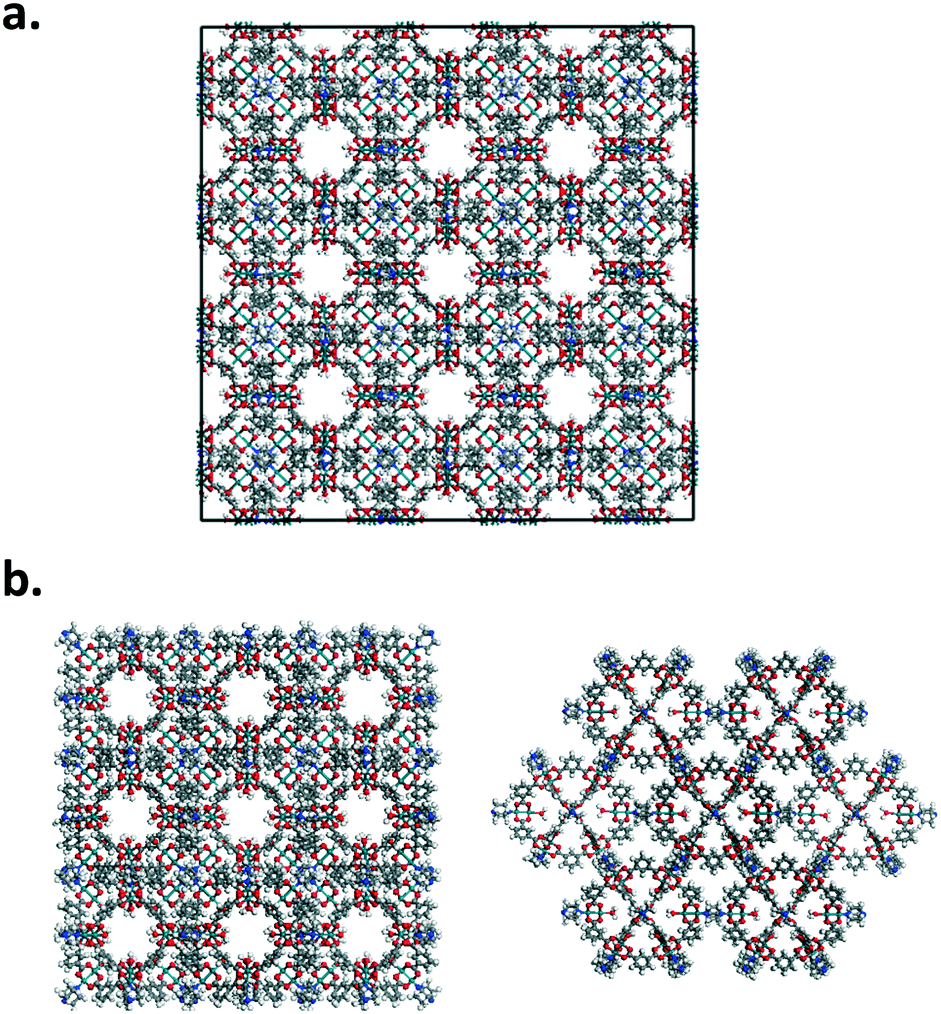 | ||
| Fig. 2 Nanoparticle generation. Panel a. shows 8 unit cells of the crystalline Rh-MOP with DABCO linkers. Bonds that cross the periodic boundaries were eliminated as well as incomplete structures that are close to the boundaries. Panel b. shows the resulting nanoparticle once the appropriate atoms have been deleted. There are 13 cages in the nanoparticle. All cages have the appropriate number of linkers. The outside of the nanoparticle contains dangling DABCO linkers. | ||
All bonded and non-bonded potentials and parameters used to describe the various structures and building blocks in this study were taken from the UFF.68 Energy minimizations were carried out using the conjugated gradient algorithm with a convergence criterion of 10−10 kcal mol−1 for the energy and 10−10 kcal mol−1 Å−1 for the force. Molecular dynamics simulations were performed in the NVT ensemble at 300 K, using the Nose–Hoover thermostat. Equations of motion were integrated using the velocity Verlet algorithm and a timestep of 1 fs. Free energy calculations were performed using the artificial neural networks (ANN) sampling method using LAMMPS and SSAGES (https://github.com/MICCoM/SSAGES-public).69,70 Free energies for the various linkers and nanoparticles were calculated using the square of their radius of gyration (Rg) as a collective variable. SSAGES employs the ANN sampling method to bias along the radius of gyration of the structures of interest and calculate the free energies. The neural network used for the enhanced sampling contained one hidden layer with 15 neurons.
Results and discussion
The soft porous coordination polymers58 of interest in this work contain 12-coordinated Rh MOP cages connected by imidazole based linkers. Since the node is 12-connected, we chose the fcu net as a proxy for these materials in this study. Crystalline structures were generated utilizing Co, Cu, Rh, or Zn MOP as nodes and DABCO, bipyridine, bix, bibPh, or bidod as linkers. The building blocks were mapped onto an fcu net using the top-down crystal generation code.38–40 The crystalline structures were then energy minimized and subjected to isotropic expansion and compression. At each interval of expansion or compression, the crystal structure was minimized holding the unit cell dimensions constant. The resulting energies were then recorded as a function of the unit cell volume and fit to the Murnaghan equation to obtain the bulk modulus of the crystal.Co, Cu, and Zn MOP cages with DABCO and bipyridine linkers have been shown experimentally to form crystalline structures53–55 while Rh MOP cages with bix, bibPh, and bidod have been shown to form amorphous systems.58,59 As such, we expect to observe significant differences in the energetic response of the volume changes in the computationally-generated crystals based on the chemical identity of the linker. We also expect the density of the resulting structure to play a role in the resulting mechanical properties.71,72
Fig. 3 reports the energy versus volume response of the Co MOP crystalline structures. DABCO shows the steepest response as volume changes, followed by bipyridine. The rest of the linkers (bix, bibPh, and bidod) show a much weaker response to the changes in the volume. The linkers bix and bibPh show a similar response while bidod shows the weakest response out of all the linkers considered.
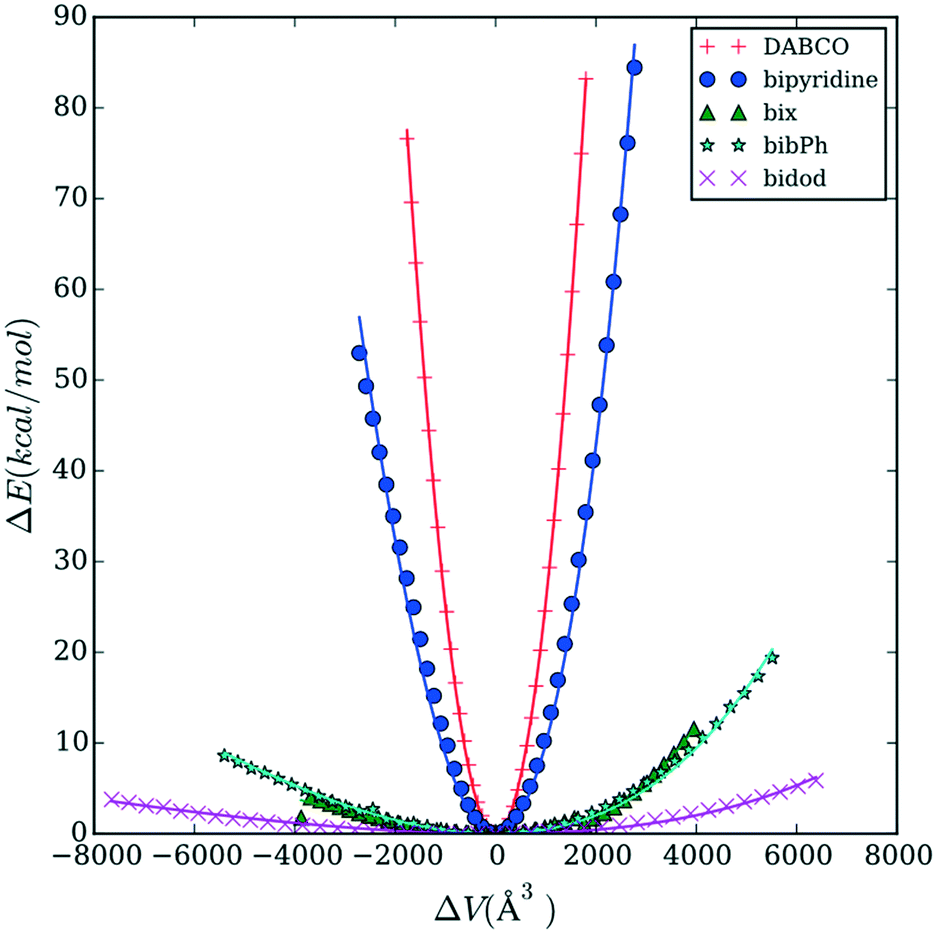 | ||
| Fig. 3 Energy versus volume response for crystalline structures containing Co cages connected by DABCO (red cross), bipyridine (blue circle), bix (green triangle), bibPh (cyan star), and bidod (magenta x). Symbols are the simulation data and lines represent the fit to the Murnaghan equation. DABCO shows the steepest change in energy followed by bipyridine. Bix, bibPh, and bidod show much weaker responses, with bidod having the weakest. | ||
The observed responses are directly related to the bulk modulus of the crystals. The bulk modulus values obtained are reported for all the combinations of MOP cages and organic linkers considered in this study in Fig. 4. Little difference is observed in the bulk modulus values for the various cages. Significant differences are observed however, when comparing the values for the various linkers. The cages that contain DABCO show the highest bulk modulus values (∼15–20 GPa) followed by the cages that contain bypiridine (∼10 GPa). The cages with the rest of the linkers show bulk modulus values close to 1 GPa or less.
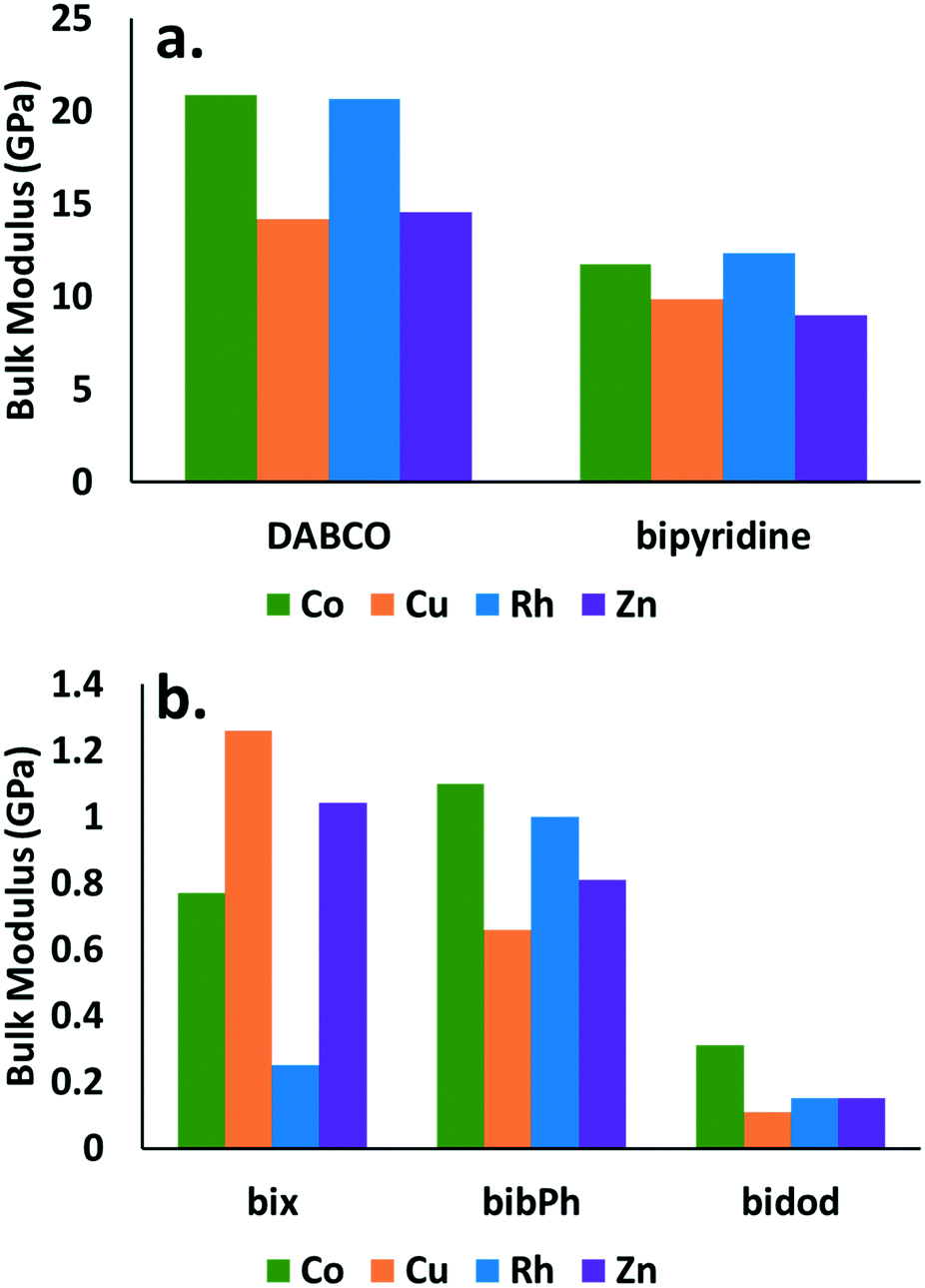 | ||
| Fig. 4 Bulk modulus for crystalline structures comprised of MOP cage and linker combinations. Panel a. shows DABCO and bipyridine while panel b. shows bix, bibPh, and bidod. Co (green), Cu (orange), Rh (blue), and Zn (purple) cages are considered. Changes in mechanical properties are attributed to the linkers, with DABCO and bipyridine showing significantly higher bulk modulus values than bix, bibPh, or bidod. | ||
Fig. 4 demonstrates significant differences in the mechanical stability between structures that contain linkers that give rise to crystalline structures (DABCO and bipyridine) and those that give rise to amorphous structures (bix, bibPh, bidod). Despite no current crystalline structures reported in the literature for Rh MOP with DABCO or bypiridine, the results suggest such a structure would share similar mechanical properties to the ones containing Co, Cu, or Zn cages. The calculations suggest that Rh MOP with DABCO or bipyridine would be crystalline. Similarly, the results suggest that Co, Cu, or Zn cages using bix, bibPh, or bidod as linkers would share similar mechanical properties to that of Rh MOP with those various linkers. One hypothesis is that the low mechanical stability of the crystalline system leads to pore collapse of the structure, excluding the MOP, resulting in the soft amorphous porous coordination polymer. Consequently, structures that have those cages with those linkers would form soft porous coordination polymers. Lastly, the differences observed for the various linkers suggest that this type of calculation can be used to determine a priori if a combination of MOP cage and linker will result in a crystalline structure or soft porous coordination polymer. Given the ease of the crystal generation and the calculation of the mechanical property, this type of approach can also lend itself well for high-throughput computational screening73 to quickly generate potential candidates for soft porous coordination polymers.
It is clear that the mechanical properties are also related to the density of the materials as shown in Fig. S1 in the ESI† and in studies in the literature.71–73 The densest materials, those that contain DABCO and bipyridine, show higher bulk modulus values than those with bix, bibPh, or bidod. The changes in density however, are not enough to explain the order of magnitude difference in the bulk moduli.
To study further the role the linkers are playing in the mechanical properties, we calculate the free energy as a function of the radius of gyration of the various linkers; Fig. 5 shows the results for DABCO and bipyridine and Fig. 6 shows the results for bix, bibPh, and bidod. Given the differences in mechanical properties observed with respect to the linkers, we expect to see free energy profiles that penalize deviations from the energy minimum for DABCO and bipyridine and free energy profiles that can potentially have multiple free energy minima for a range of values of Rg for bix, bibPh, and bidod.
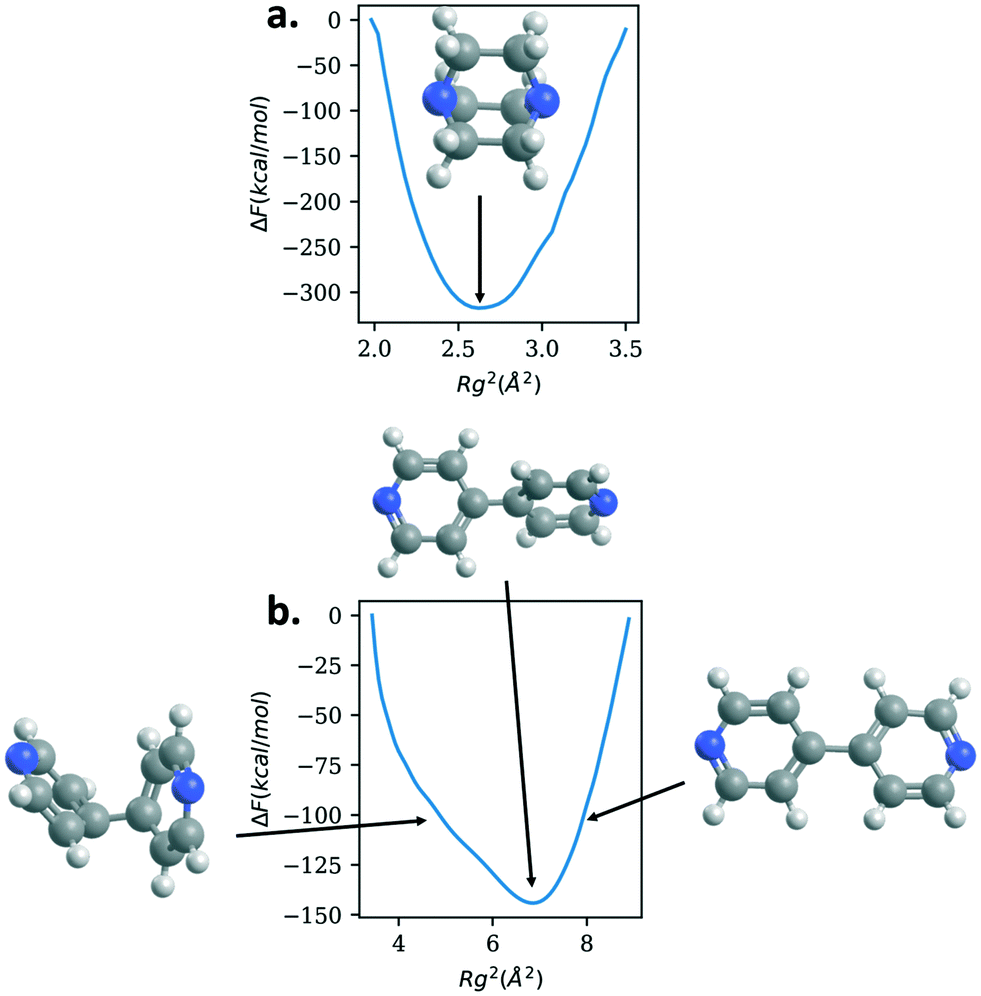 | ||
| Fig. 5 Free energy versus radius of gyration squared (Rg2) for DABCO (panel a.) and bipyridine (panel b.) linkers, with insets showing the configurations corresponding to the various values of Rg2. Both linkers show one deep free energy minimum with strong free energy penalties for changes in the Rg values. C = gray, H = white, N = blue. | ||
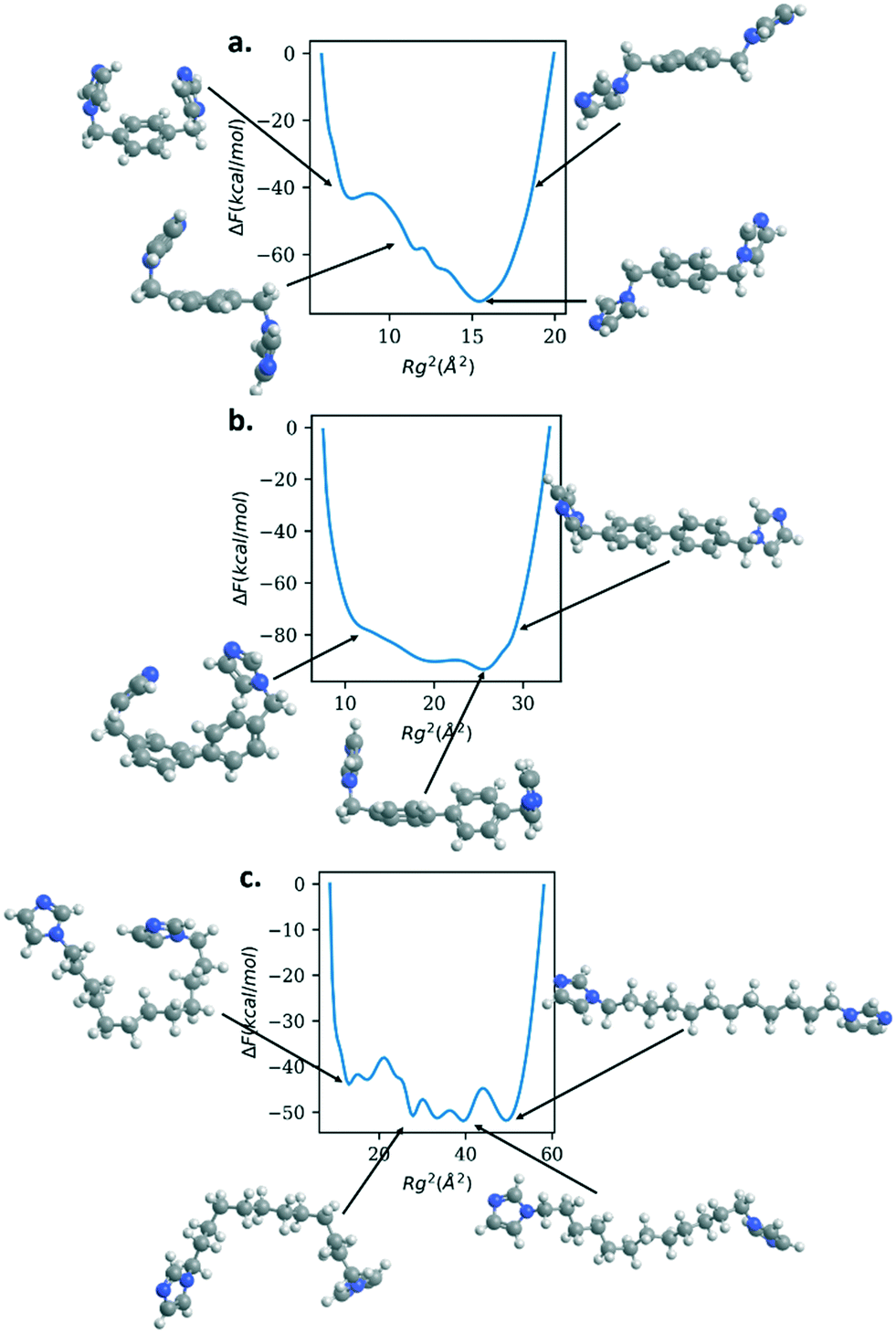 | ||
| Fig. 6 Free energy versus radius of gyration squared (Rg2) for bix (panel a.), bibPh (panel b.), and bidod (panel c.) and the corresponding configurations for the differing values of Rg2. All linkers show multiple free energy minima within 10–20 kcal mol−1 of each other. C = gray, H = white, N = blue. | ||
DABCO, the linker used in the structures in the materials with the highest bulk modulus values, shows one deep minimum in the free energy. Bipyridine, the other linker found in the crystalline MOF structures, also has one minimum. The free energy minimum shows the pyridine rings are rotated with respect to each other. As the Rg value increases so does the free energy as the rings lie in the same plane and rings are slightly stretched. The free energy also increases as the Rg value decreases but the energy increase is not as steep as with the extension. Bipyridine structures with low values of Rg show the rings facing each other bending at the carbon atoms that connect them.
Fig. 6 shows the free energy of the Rg for bix, bibPh, and bidod. All of the linkers show much shallower free energy profiles when compared to DABCO and bipyridine. The free energy profile for bix shows a minimum for the Rg2 around 15 Å2. The structure shows the imidazole rings above and below the center phenyl ring. As the linker Rg value increases, the free energy also increases and the structure reveals the imidazole rings closer to the plane of the center phenyl ring. As the Rg values decrease, the free energy rises but also shows free energy minima. The structure shows the imidazole rings bending towards the phenyl rings, above and below it. Another metastable state shows the imidazole rings bending towards the phenyl rings in the same plane.
The linker bibPh shows a range of 20 Å2 that contains structures within 20 kcal mol−1. The free energy minimum shows twisted phenyl rings, similar to the bipyridine free energy minimum, with the imidazole rings above the phenyl rings. Higher Rg values stretches the linker, aligning the phenyl rings to be in the same plane along with the imidazole rings. Smaller values of Rg compared to the free energy minimum show the imidazole rings bending towards each other as well as bending occurring at the carbon atoms that connect the phenyl rings. Lastly, the bidod linker shows a range of 40 Å2 with structures within 10 kcal mol−1. As shown in the structures highlighted in the figure, the alkane chain that connects the imidazole rings offers flexibility in the structures, from fully stretched to bends introduced along the chain that bring the imidazole rings close together. We note that for the mechanical property calculation of the crystalline system with the bidod linker, only the most stretched configuration was used in the crystalline structure. As shown, the mechanical properties also depend on the density of the structure. Therefore, using other configurations that effectively decrease the length between the connection points would also decrease the unit cell volume and increase the bulk modulus. We expect, though, the values to remain in the same order of magnitude as the ones shown in this study.
It is important to note that what imbues these linkers with the flexibility is the presence of the sp3 carbons that connect the rigid aromatic rings, phenyl rings to imidazole rings (bix and bibPh) or imidazole to imidazole rings (bidod). The free energy profiles in Fig. 6 show the configurations of the various linkers differ by the bending of the angle centered at the sp3 carbon atoms. Therefore, this is an important chemical moiety that can be exploited in linkers for future soft porous coordination polymers.
To explore the consequence of the flexibility, or lack thereof, of the linkers on nanoparticles of this class of materials, we calculate the free energy as a function of the radius of gyration of the nanoparticles. As representative structures, we chose Rh MOP with DABCO as the linker and Rh MOP with bibPh as the linker. Only the linkers were considered for the biasing of the radius of gyration. We expect the free energy profiles for the nanoparticles to mirror those of their respective linkers. For Rh MOP with DABCO we expect to see free energy penalties for deviations from the crystalline structure while we expect to see multiple free energy minima for Rh MOP with bibPh. Fig. 7 shows the results for both structures.
 | ||
| Fig. 7 Free energy versus radius of gyration squared (Rg2) of nanoparticles comprised of Rh MOP cages with DABCO (panel a.) or bibPh (panel b.) along with insets corresponding to various values of Rg2. Rh MOP with DABCO shows only one free energy minimum, where the crystalline arrangement lies. Rh MOP with bibPh instead shows multiple free energy minima showing collapsed and open structures. One of the free energy minima corresponds to the crystalline arrangement. | ||
Rh MOP with DABCO shows one free energy minimum that lies in the crystalline arrangement of the cages. Rh MOP with bibPh shows richer behavior. There are multiple free energy minima at low and high values of Rg2; the figure illustrates the structures. Two of the free energy minima show amorphous structures with different arrangement of the Rh cages. Interestingly, one of the free energy minima shows the crystalline arrangement of the cages. The simulations suggest the crystalline arrangement of the MOP cages with bibPh is a metastable state and could therefore be attained given proper conditions. These results suggest different configurations can be accessible to the soft porous coordination polymers, including the crystalline arrangement, provided a stimulus to overcome energetic barriers. One such stimulus could be heat of adsorption, as is the case for DUT-49 and its structural changes that give rise to the negative adsorption behavior. Changing the chemistry of the linker and its flexibility, offers the opportunity to tailor the mechanical properties as well as possible amorphous to crystalline transformations of the system. Their targeted manipulation could have technological advantages in adsorption and separation systems, sensing, catalysis, etc.
Conclusions
Crystalline structures were generated using topological algorithms (Co, Cu, Rh, and Zn MOP as nodes and DABCO, bipyridine, bix, bibPh, and bidod as linkers) and their mechanical properties calculated using classical molecular mechanics. Bulk modulus values were found to be dependent on the linkers and not the MOP cages. From a mechanical stability perspective, these results suggest that Rh MOP with DABCO or bipyridine would result in a crystalline material, despite not being currently reported in the literature. Similarly, the results suggest that structures with Co, Cu, or Zn MOP cages and bix, bibPh, or bidod as linkers would result in soft porous coordination polymers. The calculated bulk modulus values are related to the flexibility of the linker with DABCO and bipyridine showing significantly higher values than bix, bibPh, and bidod. Free energy calculations on the radius of gyration of the linkers reveal DABCO and bipyridine have one free energy minimum, while bix, bibPh, and bidod have multiple free energy minima. Free energy calculations on the radius of gyration of nanoparticles of Rh MOP with DABCO show one free energy minimum at its crystalline arrangement. Meanwhile, the same calculation for Rh MOP with bibPh shows free energy minima with amorphous configurations. Notably, the crystalline configuration of the MOP cages is shown to be metastable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
YJC acknowledges the computational resources at the Center for Research Computing at the University of Notre Dame. YJC would also like to thank the Melchor program and start-up funding from the University of Notre Dame. SF acknowledges JSPS KAKENHI grant number 19H04575 (Coordination Asymmetry).Notes and references
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, Methane storage in metal–organic frameworks: current records, surprise findings, and challenges, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Large-scale Screening of hypothetical metal-organic frameworks, Nat. Chem., 2012, 83–89 CrossRef CAS PubMed.
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Metal-organic frameworks for carbon dioxide capture and methane storage, Adv. Energy Mater., 2017, 7, 1601296 CrossRef.
- C. E. Wilmer, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Structure-property relationships of porous materials for carbon dioxide separation and capture, Energy Environ. Sci., 2012, 5, 9849–9856 RSC.
- N. S. Bobbitt and R. Q. Snurr, Molecular modelling and machine learning for high-throughput screening of metal-organic frameworks for hydrogen storage, Mol. Simul., 2019, 1–13 Search PubMed.
- Y. J. Colón, D. Fairen-Jimenez, C. E. Wilmer and R. Q. Snurr, High-Throughput Screening of Porous Crystalline Materials for Hydrogen Storage Capacity near Room Temperature, J. Phys. Chem. C, 2014, 118, 5383–5389 CrossRef.
- L. J. Murray, M. Dinca and J. R. Long, Hydrogen storage in metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Hydrogen Storage in Metal–Organic Frameworks, Chem. Rev., 2011, 112, 782–835 CrossRef PubMed.
- J. Sculley, D. Yuan and H.-C. Zhou, The current status of hydrogen storage in metal-organic frameworks-updated, Energy Environ. Sci., 2011, 4, 2721–2735 RSC.
- J. Liang, Z. Liang, R. Zou and Y. Zhao, Heterogeneous catalysis in zeolites, mesoporous silica, and metal–organic frameworks, Adv. Mater., 2017, 29, 1701139 CrossRef PubMed.
- A. Corma, H. Garcia and F. Llabrés i Xamena, Engineering metal organic frameworks for heterogeneous catalysis, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Metal-organic framework materials as catalysts, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- L. Ma, C. Abney and W. Lin, Enantioselective catalysis with homochiral metal-organic frameworks, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- M. C. Bernini, D. Fairen-Jimenez, M. Pasinetti, A. J. Ramirez-Pastor and R. Q. Snurr, Screening of bio-compatible metal–organic frameworks as potential drug carriers using Monte Carlo simulations, J. Mater. Chem. B, 2014, 2, 766–774 RSC.
- C. Gaudin, D. Cunha, E. Ivanoff, P. Horcajada, G. Chevé, A. Yasri, O. Loget, C. Serre and G. Maurin, A quantitative structure activity relationship approach to probe the influence of the functionalization on the drug encapsulation of porous metal-organic frameworks, Microporous Mesoporous Mater., 2012, 157, 124–130 CrossRef CAS.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Metal–Organic Frameworks as Efficient Materials for Drug Delivery, Angew. Chem., 2006, 118, 6120–6124 CrossRef.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. a. Vallet-Regí, M. Sebban, F. Taulelle and G. r. Férey, Flexible Porous Metal-Organic Frameworks for a Controlled Drug Delivery, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- J. A. Greathouse, N. W. Ockwig, L. J. Criscenti, T. R. Guilinger, P. Pohl and M. D. Allendorf, Computational screening of metal–organic frameworks for large-molecule chemical sensing, Phys. Chem. Chem. Phys., 2010, 12, 12621 RSC.
- L. Sarkisov, Toward Rational Design of Metal–Organic Frameworks for Sensing Applications: Efficient Calculation of Adsorption Characteristics in Zero Loading Regime, J. Phys. Chem. C, 2012, 116, 3025–3033 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal–Organic Framework Materials as Chemical Sensors, Chem. Rev., 2011, 112, 1105–1125 CrossRef PubMed.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, From fundamentals to applications: a toolbox for robust and multifunctional MOF materials, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Metal–organic frameworks for energy storage: Batteries and supercapacitors, Coord. Chem. Rev., 2016, 307(Part 2), 361–381 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, The Chemistry and Applications of Metal-Organic Frameworks, Science, 2013, 341(6149), 1230444 CrossRef PubMed.
- M. P. Yutkin, D. N. Dybtsev and V. P. Fedin, Homochiral porous metal-organic coordination polymers: synthesis, structure and functional properties, Russ. Chem. Rev., 2011, 80, 1009 CrossRef CAS.
- H.-L. Jiang and Q. Xu, Porous metal-organic frameworks as platforms for functional applications, Chem. Commun., 2011, 47, 3351–3370 RSC.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, Light-Harvesting Metal–Organic Frameworks (MOFs): Efficient Strut-to-Strut Energy Transfer in Bodipy and Porphyrin-Based MOFs, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed.
- A. Sturluson, M. T. Huynh, A. R. Kaija, C. Laird, S. Yoon, F. Hou, Z. Feng, C. E. Wilmer, Y. J. Colón, Y. G. Chung, D. W. Siderius and C. M. Simon, The role of molecular modelling and simulation in the discovery and deployment of metal-organic frameworks for gas storage and separation, Mol. Simul., 2019, 1–40 Search PubMed.
- S. Kitagawa, Metal–organic frameworks (MOFs), Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Supramolecular coordination: self-assembly of finite two- and three-dimensional ensembles, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, Supermolecular building blocks (SBBs) for the design and synthesis of highly porous metal-organic frameworks, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS PubMed.
- V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, A supermolecular building approach for the design and construction of metal–organic frameworks, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC.
- D. Kim, X. Liu and M. S. Lah, Topology analysis of metal–organic frameworks based on metal–organic polyhedra as secondary or tertiary building units, Inorg. Chem. Front., 2015, 2, 336–360 RSC.
- H. N. Miras, L. Vilà-Nadal and L. Cronin, Polyoxometalate based open-frameworks (POM-OFs), Chem. Soc. Rev., 2014, 43, 5679–5699 RSC.
- A. A. Korlyukov, A. V. Vologzhanina, M. I. Buzin, N. V. Sergienko, B. G. Zavin and A. M. Muzafarov, Cu(II)-Silsesquioxanes as Secondary Building Units for Construction of Coordination Polymers: A Case Study of Cesium-Containing Compounds, Cryst. Growth Des., 2016, 16, 1968–1977 CrossRef CAS.
- A. N. Bilyachenko, A. A. Korlyukov, A. V. Vologzhanina, V. N. Khrustalev, A. N. Kulakova, J. Long, J. Larionova, Y. Guari, M. S. Dronova and U. S. Tsareva, Tuning linkage isomerism and magnetic properties of bi-and tri-metallic cage silsesquioxanes by cation and solvent effects, Dalton Trans., 2017, 46, 12935–12949 RSC.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Discovery and introduction of a (3,18)-connected net as an ideal blueprint for the design of metal–organic frameworks, Nat. Chem., 2014, 6, 673–680 CrossRef CAS PubMed.
- D. A. Gomez-Gualdron, Y. J. Colon, X. Zhang, T. C. Wang, Y.-S. Chen, J. T. Hupp, T. Yildirim, O. K. Farha, J. Zhang and R. Q. Snurr, Evaluating topologically diverse metal-organic frameworks for cryo-adsorbed hydrogen storage, Energy Environ. Sci., 2016, 9, 3279–3289 RSC.
- Y. J. Colón, D. A. Gómez-Gualdrón and R. Q. Snurr, Topologically Guided, Automated Construction of Metal–Organic Frameworks and Their Evaluation for Energy-Related Applications, Cryst. Growth Des., 2017, 17, 5801–5810 CrossRef.
- R. Anderson and D. A. Gómez-Gualdrón, Increasing topological diversity during computational “synthesis” of porous crystals: how and why, CrystEngComm, 2019, 21, 1653–1665 RSC.
- J. F. Eubank, F. Nouar, R. Luebke, A. J. Cairns, L. Wojtas, M. Alkordi, T. Bousquet, M. R. Hight, J. Eckert and J. P. Embs, On demand: the singular rht net, an ideal blueprint for the construction of a metal–organic framework (MOF) platform, Angew. Chem., 2012, 124, 10246–10250 CrossRef.
- R. Luebke, J. F. Eubank, A. J. Cairns, Y. Belmabkhout, L. Wojtas and M. Eddaoudi, The unique rht-MOF platform, ideal for pinpointing the functionalization and CO2 adsorption relationship, Chem. Commun., 2012, 48, 1455–1457 RSC.
- R. Luebke, Ł. J. Weseliński, Y. Belmabkhout, Z. Chen, Ł. Wojtas and M. Eddaoudi, Microporous Heptazine Functionalized (3,24)-Connected rht-Metal–Organic Framework: Synthesis, Structure, and Gas Sorption Analysis, Cryst. Growth Des., 2014, 14, 414–418 CrossRef CAS.
- O. Delgado-Friedrichs and M. O'Keeffe, Three-periodic tilings and nets: face-transitive tilings and edge-transitive nets revisited, Acta Crystallogr., Sect. A: Found. Crystallogr., 2007, 63, 344–347 CrossRef CAS PubMed.
- Y. Zou, M. Park, S. Hong and M. S. Lah, A designed metal–organic framework based on a metal–organic polyhedron, Chem. Commun., 2008, 2340–2342 RSC.
- R. Babarao, M. Eddaoudi and J. W. Jiang, Highly Porous Ionic rht Metal−Organic Framework for H2 and CO2 Storage and Separation: A Molecular Simulation Study, Langmuir, 2010, 26, 11196–11203 CrossRef CAS PubMed.
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, Enhanced CO2 Binding Affinity of a High-Uptake rht-Type Metal−Organic Framework Decorated with Acylamide Groups, J. Am. Chem. Soc., 2010, 133, 748–751 CrossRef PubMed.
- H. Wu, K. Yao, Y. Zhu, B. Li, Z. Shi, R. Krishna and J. Li, Cu-TDPAT, an rht-type Dual-Functional Metal–Organic Framework Offering Significant Potential for Use in H2 and Natural Gas Purification Processes Operating at High Pressures, J. Phys. Chem. C, 2012, 116, 16609–16618 CrossRef CAS.
- O. K. Farha, A. O. Yazaydin, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, De Novo Synthesis of a Metal-Organic Framework Material Featuring Ultrahigh Surface Area and Gas Storage Capacities, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- D. Fairen-Jimenez, Y. J. Colón, O. K. Farha, Y.-S. Bae, J. T. Hupp and R. Q. Snurr, Understanding excess uptake maxima for hydrogen adsorption isotherms in frameworks with rht topology, Chem. Commun., 2012, 48, 10496–10498 RSC.
- U. Stoeck, S. Krause, V. Bon, I. Senkovska and S. Kaskel, A highly porous metal–organic framework, constructed from a cuboctahedral super-molecular building block, with exceptionally high methane uptake, Chem. Commun., 2012, 48, 10841–10843 RSC.
- A. J. Cairns, J. A. Perman, L. Wojtas, V. C. Kravtsov, M. H. Alkordi, M. Eddaoudi and M. J. Zaworotko, Supermolecular Building Blocks (SBBs) and Crystal Design: 12-Connected Open Frameworks Based on a Molecular Cubohemioctahedron, J. Am. Chem. Soc., 2008, 130, 1560–1561 CrossRef CAS PubMed.
- H. Chun, Low-Level Self-Assembly of Open Framework Based on Three Different Polyhedra: Metal-Organic Analogue of Face-Centered Cubic Dodecaboride, J. Am. Chem. Soc., 2008, 130, 800–801 CrossRef CAS PubMed.
- H. Chun, H. Jung and J. Seo, Isoreticular Metal-Organic Polyhedral Networks Based on 5-Connecting Paddlewheel Motifs, Inorg. Chem., 2009, 48, 2043–2047 CrossRef CAS PubMed.
- H.-N. Wang, X. Meng, G.-S. Yang, X.-L. Wang, K.-Z. Shao, Z.-M. Su and C.-G. Wang, Stepwise assembly of metal–organic framework based on a metal–organic polyhedron precursor for drug delivery, Chem. Commun., 2011, 47, 7128–7130 RSC.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, A pressure-amplifying framework material with negative gas adsorption transitions, Nature, 2016, 532, 348 CrossRef CAS PubMed.
- J. D. Evans, S. Krause, S. Kaskel, M. B. Sweatman and L. Sarkisov, Exploring the thermodynamic criteria for responsive adsorption processes, Chem. Sci., 2019, 10, 5011–5017 RSC.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Self-assembly of metal–organic polyhedra into supramolecular polymers with intrinsic microporosity, Nat. Commun., 2018, 9, 2506 CrossRef PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, V. Guillerm, K. Urayama, D. Maspoch and S. Furukawa, A Coordinative Solubilizer Method to Fabricate Soft Porous Materials from Insoluble Metal–Organic Polyhedra, Angew. Chem., 2019, 131, 6413–6416 CrossRef.
- X.-L. Lv, S. Yuan, L.-H. Xie, H. F. Darke, Y. Chen, T. He, C. Dong, B. Wang, Y.-Z. Zhang, J.-R. Li and H.-C. Zhou, Ligand Rigidification for Enhancing the Stability of Metal–Organic Frameworks, J. Am. Chem. Soc., 2019, 141, 10283–10293 CrossRef CAS.
- J. Liu, W. Duan, J. Song, X. Guo, Z. Wang, X. Shi, J. Liang, J. Wang, P. Cheng, Y. Chen, M. J. Zaworotko and Z. Zhang, Self-Healing Hyper-Cross-Linked Metal–Organic Polyhedra (HCMOPs) Membranes with Antimicrobial Activity and Highly Selective Separation Properties, J. Am. Chem. Soc., 2019, 141, 12064–12070 CrossRef CAS.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carné-Sánchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Rhodium–Organic Cuboctahedra as Porous Solids with Strong Binding Sites, Inorg. Chem., 2016, 55, 10843–10846 CrossRef CAS PubMed.
- M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, The Reticular Chemistry Structure Resource (RCSR) Database of, and Symbols for Crystal Nets, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- S. Plimpton, Fast parallel algorithms for short-range molecular dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- P. G. Boyd, S. M. Moosavi, M. Witman and B. Smit, Force-Field Prediction of Materials Properties in Metal-Organic Frameworks, J. Phys. Chem. Lett., 2017, 8, 357–363 CrossRef CAS PubMed.
- N. C. Burtch, J. Heinen, T. D. Bennett, D. Dubbeldam and M. D. Allendorf, Mechanical Properties in Metal–Organic Frameworks: Emerging Opportunities and Challenges for Device Functionality and Technological Applications, Adv. Mater., 2018, 30, 1704124 CrossRef.
- F. D. Murnaghan, The Compressibility of Media under Extreme Pressures, Proc. Natl. Acad. Sci. U. S. A., 1944, 30, 244–247 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- H. Sidky and J. K. Whitmer, Learning free energy landscapes using artificial neural networks, J. Chem. Phys., 2018, 148, 104111 CrossRef.
- H. Sidky, Y. J. Colón, J. Helfferich, B. J. Sikora, C. Bezik, W. Chu, F. Giberti, A. Z. Guo, X. Jiang, J. Lequieu, J. Li, J. Moller, M. J. Quevillon, M. Rahimi, H. Ramezani-Dakhel, V. S. Rathee, D. R. Reid, E. Sevgen, V. Thapar, M. A. Webb, J. K. Whitmer and J. J. de Pablo, SSAGES: Software Suite for Advanced General Ensemble Simulations, J. Chem. Phys., 2018, 148, 044104 CrossRef PubMed.
- J. C. Tan, T. D. Bennett and A. K. Cheetham, Chemical structure, network topology, and porosity effects on the mechanical properties of Zeolitic Imidazolate Frameworks, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9938–9943 CrossRef CAS PubMed.
- S. M. Moosavi, P. G. Boyd, L. Sarkisov and B. Smit, Improving the Mechanical Stability of Metal–Organic Frameworks Using Chemical Caryatids, ACS Cent. Sci., 2018, 4, 832–839 CrossRef CAS PubMed.
- P. Z. Moghadam, S. M. J. Rogge, A. Li, C.-M. Chow, J. Wieme, N. Moharrami, M. Aragones-Anglada, G. Conduit, D. A. Gomez-Gualdron, V. Van Speybroeck and D. Fairen-Jimenez, Structure-Mechanical Stability Relations of Metal-Organic Frameworks via Machine Learning, Matter, 2019, 1, 219–234 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00117d |
| This journal is © The Royal Society of Chemistry 2020 |