
Understanding the role of linker flexibility in soft porous coordination polymers†
 *a
and
Shuhei
Furukawa
b
*a
and
Shuhei
Furukawa
b
Abstract
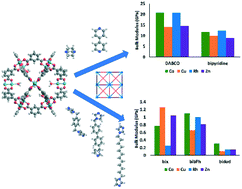
Soft porous coordination polymers are a new class of materials that have the potential to combine favorable features of metal–organic polyhedra (MOP) and frameworks (MOFs) with those of soft materials: permanent porosity and chemical specificity with processability and optical, electronic, and mechanical responses. Fundamental studies are needed to guide the future technological applications of these materials. We employ topologically-based algorithms to generate crystalline structures in the fcu topology and use classical molecular modeling to calculate their mechanical properties. The generated crystals used dimetal paddlewheel based MOP with cuboctahedral topology [M2(bdc)2]12 (M = Co, Cu, Rh, and Zn; bdc = 1,3-benzenedicarboxylate) as the nodes and 1,4-diazabicyclo[2.2.2]octane (DABCO), bipyridine, 1,4-bis(imidazol-1-ylmethyl)benzene (bix), 4,4′-bis(imidazol-1-ylmethyl)biphenyl (bibPh), and 1,1′-(1,12-dodecanediyl)bis[1H-imidazole] (bidod) as the linkers. Structures containing DABCO or bipyridine are found to have bulk moduli approximately an order of magnitude higher than those containing bix, bibPh, or bidod. The differences in mechanical properties are associated with the linker flexibility, as evidenced by free energy calculations on the radius of gyration of the linkers. We also calculate free energies as a function of radius of gyration of chosen nanoparticles.
- This article is part of the themed collection: MSDE Emerging Investigators 2020
 Please wait while we load your content...
Please wait while we load your content...

