
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigations of the generality of quaternary ammonium salts as alkylating agents in direct C–H alkylation reactions: solid alternatives for gaseous olefins†
David
Schönbauer
 a,
Manuel
Spettel
a,
Robert
Pollice
ab,
Ernst
Pittenauer
c and
Michael
Schnürch
*a
a,
Manuel
Spettel
a,
Robert
Pollice
ab,
Ernst
Pittenauer
c and
Michael
Schnürch
*a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, 1060 Wien, Austria. E-mail: michael.schnuerch@tuwien.ac.at
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1-5/10, 8093 Zürich, Switzerland
cInstitute of Chemical Technologies and Analytics, TU Wien, Getreidemarkt 9/164, 1060 Wien, Austria
First published on 28th March 2019
Abstract
C–H alkylation reactions using short chain olefins as alkylating agents could be operationally simplified on the lab scale by using quaternary ammonium salts as precursors for these gaseous reagents: Hofmann elimination delivers in situ the desired alkenes with the advantage that the alkene concentration in the liquid phase is high. In case a catalytic system did not tolerate the conditions for Hofmann elimination, a very simple spatial separation of both reactions, Hofmann elimination and direct alkylation, was achieved to circumvent possible side reactions or catalyst deactivation. Additionally, the truly catalytically active species of a rhodium(I) mediated alkylation reaction could be identified by using this approach.
Introduction
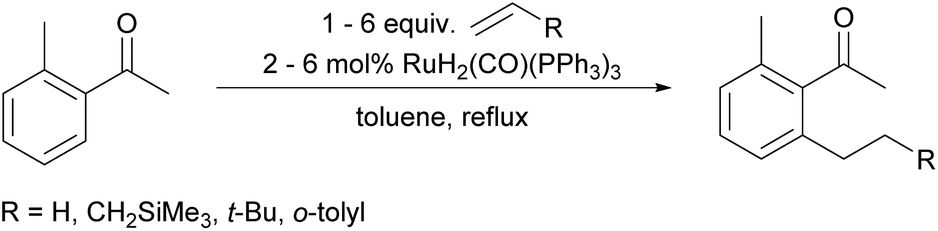
In recent years, the direct functionalization of C–H bonds has established itself as an increasingly important method for the formation of new C–C bonds.1–6 A key contribution to this field was the acetyl directed alkylation of aromatic C–H bonds disclosed by Murai in 1993![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 (Scheme 1). This paper inspired much subsequent research and can be considered as the starting point of directing group (DG) assisted C–H activation chemistry.8,9
7 (Scheme 1). This paper inspired much subsequent research and can be considered as the starting point of directing group (DG) assisted C–H activation chemistry.8,9
 | ||
| Scheme 1 Murai reaction. | ||
Among the many transformations known in metal-catalyzed direct C–H functionalization, alkylation reactions have received specific attention10–12 and have been the focus of interest of our research in recent times as well.13–16 Generally, either alkyl halides17–24 or olefins10,25,26 are used as alkylating agents. Usually, the former are attached to the catalyst by oxidative addition and coupled with the substrate. The catalyst is re-entering the catalytic cycle upon reductive elimination and the product is released (Cycle I in Scheme 2).27 In this process, alkyl halides are reduced, and an acid equivalent is generated, which is usually quenched by the addition of a base. On the other hand, the alkylation of aromatic C–H bonds with olefins includes usually a hydroarylation step of the olefin and hence no acid equivalent is formed (Cycle II).
 | ||
| Scheme 2 Schematic mechanisms for alkylation reactions with alkyl halides (Cycle I) or olefins as reagents (Cycle II). | ||
Consequently, these reactions are typically carried out in the absence of a base.10 It has to be mentioned that the application of short chain olefins is clearly underrepresented,28–30 which is certainly due to their gaseous nature and the problems of dosing gases on the nowadays typically applied mmol or sub-mmol scale. Additionally, the need for using high- or at least medium-pressure equipment is another factor contributing to the scarcity of examples with gaseous olefins. Hence, only liquid olefins (typically starting from hex-1-ene) are used,10 or polar groups are incorporated in the olefin as in acrylates and derivatives thereof. The simplest alternatives would be alkyl halides,17–24 since they are typically liquid. However, these reagents also have drawbacks because they are environmentally problematic, toxic or carcinogenic and therefore not an attractive substitute. Due to these complications, it would be ideal to find alternative solid reagents for short linear olefins. They should be cheap, bench-stable and therefore easy to handle and hence usable for small-scale synthesis (up to ∼1 mmol) in academia or also in pharmaceutical industry for instance during an initial library synthesis for subsequent screening of potential drug candidates. Within this contribution, our efforts in this direction are reported.
Results and discussion
Benzylic amines and catalytically active species
Recently, we showed that these requirements were met with tetraalkylammonium salts as surrogates for olefins.15 Upon addition of potassium hydroxide to the reaction mixture it was possible to generate the corresponding olefin in situ using Hofmann elimination.31 During that work, we demonstrated ethylation and other n-alkylations (up to n-octylation) of N-benzyl-2-aminopyridines (1) via C–H activation using the corresponding quaternary ammonium salts and with moderate to good yields. A recent contribution by Chatani and coworkers used PhMe3NI as the alkylating agent for aromatic C–H bonds as well.32 Since quaternary ammonium salts are typically prepared from the corresponding alkyl halides, we also investigated a protocol which tried to use the alkyl halides directly as olefin precursors.16 Also this transformation could be realized successfully and Scheme 3 gives a comparison between the two methods. Yields are usually 5–10% lower (Scheme 1, products 3–7) for the alkyl halide protocol but only for ethylation towards 2, a major difference between the reaction with ethyl bromide (25%) and NEt4Br (68%) was observed. It has to be mentioned that the latter reaction also gave 63% yield if the reaction was performed in air, showing that inert conditions or even handling of the reaction in a glovebox are unnecessary.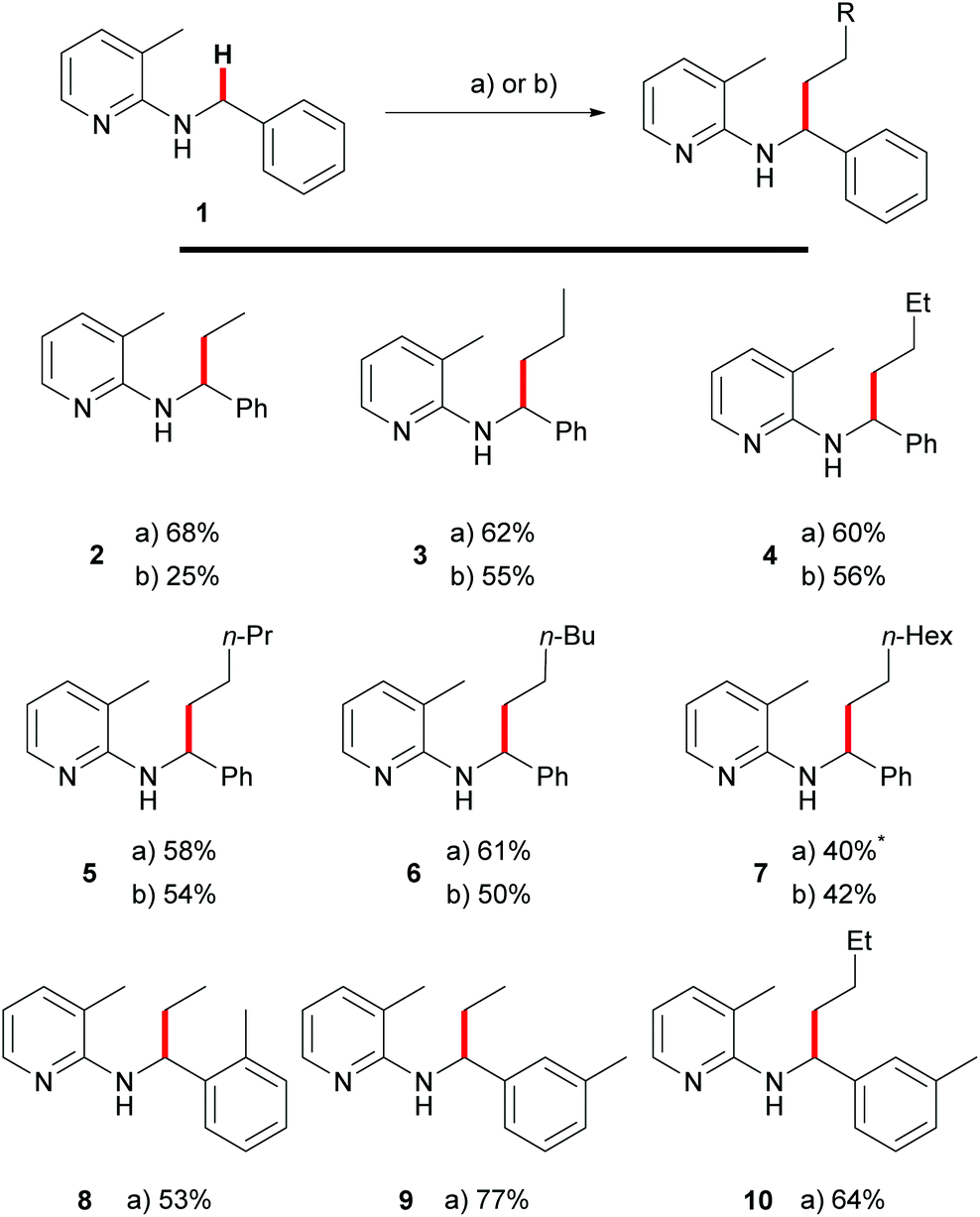 | ||
| Scheme 3 Comparison of alkylation with quaternary ammonium salt (a) or alkyl bromides as alkylating agents (b); (a) 1.0 equiv. quaternary ammonium salt, 3.0 equiv. KOH, 5 mol% [Rh(cod)Cl]2, toluene, 140 °C, overnight; (b) 3.0 equiv. alkyl bromide, 4.5 equiv. K2CO3, 5 mol% [Rh(cod)Cl]2, toluene, 160 °C, overnight; reactions were conducted on a 50 mmol scale. *84 h reaction time. | ||
Both protocols rely on the efficient generation of olefins by elimination, which is the reason for the elevated temperature employed for alkyl bromides (160 °C compared to 140 °C). In this case, the elimination requires not only higher temperatures, but also the presence of the rhodium catalyst, which was shown by control experiments (1-bromododecane or 1-bromo-2-phenylethane was heated together with K2CO3 in toluene with or without the presence of [Rh(cod)Cl]2. The corresponding elimination products, dodec-1-ene resp. styrene, were only detected by GC/MS when the catalyst was present) in our previous publication.13
Both protocols require the addition of a base to either neutralize the formed acid (alkyl halide protocol) or to enable efficient Hofmann elimination. Hence, it was expected that when using hex-1-ene instead of 1-bromohexane as the alkylating agent, the base can be avoided since this is an overall neutral transformation. However, we found that this is not the case and a base is also necessary, when using an olefin directly.13 The curious reason for this was identified when the catalytically active species were identified, which is described in the next paragraphs.
The need for three equivalents of KOH to facilitate Hofmann elimination has to be considered as a limitation for a broad applicability, due to incompatibility with certain catalytic systems since the Hofmann elimination and the alkylation occurred in a one-pot setup. Hence, we were searching for a possibility of overcoming this issue and the easiest way would be a spatial separation of olefin formation and the C–C bond forming reaction, where in one reaction compartment the gaseous olefin is produced, whereas on the other side the C–H activation reaction can take place. Such a spatial separation between the formation of a gaseous reagent and another synthetic transformation was already developed by Skrydstrup, in the context of carbonylation reactions.33–35 As a first test system we used again our model substrate 1. In chamber one of a so-called COware vial, the substrate and [Rh(cod)Cl]2 were placed, whereas the second chamber contained NEt4Br and KOH (Scheme 4). Interestingly, there was no conversion whatsoever in this experiment.
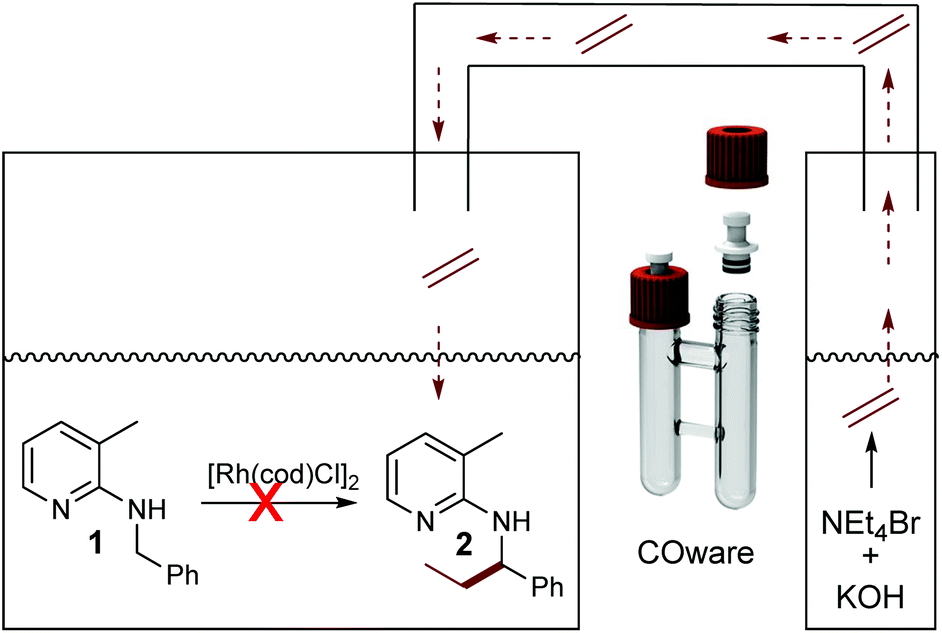 | ||
| Scheme 4 Spatial separation of Hofmann elimination and alkylation. | ||
In light of our previous efforts to determine the catalytically active species,14 this result was not totally surprising to us. There, we could associate a first order dependence for the addition of an inorganic base and an induction period in which we hypothesized that the base (in this study K2CO3) reacts with the catalyst to form the catalytically active species.14 In the course of this study some filtration experiments were conducted. The most important ones were the following: catalyst, K2CO3 and toluene were heated to 150 °C; the solids (basically K2CO3) were filtered and substrate 1 and hex-1-ene were added to the filtrate. It was found that in this reaction solution the alkylation towards 6 proceeded with essentially the same rate as compared to an experiment without filtration of solids (Scheme 5). This result shows that the presence of K2CO3 is only necessary during the first few minutes of the reaction in the formation of the catalytically active species and does not need to interact with the olefin or substrate.
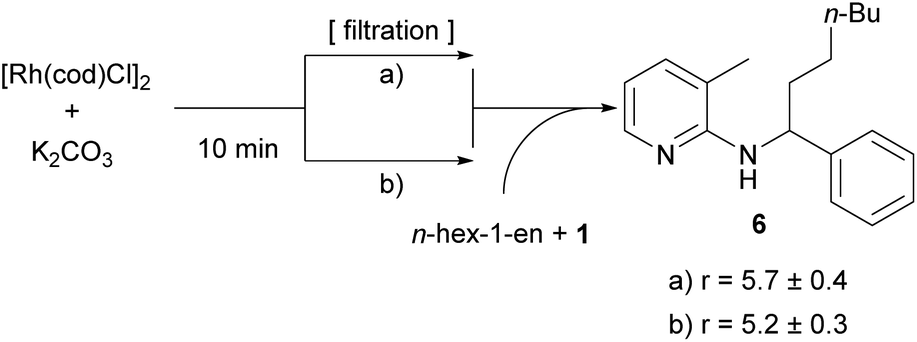 | ||
| Scheme 5 Filtration experiments: 5 mol% [Rh(cod)Cl]2, and 3.0 equiv. K2CO3 were prestirred in toluene at 150 °C for 10 min. Then, in experiment (a) the solids were removed by filtration and substrate 1 (1 equiv.) and hex-1-ene (3.0 equiv.) were added; in experiment (b), the same prestirring conditions were used and also the same amount of reagents was added but without an intermediate filtration step. Rates were determined by GC analysis of three individual runs with dodecane as an internal standard and are given in [10−5 mol L−1 s−1]. | ||
Evaporation of the filtrate before adding a substrate or olefin gave a solid, which showed absorption at 1572.6 cm−1 in ATR-IR measurements. This could be interpreted as a carbonato species, since similar values were reported for such rhodium complexes (1629 cm−1 for Cp*Rh(μ-O)2CO36 and 1586 cm−1 for Ru(NHC)2(CO)2(CO3)37) which we proposed to be the most likely catalytically active species. However, at this point we also could not rule out a rhodium hydroxide species formed from the deprotonation of H2O introduced with K2CO3.14 Unfortunately, it was not possible to confirm the identity of the obtained material by X-ray crystallography, since no crystalline material could be obtained. Now, in light of the result that our Hofmann elimination protocol is carried out in the presence of KOH and not K2CO3, a rhodium carbonato species can be ruled out. These new circumstances motivated us to investigate the nature of the catalytically active species again and we subjected the solids from filtration experiments to MALDI-MS analysis.
Interestingly, [Rh(cod)Cl]2 was not detected even in trace amounts and the only rhodium species found was [Rh(cod)(OH)]2 (for details see the ESI†). The origin of the OH groups is the adsorbed water on K2CO3, which also explains our previous finding that the reaction rate drops with the water content of K2CO3.14 In the quaternary ammonium salt protocol, KOH can naturally serve as the OH− source. This strongly suggests that this hydroxido species is the catalytically active one, which could be confirmed by simple control experiments using hex-1-ene as the olefin (Scheme 6): when [Rh(cod)(OH)]2 was used without the addition of a base, the reaction proceeded even slightly better than with the combination of [Rh(cod)Cl]2 and K2CO3.
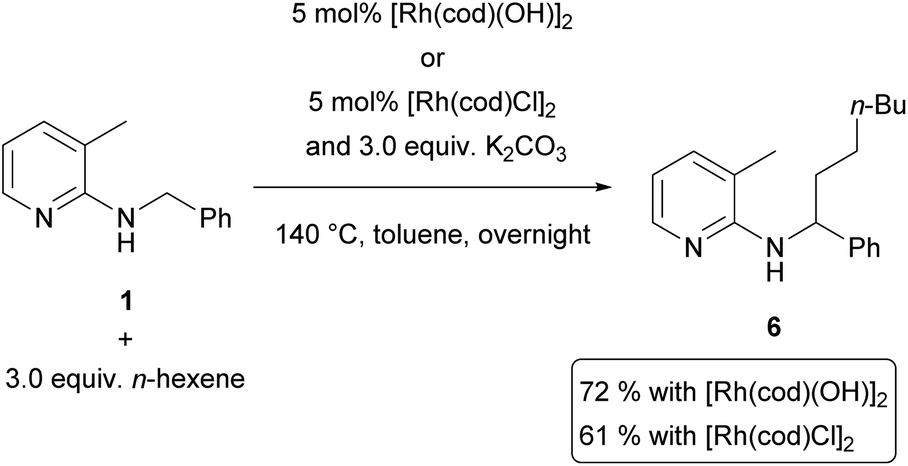 | ||
| Scheme 6 Control experiments using different rhodium catalysts. Reactions were performed on a 50 mmol scale. | ||
Having established [Rh(cod)(OH)]2 as catalytically active species when olefins are used as alkylating agents, this had to be confirmed for the Hofmann elimination protocol as well. Hence, we carried out this method with either [Rh(cod)Cl]2 or [Rh(cod)(OH)]2 and compared the kinetic profile (Fig. 1). It was found that when the hydroxido dimer was used as a catalyst, the reaction proceeds slightly faster, indicating the absence of an induction period resulting from the initial formation of the catalytically active species from [Rh(cod)Cl]2. At the end of the reaction time, the overall conversion reaches the same value of 80% after 3 hours (for [Rh(cod)(OH)]2 even after 90 minutes).
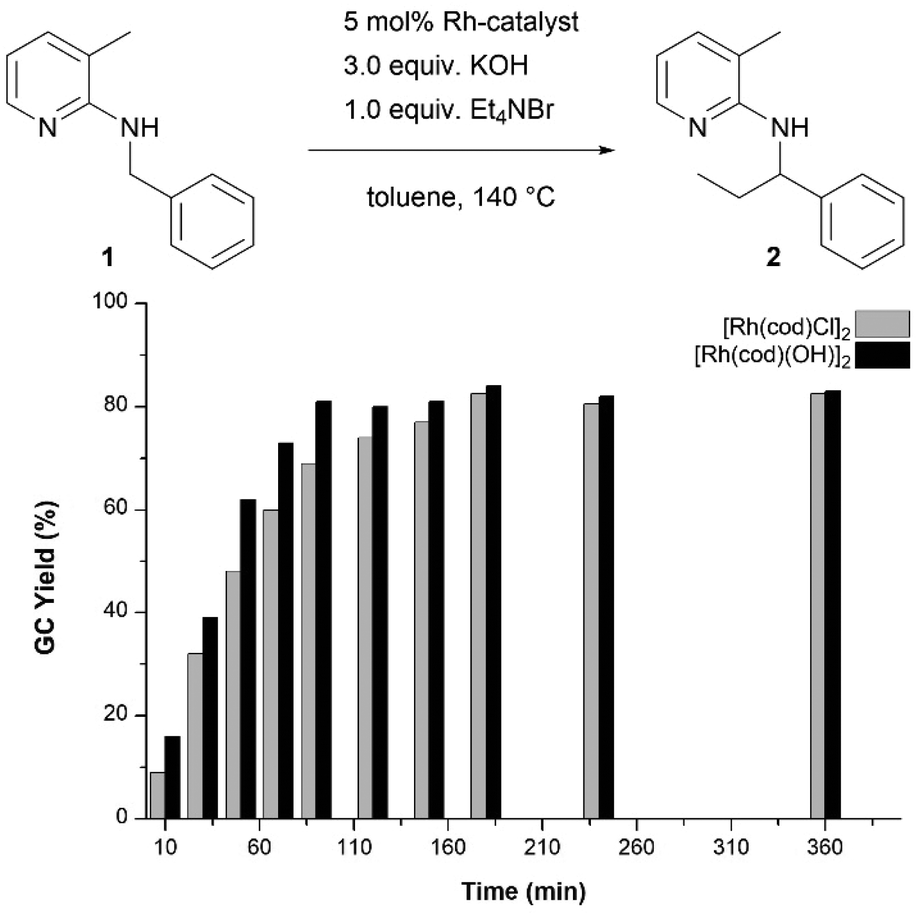 | ||
| Fig. 1 Kinetic profile and comparison of the performance between [Rh(cod)Cl]2 and [Rh(cod)(OH)]2 as catalysts in the ethylation reaction of 1. GC-yields were determined with dodecane as the internal standard. | ||
Now, the two-chamber approach via COware vials was revisited. This time, as indicated in Scheme 7, we charged chamber A with [Rh(cod)(OH)]2 and substrate and chamber B with KOH and ammonium salt. Contrary to the analogous experiment with [Rh(cod)Cl]2 as a catalyst, the reaction worked well, and 65% of 2 was isolated.
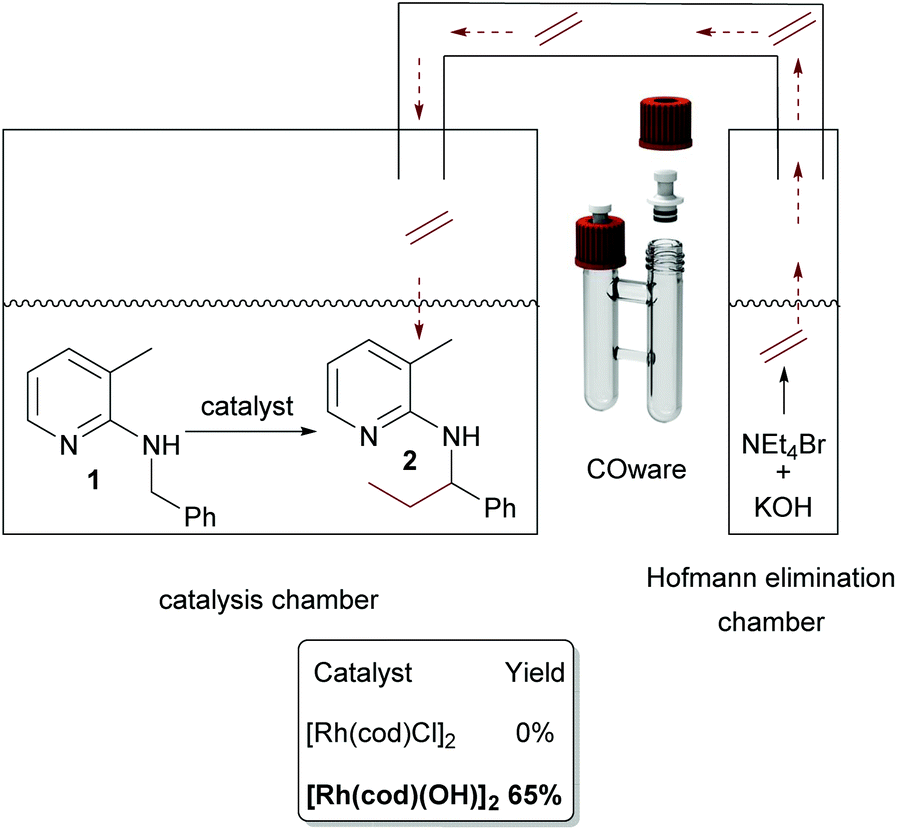 | ||
| Scheme 7 Finding of catalytically active species. Chamber A: Substrate (0.5 mmol), 5 mol% catalyst, 2.0 mL toluene; chamber B. 2.0 equiv. Et4NBr, 6.0 equiv. KOH, 2.0 mL toluene. Reaction was run at 140 °C overnight. | ||
To this point, we have demonstrated the possibility of substituting olefins with alkyl bromides or quaternary ammonium salts in direct alkylation reactions on a model substrate. Additionally, we also identified the catalytically active species in these reactions and showed that the Hofmann elimination step can be separated from the alkylation reaction, opening new possibilities regarding the catalyst systems to be applied in combination with our Hofmann elimination/alkylation method. The next task was to show that this method can be applied in a more general manner to other substrates as well.
Investigating the generality of the Hofmann elimination/alkylation protocol
We already demonstrated the possibility of using tetraethyl ammonium bromide as the ethylene precursor in single examples on two other systems with different catalysts, showing that not only [Rh(cod)(OH)]2 can be used, but also other rhodium and also ruthenium catalysts (see Scheme 8A (ref. 7) and Scheme 8B (ref. 38)).15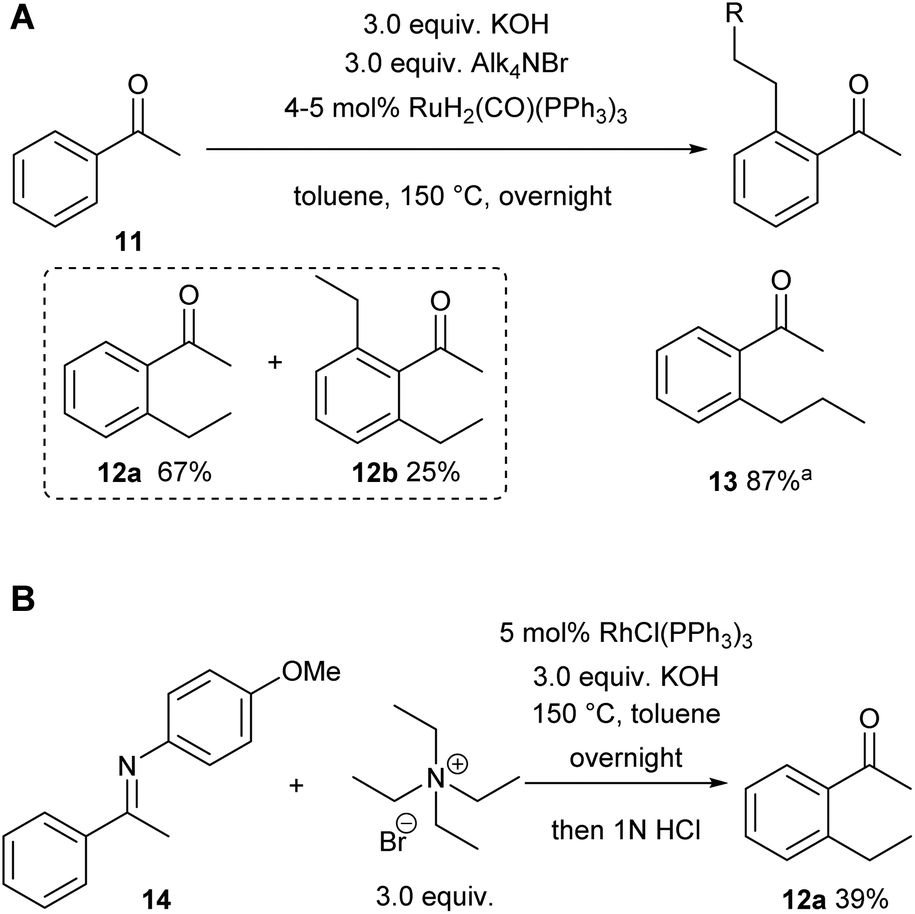 | ||
| Scheme 8 A: Alkylation with acetophenone as a substrate; 0.5 mmol scale, a9.0 equiv. KOH were used. Yield was determined out of the mixture of substrate and product. B: Imine-directed C–H alkylation reaction with tetraethylammonium bromide as an alkyl source. | ||
A natural first choice for further application was the famous Murai reaction.7 When we used acetophenone as a substrate we observed almost full conversion to a mixture of mono- (67%) and bis-ethylated (25%) product when using 3.0 equivalents of ammonium salt and base in the one-pot approach (Scheme 8A). However, when we tried to broaden the scope towards longer chain lengths a significant drop in conversion was observed under the same conditions: only 60% conversion was reached when nPr4NBr was used as a propylene precursor. Prolonged reaction times and/or higher catalyst loadings could not improve the yields. Only when a substantial excess of base was added (9.0 equiv.), the reaction commenced to almost full conversion, giving the desired product 13 now in excellent 87% yield (Scheme 8A). In this case, only small amounts of the bis-alkylated product were detected in GC/MS, indicating that mono-selectivity is brought about when longer chain lengths are introduced.
On further increasing the chain length to n-butyl, the conversion was diminished to only 40% and isolation of the product was not possible anymore due to additional by-product formation. Apparently, the ammonium salt is not the only species reacting with OH– to induce Hofmann elimination, but also the enolate of the substrate is generated forming product 15 (detected by GC/MS) upon reaction with the ammonium salt as indicated in Scheme 9.
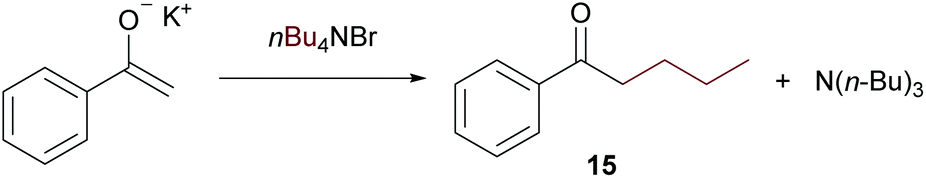 | ||
| Scheme 9 Possible side-product formation during the modified Murai–Chatani–Kakiuchi reaction. | ||
The aforementioned two chamber approach would clearly avoid this problem. However, even when applied with the more reactive propene surrogate, the conversion was lower compared to the one-pot protocol (60% for 9.0 equiv. KOH and 3.0 equiv. ammonium salt). Still, GC/MS analysis showed an essentially complete absence of side products.
The fact that the conversion decreases with increasing chain length of the ammonium salt and that the elimination to the alkene gets slower with longer chain lengths39 facilitates the occurrence of side reactions induced by the presence of KOH. Hence, additional base cannot completely solve this problem in this specific case, since it increases not only the rate of Hofmann elimination, but also of by-product formation. Why the two-chamber approach does not improve the butylation reaction significantly might be explained by a too low concentration of olefin in the solution phase, since in the originally published contribution, high amounts of unactivated olefins (5.0 equiv.) also were necessary for full conversion of the material. In such cases, a pressurized reaction with olefin gas might be the better choice since alternatively large quantities of solid quaternary ammonium salts have to be applied.7
The next substrate to be tested was 16 carrying a dihydrooxazole as a directing group. Initially, again the one-pot protocol was investigated, since it is the operationally simpler one. Originally, this transformation was published with Ru3CO12 as a catalyst and 7 atm of ethylene yielding 53% product and 20% of a CO containing by-product.40 However, in our hands Ru3CO12 proved to be unreactive towards alkylation either in a one-pot fashion or in COware vials. Hence, we tested again [Rh(cod)Cl]2 as a catalyst and now ethylation was achieved giving 17 in 29% yield (Scheme 10, 40% starting material was recovered, →48% based on the recovered starting material).
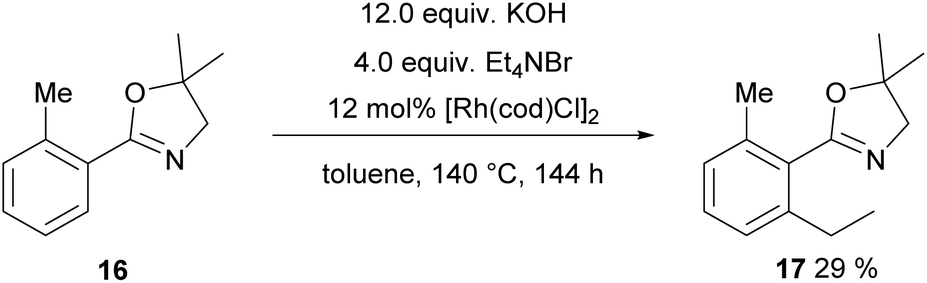 | ||
| Scheme 10 Ethylation with a dihydroxazole directing group. | ||
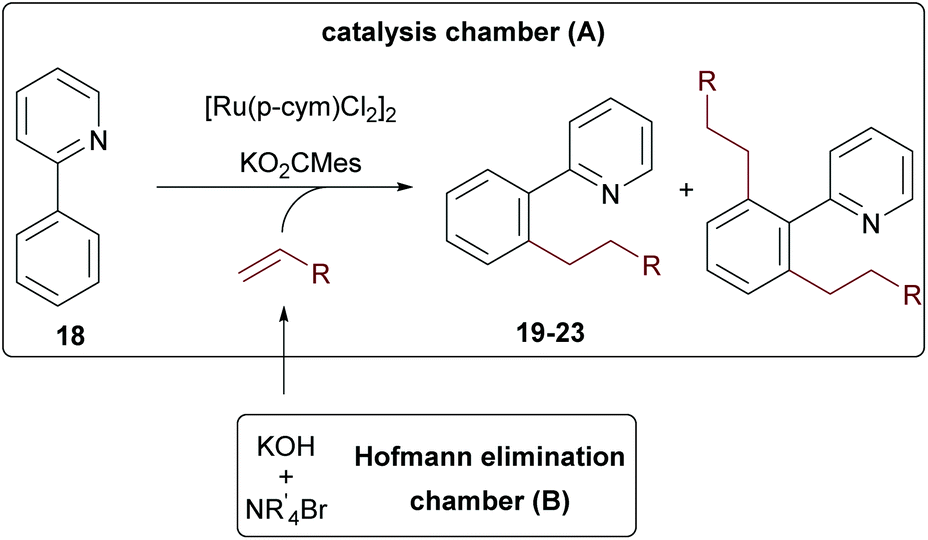
As final example, 2-phenylpyridine 18 was tested, since this substrate is often used as a model system for developing new C–H activation reactions.41–48 Intrigued by the reported high yields and mono-selectivity, we probed an alkylation protocol with terminal olefins initially published by the group of Ackermann.49
First, the reaction was tested with tetraethyl ammonium bromide in a one-pot approach. Here a maximum conversion of 80% was achieved according to GC/MS. When ammonium salts with longer alkyl chains were used, the desired products were only formed in trace amounts, indicating again a special role for ethylene. These findings motivated us to probe the reaction in the two-chamber reactor. Gratifyingly, this solved the problem and all reactions performed with good conversion (>80%) for all tested ammonium salts (see Table 1). A direct comparison to the Ackermann results is only possible for hexylation, since no shorter olefins were used in this contribution due to the already mentioned issues with the physical properties of shorter olefins. Our yield for the n-hexylated product 23 was comparable to the one achieved in the literature with the olefin with 72% of 23 in our protocol and 78% in the literature (see entry 5).49 The yields remained essentially the same when shorter olefin surrogates were used (entry 3, 21, n-butyl 75% and entry 4, 22, n-pentyl 71%). However, when Et4NBr and n-Pr4NBr were used, the bis-alkylated (19b and 20b) products were also formed. While for the propyl-derivative only 4% side product 20b was generated (entry 2), the bis-ethylated product 19b was produced in almost the same amounts as the desired product 19a (entry 1). This interesting effect suggests that the excellent mono-selectivity of the original protocol (no formation of any bis-alkylation products) is not only determined by the in situ formed sterically demanding ruthenium complex, but also by the size of the olefin. It can be expected that this specific example will not be unique in this regard.
Conclusions
In summary, it was shown that tetraalkylammonium salts can be used as solid surrogates for simple unactivated alkenes using Hofmann elimination for in situ formation of olefins. The method is especially useful for substituting olefins with up to five carbons, since these olefins are either gaseous or have very low boiling points and, hence, are difficult to handle and dose on a small scale. Even though Hofmann elimination requires excess of KOH, the protocol could also be applied for catalytic systems incompatible with strongly basic conditions. Here spatial separation via a two-vessel approach can solve the problem and delivers similar yields to the initial literature precedences using olefins. Furthermore, it was shown that the alkylation selectivity between alkenes and 2-phenylpyridine is not only depending on the catalyst and its additives, but also on the chain-length of the participating olefin: in situ formed ethylene and propylene gave mixtures of mono- and bis-alkylation products, whereas from butylene on the reaction was mono-selective. Furthermore, the two-vessel approach also allowed the identification of the catalytically active species of the originally investigated alkylation reaction of N-benzyl-2-aminopyridines, by showing that [Rh(cod)Cl]2 is catalytically inactive and needs to be transformed to [Rh(cod)(OH)]2 first. Efforts for further generalization of our protocol are ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Roudesly, J. Oble and G. Poli, J. Mol. Catal. A: Chem., 2017, 426, 275–296 CrossRef CAS.
- J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS PubMed.
- J. Wencel-Delord, F. W. Patureau and F. Glorius, Top. Organomet. Chem., 2016, 55, 1–27 CAS.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- J. Q. Yu, Adv. Synth. Catal., 2014, 356, 1393–1393 CrossRef CAS.
- X. S. Xue, P. Ji, B. Zhou and J. P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- Z. K. Chen, B. J. Wang, J. T. Zhang, W. L. Yu, Z. X. Liu and Y. H. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- F. Kakiuchi, Top. Organomet. Chem., 2007, 24, 1–33 CrossRef CAS.
- C. Bruneau and P. H. Dixneuf, Top. Organomet. Chem., 2016, 55, 137–188 CrossRef CAS.
- R. Pollice, N. Dastbaravardeh, N. Marquise, M. D. Mihovilovic and M. Schnürch, ACS Catal., 2015, 5, 587–595 CrossRef CAS PubMed.
- R. Pollice and M. Schnürch, J. Org. Chem., 2015, 80, 8268–8274 CrossRef CAS PubMed.
- M. Spettel, R. Pollice and M. Schnürch, Org. Lett., 2017, 19, 4287–4290 CrossRef CAS PubMed.
- M. Anschuber, R. Pollice and M. Schnurch, Monatsh. Chem., 2019, 150, 127–138 CrossRef PubMed.
- R. Y. Zhu, J. He, X. C. Wang and J. Q. Yu, J. Am. Chem. Soc., 2014, 136, 13194–13197 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 9279–9282 CrossRef CAS PubMed.
- B. Xiao, Z. J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 616–619 CrossRef CAS PubMed.
- S. Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS PubMed.
- K. Chen and B. F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950–11954 CrossRef CAS PubMed.
- Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS PubMed.
- W. Song, S. Lackner and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 2477–2480 CrossRef CAS PubMed.
- B. M. Monks, E. R. Fruchey and S. P. Cook, Angew. Chem., Int. Ed., 2014, 53, 11065–11069 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- L. Ackermann, Chem. Commun., 2010, 46, 4866–4877 RSC.
- L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS.
- M. Lail, B. N. Arrowood and T. B. Gunnoe, J. Am. Chem. Soc., 2003, 125, 7506–7507 CrossRef CAS PubMed.
- S. A. Burgess, E. E. Joslin, T. B. Gunnoe, T. R. Cundari, M. Sabat and W. H. Myers, Chem. Sci., 2014, 5, 4355–4366 RSC.
- A. W. von Hofmann, Ann. Chem. Pharm., 1851, 78, 253–286 CrossRef.
- T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162–3165 CrossRef CAS PubMed.
- S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594–605 CrossRef CAS PubMed.
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS PubMed.
- P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061–6071 CrossRef CAS PubMed.
- W. G. Jia, Y. F. Han and G. X. Jin, Organometallics, 2008, 27, 6035–6038 CrossRef CAS.
- C. E. Ellul, O. Saker, M. F. Mahon, D. C. Apperley and M. K. Whittlesey, Organometallics, 2008, 27, 100–108 CrossRef CAS.
- C. H. Jun, J. B. Hong, Y. H. Kim and K. Y. Chung, Angew. Chem., Int. Ed., 2000, 39, 3440–3442 CrossRef CAS PubMed.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- F. Kakiuchi, T. Sato, M. Yamauchi, N. Chatani and S. Murai, Chem. Lett., 1999, 28, 19–20 CrossRef.
- J. H. Chu, S. L. Tsai and M. J. Wu, Synthesis, 2009, 3757–3764 CAS.
- B. Punji, W. Song, G. A. Shevchenko and L. Ackermann, Chem. – Eur. J., 2013, 19, 10605–10610 CrossRef CAS PubMed.
- M. E. Tauchert, C. D. Incarvito, A. L. Rheingold, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 1482–1485 CrossRef CAS PubMed.
- D. Zhao, J. H. Kim, L. Stegemann, C. A. Strassert and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 4508–4511 CrossRef CAS PubMed.
- K. Parthasarathy, A. R. Azcargorta, Y. Cheng and C. Bolm, Org. Lett., 2014, 16, 2538–2541 CrossRef CAS PubMed.
- B. Zhou, J. Du, Y. Yang, H. Feng and Y. Li, Org. Lett., 2013, 15, 6302–6305 CrossRef CAS PubMed.
- A. Tlili, J. Schranck, J. Pospech, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 6293–6297 CrossRef CAS PubMed.
- S. L. Yedage and B. M. Bhanage, Synlett, 2015, 2161–2169 CAS.
- M. Schinkel, I. Marek and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 3977–3980 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and copies of spectra. See DOI: 10.1039/c9ob00243j |
| This journal is © The Royal Society of Chemistry 2019 |