Unveiling the role of water in the coupling of pyrroles and isocyanates to amidopyrroles inside a hexameric resorcinarene capsule
Received
6th May 2025
, Accepted 9th February 2026
First published on 11th February 2026
Abstract
DFT calculations uncovered the crucial role of non-structural water molecules trapped inside the cavity of a self-assembled hexameric resorcinarene capsule in the reaction between N-methylpyrrole and phenyl isocyanate to generate an amidopyrrole through two crucial steps: C–C coupling and a 1,3C → N proton wire mechanism. Incidentally, the reaction in the bulk solvent is energetically demanding and underscores the role of encapsulation. In fact, non-structural incorporation of water molecules modulates the acidity of the hexameric resorcinarene capsule through hydrogen-bonded networks. This results in a shift in the rate-determining transition state to the initial C–C coupling step inside the capsule, as opposed to the energy intensive proton-wire mechanism in solution. Thus, the incorporated water molecules within the supramolecular cavity helps in accelerating the overall rate of the acid-catalyzed transformation. Our work, thus, indicates the advantage of the ubiquitous presence of water-enriched local domains in providing utmost control to the supramolecule in the catalytic process. These local hydrophilic domains with water clusters, encapsulated within organic molecules, emulate the kinetics, selectivity and mass transfer observed in some natural processes. The enhanced catalytic activity is primarily attributed to water molecule’s ability to stabilize the reactants, products, intermediates or transition states through remote bond polarization, or proton shuttling, or ability of water to act as a co-catalyst.
Introduction
Supramolecular self-assembled capsules and cages are fascinating chemical discoveries that allow well-defined control over the stereo- and regio-selective outcomes of the chemical processes occurring inside their host cavities.1 These simple yet architecturally complex assemblies possess unique shapes and rigid frameworks to host diverse substrates of various sizes, with specific recognition of guest molecules, in a solvent-free microenvironment. Interestingly, the trapped molecules display modified chemical properties compared with those in the bulk solvent due to the pre-organization of substrates to a specific conformation within the supramolecular cavities, which leads to the desired chemical conversions.2 Notably, chemical transformations within the confined chambers of supramolecules draw inspiration from the active site of enzymes (Scheme 1),2b where the substrates are surrounded by the enzymatic pocket and the reactions are controlled by the collective effort of the optimum fitting of the substrate and non-covalent interactions exerted by the secondary coordination spheres,3 resulting in catalytic activity under mild conditions. Similarly, supramolecular capsules and cages accommodate guest molecules in a locally electrostatic and dispersion-dominated chemical environment,4 suppressing undesired alternative transformations in bulk solvent. Hence, these supramolecular catalysts offer atom-economic reactions and hold excellent promise as highly sustainable bio-mimicking materials for achieving accelerated reaction kinetics.5 However, a major limitation in reducing the gap between enzyme catalysis and biomimicking supramolecular catalysis for large-scale organic synthesis lies in the understanding of the principles for substrate activation in nanoconfined environments, where both steric effects and attractive van der Waals interactions are expected to intervene. The major challenge stems from the arduous identification and isolation of reactive intermediates during the reaction of host–guest complexes, by kinetic or dynamic NMR measurements, which are precluded by complex self-assembly and reversible encapsulation thermodynamics on the timescales of seconds6a or minutes.6b Hence, understanding the key mechanistic aspects would offer enzyme-like precise reactivity controls and the possibility of designing systems that can achieve high-throughput synthesis of value-added chemicals.
 |
| | Scheme 1 Amidopyrrole formation from pyrrole and isocyanate in the hexameric resorcinarene capsule.7b | |
Recently, several supramolecular research groups have unfolded key reactions utilizing the self-assembled hexameric resorcinarene capsule C (Scheme 1).7 Particularly interesting is the coupling of pyrroles and isocyanates to yield amidopyrroles in water-saturated chloroform in the mandatory presence of the capsule (Scheme 2), as reported by La Manna and co-workers in a recent study.7b This is extremely relevant in the context of druggable targets and inhibitory activities by molecular cage systems.8 Originally reported by Atwood and MacGillivray,9 capsule C in its solid state consists of a chiral spherical molecular assembly of six resorcinarene molecules and eight water molecules, held together by 60 hydrogen bonds. In an apolar solvent medium, the capsule features a well-defined cavity with an internal volume of about 1375 Å3 and elevated Brønsted acidity (pKa = 5.6 as referenced by protonation of pyridine)7d compared with the monomeric units due to the optimal existence of the H-bond network. Furthermore, a combined NMR spectroscopy and classical molecular dynamics (MD) simulation study identified two distinct conformers of the hexameric undecyl-resorcin[4]arene capsule (CA and CB), featuring 8 and 15 hydrogen-bonded water molecules.10,11 In fact, peripheral structural changes in the capsule effectuated by water molecules are believed to modulate the acidity of the supramolecular assembly, resulting in a 10-fold enhancement in the catalytic rate of an acid-catalysed Diels–Alder reaction by CB as compared to CA.10 Additionally, NMR diffusion studies detected several chloroform molecules inside the cavity of this superstructure, further adding layers of complexity to the self-assembly strategy.12 Indeed, the capsule is surmised to have a strong affinity towards zwitterionic tetrahexylammonium bromide over chloroform molecules, indicating a strongly electrostatic host environment.12 Hence, during self-assembly in H2O-saturated chloroform (Scheme 2), one cannot rule out the trapping of activated non-structural water molecules in addition to the structural water molecules in the H-bonded networks. This concept is in line with the discovery of isolated water droplets inside a Ga4L612− supramolecular tetrahedral cage employing THz absorption spectroscopy and ab initio molecular dynamics.13 Although mechanistic studies on cavity-controlled selectivity are reported in the literature,4,10,14 to date, an understanding of the role of trapped water molecules in reactivity is underexplored and warrants a thorough investigation. This, we believe, will open new channels to achieve highly efficient organic transformations in locally water-enriched media, as pervasive and regulated in Nature.
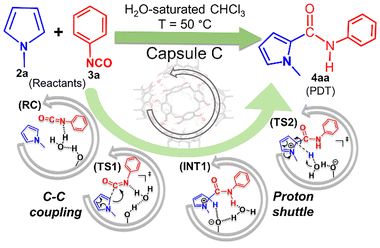 |
| | Scheme 2 Reaction conditions for the coupling of 2a and 3a to yield 4aa in the presence of the capsule C, with snapshots of the proposed reaction species. | |
In the present work, therefore, we undertake a DFT study to explore the binding of additional non-structural water molecules, which have greater geometrical flexibility than the water molecules in the hydrogen-bonded networks of capsule C, and unravel the mechanistic intricacies of the coupling between N-methylpyrrole (2a) and phenyl isocyanate (3a) (Scheme 2). We hypothesize two crucial steps: C–C bond formation through nucleophilic attack of the pyrrole on the carbonyl of the isocyanate and a 1,3C → N proton wire mechanism through acid–base catalysis (Scheme 2).
Computational details
Density functional theory calculations were performed within the Gaussian 09 and ORCA 5.0.3 suites of quantum chemical packages.15 To compensate for the high computational cost, the bulky undecyl alkyl group of the capsule (the so-called “feet”) was replaced with methyl substituents. Modelling the formation of amidopyrrole inside the capsule with an additional water molecule results in a system containing a total of 486 atoms. Geometry optimizations and identification of relevant stationary states of such a large system with DFT are impractical. Hence, an in silico investigation was conducted using Morokuma and co-workers’ ONIOM method,16 which builds upon the subtractive or extrapolative QM/MM formalism such that the total energy is given by eqn (1).| | | EONIOM(QM/MM) = EQM,model + EMM,real − EMM,model, | (1) |
i.e. the total energy of the whole (“real”) system is evaluated as the QM energy of the model system (EQM,model), in addition to the MM energy of the real system (EMM,real), minus the MM energy of the model system (EMM,model). In our approach, during geometry optimizations, the substrates 2a and 3a, together with one monomer of the cavitand, strongly interacting through H-bonds and the additional water molecule, comprising 102 atoms, were treated with DFT(M06-2X/6-31G(d,p)) and the rest of the supramolecular assembly by the semi-empirical PM6 method. This is a standard practice in some of the recent literature.7a,c Notably, in most of the earlier mechanistic studies with DFT, only two units of the host molecules were envisioned to form a capsule.4,14 Since conventional DFT calculations scale cubically (O(N3)), the computational cost for ∼500 atoms is enormous; thus, relatively cheaper alternatives are required for geometry optimizations and vibrational analysis. Hence, we utilized a two-layer ONIOM2 (QM1:QM2) method for optimizations of large systems with the M06-2X/6-31G(d,p)//PM6 level of theory (see Fig. S4), where the target energy of the real system, Ehigh,real, is given by16| | | Ehigh,real ≈ EONIOM2(high:low) = Ehigh,model + Elow,real – Elow,model. | (2) |
An S-value test was conducted to diagnose the accuracy of the low-level method (PM6) to describe the electronic effect or the “substituent effect” of the large “real” system (Table S5) as compared to the high-level method (M062X/6-31G(d,p)).16 A ΔS-value of ∼0 suggested that the low-level method PM6 can be appropriately used in combination with the present high-level method, M06-2X/6-31(d,p) (Table S5). Conformational searches were performed manually by evaluating several conformers of the minimum and maximum structures, and the lowest energy geometries are reported. However, in the absence of the capsule, i.e. in implicit solvent (S) and assisted by a resorcin-arene monomer (M), the optimizations were computed at the M06-2X/6-31G(d,p) level. The structures were always optimized in implicit solvent by employing the polarizable continuum model (CPCM) with chloroform (ε = 4.71) as the solvent. Harmonic vibrational frequencies were computed at the same level of theory to distinguish transition states (with one negative Hessian index) from minima (with all positive Hessian indices). The non-thermal zero-point energy (ZPE) correction and the thermal corrections to enthalpy and Gibbs free energy were obtained from frequency calculations, using standard approximations at 298.15 K and 1 atm. pressure. Moreover, the electronic energies were refined by single-point calculations with full QM(DFT) calculations over all atoms (including 486 atoms for the reactions inside capsule) in the solvent phase with the B3LYP functional,17 overlaid with Grimme's empirical dispersion (D3BJ) term,18 employing the IOP (3/124 = 40) keyword, in conjugation with the larger triple-ξ Def2-TZVP basis set on all atoms. To mimic the mixed solvent of water and chloroform at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio within the domain of the CPCM solvation model, the following dielectric parameters of water were used: static dielectric constant (ε = 78.35), dynamic dielectric constant (ε∞ = 1.77), hydrogen-bond acidity (1.17) and basicity (0.4), the surface tension of the solvent at interface (23.23), carbon aromaticity (0.0) and electronegative halogenicity (0.0), while the following parameters of chloroform were considered: static dielectric constant (ε = 4.7), dynamic dielectric constant (ε∞ = 2.09), hydrogen-bond acidity (0.15) and basicity (0.02), the surface tension of the solvent at interface (38.39), carbon aromaticity (0.0) and electronegative halogenicity (0.75), following Paton and co-workers’ methodology.19 Relative Gibbs free energies are evaluated by adding thermal corrections obtained from vibrational analyses to the solvent-phase electronic energies. Additionally, a correction term of 1.89 kcal mol−1 (at 298.15 K) was included as necessary to account for the standard-state concentration of 1 M, especially to account for the solvation of many species, including micro-solvated water molecules, taking part in the transition state.20 Thus, the discussions in the main text are provided based on Gibbs free energies (kcal mol−1) at the B3LYP-D3(BJ)/CPCM {H2O:CHCl3 = 1:1}/Def2-TZVP//ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6] level of theory. Furthermore, to evaluate the dependence on the DFT functional, energies are computed at B3LYP/CPCM {H2O:CHCl3 = 1:1}/Def2-TZVP//ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6] and M06-2X/CPCM {H2O:CHCl3 = 1:1}/Def2-TZVP//ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6] (see Table S2). To check for dependence on solvent dielectric constant, single-point energies are also conducted in chloroform as solvent with B3LYP-D3(BJ), M06-2X and B3LYP functionals (see Table S3).
:1 ratio within the domain of the CPCM solvation model, the following dielectric parameters of water were used: static dielectric constant (ε = 78.35), dynamic dielectric constant (ε∞ = 1.77), hydrogen-bond acidity (1.17) and basicity (0.4), the surface tension of the solvent at interface (23.23), carbon aromaticity (0.0) and electronegative halogenicity (0.0), while the following parameters of chloroform were considered: static dielectric constant (ε = 4.7), dynamic dielectric constant (ε∞ = 2.09), hydrogen-bond acidity (0.15) and basicity (0.02), the surface tension of the solvent at interface (38.39), carbon aromaticity (0.0) and electronegative halogenicity (0.75), following Paton and co-workers’ methodology.19 Relative Gibbs free energies are evaluated by adding thermal corrections obtained from vibrational analyses to the solvent-phase electronic energies. Additionally, a correction term of 1.89 kcal mol−1 (at 298.15 K) was included as necessary to account for the standard-state concentration of 1 M, especially to account for the solvation of many species, including micro-solvated water molecules, taking part in the transition state.20 Thus, the discussions in the main text are provided based on Gibbs free energies (kcal mol−1) at the B3LYP-D3(BJ)/CPCM {H2O:CHCl3 = 1:1}/Def2-TZVP//ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6] level of theory. Furthermore, to evaluate the dependence on the DFT functional, energies are computed at B3LYP/CPCM {H2O:CHCl3 = 1:1}/Def2-TZVP//ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6] and M06-2X/CPCM {H2O:CHCl3 = 1:1}/Def2-TZVP//ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6] (see Table S2). To check for dependence on solvent dielectric constant, single-point energies are also conducted in chloroform as solvent with B3LYP-D3(BJ), M06-2X and B3LYP functionals (see Table S3).
To verify the dependence of the low-level method on predicting the structures and thereby affecting the overall relative energies, we have conducted additional geometry optimizations at ONIOM[B3LYP-D3(BJ)/ALPB(CHCl3)/Def2-SVP:XTB] with ORCA 5.0.315b and further single-point energy refinements at the B3LYP-D3(BJ)/ALPB(CHCl3)/Def2-TZVP level of theory. For all geometry optimizations, the following thresholds were used: convergence tolerance of 5 × 10−6Eh for energy changes, 3 × 10−4Eh per Bohr for maximum gradients, 1 × 10−4Eh per Bohr for root-mean-square (RMS) gradients, 4 × 10−3 Bohr for maximum displacements, and 2 × 10−3 Bohr for RMS displacements. The numerical integration Grid6 was used throughout. Tight convergence criteria with an energy tolerance of 1 × 10−8Eh for self-consistent field (SCF) achievement were utilized for all ORCA computations.
To gather an in-depth analysis of the key interactions responsible for relative energies, local energy decomposition (LED) partitioning of DLPNO-CCSD(T)/def2-TZVPP was utilized.21 The theoretical background of this useful tool has been detailed elsewhere.21 In principle, the binding energy (ΔE) between a pair of fragments X and Y is hypothesized to consist of “geometric preparation energy” (ΔEgeo-prep) or the amount of strain required to distort them from their equilibrium structures to an optimal point of interaction and the inter-fragment interaction energy (ΔEint). The latter is further decomposed into various physical components, such as electrostatic interaction (ΔEelstat), quantum mechanical exchange (ΔEexchange), charge-transfer correlation (ΔECT), and the London dispersion energy (ΔEdisp). In short, ΔEint is summed up into dispersive (ΔEdisp) and non-dispersive (ΔEnon-disp) components. Hence, according to this scheme, the relative free energy of activation for a transition state is given by:
| |  | (3) |
where Δ
G‡corr incorporates solvent, thermal, and entropy corrections to Gibbs free energy.
NBO second-order perturbation theory analysis and Wiberg bond indices were calculated at the B3LYP-D3(BJ)/6-311+G(d,p) level, taking only the fragments of the substrates out of the relevant optimized geometries (Fig. S2). Within the context of conceptual DFT, Parr et al. defined the global electrophilicity index ω as:
| | 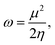 | (4) |
where
µ is the electronic chemical potential, and
η is the chemical hardness.
22a Furthermore, the electrophilic character of a local reactive site by a local electrophilicity index
ω+(
r) is given by:
where
f+(
r) is the Fukui function for nucleophilic attack obtained by using atomic populations. Similarly, nucleophilicity or electron-donating power
ω− is obtained from the formalism of Gazquez
et al.,
22b such that:
| | 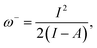 | (6) |
where
I and
A are the vertical ionization energy and electron affinity, respectively.
Results and discussion
We first pursued the reaction in the absence of the capsule to evaluate the role of encapsulation. As shown in Fig. 1, we hypothesize the formation of pre-reaction complexes, starting from separated molecules. Hence, separated reactants, water molecules or a single resorcin arene monomer are considered as the reference point for the free-energy profiles in Fig. 1 to properly address the entropic penalty. Next, C–C bond formation between the pyrrole and isocyanate substrates in the solvent medium takes place through TS1@SU at a calculated energy barrier of 22.5 kcal mol−1. Once the C–C coupled intermediate, INT1@SU, is formed in a highly endergonic manner (ΔG = 22.1 kcal mol−1), a 1,3C → N proton transfer takes place viaTS2@SU at a predominantly high barrier of 45.5 kcal mol−1. Interestingly, in the presence of explicit water molecules, the energetic expense for both C–C coupling through TS1@SWA and the proton-wire step through TS2@SWA is reduced by ∼1 and 15 kcal mol−1, respectively, indicating the benefit of accommodating water molecules in the vicinity.20a Importantly, it was reported by La Manna et al. that the reactions of pyrroles and isocyanates under the standard conditions, but in the absence of the capsule, did not lead to completion even after prolonged reaction time (40 hours).7b The computed reaction barriers agree to the observation by La Manna and others. In fact, conversion of the rate-determining barrier of TS2@SWA (ΔG‡ = 26.0 kcal mol−1) to an Eyring rate-constant (k) of 5.4 × 10−7 s−1 and a half-life of 14.8 days, further intensifying the need for encapsulation.
 |
| | Fig. 1 Gibbs free energy profiles in kcal mol−1 at B3LYP-D3BJ/Def2-TZVP/CPCM(CHCl3:H2O = 1:1) on the M06-2X/CPCM(CHCl3)/6-31G(d,p)-optimized geometries for the coupling of 2a and 3a to yield 4aa in implicit solvent (S) and assisted by a resorcin-arene monomer (M). @MWA = water-assisted monomer; @MU = unassisted monomer; @SWA = water-assisted implicit solvent; @SU = unassisted implicit solvent. Distances shown are in the units of Å. | |
Next, we consider the assistance of a single monomeric resorcin-arene unit to facilitate the coupling. However, here, the optimum anchorage of the monomer to the substrates through dispersion interactions enables the reactant complexes as the thermodynamic reference to be used to compute the activation-free energy barrier (see Fig. 1). Interestingly, the rearrangement of the two reactants into the intermediate is a mildly endergonic process in the presence (INT1@MWA, ΔG = 1.1 kcal mol−1) or absence (INT1@MU, ΔG = 2.6 kcal mol−1) of water assistance with the monomer (M) and features a new amido-NH bond. Furthermore, strong, attractive H-bonds stabilize the preceding (TS1@MWA and TS1@MU) and succeeding (TS2@MWA and TS2@MU) transition states as compared to the significantly energy-intensive reaction in solution (Fig. 1). In fact, encapsulation of the substrates in the single resorcin-arene moiety is impacted by a change in the rate-determining barrier to the initial nucleophilic addition of pyrrole to isocyanate (TS1@MWA = 14.2 kcal mol−1).
At this stage, our analysis reveals that a large geometric distortion is required to constrain the pyrrole and isocyanate in TS1, particularly the angular bending of the N–C–O moiety (Fig. 1, inset). Notably, bifurcation of the activation energy (ΔE‡) into distortion energy (ΔEdist), due to geometric rearrangement, and interaction energy (ΔEint), due to overlapping orbitals of the two substrates, provides meaningful insights into the C–C coupling transition states (Fig. 2).23 As shown in Fig. 2, ΔEdist dictates the extent of ΔE‡. A detailed investigation shows that TS1@SU suffers the greatest geometric distortion in the substrates (46.2 kcal mol−1), while TS1@MWA experiences the least (25.3 kcal mol−1). This observation is in line with the trend in second-order perturbation analyses, E(2), between the donor and the acceptor orbitals24 in transition states as follows: TS1@SU (29.0 kcal mol−1) > TS1@SWA (37.1 kcal mol−1) > TS1@MU (71.3 kcal mol−1) > TS1@MWA (97.9 kcal mol−1) (Fig. S1), emphasizing the crucial role of anchorage through non-covalent interactions.
 |
| | Fig. 2 Energy decomposition analysis TS1@SU, TS1@SWA, TS1@MU, and TS1@MWA at B3LYP-D3BJ/Def2-TZVP/CPCM in kcal mol−1. | |
To further understand the factors responsible for the unusual C–C coupling in bulk implicit solvent as compared to monomeric confinement, we analysed the various physical interactions between the substrates and the resorcin-arene monomer using the DLPNO-CCSD(T)/LED methodology. As observed in Table 1, for all the transition states, a large amount of energy is invested in geometric preparation. As expected, detailed investigation demonstrates that ΔE‡geo-prep-3a for substrate 3a (isocyanate) is the largest in TS1@SU, followed by TS1@SWA, TS1@MWA, and TS1@MU, arising from the addition of pyrrole to the bent, congested orientation of isocyanate. Hence, a comparison of TS1@SU and TS1@SWA shows that the former is hugely destabilizing. Decomposition of ΔE‡int into dispersive and nondispersive components reveals that London dispersion forces preferentially stabilize the transition states, further emphasizing the crucial role of H-bonding networks.21c The attractive dispersion forces are significantly larger in TS1@MWA compared to its competing transition state, TS1@MU, reversing the trend of ΔE‡geo-prep-3a, and suggesting that a balance of repulsive steric interactions and dispersive interactions is responsible for the desired outcome (Table 1).
Table 1 Decomposition of the reaction barriers (ΔG‡) in kcal mol−1 calculated by the DLPNO-CCSD(T)/Def2-TZVPP/LED scheme for the C–C coupling in TS1@SU, TS1@SWA, TS1@MU, and TS1@MWA
|
|
TS1@SU |
TS1@SWA |
TS1@MU |
TS1@MWA |
| ΔG‡ |
41.6 |
25.6 |
23.1 |
18.2 |
| ΔE‡ |
38.9 |
23.4 |
23.0 |
15.0 |
| ΔE‡geo-prep |
45.7 |
33.0 |
25.4 |
23.7 |
| ΔE‡geo-prep-3a |
35.0 |
27.2 |
18.8 |
21.4 |
| ΔE‡int |
−6.8 |
−9.6 |
−2.4 |
−8.8 |
| ΔE‡disp |
−8.4 |
−7.8 |
−0.1 |
−7.5 |
| ΔE‡no-disp |
1.6 |
−1.8 |
−2.2 |
−1.2 |
Thereby, we diverge on the reactivity inside the capsules, which are inherently reaction vessels, bio-mimicking the active site of several (metallo)enzymes.25 Notably, for reactions within the confined environment, the self-assembly binding enthalpy is expected to overshadow the entropic cost; hence, reactant complexes would be the reference point necessary to channelize the reaction. Analogous to RC@CU (Fig. 3), in the experimental study, the authors propose the formation of a heterocomplex 2a + 3a@C, on the basis of reactivity inhibition in the presence of competitive hosts such as [NEt3]+ and during H-bond disruption by DMSO.7b The optimized geometries of RC@CU and RC@CWA show the guest molecules align in a T-shape, such that any susceptible steric repulsions within the cavitand are judiciously avoided. The energy barriers for the C–C coupling step in the presence (viaTS1@CWA, ΔG‡ = 14.2 kcal mol−1) or absence (viaTS1@CU, ΔG‡ = 18.3 kcal mol−1) of the non-structural water molecule inside the capsule are comparable to the reactions with M, highlighting similar dispersion effects imposed by the cavity. It may be presumed that intermediate INT1@MWA is formed through monomer assistance,14c and is trapped inside the capsule, facilitated by the capsule's acidity, where it undergoes the proton-wire mechanism (Fig. 3). Indeed, our calculations reveal an enhancement in the average pKa values of the alcoholic –OH groups on going from MU(8.2) and MWA(8.0) to CU(3.2) and CWA(2.3). Thus, while TS2@MWA and TS2@MU display activation energies in the order of 7.9–8.6 kcal mol−1, the corresponding barriers for TS2@CWA and TS2@CU are significantly curtailed to 2.6 and 5.6 kcal mol−1, respectively. Notably, the formation of product 4aa@CWA is calculated to be highly exergonic compared to all the other cases studied.
 |
| | Fig. 3 Gibbs free energy profiles in kcal mol−1 at B3LYP-D3BJ/Def2-TZVP/CPCM(CHCl3:H2O = 1:1) on ONIOM[M06-2X/CPCM(CHCl3)/6-31G(d,p):PM6]-optimized geometries for the coupling of 2a and 3a to yield 4aa inside capsule C. @CWA = water-assisted capsule; @CU = unassisted capsule. Optimized geometries of RC@CU and RC@CWA (one M is omitted for clarity). Colour code: C (grey), O (red), H (white), N (blue). | |
Furthermore, molecular electrostatic potential (MEP) plots of the catalytic frameworks in INT1 show an electropositive environment around the –OH groups at the rim, while a distinct electronegative cloud emerges surrounding the phenoxide (O–) moiety (Fig. 4). Additionally, the calculated high nucleophilicity (ω− = −7.8 eV) on the catalytic framework in INT1@CWA can be directly correlated to the capsule's augmented Brønsted acidity in the presence of the additional hydrogen-bonded water molecule. In fact, the electrophilicity (ω+ = 15.1 eV, Table S4) of the pyrrolic C–H unit and the low Wiberg bond index (0.75, Fig. S3) in INT1@CWA highlight that an electronic balance is achieved in TS2@CWA for the synchronous C–H activation coupled to the proton shuttle. Thus, our results validate that the water-assisted capsule is the active catalyst. Reduced density gradient (RDG) plots further support our understanding of augmented NCIs in water-assisted transition states for C–C coupling (Fig. S5).
 |
| | Fig. 4 MEP plot and computed nucleophilicity of the catalytic framework in INT1. The colour spectrum ranges from negative (blue) to positive (red) potential within ±0.01 au. | |
Nevertheless, our work also hints towards the advantage of the ubiquitous presence of water-enriched local domains, which regulate reaction kinetics, selectivity, and mass transfer in natural processes, primarily due to the unique properties of water molecules, such as remote bond polarization, proton shuttling, and the ability to act as co-catalysts.26 Interestingly, such a natural phenomenon is laterally existent in several industrial-grade processes, working in confined or unconfined environments.26 Recently, the promotional role of water as an active mediator in C–C coupling reactions has been verified by isotope effect experiments and molecular dynamics simulations.26b Indeed, a comparison with one-, two- and three-water molecule-assisted pathways in confinement of the capsule together with an unassisted mechanism strongly support our argument that non-structural water molecules form a bridge between the catalyst and the substrates, polarizing the neighbouring terminals, discharging the steric constrains associated with the optimally oriented substrates, and thereby promoting the C–C coupling step with predominantly low-barriers ranging between 12.7 and 16.4 kcal mol−1 as opposed to the unassisted mechanism with a 26.6 kcal mol−1 reaction barrier predicted by B3LYP-D3BJ/Def2-TZVP/ALPB(CHCl3) calculations on ONIOM[B3LYP-D3(BJ)/ALPB(CHCl3)/def2-SVP:XTB] optimized geometries (Fig. 5).
 |
| | Fig. 5 Gibbs free-energy profiles in kcal mol−1 at B3LYP-D3BJ/Def2-TZVP/ALPB(CHCl3) on ONIOM[B3LYP-D3(BJ)/ALPB(CHCl3)/def2-SVP:XTB]15b optimized geometries for the coupling of 2a and 3a to yield 4aa inside capsule C. @CWA = water-assisted capsule (solid black line); @C2WA = two-water-assisted capsule (dashed black line); @C3WA = three-water-assisted capsule (dotted black line) @CU = unassisted capsule (shown by red line). Subscript O refers to reactions studied with ORCA 5.0.3. | |
Conclusions
In conclusion, we unravelled an unprecedented water-assisted reaction avenue for the coupling of pyrroles and isocyanates to yield amidopyrroles inside a hexameric resorcin-arene capsule. Our results underscore the role of encapsulation together with assistance from non-structurally incorporated water molecules to expedite C–C coupling through nucleophilic addition and synchronous C–H activation and a proton-shuttle mechanism in the self-assembled capsule, as compared to the bulk solvent. Our results strongly suggest the role of a water-rich local domain in providing a balanced hydrophilic encapsulation within a hydrophobic suprastructure, thereby providing the desired stabilization of the micro-solvated transition states. We envisage that our present work will provide new directions towards nanoconfinement-driven chemistry, to achieve high-efficiency and selectivity of organic transformations in hydrophilic encapsulations27 and diversify the role of water as a co-solvent.
Author contributions
SKM, BG and LR performed all the calculations. LR wrote the manuscript and made the graphics and figures. All the authors gave consent for the final form of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: Fig. S1–S3 and Tables S1–S4, XYZ coordinates and additional computational details. See DOI: https://doi.org/10.1039/d5cp01713k.
Acknowledgements
L.R thanks CSIR, India (01WS(001)/2023-24/EMR-II/ASPIRE), ANRF, India (ANRF/ARG/2025/007177/CS), and SRIC, IIT Kharagpur (IIT/SRIC/FSRG/2025/06) for funding. SKM thanks ICT-IOCB, and BG thanks CSIR-ASPIRE for the research fellowship. This study used the resources of the ParamShakti supercomputing facility of IIT Kharagpur, established under the National Supercomputing Mission of the Government of India and supported by CDAC, Pune.
Notes and references
-
(a) D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445 Search PubMed;
(b) M. Mastalerz, Angew. Chem., Int. Ed., 2010, 49, 5042 CrossRef CAS PubMed;
(c) W. Wang, Y.-X. Wang and H.-B. Yang, Chem. Soc. Rev., 2016, 45, 2656 RSC.
-
(a) B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem. – Eur. J., 2020, 26, 15446 CrossRef CAS PubMed;
(b) Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107 Search PubMed.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360 RSC.
- H. Daver, J. N. Harvey, J. Rebek Jr and F. Himo, J. Am. Chem. Soc., 2017, 139, 15494 CrossRef CAS PubMed.
- L. Catti, Q. Zhang and K. Tiefenbacher, Chem. – Eur. J., 2016, 22, 9060 CrossRef CAS PubMed.
-
(a) S. L. Craig, S. Lin, J. Chen and J. Rebek, J. Am. Chem. Soc., 2002, 124, 8780 CrossRef CAS PubMed;
(b) T. Heinz, D. M. Rudkevich and J. Rebek Jr, Nature, 1998, 394, 764 Search PubMed.
-
(a) P. La Manna, M. De Rosa, C. Talotta, A. Rescifina, G. Floresta, A. Soriente, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2020, 59, 811 CrossRef CAS PubMed;
(b) P. La Manna, C. Talotta, M. De Rosa, A. Soriente, C. Gaeta and P. Neri, Org. Lett., 2020, 22, 2590 CrossRef CAS PubMed;
(c) P. La Manna, C. Talotta, G. Floresta, M. De Rosa, A. Soriente, A. Rescifina, C. Gaeta and P. Neri, Angew. Chem., Int. Ed., 2018, 57, 5423 CrossRef CAS PubMed;
(d) C. Gaeta, C. Talotta, M. De Rosa, P. La Manna, A. Soriente and P. Neri, Chem. – Eur. J., 2019, 25, 4899 CrossRef CAS PubMed.
-
(a) R. Silvestri, G. La Regina, G. De Martino, M. Artico, O. Befani, M. Palumbo, E. Agostinelli and P. Turini, J. Med. Chem., 2003, 46, 917 CrossRef CAS PubMed;
(b) G. Montà-González, D. Bastante-Rodríguez, A. García-Fernández, P. Lusby, R. Martínez-Máñez and V. Martí-Centelles, Chem. Sci., 2024, 15, 10010 RSC.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469 CrossRef CAS.
- D. A. Poole III, S. Mathew and J. N. H. Reek, J. Am. Chem. Soc., 2021, 143, 16419 CrossRef CAS PubMed.
- A. Katiyar, J. C. F. Sovierzoski, P. B. Calio, A. A. Vartia and W. H. Thompson, Chem. Commun., 2019, 55, 6591 RSC.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148 CrossRef CAS PubMed.
- F. Sebastiania, T. A. Bender, S. Pezzottia, W.-L. Li, G. Schwaab, R. G. Bergman, K. N. Raymond, F. Dean Toste, T. Head-Gordon and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32954 CrossRef PubMed.
-
(a) V. Vaissier Welborn, W.-L. Li and T. Head-Gordon, Nat. Commun., 2020, 11, 415 CrossRef PubMed;
(b) H. Daver, J. Rebek Jr and F. Himo, Chem. – Eur. J., 2020, 26, 10861 Search PubMed;
(c) P. La Manna, C. Talotta, C. Gaeta, Y. Cohen, S. Slovak, A. Rescifina, P. Della Sala, M. De Rosa, A. Soriente and P. Neri, Org. Chem. Front., 2022, 9, 2453 Search PubMed.
-
(a)
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al., Gaussian 09 (Revision A.02), Gaussian, Inc., Wallingford CT, 2009 Search PubMed;
(b) F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 Search PubMed;
(b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
-
(a) S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed;
(b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 Search PubMed.
- X. Zhang and R. S. Paton, Chem. Sci., 2020, 11, 9309 RSC.
-
(a) P. L. da Silva, L. Guimaraes and J. R. Pliego Jr, J. Phys. Chem. B, 2013, 117, 6487 CrossRef PubMed;
(b) M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Angew. Chem., 2022, 134, e202205735 Search PubMed;
(c) A. K. Singh and L. Roy, Eur. J. Inorg. Chem., 2023, e202300412 CrossRef CAS.
-
(a) D. Yepes, F. Neese, B. List and G. Bistoni, J. Am. Chem. Soc., 2020, 142, 3613 CrossRef CAS PubMed;
(b) G. Bistoni, A. Altun, Z. Wang and F. Neese, Acc. Chem. Res., 2024, 57, 1411 CrossRef CAS PubMed;
(c) S. K. Mahapatra, B. Ghosh and L. Roy, ChemCatChem, 2025, 17, e01069 Search PubMed.
-
(a) R. G. Parr, L. Von Szentpaly and S. B. Liu, J. Am. Chem. Soc., 1999, 121, 1922 Search PubMed;
(b) J. L. Gazquez, A. Cedillo and A. Vela, J. Phys. Chem. A, 2007, 111, 1966 Search PubMed.
-
(a) M. Baidya, D. Maiti, L. Roy and S. De Sarkar, Angew. Chem., Int. Ed., 2022, 61, e202111679 CrossRef CAS PubMed;
(b) L. Roy, B. Ghosh and A. Paul, J. Phys. Chem. A, 2017, 121, 5204–5216 Search PubMed;
(c) W. J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC;
(d) R. Khuntia, S. K. Mahapatra, L. Roy and S. C. Pan, Chem. Sci., 2023, 14, 10768 RSC.
- A. K. Singh and L. Roy, Chem. Phys. Chem., 2024, 25, e202400533 CrossRef PubMed.
-
(a) H. S. Ali and S. P. de Visser, Chem. – Eur. J., 2022, 28, e202104167 CrossRef CAS PubMed;
(b) R. N. Manna, T. Malakar, B. Jana and A. Paul, ACS Catal., 2018, 8, 10043 CrossRef CAS;
(c) L. Roy, ChemCatChem, 2018, 10, 3683 CrossRef CAS.
-
(a) G. Li, B. Wang and D. E. Resasco, ACS Catal., 2020, 10, 1294 CrossRef CAS and references therein;
(b) D. T. Ngo, Q. Tan, B. Wang and D. E. Resasco, ACS Catal., 2019, 9, 2831–2841 Search PubMed.
- Shashi, S. Das, A. Datta and J. Dasgupta, J. Phys. Chem. Lett., 2025, 16, 11464–11471 Search PubMed.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Bijoy
Ghosh
a,
Bijoy
Ghosh









