DOI:
10.1039/D4TC05445H
(Paper)
J. Mater. Chem. C, 2025,
13, 8671-8681
Synergistic effects of triplet–triplet annihilation and reverse intersystem crossing in a platinum-based electrochemiluminescent metallopolymer emitter†
Received
26th December 2024
, Accepted 5th March 2025
First published on 5th March 2025
Abstract
This work presents the synthesis, electrochemical behavior, and photophysical properties of a novel platinum acetylide metallopolymer (p-PtBTD), designed for enhanced electrogenerated chemiluminescence (ECL) performance. The metallopolymer features alternating π-conjugated segments of 2,1,3-benzothiadiazole (BTD) and trans-Pt(PBu3)2 units. The inclusion of platinum centers facilitates efficient intersystem crossing (ISC), allowing the radiative decay of triplet excitons, which is typically challenging in conventional ECL systems dominated by singlet emission. Photoluminescence (PL) studies reveal dual emission, with a prominent fluorescence peak at 584 nm and a weak phosphorescence peak near 800 nm. Electrochemical investigations demonstrate quasi-reversible oxidation and irreversible reduction waves for p-PtBTD, while transient ECL studies reveal instability in the radical cation, which has been successfully addressed using tripropylamine (TPrA) as a co-reactant. The ECL spectrum shows dual emission arising from both singlet and triplet states, facilitated by triplet–triplet annihilation (TTA) and reverse intersystem crossing (RISC) due to a small energy gap (∼0.5 eV) between these states. This dual emission mechanism, involving both fluorescence and phosphorescence, highlights the potential of p-PtBTD for advanced ECL applications, particularly in sensing and optoelectronics. These findings underscore the utility of metallopolymers in overcoming the limitations of traditional ECL systems, paving the way for more efficient and versatile luminescent materials.
Introduction
Organometallic π-conjugated materials have garnered significant attention in recent decades due to their promising applications, particularly in fields like electrogenerated chemiluminescence (ECL) and optoelectronics.1–7 These materials, especially π-conjugated polymers, can achieve full-color ECL by sequentially injecting holes and electrons into their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), leading to exciton formation and light emission through radiative decay. Early studies on fluorescent polymers, which utilized mechanisms like intrachain resonance energy transfer and aggregation-induced emission, aimed to enhance ECL efficiency and explore sensor applications.8–11 However, these polymers typically suffer from spin-forbidden transitions, which restrict ECL emission to singlet states, causing up to 75% of the energy stored in triplet states to be lost.12
Our research focuses on overcoming these limitations by investigating triplet-excited states in ECL, utilizing a more energy-efficient singlet–triplet/triplet–singlet intersystem crossing route.13 Specifically, we designed platinum acetylide metallopolymers that harness the radiative decay of electrochemically generated triplet excitons. The inclusion of platinum, a heavy metal, within the π-conjugated polymer chain is expected to significantly enhance intersystem crossing (ISC) yields, enabling more effective utilization of triplet states.14
Recently, time-activated delayed fluorescence (TADF) polymers (i.e., PCzAPT10) have been reported in the context of ECL, representing a breakthrough in the field.7 Inspired by these developments, we synthesized a platinum acetylide metallopolymer (p-PtBTD) incorporating alternating π-conjugated segments of a 2,1,3-benzothiadiazole (BTD) acceptor moiety and trans-Pt(PBu3)2 units. BTD is a well-known electron acceptor, frequently used in electrochemical and ECL applications.15–20 Its incorporation was intended to fine-tune the polymer's electronic properties, narrow the energy gap, and improve the stability and electrochemical activity of the resulting polymer.21–26
This study presents the synthesis, electrochemical characterization, photophysical properties, and ECL behavior of p-PtBTD, a unique ECL-active TADF p-PtBTD (Fig. 1). Our investigations reveal the presence of both fluorescence and phosphorescence in the PL spectrum of p-PtBTD. Electrochemical analyses show quasi-reversible oxidation and irreversible reduction behavior, while transient ECL studies highlight instabilities in cation formation, which have been mitigated through the introduction of TPrA as a co-reactant. The dual ECL emission has been confirmed through a triplet–triplet annihilation (TTA) and triplet–singlet Förster resonance energy transfer (FRET) mechanism, which was facilitated by a small energy gap (∼0.49 eV) between the singlet and triplet excited states.
 |
| | Fig. 1 The chemical structure of the p-PtBTD metallopolymer. | |
Results and discussion
Photophysics
Fig. 2a shows the UV-Vis absorption and the steady-state PL spectra of p-PtBTD in 1 mM DCM. The platinum-acetylide backbone's π–π* transition, polarized along the long axis of the polymer, produces a broad, split absorption spectrum with maxima at 354 nm and 451 nm.27 The PL spectrum shows a strong fluorescence peak at 584 nm with a short lifetime of 2.42 ns (Fig. S10, ESI†) and a minor phosphorescence peak around 800 nm, consistent with previous reports.28–30 However, the phosphorescence lifetime at 800 nm could not be measured due to insufficient signal intensity.30–32 This observation may appear to contradict the expected enhancement of the triplet excited state due to the presence of the platinum atom (heavy atom effect). A plausible explanation is the increased delocalization of the singlet π–π* state along the platinum-acetylide polymer chain. Liu et al. reported that as the chain length of platinum-acetylide polymers increases, the ratio of the 0–1 to 0–0 vibronic bands decreases, indicating a reduction in electron-vibrational coupling.33 This suggests that the singlet excited state becomes more delocalized as the polymer chain grows longer. This delocalization can lead to enhanced fluorescence due to better orbital overlaps and stronger radiative transitions. The observed weaker phosphorescence in our Pt-acetylide polymer can be attributed to the localization of the triplet exciton, as symmetry breaking in the excited state leads to confinement of the triplet state on specific segments of the polymer. Similar behavior has been reported in [Pt(P(nBu)3)2(ethynylbenzene)2] complexes, where the triplet exciton becomes localized on a single ethynylbenzene ligand due to symmetry breaking and exhibits an activated hopping mechanism for transport.34 This localization reduces the efficiency of radiative decay (T1 → S0), contributing to weaker phosphorescence in p-PtBTD. The decay profiles at 310 K, 190 K, and 100 K are shown that lower temperatures reduce nonradiative decay lifetime (Fig. 2b), thereby demonstrating the occurrence of RISC.
 |
| | Fig. 2 (a) UV-Vis absorption spectra (cyan solid line), PL spectra (purple solid line) and ECL spectra (red solid line). Excitation wavelength for PL: 354 nm. ECL spectra of 1 mM p-PtBTD without TPrA in 0.1 M TBAPF6 in DCM is obtained by pulsing between 80 mV past the oxidation peak potential and 2nd reduction potential. (b) Normalized transient PL decay of p-PtBTD monitored at 584 nm at 300 K (cyan curve), 190 K (purple curve) and 100 K (pink curve). (c) Ultrafast transient absorption spectrum of p-PtBTD. (d) Kinetic trace of decay of p-PtBTD probed at 514 nm (cyan curve) and 690 nm (red curve). Excitation wavelength is 360 nm. | |
Exciton dynamics of p-PtBTD
Fig. 2c presents the transient absorption (TA) spectra of p-PtBTD in solution, providing insights into the electronic properties and dynamic profiles of transient states generated by photoexcitation. Also, it can represent the excited state dynamic of the PtBTD after electrochemical excitation. The fs-TA spectrum is characterized by strong bleaching in the near-UV region, attributed to ground-state depletion, along with broad, moderately intense excited-state absorption extending throughout the visible region, with a maximum at 690 nm. The visible absorption band at 690 nm is attributed to ISC in the p-PtBTD oligomer, with a time constant of approximately 900 fs (Fig. S11b, ESI†), which is consistent with previously reported ISC rates in platinum-acetylide oligomers.14 Meanwhile, the transient signal at 514 nm shows growth with a time constant of 2.32 ps (Fig. S11a, ESI†), corresponding to the ground-state recovery process via relaxation from the excited states to the singlet ground state (Fig. 2d). This kinetic data supports the existence of the triplet-excited state in p-PtBTD.
Temperature-dependent time-resolved photoluminescence (TRPL) spectroscopy was employed to investigate the exciton dynamics of the p-PtBTD polymer (Fig. 3a). The decay kinetics were fitted to a bi-exponential decay model, revealing two distinct temporal decay processes, including short lifetime (τ1) and long lifetime (τ2). As listed in Table S1 (ESI†), the TRPL decay curves predominantly feature a fast decay component with τ1 ranging from 2.0 to 4.3 ns and a slower decay component with τ2 ranging from 0.48 to 1.11 μs. Furthermore, the average lifetimes (τavg) are temperature-dependent, increasing from 0.48 μs at 100 K to 1.11 μs at 340 K. Further evidence of delayed PL is observed in the large amplitude component (81.67% to 98.51%) and the small amplitude component (1.49% to 18.33%) as the temperature increases (Table S1, ESI†). These features indicate the presence of thermally activated delayed fluorescence (TADF) in p-PtBTD.
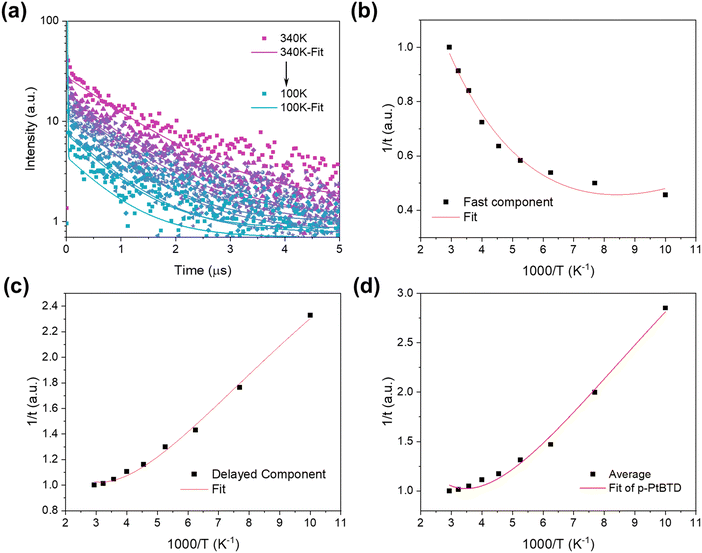 |
| | Fig. 3 (a) Temperature-dependent time-resolved photoluminescence (TRPL) decay curves at 584 nm for p-PtBTD, measured at various temperatures (100 K to 340 K). (b) Arrhenius plot of the fast decay component, (c) Arrhenius plot of the slow (delayed) decay component, and (d) Arrhenius plot of the average decay lifetime for p-PtBTD. The red lines in panels (b)–(d) represent fits to the equation  , where Ea is the activation energy and τ is the lifetime. , where Ea is the activation energy and τ is the lifetime. | |
Marcus theory has been broadly used to explain the electron transfer in distinct molecular systems and illustrate the transfer of electron–hole pairs.35 Marcus theory describes how the rate of electron transfer depends on the free energy change (ΔG) of the reaction and the reorganization energy (λ) of p-PtBTD. Based on our previous investigation,36–39 we adopted Marcus theory to describe the photoinduced charge-carrier dynamics of p-PtBTD emitters. The calculation of activation energies for the fast and slow decay, which are 5.13 meV and 13.72 meV, respectively, was performed. These values were obtained by fitting the PL decay curves with the equation  (Fig. 3b–d and Table 1). The pre-exponential factor from the fit-to equation yields temperature-independent electronic coupling matrix elements |HAB|, whose values represent the overlap of the excited-state wave functions between the initial and final sites, and are determined to be 6.34 × 10−3 cm−1 and 6.68 × 10−3 cm−1. The extracted activation energies provide evidence for the localization of the singlet excited state. These values reinforce the role of these processes in the dual-emission behavior of p-PtBTD.
(Fig. 3b–d and Table 1). The pre-exponential factor from the fit-to equation yields temperature-independent electronic coupling matrix elements |HAB|, whose values represent the overlap of the excited-state wave functions between the initial and final sites, and are determined to be 6.34 × 10−3 cm−1 and 6.68 × 10−3 cm−1. The extracted activation energies provide evidence for the localization of the singlet excited state. These values reinforce the role of these processes in the dual-emission behavior of p-PtBTD.
Table 1 The activation energy and electronic coupling of p-PtBTD
|
|
Activation energy (Ea)/meV |
Electronic coupling (|HAB|)/cm−1 |
|
τ
1 (fast) |
5.60 |
6.91 × 10−3 |
|
τ
2 (slow/delayed) |
13.72 |
6.68 × 10−3 |
| 〈τ〉 |
12.54 |
8.46 × 10−3 |
The low activation energy (5.60 meV) and stronger electronic coupling (6.91 × 10−3 cm−1) associated with the τ1 suggest that the singlet exciton is delocalized across the polymer backbone, facilitating efficient radiative decay and enhanced fluorescence. In contrast, the higher activation energy (13.72 meV) and weaker electronic coupling (6.68 × 10−3 cm−1) for the τ2 indicate that the triplet exciton is localized, likely confined to specific regions of the polymer, resulting in slower decay and weaker phosphorescence. These results are consistent with the general photophysical behavior of platinum-acetylide polymers, where singlet states tend to be delocalized and triplet states localized due to symmetry breaking and strong spin–orbit coupling.
Electrochemistry and heterogeneous electron transfer kinetics
CV experiments provide insights into the electrochemical dynamics of p-PtBTD, including electrochemical reversibility, the diffusion coefficient, the number of electrons transferred, and the stability of radical cations and anions. Generally, the CV of p-PtBTD in DCM showed one reduction and two oxidation waves (Fig. 4a). The CV graph showed irreversible reduction and oxidation waves at the potential around −0.44 V vs. SCE, −1.07 V vs. SCE, and +0.79 V vs. SCE. The irreversible waves of p-PtBTD indicate that the p-PtBTD radical anion and radical cation are unstable. All observed waves are associated with the oxidation and reduction of the π-conjugated segments within benzo[1,2,5]thiadiazole. The irreversible character of these waves can be explained by the charge diffusion on the π-conjugated chain along the metallopolymer, leading to the disappearance of the charge during the electrochemical process.
 |
| | Fig. 4 (a) Standard cyclic voltammograms (CV) of p-PtBTD at scan rate 100 mV s−1. (b) Reduction voltammogram of p-PtBTD at various scan rates. (c) Oxidation voltammogram of p-PtBTD at various scan rates. (d) 1st reduction peak current versus v1/2. (e) 2nd reduction peak current versus v1/2. (f) Oxidation peak current versus v1/2. All CVs were performed with 1 mM p-PtBTD in 100 mM TBAPF6 using DCM as a solvent. | |
The CVs for reduction and oxidation from 10 mV s−1 to 1 V s−1 (Fig. 4b and c) were conducted to confirm the electrochemical reversibility of the reduction and oxidation of p-PtBTD. Fig. 4d–f shows the scan-rate-dependent CVs for the oxidation, 1st reduction, and 2nd reduction peaks, respectively. The peak currents vary linearly with the square root of the scan rate for the 1st oxidation wave (ip,ox), the 1st reduction wave (ip,red1), and the 2nd reduction wave (ip,red2), indicating that the reactions are diffusion-controlled. The diffusion coefficient (d) and the transfer coefficient (α) are estimated so that the heterogeneous electron-transfer rate constant (k0) can be determined. Eqn (1) shows the Randles–Sevcik equation for reversible systems,40
| |  | (1) |
For an irreversible system, a modified equation is given by
eqn (2)| |  | (2) |
In eqn (1) and (2), I is the peak current (A), n is the number of exchanged electrons, n′ is the total number of electrons transferred before reaching the rate-determining step, α is the transfer coefficient, A is the active surface area of the working electrode (cm2), D is the diffusion coefficient (cm2 s−1), and C is the bulk concentration of the participating species (mol cm−3), and ν is the voltage scan rate (V s−1).
The Gileadi method determines the heterogeneous electron-transfer rate constant (k0), determining the critical scan rate with reversible to irreversible or quasi-reversible to irreversible reaction transition.41 The two asymptotes of Epvs. the log(ν) at low and high scan rates estimate the critical scan rate (Vc) (Fig. S12d–f, ESI†), and then the k0 can be determined by eqn (3).42
| | 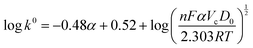 | (3) |
The Tafel plot based on the descending parts of the cathodic and anodic peaks of the cyclic voltammogram, the slope of the plot of log(i) versus potential determines the α by eqn (4).40
| |  | (4) |
From the scan rate studies, as shown in Fig. S12a–c (ESI†), the peak current varied linearly with the square root of the scan rate for the oxidation wave (ip,o) and the reduction waves (ip,r), showing the diffusion control features of the current. The diffusion coefficients, D, were determined by the Randles–Sevcik equation and are listed in Table 2. The single electron-transfer step in each wave, as well as the experiment condition at 25 °C, was assumed for the calculation. Similar k0 were found at the oxidation potential and 1st reduction potential. The 1st reduction potential showed lower electron transfer rates than the 2nd reduction potential, which are 3.4 × 10−3 and 5.7 × 10−3 cm s−1, respectively. Interestingly, the CV analysis showed that p-PtBTD more easily accepts additional electrons after the 1st reduction happened on the electrode surface.
Table 2 Kinetic parameters of the p-PtBTD metallopolymer. Kinetic parameters including E°, D, k°, and α for p-PtBTD in DCM/0.1 M TBAPF6 at room temperature
|
|
1st reduction |
2nd reduction |
Oxidation |
|
E
pa/pc/V vs. SCE |
−0.44 |
−1.07 |
0.79 |
| 10−5D/cm2 s−1 |
1.04 |
2.26 |
0.85 |
|
α
|
0.07 |
0.10 |
0.16 |
|
k°/cm s−1 |
0.0034 |
0.0057 |
0.0035 |
Electrogenerated chemiluminescence
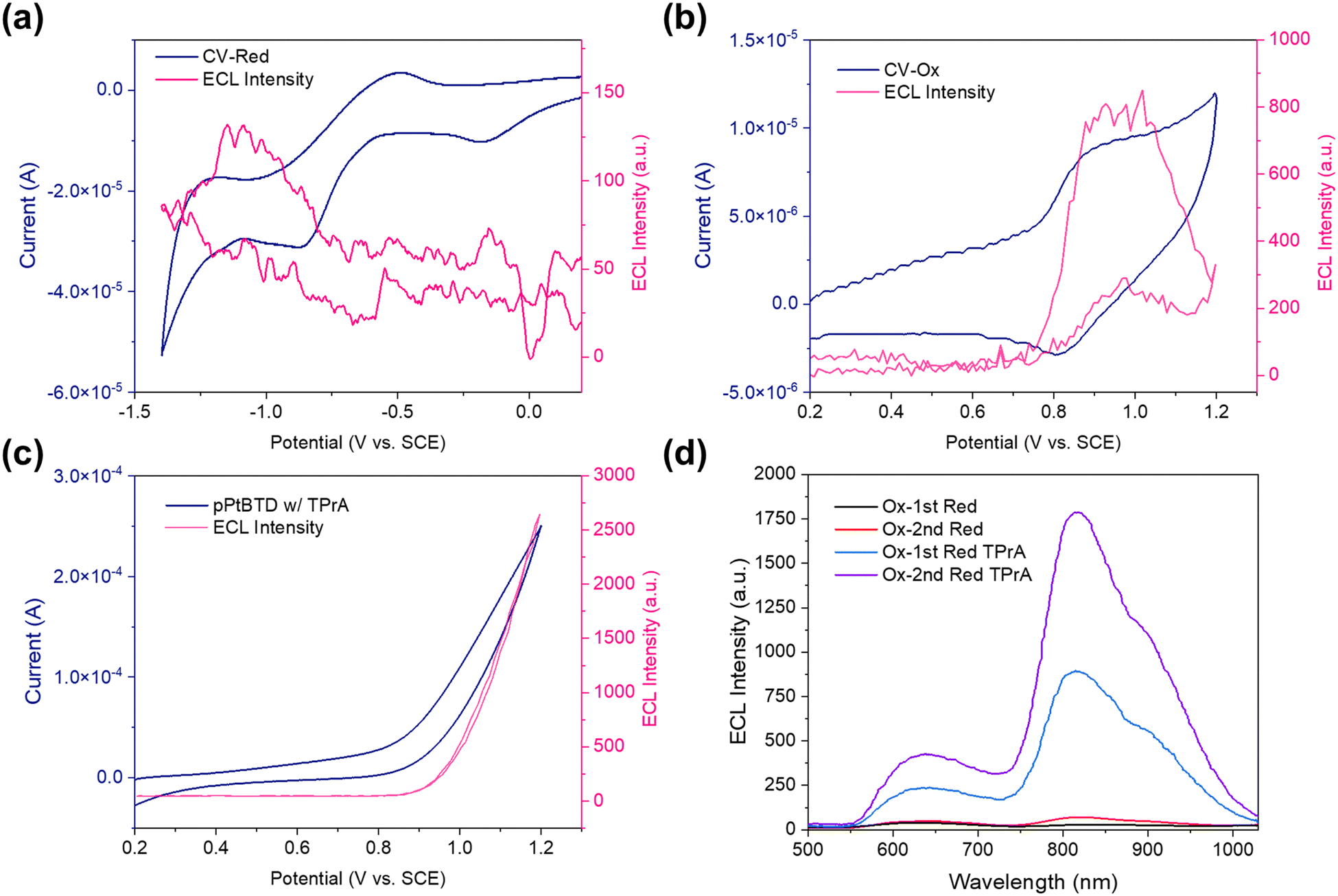
To investigate the electrochemical and ECL behavior of the p-PtBTD polymer, CV–ECL measurements were conducted for both oxidation and reduction processes, as well as in the presence of TPrA as a co-reactant. Fig. 5a presents the simultaneous CV (blue curve) and ECL (pink curve) measurement during the reduction of p-PtBTD. The reduction wave shows an onset near −0.4 V vs. SCE, with a corresponding rise in ECL intensity observed between −0.5 V and −1.0 V. The ECL signal intensity reaches a maximum near −1.2 V. Fig. 5b shows the oxidation CV–ECL data, where the CV curve exhibits a distinct oxidation peak at around +0.8 V vs. SCE. A significant increase in ECL intensity is observed in this region, with the maximum ECL emission occurring beyond +1.0 V. In the presence of TPrA as a co-reactant (Fig. 5c), both the oxidation and ECL intensity are significantly enhanced. The CV curve shows a sharp oxidation peak at +1.0 V vs. SCE, while the ECL emission intensity increases substantially compared to the measurements without TPrA, reaching values over 2500 a.u. This enhancement can be attributed to the co-reactant mechanism, where the TPrA radical cations generated during the oxidation process facilitate the formation of the excited states responsible for the ECL emission.43
 |
| | Fig. 5 CV–ECL simultaneous measurements by the pulsing potential from (a) approximately 0.0 V to 1.2 V vs. SCE and (b) approximately 0.0 V to −1.5 V vs. SCE, (c) 1 mM p-PtBTD in the presence of 20 mM TPrA as the coreactant for pulsing potential from ≈ +0.8 to −0.1 V vs. SCE. (d) ECL spectrum of 1st reduction and oxidation potential and 2nd reduction and oxidation potential with TPrA as a co-reactant. 1 mM p-PtBTD pulsed in DCM between 80 mV past the reduction peak and at 80 mV past the 1st oxidation potential, respectively. Pulse width is 1 second. | |
Fig. 5d compares the ECL spectra for the first and second reduction–oxidation cycles, with and without TPrA. The ECL emission intensity increased slightly depending on the redox process, where the second reduction–oxidation cycle produced a more intense ECL signal, particularly in the presence of TPrA. This observation highlights the critical role of the co-reactant in stabilizing the radical ions and boosting ECL efficiency, making TPrA an effective agent for enhancing ECL in the p-PtBTD system.
During transient ECL, the potential was stepped from −80 mV relative to the cathodic peak potential (Epc) to +80 mV relative to the anodic peak potential (Epa) to produce a p-PtBTD cation and anion. The fast-responsive emissions at different ECL intensities were observed in the cathodic and anodic pulses (Fig. 6). Asymmetric ECL transients were more pronounced during the cathodic pulses compared to the anode pulses, attributed to instability of the radical anion (p-PtBTD−˙) as demonstrated in CV studies.44 Furthermore, during the cathodic pulse, the radical cation (p-PtBTD+˙) possibly diffuses away from the annihilation zone but returns during subsequent pulses, leading to considerable decay after the first anodic pulse.45 The inconsistency of ECL intensities across different potentials suggests instability in forming stable radical anions.
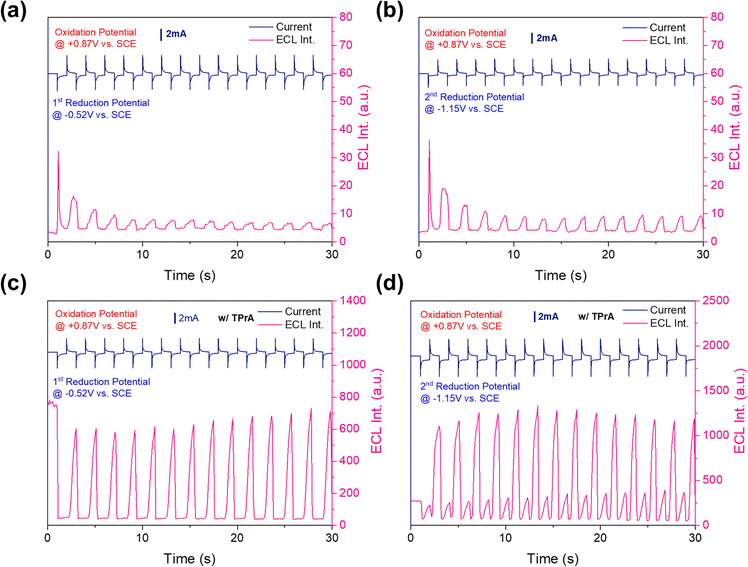 |
| | Fig. 6 Current (dark blue) and ECL transient (dark pink) measurements by the pulsing potential from (a) 1st reduction and oxidation potential and (b) 2nd reduction and oxidation potential, (c) 1st reduction and oxidation potential and (d) 2nd reduction and oxidation potential with TPrA as a co-reactant for 1 mM p-PtBTD pulsed in DCM between 80 mV past the reduction peak and at 80 mV past the 1st oxidation potential, respectively. Pulse width is 1 second. | |
To enhance the ECL performance, TPrA was used as a co-reactant (Fig. 6c and d). Initially oxidized to form a short-lived TPrA radical cation (TPrA˙+), TPrA undergoes deprotonation at an α-carbon to produce a strongly reducing intermediate (TPrA˙),46 which lastly is reduced by the oxidized p-PtBTD cations, giving rise to the stronger ECL emission. The ECL intensity of the 1st reduction potential is weaker than that of the 2nd reduction potential. The TPA co-reactant has demonstrated its efficacy in reducing p-PtBTD, which can be seen as a cooperative action under specified circumstances. This effectiveness is likely due to the increased reduction potential at the electrode during the 2nd reduction potential stage. Such an increase allows for more efficient energy utilization, generating a higher number of populated radical anions (p-PtBTD−˙). This results in a boost in the ECL intensity.
ECL mechanism
Fig. 2a shows a 41-nm red shift of the ECL spectrum compared to the fluorescence spectrum. Additionally, a new ECL peak appears at around 850 nm, which is assigned to the triplet state emission. The ECL emission wavelength under different conditions (i.e., 1 mM p-PtBTD with and without TPrA co-reactant under 1st reduction/2nd reduction and oxidation pulses) remain consistent (Fig. 5d). The energy of the excited singlet state Es (in eV) was determined using equation Es = 1239.81/λ (in nm), where λ is the maximum emission wavelength (i.e., 584 nm).12 The corresponding excited singlet-state energy was calculated to be 2.12 eV.
The energy of the annihilation reaction was estimated from the equation of  , where
, where  , and
, and  Considering the entropy effect (estimated at 0.1 eV), the corrected values are 1.13 eV and 1.76 eV, respectively. In the case of the co-reactant TPrA, the potential of the TPrA˙ radical is 1.5 V vs. NHE,47 yielding a thermodynamic potential of 2.03 eV. The enthalpies of the radical ion annihilation reactions (i.e., 1.13 eV, 1.76 eV, and 2.03 eV) are all smaller than the energy required to populate the singlet excited state (2.12 eV), suggesting that the ECL of p-PtBTD is likely processed via the T-route (energy-deficient system).
Considering the entropy effect (estimated at 0.1 eV), the corrected values are 1.13 eV and 1.76 eV, respectively. In the case of the co-reactant TPrA, the potential of the TPrA˙ radical is 1.5 V vs. NHE,47 yielding a thermodynamic potential of 2.03 eV. The enthalpies of the radical ion annihilation reactions (i.e., 1.13 eV, 1.76 eV, and 2.03 eV) are all smaller than the energy required to populate the singlet excited state (2.12 eV), suggesting that the ECL of p-PtBTD is likely processed via the T-route (energy-deficient system).
To explain the occurrence of dual ECL in the p-PtBTD polymer, we propose that the process involves both triplet–triplet annihilation (TTA) and reverse intersystem crossing (RISC). The mechanism can be described as follows:
| | | PtBTD + e− → PtBTD− (formation of anion) | (5) |
| | | PtBTD − e− → PtBTD+ (formation of cation) | (6) |
| | | PtBTD+ + PtBTD− → 3PtBTD* + PtBTD (generation of triplet excited state) | (7) |
| | | 3PtBTD* → PtBTD + hv (phosphorescence relaxation) | (8) |
| | | 3PtBTD* + 3PtBTD* → 1PtBTD* + PtBTD (triplet–triplet annihilation, TTA) | (9) |
| | | 1PtBTD* → PtBTD + hv (florescence relaxation) | (10) |
In the presence of TPrA as a co-reactant, the ECL mechanism involves the following steps:
| | | PtBTD − e− → PtBTD+ (formation of cation) | (11) |
| | | TPrA − e− → [TPrA˙]+ (formation of radical) | (12) |
| | | [TPrA˙]+ → TPrA˙ + H+ (homogeneous chemical reaction) | (13) |
| | | PtBTD + TPrA˙ → 3PtBTD* + products (excited state formation) | (14) |
| | | 3PtBTD* → PtBTD + hv (Phosphorescence) | (15) |
The triplet excited state 3PtBTD* formed via the T-route primarily relaxes through phosphorescence, which relaxes primarily via phosphorescence in eqn (8) (Fig. 5d). However, under certain conditions, 3PtBTD* can collide to undergo TTA, generating singlet excited states (1PtBTD*), which emit fluorescence. The ratio of fluorescence and phosphorescence varies with the pulsing condition. For instance, the 1st reduction potential pulsing exhibits that the diffusion coefficient of p-PtBTD is lower (1.04 × 10−5 cm2 s−1), suggesting slower mass transfer and potential aggregation on the electrode surface, which increases the probability of TTA and enhances fluorescence (Fig. S13, ESI†). Conversely, during the 2nd reduction pulsing, the higher diffusion coefficient (2.26 × 10−5 cm2 s−1) reduces aggregation, making phosphorescence more dominant. This shift in the fluorescence-to-phosphorescence ratio reflects the interplay of TTA and RISC in the ECL process.
Additionally, the combined temperature-dependent TRPL results in Fig. 3 and Marcus theory analysis provide strong evidence that both RISC and TTA are feasible pathways contributing to the dual ECL emission in the p-PtBTD system. These findings highlight the compliance with thermally activated processes and triplet exciton dynamics in driving the observed photophysical behavior. The fast decay component (τ1) exhibits a relatively low activation energy (Ea = 5.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) meV) for exciton transfer between molecules. Furthermore, previous studies have shown that when the energy gap between singlet and triplet excited states is small (∼0.5 eV), delayed fluorescence via RISC becomes feasible.48,49 For example, the energy gap of p-PtBTD between 1PtBTD* and 3PtBTD* is ∼0.64 eV, which decreases further to ∼0.49 eV under ECL conditions (Fig. 5d). The low Ea could reflect the minimal energy barrier needed for interchain charge transfer or exciton coupling. If such coupling enhances spin–orbit interactions or facilitates interconversion between T1 and S1, it indirectly supports the feasibility of RISC. The slower, delayed decay component (τ2) has a higher activation energy (Ea = 13.72 meV) which is two times greater than that of τ1, which aligns with mechanisms involving triplet exciton diffusion and annihilation (TTA).50 Consequently, dual ECL emission arises from both TTA and RISC mechanisms, producing fluorescence and phosphorescence.
meV) for exciton transfer between molecules. Furthermore, previous studies have shown that when the energy gap between singlet and triplet excited states is small (∼0.5 eV), delayed fluorescence via RISC becomes feasible.48,49 For example, the energy gap of p-PtBTD between 1PtBTD* and 3PtBTD* is ∼0.64 eV, which decreases further to ∼0.49 eV under ECL conditions (Fig. 5d). The low Ea could reflect the minimal energy barrier needed for interchain charge transfer or exciton coupling. If such coupling enhances spin–orbit interactions or facilitates interconversion between T1 and S1, it indirectly supports the feasibility of RISC. The slower, delayed decay component (τ2) has a higher activation energy (Ea = 13.72 meV) which is two times greater than that of τ1, which aligns with mechanisms involving triplet exciton diffusion and annihilation (TTA).50 Consequently, dual ECL emission arises from both TTA and RISC mechanisms, producing fluorescence and phosphorescence.
In summary, the dual ECL emission of p-PtBTD results from the coexistence of TTA and RISC. TTA contributes to fluorescence by converting triplet excitons into singlet states, while RISC provides an efficient mechanism for triplet-to-singlet conversion due to the small energy gap. Together, these processes explain the observed fluorescence and phosphorescence in the ECL spectrum.
Conclusions
In summary, the photophysical and electrochemical properties of the platinum-acetylide polymer p-PtBTD were thoroughly investigated using a combination of spectroscopic techniques, cyclic voltammetry, and electrochemiluminescence measurements. The UV-Vis absorption and PL spectra revealed strong fluorescence at 584 nm, with a minor phosphorescence peak observed near 800 nm. Transient absorption studies provided insights into exciton dynamics, suggesting rapid intersystem crossing (ISC) and the presence of triplet excited states. Temperature-dependent TRPL studies further confirmed the TADF behavior of p-PtBTD, with distinct fast and slow decay components attributed to singlet and triplet excitons, respectively. Electrochemical studies demonstrated the irreversible oxidation and reduction processes, with diffusion-controlled electron transfer kinetics. The ECL studies revealed dual emission peaks corresponding to singlet and triplet states, with enhancement in the presence of the co-reactant TPrA. The dual ECL emission is attributed to the combination of phosphorescence from triplet states and fluorescence from singlet states, likely facilitated by both TTA from the T-route mechanism and RISC due to the small energy gap (∼0.5 eV) between the singlet and triplet excited states. This work provides a comprehensive understanding of the excited-state dynamics and electron-transfer mechanisms in p-PtBTD, highlighting its potential for applications in optoelectronic devices and ECL-based sensing technologies.
Data availability
Data available on request from the authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was partially supported by the Research Grants Council of Hong Kong (grant no. CityU 21203518 and F-CityU106/18), Innovation and Technology Commission (grant no. MHP/104/21), Shenzhen Science Technology and Innovation Commission (grant no. JCYJ20210324125612035, R-IND12303, and R-IND12304), and City University of Hong Kong (grant no. 7005289, 7005580, 7005720, 9667213, 9667229, 9680331 and 9678291). W. N. acknowledges financial support from National Natural Science Foundation of China (grant no. 21974131). Z. H. thanks the National Key Research and Development Program of China (no. 2021YFA1500900) and the National Natural Science Foundation of China (no. 52071174). Y. T. acknowledges the financial support from the Start-up fund of SUSTech (grant no. Y01256114). X. X. thanks the support from Guangdong Provincial Science and Technology Plan Project (grant no. 2023A0505010003) and Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDA 21061001). C. H. M. is thankful for the financial support from the Hong Kong Jockey Club under the research work Hong Kong JC STEM Lab for Circular Bio-economy (Project No. 2023-0078).
References
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Highly efficient phosphorescent emission from organic electroluminescent devices, Nature, 1998, 395(6698), 151–154 Search PubMed.
- G. G. Dubinina, R. S. Price, K. A. Abboud, G. Wicks, P. Wnuk, Y. Stepanenko, M. Drobizhev, A. Rebane and K. S. Schanze, Phenylene vinylene platinum(II) acetylides with prodigious two-photon absorption, J. Am. Chem. Soc., 2012, 134(47), 19346–19349 Search PubMed.
- M. Du, L. Zhang, Y. Meng, J. Chen and F. Liu, Impact of Surface Chemistry on Emulsion–Electrode Interactions and Electron-Transfer Kinetics in the Single-Entity Electrochemistry, Anal. Chem., 2024, 96(3), 1038–1045, DOI:10.1021/acs.analchem.3c03462.
- Y. Peng, Q. Ruan, C. H. Lam, F. Meng, C.-Y. Guan, S. P. Santoso, X. Zou, T. Y. Edward, P. K. Chu and H.-Y. Hsu, Plasma-implanted Ti-doped hematite photoanodes with enhanced photoelectrochemical water oxidation performance, J. Alloys Compd., 2021, 870, 159376 Search PubMed.
- L. Fu, K. Fu, X. Gao, S. Dong, B. Zhang, S. Fu, H.-Y. Hsu and G. Zou, Enhanced Near-Infrared Electrochemiluminescence from Trinary Ag–In–S to Multinary Ag–Ga–In–S Nanocrystals via Doping-in-Growth and Its Immunosensing Applications, Anal. Chem., 2021, 93(4), 2160–2165, DOI:10.1021/acs.analchem.0c03975.
- F. Rizzo, F. Polo, G. Bottaro, S. Fantacci, S. Antonello, L. Armelao, S. Quici and F. Maran, From Blue to Green: Fine-Tuning of Photoluminescence and Electrochemiluminescence in Bifunctional Organic Dyes, J. Am. Chem. Soc., 2017, 139(5), 2060–2069 Search PubMed.
- P. Huang, B. Zhang, Q. Hu, B. Zhao, Y. Zhu, Y. Zhang, Y. Kong, Z. Zeng, Y. Bao and W. Wang, Polymer electrochemiluminescence featuring thermally activated delayed fluorescence, ChemPhysChem, 2021, 22(8), 726–732 Search PubMed.
- N. Wang, Z. Wang, L. Chen, W. Chen, Y. Quan, Y. Cheng and H. Ju, Dual resonance energy transfer in triple-component polymer dots to enhance electrochemiluminescence for highly sensitive bioanalysis, Chem. Sci., 2019, 10(28), 6815–6820 RSC.
- S. Rao, X. Zou, S. Wang, Y. Lu, T. Shi, H.-Y. Hsu, Q. Xu and X. Lu, Electrodeposition of Ni-Cu alloy films from nickel matte in deep eutectic solvent, Mater. Chem. Phys., 2019, 232, 6–15 Search PubMed.
- X. Yang, J. Yang, M. I. Ullah, Y. Xia, G. Liang, S. Wang, J. Zhang, H.-Y. Hsu, H. Song and J. Tang, Enhanced Passivation and Carrier Collection in Ink-Processed PbS Quantum Dot Solar Cells via a Supplementary Ligand Strategy, ACS Appl. Mater. Interfaces, 2020, 12(37), 42217–42225 Search PubMed.
- L. Cui, S. Yu, W. Gao, X. Zhang, S. Deng and C.-Y. Zhang, Tetraphenylenthene-based conjugated microporous polymer for aggregation-induced electrochemiluminescence, ACS Appl. Mater. Interfaces, 2020, 12(7), 7966–7973 Search PubMed.
-
A. J. Bard, Electrogenerated chemiluminescence, CRC Press, 2004 Search PubMed.
- C. H. Mak, R. Liu, X. Han, Y. Tang, X. Zou, H. H. Shen, Y. Meng, G. Zou and H. Y. Hsu, Thermally Activated Delayed Phosphorescence and Interchromophore Exciton Coupling in a Platinum-Based Organometallic Emitter, Adv. Opt. Mater., 2020, 8(20), 2001023 Search PubMed.
- G. Ramakrishna, T. Goodson III, J. E. Rogers-Haley, T. M. Cooper, D. G. McLean and A. Urbas, Ultrafast intersystem crossing: excited state dynamics of platinum acetylide complexes, J. Phys. Chem. C, 2009, 113(3), 1060–1066 Search PubMed.
- B. A. Neto, A. A. Lapis, E. N. da Silva Júnior and J. Dupont, 2,1,3-Benzothiadiazole and Derivatives: Synthesis, Properties, Reactions, and Applications in Light Technology of Small Molecules, Eur. J. Org. Chem., 2013, 228–255 CrossRef CAS.
- H.-Y. Hsu, L. Ji, C. Zhang, C. H. Mak, R. Liu, T. Wang, X. Zou, S.-Y. Leu and E. T. Yu, Ultra-stable 2D layered methylammonium cadmium trihalide perovskite photoelectrodes, J. Mater. Chem. C, 2018, 6(43), 11552–11560, 10.1039/C8TC02153H.
- J. Yuan, C. Zhang, Y. Zhen, Y. Zhao and Y. Li, Enhancing the performance of an all-organic non-aqueous redox flow battery, J. Power Sources, 2019, 443, 227283 Search PubMed.
- Y. Tang, C. H. Mak, R. Liu, Z. Wang, L. Ji, H. Song, C. Tan, F. Barrière and H. Y. Hsu,
In situ formation of bismuth-based perovskite heterostructures for high-performance cocatalyst-free photocatalytic hydrogen evolution, Adv. Funct. Mater., 2020, 2006919 CrossRef CAS.
- H.-Y. Hsu, L. Ji, M. Du, J. Zhao, T. Y. Edward and A. J. Bard, Optimization of Lead-free Organic–inorganic Tin (II) Halide Perovskite Semiconductors by Scanning Electrochemical Microscopy, Electrochim. Acta, 2016, 220, 205–210 Search PubMed.
- R. Liu, C. H. Mak, X. Han, Y. Tang, G. Jia, K.-C. Cheng, H. Qi, X. Zou, G. Zou and H.-Y. Hsu, Efficient electronic coupling and heterogeneous charge transport of zero-dimensional Cs4PbBr6 perovskite emitters, J. Mater. Chem. A, 2020, 8(45), 23803–23811, 10.1039/D0TA06076C.
- M. Shen, J. N. Rodriguez-Lopez, J. Huang, Q. Liu, X.-H. Zhu and A. J. Bard, Electrochemistry and electrogenerated chemiluminescence of dithienylbenzothiadiazole derivative. differential reactivity of donor and acceptor groups and simulations of radical cation–anion and dication–radical anion annihilations, J. Am. Chem. Soc., 2010, 132(38), 13453–13461 CrossRef CAS PubMed.
- Y. Peng, C. H. Mak, J.-J. Kai, M. Du, L. Ji, M. Yuan, X. Zou, H.-H. Shen, S. P. Santoso and J. C. Colmenares,
et al., Recent progress on post-synthetic treatments of photoelectrodes for photoelectrochemical water splitting, J. Mater. Chem. A, 2021, 9(47), 26628–26649, 10.1039/D1TA05935A.
- Y. Tang, C. H. Mak, G. Jia, K.-C. Cheng, J.-J. Kai, C.-W. Hsieh, F. Meng, W. Niu, F.-F. Li and H.-H. Shen,
et al., Lead-free hybrid perovskite photocatalysts: surface engineering, charge-carrier behaviors, and solar-driven applications, J. Mater. Chem. A, 2022, 10(23), 12296–12316, 10.1039/D2TA01170K.
- Y. Peng, M. Du, X. Zou, G. Jia, S. Permatasari Santoso, X. Peng, W. Niu, M. Yuan and H.-Y. Hsu, Suppressing photoinduced charge recombination at the BiVO4||NiOOH junction by sandwiching an oxygen vacancy layer for efficient photoelectrochemical water oxidation, J. Colloid Interface Sci., 2022, 608, 1116–1125, DOI:10.1016/j.jcis.2021.10.063.
- K. M. Omer, S.-Y. Ku, J.-Z. Cheng, S.-H. Chou, K.-T. Wong and A. J. Bard, Electrochemistry and electrogenerated chemiluminescence of a spirobifluorene-based donor (triphenylamine)− acceptor (2, 1, 3-benzothiadiazole) molecule and its organic nanoparticles, J. Am. Chem. Soc., 2011, 133(14), 5492–5499 Search PubMed.
- K. M. Omer, S.-Y. Ku, K.-T. Wong and A. J. Bard, Green electrogenerated chemiluminescence of highly fluorescent benzothiadiazole and fluorene derivatives, J. Am. Chem. Soc., 2009, 131(30), 10733–10741 CrossRef CAS.
- Y. Liu, S. J. Jiang, K. Glusac, D. H. Powell, D. F. Anderson and K. S. Schanze, Photophysics of monodisperse platinum-acetylide oligomers: Delocalization in the singlet and triplet excited states, J. Am. Chem. Soc., 2002, 124(42), 12412–12413 CrossRef CAS PubMed.
- M. S. Khan, M. K. Al-Suti, M. R. Al-Mandhary, B. Ahrens, J. K. Bjernemose, M. F. Mahon, L. Male, P. R. Raithby, R. H. Friend and A. Köhler, Synthesis and characterisation of new acetylide-functionalised aromatic and hetero-aromatic ligands and their dinuclear platinum complexes, Dalton Trans., 2003, 65–73 RSC.
- Y. Tang, C. H. Mak, J. Zhang, G. Jia, K.-C. Cheng, H. Song, M. Yuan, S. Zhao, J.-J. Kai and J. C. Colmenares,
et al., Unravelling the Interfacial Dynamics of Bandgap Funneling in Bismuth-Based Halide Perovskites, Adv. Mater., 2023, 35(2), 2207835, DOI:10.1002/adma.202207835.
- A. Lapprand, N. Khiri, D. Fortin, S. Jugé and P. D. Harvey, Organometallic Oligomers Based on Bis (arylacetylide) bis (p-chirogenic phosphine) platinum(II) Complexes: Synthesis and Photonic Properties, Inorg. Chem., 2013, 52(5), 2361–2371 CrossRef CAS PubMed.
- J. Feng, C. H. Mak, G. Jia, B. Han, H.-H. Shen, S. P. Santoso, J.-J. Kai, M. Yuan, H. Song and J. C. Colmenares,
et al., Unlocking Interfacial Interactions of In Situ Grown Multidimensional Bismuth-Based Perovskite Heterostructures for Photocatalytic Hydrogen Evolution, Adv. Energy Mater., 2024, 14(43), 2402785, DOI:10.1002/aenm.202402785.
- Y. Tang, C. H. Mak, C. Wang, Y. Fu, F.-F. Li, G. Jia, C.-W. Hsieh, H.-H. Shen, J. C. Colmenares and H. Song,
et al., Bandgap Funneling in Bismuth-Based Hybrid Perovskite Photocatalyst with Efficient Visible-Light-Driven Hydrogen Evolution, Small Methods, 2022, 6(8), 2200326, DOI:10.1002/smtd.202200326.
- Y. Liu, S. Jiang, K. Glusac, D. H. Powell, D. F. Anderson and K. S. Schanze, Photophysics of monodisperse platinum-acetylide oligomers: Delocalization in the singlet and triplet excited states, J. Am. Chem. Soc., 2002, 124(42), 12412–12413 Search PubMed.
- E. R. Batista and R. L. Martin, Exciton Localization in a Pt− Acetylide Complex, J. Phys. Chem. A, 2005, 109(43), 9856–9859 Search PubMed.
- R. A. Marcus, On the theory of oxidation-reduction reactions involving electron transfer. I, J. Chem. Phys., 1956, 24(5), 966–978 Search PubMed.
- H.-Y. Hsu, J. H. Vella, J. D. Myers, J. Xue and K. S. Schanze, Triplet exciton diffusion in platinum polyyne films, J. Phys. Chem. C, 2014, 118(42), 24282–24289 Search PubMed.
- R. Liu, C. H. Mak, X. Han, Y. Tang, G. Jia, K.-C. Cheng, H. Qi, X. Zou, G. Zou and H.-Y. Hsu, Efficient electronic coupling and heterogeneous charge transport of zero-dimensional Cs 4 PbBr 6 perovskite emitters, J. Mater. Chem. A, 2020, 8(45), 23803–23811 Search PubMed.
- J. Feng, C. H. Mak, G. Jia, B. Han, H. H. Shen, S. P. Santoso, J. J. Kai, M. Yuan, H. Song and J. C. Colmenares, Unlocking Interfacial Interactions of In Situ Grown Multidimensional Bismuth-Based Perovskite Heterostructures for Photocatalytic Hydrogen Evolution, Adv. Energy Mater., 2024, 2402785 Search PubMed.
- Z. Chen, H.-Y. Hsu, M. Arca and K. S. Schanze, Triplet energy transport in platinum-acetylide light harvesting arrays, J. Phys. Chem. B, 2015, 119(24), 7198–7209 Search PubMed.
-
J. Wang, Analytical Electrochemistry, John Willey and Sons. Inc., New York, 2000, pp. 81–84 Search PubMed.
- N. K. Bhatti, M. S. Subhani, A. Y. Khan, R. Qureshi and A. Rahman, Heterogeneous electron transfer rate constants of viologen at a platinum disk electrode, Turk. J. Chem., 2006, 29(6), 659–668 Search PubMed.
- N. K. Bhatti, M. S. Subhani, A. Y. Khan, R. Qureshi and A. Rahman, Heterogeneous electron transfer rate constants of viologen monocations at a platinum disk electrode, Turk. J. Chem., 2006, 30(2), 165–180 Search PubMed.
- W. Miao, Electrogenerated chemiluminescence and its biorelated applications, Chem. Rev., 2008, 108(7), 2506–2553 Search PubMed.
- H. Qi, Y.-H. Chen, C.-H. Cheng and A. J. Bard, Electrochemistry and Electrogenerated Chemiluminescence of Three Phenanthrene Derivatives, Enhancement of Radical Stability, and Electrogenerated Chemiluminescence Efficiency by Substituent Groups, J. Am. Chem. Soc., 2013, 135(24), 9041–9049 CAS.
- T. Nobeshima, T. Morimoto, K. Nakamura and N. Kobayashi, Advantage of an AC-driven electrochemiluminescent cell containing a Ru (bpy) 32+ complex for quick response and high efficiency, J. Mater. Chem., 2010, 20(47), 10630–10633 RSC.
- P. J. Smith and C. K. Mann, Electrochemical dealkylation of aliphatic amines, J. Org. Chem., 1969, 34(6), 1821–1826 CAS.
- R. Y. Lai and A. J. Bard, Electrogenerated chemiluminescence. 70. The application of ECL to determine electrode potentials of tri-n-propylamine, its radical cation, and intermediate free radical in MeCN/benzene solutions, J. Phys. Chem. A, 2003, 107(18), 3335–3340 CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492(7428), 234 CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Triplet harvesting with 100% efficiency by way of thermally activated delayed fluorescence in charge transfer OLED emitters, Adv. Mater., 2013, 25(27), 3707–3714 CAS.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Towards efficient solid-state triplet–triplet annihilation based photon upconversion: Supramolecular, macromolecular and self-assembled systems, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Yaojia
Ai
ab,
Yaojia
Ai



 , where Ea is the activation energy and τ is the lifetime.
, where Ea is the activation energy and τ is the lifetime.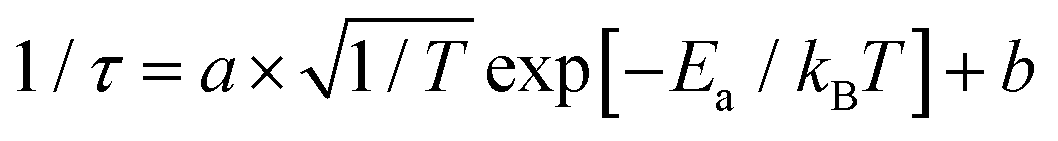 (Fig. 3b–d and Table 1). The pre-exponential factor from the fit-to equation yields temperature-independent electronic coupling matrix elements |HAB|, whose values represent the overlap of the excited-state wave functions between the initial and final sites, and are determined to be 6.34 × 10−3 cm−1 and 6.68 × 10−3 cm−1. The extracted activation energies provide evidence for the localization of the singlet excited state. These values reinforce the role of these processes in the dual-emission behavior of p-PtBTD.
(Fig. 3b–d and Table 1). The pre-exponential factor from the fit-to equation yields temperature-independent electronic coupling matrix elements |HAB|, whose values represent the overlap of the excited-state wave functions between the initial and final sites, and are determined to be 6.34 × 10−3 cm−1 and 6.68 × 10−3 cm−1. The extracted activation energies provide evidence for the localization of the singlet excited state. These values reinforce the role of these processes in the dual-emission behavior of p-PtBTD.





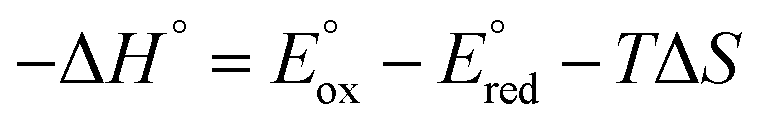 , where
, where  , and
, and  Considering the entropy effect (estimated at 0.1 eV), the corrected values are 1.13 eV and 1.76 eV, respectively. In the case of the co-reactant TPrA, the potential of the TPrA˙ radical is 1.5 V vs. NHE,47 yielding a thermodynamic potential of 2.03 eV. The enthalpies of the radical ion annihilation reactions (i.e., 1.13 eV, 1.76 eV, and 2.03 eV) are all smaller than the energy required to populate the singlet excited state (2.12 eV), suggesting that the ECL of p-PtBTD is likely processed via the T-route (energy-deficient system).
Considering the entropy effect (estimated at 0.1 eV), the corrected values are 1.13 eV and 1.76 eV, respectively. In the case of the co-reactant TPrA, the potential of the TPrA˙ radical is 1.5 V vs. NHE,47 yielding a thermodynamic potential of 2.03 eV. The enthalpies of the radical ion annihilation reactions (i.e., 1.13 eV, 1.76 eV, and 2.03 eV) are all smaller than the energy required to populate the singlet excited state (2.12 eV), suggesting that the ECL of p-PtBTD is likely processed via the T-route (energy-deficient system).