
Synthesis of semi-rigid-biobased polyesters from renewable furanic cyclobutane diacid
Luan Moreira
Grilo
ab,
Sara
Faoro
 ac,
Beatriz
Agostinho
d,
Andreia F.
Sousa
d,
Nathanael
Guigo
e,
Katja
Loos
a,
Dina
Maniar
*a and
Talita Martins
Lacerda
*b
ac,
Beatriz
Agostinho
d,
Andreia F.
Sousa
d,
Nathanael
Guigo
e,
Katja
Loos
a,
Dina
Maniar
*a and
Talita Martins
Lacerda
*b
aMacromolecular Chemistry and New Polymeric Materials, Zernike Institute for Advanced Materials, Faculty of Science and Engineering, University of Groningen, The Netherlands. E-mail: d.maniar@rug.nl
bBiotechnology Department, Lorena School of Engineering, University of São Paulo, Brazil. E-mail: talitalacerda@usp.br
cCenter for Education and Research on Macromolecules, CESAM Research Unit, University of Liege, Belgium
dCICECO-Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Portugal
eMines Paris, PSL University, CNRS, CEMEF (UMR 7635), 06904 Sophia Antipolis Cedex, France
First published on 12th August 2025
Abstract
The pursuit of novel sustainable materials is driving advancements in polymer science, with the consolidation of furfural and hydroxymethylfurfural derivatives as key renewable building blocks. 3,4-Di(furan-2-yl)cyclobutane-1,2-dicarboxylic acid (CBDA) is a highly promising rigid-structure biobased monomer that is readily synthesized from furfural. In this study, we expanded the scope of CBDA-based polymers by synthesizing this platform molecule and investigating its polymerization with a series of aliphatic diols of varying chain lengths. CBDA was successfully synthesized from furfural-derived 3-(2-furyl)acrylic acid through a green and efficient UV-mediated solid-state dimerization reaction. Subsequent polymerization was carried out in bulk via a two-step method. The success of the polymerization was confirmed through ATR-FTIR, 1H NMR, and 13C CP/MAS NMR spectroscopy. The resulting polymers presented average molecular weights ( ) of up to 11
) of up to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1. Thermogravimetric analysis (TGA) revealed good thermal stability, with a Td10% ranging from 263 to 284 °C and 50% weight retention observed up to 388 °C. Furthermore, DSC analysis indicated that the glass transition temperature of the polymers could be tailored, varying from 6 to 52 °C depending on the chain length of the utilized diol. These results underscore the potential of CBDA as a renewable rigid monomer for the development of sustainable biobased materials.
200 g mol−1. Thermogravimetric analysis (TGA) revealed good thermal stability, with a Td10% ranging from 263 to 284 °C and 50% weight retention observed up to 388 °C. Furthermore, DSC analysis indicated that the glass transition temperature of the polymers could be tailored, varying from 6 to 52 °C depending on the chain length of the utilized diol. These results underscore the potential of CBDA as a renewable rigid monomer for the development of sustainable biobased materials.
Introduction
Humans’ dependence on fossil resources as feedstocks for fuels, chemicals, and materials has led to numerous environmental concerns, such as the direct relationship between the exploitation of such resources and greenhouse gas emissions, which severely affect the global climate.1,2 Furthermore, the sources of these raw materials, namely petroleum and gas reserves, are unequally distributed worldwide, and their supply and price are volatile and subjected to political speculation.3 Thus, lignocellulosic biomass has become an increasingly attractive feedstock, owing to its widespread availability from agricultural, forestry, and certain industrial waste streams, with an estimated annual global production exceeding 200 billion metric tons.4,5 In addition to being inexpensive and abundant, its renewable nature plays a crucial role in promoting a more circular and sustainable economy.6 As a result, research on biomass valorization has flourished in recent decades, driven by the growing importance of developing strategies to convert this complex feedstock into value-added chemicals.7In this context, the search for biobased platform chemicals to develop innovative renewable polymers is progressing rapidly. Notably, furfural (F) and hydroxymethylfurfural (HMF), derived from lignocellulosic feedstocks, stand out as versatile platform molecules with broad applications in the synthesis of high-value biobased compounds.8–13 With the advancement of a new generation of biobased polymers to potentially replace their fossil-based counterparts, certain F and HMF derivatives have gained popularity as renewable building blocks. These include molecules such as furfuryl alcohol, employed in resins and composites;14–17 furfurylamine, used in polyamides and polyureas;18–21 furfuryl methacrylate, utilized in polymers and composites;22–24 2,5-bis(hydroxymethyl)furan (BHMF), applied in polyesters;25–28 and 2,5-furandicarboxylic acid (FDCA), used in polyamides and polyesters, most notably poly(ethylene furanoate), a renewable alternative to poly(ethylene terephthalate) (PET).29–35 These relevant furan building blocks are depicted in Fig. 1.
 | ||
| Fig. 1 Common furan building blocks for biobased materials. | ||
3-(2-Furyl)acrylic (FAA) acid is another furfural derivative with interesting potential applications. As a photoactive molecule, it can easily undergo a [2 + 2] photocycloaddition reaction under UV light to produce its dimer 3,4-di(furan-2-yl)cyclobutane-1,2-dicarboxylic acid (CBDA).36 This dimerization phenomenon is also observed in other molecules, such as cinnamic acid and its analogs, including 3-(2-thienyl)acrylic acid and 3-(3-pyridyl)acrylic acid, as well as in heteroarylene-vinylenes and related compounds.37–40 These reactions consistently yield substituted cyclobutane derivatives, which are of significant interest because of their versatile applications. The cyclobutane structure, in particular, has found unique utility across a wide range of research fields, including materials science, fuel technology, and pharmaceutical synthesis.41–45 CBDA has relevant potential to serve as a biobased rigid diacid building block, with previous studies demonstrating its suitability as a polycondensing monomer.36,46 Moreover, when used as hardener with a flexible epoxidized vegetable oil, it formed a crosslinked network that exhibited high glass transition temperature, high strength, and relatively good toughness.47,48 Additionally, in many of these cyclobutane molecules, the ring-forming bonds are reversible and can be cleaved under specific photochemical, mechanical, and/or thermal stimuli.49–53 These characteristics make them promising candidates as platform molecules for the design of degradable or recyclable polymers, which are in growing demand as part of strategies to achieve more sustainable end-of-life management of polymeric waste.54–57
Therefore, this work further explores CBDA as a platform molecule for polymer synthesis, expanding the library of linear CBDA-based polyesters by pairing it with an assortment of aliphatic diols and obtaining polymers with tunable thermal properties. CBDA was synthesized via the solid-sated suspension photocycloaddition of FAA, using commercially available UVA LEDs and recycling the reaction medium of the process. Then, CBDA was polymerized in bulk with different diols to avoid the excess use of organic solvents, using titanium(IV) isopropoxide (TTIP) as catalyst. Enzymatic polymerization of the ethyl ester of CBDA was also attempted, employing immobilized Candida antarctica lipase B (CALB) as catalyst. ATR-FTIR, 1H NMR, 13C CP/MAS NMR, TGA, DSC, and WAXD were used to characterize the synthesized polymers.
Experimental section
Materials
3-(2-Furyl) acrylic acid (FAA, 99%), 1,4-butanediol (99%), 1,6-hexanediol (99%), 1,8-octanediol (98%), 1,10-decanediol (98%), titanium(IV) isopropoxide (TTIP, 99.999%), trifluoroacetic acid (TFA, 99%), triethylamine (TEA, 99.5%), oxalyl chloride (99%), Novozyme 435 (N435, Candida antarctica lipase B immobilized in acrylic resin, recombinant, expressed in Aspergillus niger, ≥5000 U g−1), molecular sieves (4 Å), deuterated dimethyl sulfoxide (DMSO-d6, 99.9%), and deuterated chloroform (CDCl3, 99.8%), were purchased from Sigma-Aldrich. N-Hexane (99%), chloroform (CHCl3, 99.5%), methanol (anhydrous, 99.8%), ethanol (anhydrous, 99.8%), tetrahydrofuran (THF, 99.5%), toluene (anhydrous, 99.8%), N,N-dimethylformamide (DMF, 99.8%), and ethyl acetate (99%) were purchased from Avantor. N435 was pre-dried in the presence of phosphorus pentoxide in a desiccator at room temperature under a high vacuum for 12 h. The molecular sieves (4 Å) were pre-activated at 250 °C under vacuum. The other chemicals were used as received without further purification.Analytical methods
Proton nuclear magnetic resonance (1H NMR) spectra were acquired at 25 °C using a Bruker Avance NMR spectrometer operating at 600 MHz. Chemical shifts were recorded in parts per million (ppm) relative to the solvent peak. Spectral analysis was performed using MestreNova 14 software.Solid-state carbon-13 cross-polarization/magic angle spinning nuclear magnetic resonance (13C CP/MAS NMR) spectra were acquired at 25 °C via a Bruker Avance NMR spectrometer operating at 400 MHz. Spectral analysis was performed via MestreNova 14 software.
Attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectra were recorded via a Bruker VERTEX 70 spectrometer equipped with a Platinum-ATR diamond single-reflection accessory. Measurements were conducted with a resolution of 4 cm−1, and spectra were collected over a 4000–400 cm−1 range, with 16 scans per sample. Atmospheric compensation and baseline correction were applied to the spectra using OPUS spectroscopy software (v7.0, Bruker Optics).
Thermogravimetric analysis (TGA) was conducted via a TA Instruments D2500 TGA instrument. The samples were heated from 25 °C to 700 °C at a rate of 10 °C min−1 under a nitrogen atmosphere (at a N2 flow rate of 50 mL min−1). Prior to the TGA measurement, each sample was preheated to 100 °C and held at that temperature for 20 min to remove residual moisture and solvents.
Differential scanning calorimetry (DSC) was employed to assess the thermal transitions of the synthesized polyesters. Measurements were carried out using a TA Instruments Q1000 DSC. Samples weighing 10–15 mg were placed in nonhermetically sealed aluminum pans. Heating–cooling–heating cycles were conducted from −50 °C to 200 °C at a rate of 10 °C min−1, under a mild nitrogen flow rate of 25 mL min−1.
Wide-angle X-ray diffraction (WAXD) patterns of the synthesized polyesters were recorded using a Bruker D8 Advance diffractometer with CuKα radiation (λ = 0.1542 nm). Data were collected at room temperature in the angular range of 5–50° (2θ).
Synthesis of 3,4-di(furan-2-yl)cyclobutane-1,2-dicarboxylic acid (CBDA)
CBDA was synthesized as previously reported.36,48 3-(2-furyl) acrylic acid powder (FAA, 12 g, 86.9 mmol) was dispersed in 250 mL of hexane and transferred to a quartz Erlenmeyer flask. The suspension was stirred and irradiated with three consumer-grade LED UV lamps (365 nm, 135 W) for 48 h. Occasionally, the walls of the flask were scraped off to remove some attached solid material, and extra hexane was added to keep the volume approximately constant, considering its evaporation. To confirm that the FAA conversion exceeded 95% and determine when to stop the reaction, 1 mL samples were collected after 24 h and 48 h for 1H NMR analysis. The product was isolated via filtration and dried under vacuum. CBDA was obtained as a pale-beige solid, and the remaining hexane was recovered for reuse.Melt polymerization of CBDA
CDBA was bulk polymerized with different diols—1,4-butanediol, 1,6-hexanediol, 1,8-octanediol, or 1,10-decanediol—in a two-step process, producing the polymers PBCB, PHCB, POCB, and PDCB, respectively. Firstly, 2 g (7.24 mmol) of CBDA, 7.24 mmol of diol, and a magnetic stirrer were added to a two-neck round bottom flask attached to a short path vacuum distillation setup, and the whole system was flushed with N2. The temperature was gradually increased to 160 °C at a rate of about 4 °C min−1, after which 0.09 mmol of TTIP was added. The mixture was kept under N2 atmosphere at 160 °C for 4 h with constant stirring. Then, the temperature was increased to 180 °C at approximately 2 °C min−1, followed by vacuum application for 2 h. After the second reaction step ended and the mixture had cooled down to room temperature, approximately 25 mL of chloroform and approximately 5 drops of TFA were added to the reaction flask. The mixture was stirred vigorously and left to dissolve overnight. However, in all the cases, the polymers did not fully dissolve in the solvent mixture, resulting primarily in a suspension of small polymer particles in the chloroform/TFA solution. This suspension was subsequently precipitated in cold methanol. Then, the polymers were separated from the supernatant by centrifugation and were left to dry overnight in a vacuum oven at 45 °C.Enzymatic polymerization of CBDA-based diester
First, CBDA was converted to its diethyl ester (CBDE), as previously reported.36 In a round-bottom flask, 2.5 g of CBDA (9.1 mmol) was dissolved in 90 mL of dry THF and purged with nitrogen. Then, 3.11 mL of oxalyl chloride (36.4 mmol) and two drops of DMF were added to the flask, and the solution was stirred for 1 h under an inert atmosphere. After this period, the THF and excess oxalyl chloride were removed under vacuum to obtain CBDA acyl chloride. TEA (2.86 mL, 20.5 mmol) was dissolved in 46 mL of anhydrous ethanol (0.78 mol) and slowly added, via a dropping funnel, into the flask containing CBDA acyl chloride over an ice bath. After this addition, the reaction mixture was stirred for 2 h at room temperature. The mixture was then filtered to remove any solid impurities, and the solvent in the filtrate was evaporated under vacuum, yielding CBDE as a viscous brownish-yellow oil. Flash column chromatography (ethyl acetate:hexane = 2:98) was used to purify the diester monomer further.
Following the CBDE synthesis, its enzymatic polymerization with aliphatic diols was carried out based on the one-step procedure reported by Jiang et al. (2016).33 First, in a 25 mL round-bottom flask, 0.5 g of CBDE (1.5 mmol) and 1.5 mmol of a diol (1,4-butanediol, 1,6-hexanediol, 1,8-octanediol, or 1,10-decanediol) were dissolved in anhydrous toluene (500 wt% relative to all monomers). Then, pre-dried N435 (20 wt%) and pre-activated 4 Å molecular sieves (200 wt%) were added, and the flask was purged with nitrogen. The flask was sealed and placed in an oil bath at 90 °C for 72 h under magnetic stirring. After this period, the toluene was evaporated under a stream of air at room temperature, and the resulting mixture was dissolved in 15 mL of chloroform. Next, N435 and molecular sieves were removed by filtration, and the filter paper and its contents were washed three times, with 10 mL of chloroform each. The excess chloroform in the filtrate was removed under vacuum, and the concentrated solution was added dropwise into an excess of cold methanol to precipitate the synthesized polymers. However, no precipitation occurred, so the crude reaction mixture was kept for 1H NMR analysis.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations), 1414 (O–H bending vibrations).
O stretching vibrations); 1163–1161 (C–O stretching vibrations); 1012 (C–O–C ring vibrations); 796–800, 733–727 (C–H out-of-plane deformation vibrations).
O stretching vibrations), 1414 (O–H bending vibrations).
O stretching vibrations); 1163–1161 (C–O stretching vibrations); 1012 (C–O–C ring vibrations); 796–800, 733–727 (C–H out-of-plane deformation vibrations).
Results and discussion
CBDA was successfully synthesized via photocycloaddition of FAA (Scheme 1) based on a previously reported procedure.36,48 The reaction conversion was determined to be approximately 95% (calculated by 1H NMR – Fig. S1). It is important to highlight the green aspects of this process, including the recyclability of the reaction medium and the use of commercially available UVA LEDs. These LEDs can achieve an electrical efficiency of 40–50%, surpassing that of traditional mercury lamps.58 Furthermore, LEDs have a significantly lower environmental impact than mercury lamps do,59 mainly due to the impacts of the production of the latter.60 | ||
| Scheme 1 Synthesis of CBDA. | ||
Polyesters were synthesized from CBDA and different diols in a two-step bulk polycondensation process based on literature reports (Scheme 2).29,61–63 In the first step, consisting on a prepolymerization, a catalyst is introduced to promote the esterification reaction, forming low-molecular-weight products (oligomers). This step is critical as it enables the monomers to react under milder conditions, producing short chains with higher boiling points and improved thermal stability compared to the starting monomers. Subsequently, elevated temperatures and reduced pressure are typically applied to achieve higher molecular weight polymers. Since esterification is an equilibrium reaction, removing the byproduct—water, in this case—is crucial to shift the equilibrium forward and drive the formation of polymers. The duration of the second step was determined by visually monitoring the viscosity of the reaction mixture in the PBCB sample, stopping the reaction when the mixture solidified. This timeframe was then applied consistently across the other samples. At the end of each polymerization reaction, the recovered polymers were weighed, and the yields were calculated (Table 1). Notably, the obtained polymers exhibited partial solubility in the standard CHCl3/TFA mixture used for recovery and were insoluble in other common organic solvents, including DMSO, DMF, and THF.
 | ||
| Scheme 2 Two-step bulk polycondensations of CBDA and different diols. | ||
| Polyester | Abbreviation | Average yielda (%) |
|
|
|---|---|---|---|---|
a These values are the average of two experimental results.
b These values were estimated through 1H NMR analysis via the following equations:  and and  , where IC is the integral intensity of the peak assigned to the furanic protons of the repeating unit (C, at 5.93 ppm), II is the integral intensity of the peak assigned to the methylene protons of the hydroxymethyl end groups (I, approximately 3.45 ppm), mC is the number of protons C present in the repeating unit, mI is the number of protons I, nI is the number of hydroxymethyl end groups, nR indicates the number of repeating units , where IC is the integral intensity of the peak assigned to the furanic protons of the repeating unit (C, at 5.93 ppm), II is the integral intensity of the peak assigned to the methylene protons of the hydroxymethyl end groups (I, approximately 3.45 ppm), mC is the number of protons C present in the repeating unit, mI is the number of protons I, nI is the number of hydroxymethyl end groups, nR indicates the number of repeating units  , MR is the molecular weight of one repeating unit, ME is the combined molecular weight of the end groups. Here, we assumed that all polyesters were terminated with one hydroxymethyl group and one carboxylic acid group.68 , MR is the molecular weight of one repeating unit, ME is the combined molecular weight of the end groups. Here, we assumed that all polyesters were terminated with one hydroxymethyl group and one carboxylic acid group.68
|
||||
| Poly(butylene cyclobutane-1,2-dicarboxylate) | PBCB | 64.1 | 7 | 2700 |
| Poly(hexamethylene cyclobutane-1,2-dicarboxylate) | PHCB | 43.1 | 10 | 4000 |
| Poly(octamethylene cyclobutane-1,2-dicarboxylate) | POCB | 66.3 | 28 | 11200 |
| Poly(decamethylene cyclobutane-1,2-dicarboxylate) | PDCB | 61.6 | 12 | 5400 |


As previously mentioned, the enzymatic polymerization of the diethyl ester of CBDA (CBDE) and aliphatic diols was also investigated. The enzyme chosen for this experiment was Candida antarctica lipase B (CALB), specifically the commercially available Novozyme 435, which is immobilized in acrylic resin. This widely used biocatalyst has been reported to effectively catalyze the synthesis of FDCA- and BHMF-based polymers.26–28,33,34 First, the esterification of CBDA was performed as described by Wang et al. (2018).36 CBDE was chosen over CBDA for this reaction because of its superior solubility in the reaction medium (toluene).34,64 The use of CBDA was also avoided because certain acidic substrates have been shown to deactivate the enzyme, thereby hindering the esterification process.65 The one-step solution polymerization of CBDE with aliphatic diols was carried out following the method outlined by Jiang et al. (2016).33 However, no polymeric product was recovered at the end of the reactions. 1H NMR analysis of the reaction mixture confirmed this, revealing only the signals corresponding to the starting reagents, as illustrated in Fig. S2. The primary hypothesis for the failure of the enzymatic polymerization centers on the specificity of the enzyme. The active-site cavity of CALB is reportedly shaped like a tunnel, which restricts the steric positioning of bulky or branched substrates.66,67 Given the cyclobutane structure of CBDE, with its pendant furan groups, the substrate is likely too sterically demanding for CALB's active site, preventing effective catalysis of the transesterification reaction with the aliphatic diols.
The synthesized polyesters were first analyzed via ATR-FTIR, and their spectra, along with those acquired from CBDA, are presented in Fig. 2. The shift of the carbonyl CO stretching band from 1695 cm−1 to 1725 cm−1 and the C–O stretching peak at 1162 cm−1 present in all the spectra of the polymers demonstrate the formation of ester bonds between CBDA and the diols. The disappearance of the O–H stretching band at 3050 cm−1 and the O–H bending band at 1414 cm−1 are also consistent with the formation of ester bonds. Additionally, the intensities of the asymmetric and symmetric C–H stretching bands at approximately 2942 cm−1 and 2874 cm−1, respectively, increased progressively in the polymer samples as the aliphatic chain of the employed diol increased, as expected. Furthermore, furan ring bands can be observed in the fingerprint region of the polymers and CBDA spectra, specifically the C–O–C ring vibrations at 1012 cm−1 and the C–H out-of-plane vibrations at approximately 798 and 730 cm−1.
 | ||
| Fig. 2 ATR-FTIR spectra of CBDA and its corresponding polyesters. | ||
To further assess the molecular structures of the synthesized polymers, the polyesters were analyzed through 1H NMR in CDCl3. Owing to the material's low solubility, it was allowed to dissolve overnight in CDCl3 with a few drops of TFA. Before analysis, the insoluble fraction was filtered off. The resulting spectra are depicted in Fig. 3. The signals labeled A, B, and C – at 7.22, 6.19, and 5.93 ppm, respectively – are attributed to the protons from the furan ring (–CH). The protons of the cyclobutane core generate signals D and E (4.21 and 3.83 ppm). The protons from the methylene units of the aliphatic diols (–CH2–) give rise to signals at 4.11 ppm (F), 1.61–1.67 ppm (G), and 1.26–1.40 ppm (H). The presence of peak F, in particular, indicates ester bond formation, providing additional evidence for the formation of polymers/oligomers. The peaks at 0.83 and 1.25 ppm, which are mainly detectable in the spectrum of PBCB, arise from impurities, most probably due to vacuum grease contamination of the samples. The low-intensity signals at 6.22–7.10 ppm and 7.27–7.49 ppm may come from minor impurities that are also present in the CBDA 1H NMR spectrum (see the SI).
 | ||
| Fig. 3 1H NMR spectra of the polyesters soluble fractions. | ||
The small peak I at 3.45 ppm can be attributed to the methylene protons of the hydroxymethyl end-groups (HO–CH2–). In addition to peak C—the most well-defined signal originating from the repeating unit structure—this peak was used to estimate the number-average degree of polymerization  and the number-average molecular weight
and the number-average molecular weight  of the soluble portion of the polymers (Table 1).68 Naturally, an increasing trend in the
of the soluble portion of the polymers (Table 1).68 Naturally, an increasing trend in the  of the samples is expected, which is simply due to the differences in the molecular weights of the diols used. Moreover,
of the samples is expected, which is simply due to the differences in the molecular weights of the diols used. Moreover, 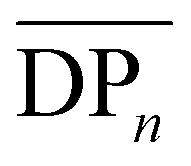 slightly increases with increasing diol chain length, as observed in the progression from PBCB to PHCB to PDCB, with average repeating units per chain of 7, 10, and 12, respectively. However, POCB exhibits a significantly higher
slightly increases with increasing diol chain length, as observed in the progression from PBCB to PHCB to PDCB, with average repeating units per chain of 7, 10, and 12, respectively. However, POCB exhibits a significantly higher  of 28. This deviation could stem from a limitation in the estimation method or may suggest that a diol chain length of 8 carbons is more favorable for the reaction than a length of 4, 6, or 10 carbons. This preference could be due to factors such as catalyst selectivity or improved miscibility in the reaction mixture.
of 28. This deviation could stem from a limitation in the estimation method or may suggest that a diol chain length of 8 carbons is more favorable for the reaction than a length of 4, 6, or 10 carbons. This preference could be due to factors such as catalyst selectivity or improved miscibility in the reaction mixture.
Considering that the highest obtained  was 28, it is evident that all the samples consisted primarily of oligomers. Wang et al. reported the synthesis and further polymerization of a similar cyclobutane monomer with ethylene glycol, obtaining both oligomers and polymers with
was 28, it is evident that all the samples consisted primarily of oligomers. Wang et al. reported the synthesis and further polymerization of a similar cyclobutane monomer with ethylene glycol, obtaining both oligomers and polymers with  values of 1968 and 22039, respectively.61 Furthermore, their polymer exhibited low solubility in common organic solvents. On this basis, it can be inferred that the insoluble fraction of the materials synthesized in this study likely consists of molecules with longer chain lengths than those detected by 1H NMR analysis. This increased chain length likely correlates with the observed insolubility.
values of 1968 and 22039, respectively.61 Furthermore, their polymer exhibited low solubility in common organic solvents. On this basis, it can be inferred that the insoluble fraction of the materials synthesized in this study likely consists of molecules with longer chain lengths than those detected by 1H NMR analysis. This increased chain length likely correlates with the observed insolubility.
Another possible explanation for the low solubility of these polyesters in common organic solvents is the occurrence of crosslinking during the polymerization reaction. The C5 carbon in the furan ring is fairly reactive and capable of undergoing electrophilic substitution, and the possibility of it reacting with the diols and producing interchain bonds is not null (Scheme 3).69,70 Therefore, solid-state 13C CP/MAS NMR analysis was employed to obtain more detailed structural data and evaluate the degree of such crosslinking in the material.
 | ||
| Scheme 3 Possible (a) mechanism of C5 electrophilic substitution, and (b) resulting crosslinked polymer structure for PBCB. | ||
The 13C CP/MAS NMR spectra of the synthesized polymers are depicted in Fig. 4. For all samples, the carbons in the furan ring—labeled A, B, C, and D—give rise to signals at approximately 142.0, 110.4, 107.5, and 152.0 ppm, respectively, with signals for carbons B and C partially overlapping. The carbons in the cyclobutane structure (E and F) produced partially overlapping peaks between 43.0 and 38.8 ppm, and the carbonyl carbon (G) appeared as a signal at 171.4 ppm. The carbon at the ester linkage (H) generates a peak at 65.2 ppm, whereas the remaining aliphatic carbons (I) yield broad signals in the range of 29.3–25.4 ppm. Thus, no new signals indicating covalent crosslinking were detected, suggesting that low solubility might be an innate feature of these polymers. Additionally, as previously mentioned, an analogous study reported the low solubility of similar structured polyesters in common organic solvents without the occurrence of crosslinks.61 Nonetheless, the analysis results should be considered carefully, given the technique's resolution limitations.
 | ||
| Fig. 4 Polyesters’ 13C solid-state NMR spectra. | ||
The thermal stability of the polymers was assessed via TGA, and the resulting thermograms are presented in Fig. 5. The very small weight loss (up to 0.96%) at approximately 100 °C, which is more evident in Fig. 5b, can be attributed to the elimination of residual solvent/moisture. The polymers showed overall good thermal stability, with Td5% values between 250 and 267 °C and Td10% values between 263 and 284 °C, with 50% weight retention observed up to 388 °C. All the polyester samples exhibited two main degradation steps, with the derivative of weight loss peaking between 277–310 °C and 390–405 °C. The first degradation step corresponded to a weight loss of 38–49%, whereas the second step resulted in a weight loss of 29–43%. These observations suggest that two distinct thermal decomposition phenomena occur. Additionally, the char yield of the samples ranged between 18–22%.
 | ||
| Fig. 5 (a) Weight (%) vs. temperature (°C); (b) derivative weight vs. temperature of the synthesized polymers. | ||
The thermal properties of the synthesized polyesters were analyzed via DSC. The first heating and cooling cycle was employed to eliminate the samples’ thermal history, and the second heating curves are depicted in Fig. 6. The glass transition temperature (Tg) of the polymers were determined to be 52, 18, 10, and 6 °C for PBCB, PHCB, POCB, and PDCB, respectively, indicating a decreasing trend as the length of the linear monomer increased. This highlights that, as expected, the diol chain length can be strategically selected to tailor the Tg of the resulting materials. Additionally, the influence of the Tg could be observed during polymer handling, as PBCB was brittle at room temperature, whereas the other samples were increasingly rubbery at room temperature. Furthermore, none of the DSC curves showed a melting endotherm, which is an evidence that all the polymers were amorphous. This was further substantiated by WAXD analysis, which did not reveal the presence of crystalline domains (Fig. S3).
 | ||
| Fig. 6 Polymers’ DSC second heating curve. | ||
Conclusion
Novel polyesters from furfural-derived CBDA and aliphatic diols were successfully synthesized, achieving yields of up to 66%. The reactions were confirmed via multiple analytical techniques, including ATR-FTIR, 1H NMR, and 13C CP/MAS NMR. The resulting polymers exhibited limited solubility in conventional organic solvents, which prevented a thorough assessment of their molecular weights. However, 1H NMR analysis allowed for an estimation of the of the soluble polymer fraction, with values reaching up to 11200 g mol−1. One hypothesis for the limited solubility was the potential crosslinking of the materials. However, detailed analyses, particularly 13C CP/MAS NMR, did not provide any evidence to support this. This finding indicates that low solubility in conventional organic solvents may be an inherent property of the synthesized polymers.
of the soluble polymer fraction, with values reaching up to 11200 g mol−1. One hypothesis for the limited solubility was the potential crosslinking of the materials. However, detailed analyses, particularly 13C CP/MAS NMR, did not provide any evidence to support this. This finding indicates that low solubility in conventional organic solvents may be an inherent property of the synthesized polymers.
Thermogravimetric analysis of the synthesized polymers demonstrated good thermal stability, with Td10% ranging from 261 to 282 °C and 50% weight retention observed up to 386 °C. Moreover, DSC revealed tunable glass transition temperatures varying from 6 to 52 °C depending on the length of the diol used. Additionally, there were no observable melting or crystallization peaks, and wide-angle X-ray diffraction (WAXD) analysis confirmed that the polyesters were 100% amorphous.
Further studies are necessary to obtain a deeper understanding of the physical and mechanical properties of the materials. Such investigations should provide a comprehensive characterization of these biobased polymers and their potential applications. Nevertheless, this work offers valuable insights into the potential of CBDA as a renewable rigid monomer, contributing to the advancement of knowledge in the field of sustainable biobased materials.
Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the SI.Supplementary information containing the 1H NMR spectra of CBDA in DMSO-d6, the 1H NMR spectra of the CBDE and 1,4-butanediol enzymatic polycondensation reaction mixture in CDCl3, and the WAXD patterns of PBCB, PHCB, and POCB is available. See DOI: https://doi.org/10.1039/d5py00485c.
Acknowledgements
LMG acknowledges the financial support from the RUG and USP double-degree PhD program and from FAPESP (Process No. 2021/02903-4).This work was supported by COST Action FUR4Sustain, CA18220, supported by COST (European Cooperation in Science and Technology). This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (DOI: 10.54499/UIDB/50011/2020), UIDP/50011/2020 (DOI: 10.54499/UIDP/50011/2020) & LA/P/0006/2020 (DOI: 10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). The FCT is acknowledged for the research contract to AFS (CEECINSTLA/00002/2022) and a doctorate grant to BA 2020.04495.BD (DOI: 10.54499/2020.04495.BD). LMG and SF thank COST for the short-term scientific mission (STSM) grant under the framework of COST Action FUR4Sustain (CA18220).
The authors thank Jur van Dijken for the TGA analysis.
References
- P. T. Benavides, J. B. Dunn, J. Han, M. Biddy and J. Markham, Exploring Comparative Energy and Environmental Benefits of Virgin, Recycled, and Bio-Derived PET Bottles, ACS Sustainable Chem. Eng., 2018, 6, 9725–9733 CrossRef.
- S. R. Nicholson, N. A. Rorrer, A. C. Carpenter and G. T. Beckham, Manufacturing energy and greenhouse gas emissions associated with plastics consumption, Joule, 2021, 5, 673–686 CrossRef.
- Y. Yang, Z. Liu, H. B. Saydaliev and S. Iqbal, Economic impact of crude oil supply disruption on social welfare losses and strategic petroleum reserves, Resour. Policy, 2022, 77, 102689 CrossRef.
- A. Pandey and Y. C. Sharma, Advancements in biomass valorization in integrated biorefinery systems, Biofuels, Bioprod. Biorefin., 2024, 18, 2078–2090 CrossRef CAS.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Catalytic conversion of lignocellulosic biomass into chemicals and fuels, Green Energy Environ., 2023, 8, 10–114 CrossRef CAS.
- E. J. Cho, L. T. P. Trinh, Y. Song, Y. G. Lee and H.-J. Bae, Bioconversion of biomass waste into high value chemicals, Bioresour. Technol., 2020, 298, 122386 CrossRef CAS PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, A sustainable wood biorefinery for low–carbon footprint chemicals production, Science, 2020, 367, 1385–1390 Search PubMed.
- J. Zhu and G. Yin, Catalytic Transformation of the Furfural Platform into Bifunctionalized Monomers for Polymer Synthesis, ACS Catal., 2021, 11, 10058–10083 Search PubMed.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Recent catalytic routes for the preparation and the upgrading of biomass derived furfural and 5-hydroxymethylfurfural, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- V. P. Kashparova, D. V. Chernysheva, V. A. Klushin, V. E. Andreeva, O. A. Kravchenko and N. V. Smirnova, Furan monomers and polymers from renewable plant biomass, Russ. Chem. Rev., 2021, 90, 750–784 Search PubMed.
- A. Jaswal, P. P. Singh and T. Mondal, Furfural – a versatile, biomass-derived platform chemical for the production of renewable chemicals, Green Chem., 2022, 24, 510–551 RSC.
- A. Gandini and T. M. Lacerda, in Furan Polymers and their Reactions, Wiley, 2023 Search PubMed.
- M. Annatelli, J. E. Sánchez-Velandia, G. Mazzi, S. V. Pandeirada, D. Giannakoudakis, S. Rautiainen, A. Esposito, S. Thiyagarajan, A. Richel, K. S. Triantafyllidis, T. Robert, N. Guigo, A. F. Sousa, E. García-Verdugo and F. Aricò, Beyond 2,5-furandicarboxylic acid: status quo, environmental assessment, and blind spots of furanic monomers for bio-based polymers, Green Chem., 2024, 26, 8894–8941 Search PubMed.
- J. Ye, S. Ma, B. Wang, Q. Chen, K. Huang, X. Xu, Q. Li, S. Wang, N. Lu and J. Zhu, High-performance bio-based epoxies from ferulic acid and furfuryl alcohol: synthesis and properties, Green Chem., 2021, 23, 1772–1781 RSC.
- D. C. Odiyi, T. Sharif, R. S. Choudhry, S. Mallik and S. Z. H. Shah, A review of advancements in synthesis, manufacturing and properties of environment friendly biobased Polyfurfuryl Alcohol Resin and its Composites, Composites, Part B, 2023, 267, 111034 Search PubMed.
- A. O. C. Iroegbu and S. S. Ray, On the chemistry of furfuryl alcohol polymerization: A review, J. Polym. Sci., 2024, 62, 1044–1060 Search PubMed.
- H. A. S. Valentino, P. de Tarso Laia dos Reis e Silva Pupi, A. Gandini and T. M. Lacerda, Furfuryl alcohol/tung oil matrix-based composites reinforced with bacterial cellulose fibres, Cellulose, 2021, 28, 7109–7121 CrossRef CAS PubMed.
- Y. Jiang, S. Yang and X. Pan, Biomass-Based Polyureas Derived from Rigid Furfurylamine and Isomannide, ACS Appl. Polym. Mater., 2022, 4, 2197–2204 CrossRef CAS.
- M.-L. Yang, Y.-X. Wu, Y. Liu, J.-J. Qiu and C.-M. Liu, A novel bio-based AB2 monomer for preparing hyperbranched polyamides derived from levulinic acid and furfurylamine, Polym. Chem., 2019, 10, 6217–6226 RSC.
- H.-H. Shu, Y. Liu, S.-L. Han, X.-Q. Fang, C. Wang and C.-M. Liu, A novel amine-first strategy suitable for preparing both functional and engineering bio-polyamides: furfurylamine as the sole furan source for bisfuranic diamine/diacid monomers, Polym. Chem., 2024, 15, 4433–4446 RSC.
- E. Trovatti, I. G. Gonçalves, A. J. Carvalho and A. Gandini, The contribution of bisfurfurylamine to the development and properties of polyureas, Polym. Int., 2020, 69, 688–692 Search PubMed.
- Z. Cao and X. Zuo, Bio-Based Self-Healing Polymeric Materials Derived from Furfuryl Alcohol Based on the Diels-Alder Reversible Reaction, Polym. Sci., Ser. B, 2023, 65, 450–456 CrossRef.
- S. Raza, J. Zhang, M. Raza, X. Li, H. Wen and C. Liu, Biomass furfural-derived green polymer microspheres: Synthesis and applications for the removal of environmental pollutants from wastewater, Microporous Mesoporous Mater., 2021, 318, 110966 Search PubMed.
- R. Techie-Menson, C. K. Rono, A. Etale, G. Mehlana, J. Darkwa and B. C. E. Makhubela, New bio-based sustainable polymers and polymer composites based on methacrylate derivatives of furfural, solketal and lactic acid, Mater. Today Commun., 2021, 28, 102721 CrossRef CAS.
- C. Post, D. Maniar, V. S. D. Voet, R. Folkersma and K. Loos, Biobased 2,5-Bis(hydroxymethyl)furan as a Versatile Building Block for Sustainable Polymeric Materials, ACS Omega, 2023, 8, 8991–9003 Search PubMed.
- D. Maniar, Y. Jiang, A. J. J. Woortman, J. van Dijken and K. Loos, Furan-Based Copolyesters from Renewable Resources: Enzymatic Synthesis and Properties., ChemSusChem, 2019, 12, 990–999 Search PubMed.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein, D. M. Petrović and K. Loos, Enzymatic Synthesis of Biobased Polyesters Using 2,5-Bis(hydroxymethyl)furan as the Building Block, Biomacromolecules, 2014, 15, 2482–2493 CrossRef CAS PubMed.
- C. Post, D. Maniar, J. A. Jongstra, D. Parisi, V. S. D. Voet, R. Folkersma and K. Loos, Enzymatic bulk synthesis, characterization, rheology, and biodegradability of biobased 2,5-bis(hydroxymethyl)furan polyesters, Green Chem., 2024, 26, 8744–8757 Search PubMed.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Biobased furandicarboxylic acids (FDCAs): effects of isomeric substitution on polyester synthesis and properties, Green Chem., 2014, 16, 1957–1966 RSC.
- S. Singhal, S. Agarwal, M. P. Mudoi, N. Singhal and R. Singh, Chemical conversion of furan dicarboxylic acid to environmentally benign polyesters: an overview, Biomass Convers. Biorefin., 2023, 13, 15619–15636 CrossRef CAS.
- M. R. Miah, Y. Dong, J. Wang and J. Zhu, Recent Progress on Sustainable 2,5-Furandicarboxylate-Based Polyesters: Properties and Applications, American Chemical Society, 2024, preprint, DOI:10.1021/acssuschemeng.3c06878.
- M. Kamran, M. G. Davidson, S. de Vos, V. Tsanaktsis and B. Yeniad, Synthesis and characterisation of polyamides based on 2,5-furandicarboxylic acid as a sustainable building block for engineering plastics, Polym. Chem., 2022, 13, 3433–3443 Search PubMed.
- Y. Jiang, D. Maniar, A. J. J. Woortman and K. Loos, Enzymatic synthesis of 2,5-furandicarboxylic acid-based semi-aromatic polyamides: enzymatic polymerization kinetics, effect of diamine chain length and thermal properties, RSC Adv., 2016, 6, 67941–67953 RSC.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, A biocatalytic approach towards sustainable furanic–aliphatic polyesters, Polym. Chem., 2015, 6, 5198–5211 Search PubMed.
- K. Loos, R. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, A Perspective on PEF Synthesis, Properties, and End-Life, Front. Chem., 2020, 8, 585 Search PubMed.
- Z. D. Wang, Q. Elliott, Z. Wang, R. A. Setien, J. Puttkammer, A. Ugrinov, J. Lee, D. C. Webster and Q. R. Chu, Furfural-Derived Diacid Prepared by Photoreaction for Sustainable Materials Synthesis, ACS Sustainable Chem. Eng., 2018, 6, 8136–8141 CrossRef CAS.
- B. B. Yagci, B. Munir, Y. Zorlu and Y. E. Türkmen, Access to Symmetrical and Unsymmetrical Cyclobutanes via Template-Directed [2 + 2]-Photodimerization Reactions of Cinnamic Acids, Synthesis, 2023, 3777–3792 Search PubMed.
- C. Body, G. Wery, L. Gubbels, K. Robeyns and T. Leyssens, Control of Regioselectivity in the Dimerization of trans-Cinnamic Acid and Its Derivatives Using Cocrystal Engineering, Cryst. Growth Des., 2024, 24, 2117–2125 CrossRef CAS.
- M. Lahav and G. M. J. Schmidt, Topochemistry. Part XVIII. The solid-state photochemistry of some heterocyclic analogues of trans-cinnamic acid, J. Chem. Soc. B, 1967, 239–243 RSC.
- V. Baret, A. Gandini and E. Rousset, Photodimerization of heteroarylene-vinylenes, J. Photochem. Photobiol., A, 1997, 103, 169–175 CrossRef CAS.
- M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, Cyclobutanes in Small-Molecule Drug Candidates, John Wiley and Sons Ltd, 2022, preprint, DOI:10.1002/cmdc.202200020.
- G. K. Kole and M. H. Mir, Isolation of elusive cyclobutane ligands via a template-assisted photochemical [2 + 2] cycloaddition reaction and their utility in engineering crystalline solids, Royal Society of Chemistry, 2022, preprint, 10.1039/d2ce00277a.
- J. Zhang, E. Xiu-Tian-Feng, B. Yang, X. Ma, X. Wang, N. Chang, Q. Zhang, J. J. Zou and J. Xie, Synthesis of Highly Strained Cyclobutane Fuels via Room-Temperature-Integrated Oxidation/[2 + 2] Cycloaddition of Lignocellulose-Based Cyclic Ketones and Cyclic Alcohols, ACS Sustainable Chem. Eng., 2024, 12(49), 17647–17655 CrossRef CAS.
- E. Lee-Ruff and G. Mladenova, Enantiomerically pure cyclobutane derivatives and their use in organic synthesis, Chem. Rev., 2003, 103, 1449–1483 CrossRef CAS PubMed.
- A. Sokolova, A. Pavlova, N. Komarova, O. Ardashov, A. Shernyukov, Y. Gatilov, O. Yarovaya, T. Tolstikova and N. Salakhutdinov, Synthesis and analgesic activity of new α-truxillic acid derivatives with monoterpenoid fragments, Med. Chem. Res., 2016, 25, 1608–1615 CrossRef CAS.
- Z. Wang, M. Scheuring, M. Mabin, R. Shahni, Z. D. Wang, A. Ugrinov, J. Butz and Q. R. Chu, Renewable Cyclobutane-1,3-dicarboxylic Acid (CBDA) Building Block Synthesized from Furfural via Photocyclization, ACS Sustainable Chem. Eng., 2020, 8, 8909–8917 CrossRef CAS.
- J. Tellers, N. Sbirrazzuoli and N. Guigo, A rigid plant oil-based thermoset with a furfural-derived cyclobutane cross-linker, Green Chem., 2021, 23, 8053–8060 RSC.
- M. Jamali Moghadam Siahkali, N. Guigo and N. Sbirrazzuoli, Cross-Linking Mechanisms of a Rigid Plant Oil-Based Thermoset from Furfural-Derived Cyclobutane, Macromolecules, 2023, 56, 404–415 CrossRef CAS.
- H. Amjaour, Z. Wang, M. Mabin, J. Puttkammer, S. Busch and Q. R. Chu, Scalable preparation and property investigation of a cis-cyclobutane-1,2-dicarboxylic acid from β-trans-cinnamic acid, Chem. Commun., 2019, 55, 214–217 RSC.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Programming a crystalline shape memory polymer network with thermo- and photo-reversible bonds toward a single-component soft robot, Sci. Adv., 2018, 4(1), 3865 CrossRef PubMed.
- D. Tunc, C. Le Coz, M. Alexandre, P. Desbois, P. Lecomte and S. Carlotti, Reversible Cross-Linking of Aliphatic Polyamides Bearing Thermo- and Photoresponsive Cinnamoyl Moieties, Macromolecules, 2014, 47, 8247–8254 CrossRef CAS.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, Mechanically Gated Degradable Polymers, J. Am. Chem. Soc., 2020, 142, 2105–2109 Search PubMed.
- G. Kaur, P. Johnston and K. Saito, Photo-reversible dimerisation reactions and their applications in polymeric systems, Polym. Chem., 2014, 5, 2171–2186 RSC.
- A. Rahimi and J. M. García, Chemical recycling of waste plastics for new materials production, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Designing Biobased Recyclable Polymers for Plastics, Elsevier Ltd, 2020, 38, 1, pp. 50–67, DOI:10.1016/j.tibtech.2019.04.011.
- N. Singh and T. R. Walker, Plastic recycling: A panacea or environmental pollution problem, npj Mater. Sustain., 2024, 2(1), 17 Search PubMed.
- S. Kumar, S. Kumar, S. Kumar, T. Yadav, P. S. Dhapola and P. K. Singh, Reducing Environmental Plastic Pollution by Designing Polymer Materials for Managed End-of-Life, Macromol. Symp., 2024, 413(1), 2300146 CrossRef.
- M. Martín-Sómer, C. Pablos, C. Adán, R. van Grieken and J. Marugán, A review on LED technology in water photodisinfection, Sci. Total Environ., 2023, 885, 163963 CrossRef PubMed.
- R. Pizzichetti, M. Martín-Gamboa, C. Pablos, K. Reynolds, S. Stanley, J. Dufour and J. Marugán, Environmental life cycle assessment of UV-C LEDs vs. mercury lamps and oxidant selection for diclofenac degradation, Sustainable Mater. Technol., 2024, 41, e01002 CrossRef.
- J. J. Rytuba, Mercury from mineral deposits and potential environmental impact, Environ. Geol., 2003, 43, 326–338 Search PubMed.
- Z. Wang, M. Scheuring, M. Mabin, R. Shahni, Z. D. Wang, A. Ugrinov, J. Butz and Q. R. Chu, Renewable Cyclobutane-1,3-dicarboxylic Acid (CBDA) Building Block Synthesized from Furfural via Photocyclization, ACS Sustainable Chem. Eng., 2020, 8, 8909–8917 CrossRef.
- R. K. Shahni, M. Mabin, Z. Wang, M. Shaik, A. Ugrinov and Q. R. Chu, Synthesis and characterization of BPA-free polyesters by incorporating a semi-rigid cyclobutanediol monomer, Polym. Chem., 2020, 11, 6081–6090 RSC.
- S. Zaidi, S. Thiyagarajan, A. Bougarech, F. Sebti, S. Abid, A. Majdi, A. J. D. Silvestre and A. F. Sousa, Highly transparent films of new copolyesters derived from terephthalic and 2,4-furandicarboxylic acids, Polym. Chem., 2019, 10, 5324–5332 RSC.
- H. Azim, A. Dekhterman, Z. Jiang and R. A. Gross, Candida antarctica Lipase B-Catalyzed Synthesis of Poly(butylene succinate): Shorter Chain Building Blocks Also Work, Biomacromolecules, 2006, 7, 3093–3097 CrossRef PubMed.
- F. Hollmann, P. Grzebyk, V. Heinrichs, K. Doderer and O. Thum, On the inactivity of Candida antartica lipase B towards strong acids, J. Mol. Catal. B: Enzym., 2009, 57, 257–261 CrossRef.
- Y. L. de los Santos, Y. L. Chew-Fajardo, G. Brault and N. Doucet, Dissecting the evolvability landscape of the CalB active site toward aromatic substrates, Sci. Rep., 2019, 9(1), 15588 CrossRef PubMed.
- P. B. Juhl, K. Doderer, F. Hollmann, O. Thum and J. Pleiss, Engineering of Candida antarctica lipase B for hydrolysis of bulky carboxylic acid esters, J. Biotechnol., 2010, 150, 474–480 CrossRef CAS PubMed.
- J. U. Izunobi and C. L. Higginbotham, Polymer molecular weight analysis by 1H NMR spectroscopy, J. Chem. Educ., 2011, 88, 1098–1104 CrossRef CAS.
- A. Gandini and M. N. Belgacem, in Monomers, Polymers and Composites from Renewable Resources, Elsevier, 2008, pp. 115–152 Search PubMed.
- L. I. Belen'kii, T. G. Kim, I. A. Suslov and N. D. Chuvylkin, Substrate and positional selectivity in electrophilic substitution reactions in pyrrole, furan, thiophene, and selenophene derivatives and related benzoannelated systems, Russ. Chem. Bull., 2005, 54, 853–863 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |