
Block copolymers from coordinative chain transfer (co)polymerization (CCT(co)P) of olefins and 1,3-dienes and mechanical properties of the resulting thermoplastic elastomers
Samy
Alioui
ab,
Damien
Montarnal
 ab,
Franck
D'Agosto
ab and
Christophe
Boisson
*ab
ab,
Franck
D'Agosto
ab and
Christophe
Boisson
*ab
aUniversité Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5128, Catalysis, Polymerization, Processes and Materials (CP2M), Villeurbanne F-69616, France. E-mail: christophe.boisson@univ-lyon1.fr
bChemistLab, Michelin CP2M ICBMS joint Laboratory, 69616 Villeurbanne, France
First published on 2nd August 2025
Abstract
Coordinative chain transfer (co)polymerization (CCT(co)P) is a degenerative chain transfer polymerization mechanism that allows for rapid and efficient chain exchanges between a main group metal/zinc (dormant site) and a transition metal (active site). The ability to generate multiple chains per catalyst molecule during this process leads to significant atom economy, addressing both economic and ecological concerns. In addition to enabling the polymerization of olefins, styrene, and conjugated dienes with high stereospecificity, this technique also provides access to highly reactive polymeryl–metal chain ends. Such extremities can serve as initiation sites for other polymerization techniques, either directly or after an exchange of functional end-group, and may lead in this way to linear block copolymers (BCPs) with a large variety of compositions. Bulk self-assembly of BCPs with adequate architectures enables the ingenious combination of the characteristics of different domains, imparting unique properties that make them suitable for a wide range of applications, such as adhesives, compatibilizers for polymer blends, and thermoplastic elastomers (TPEs). The latter are particularly attractive in the context of a sustainable circular economy: TPEs exhibit at service temperature the elastic properties of crosslinked elastomers, yet they can be molded and extruded like thermoplastics at higher temperatures. This review describes the state-of-the-art synthesis strategies employing CCT(co)P and chain shuttling polymerization (CSP) of olefins and conjugated dienes, either independently or combined with other controlled polymerization techniques, for the synthesis of linear BCPs and multiblock copolymers (MBCs). Some of these polymers constitute a new class of TPEs. In a second section, the control of the morphology of these materials by the architectures of the BCPs and crystallization-driven self-assembly and their thermomechanical properties are discussed.
 Samy Alioui | Samy Alioui, after completing his Master's degree in Molecular Chemistry and Functional Polymers at Sorbonne University and the University of Créteil, respectively, in 2021, joined ChemisLab, a joint laboratory between CNRS and Michelin, to pursue his PhD on the synthesis and characterization of elastomeric thermoplastics (TPEs), under the supervision of Christophe Boisson, Franck D'Agosto and Damien Montarnal. In 2024, after completing his PhD, he joined LVMH (LVMH Gaïa) to work on the development of new sustainable polymers. His research interests focus on the synthesis of block copolymers via anionic polymerization and coordination catalysis, as well as the structural, morphological, and mechanical analyses of polymer materials. |
 Damien Montarnal | Damien Montarnal obtained his PhD from ESPCI ParisTech, France in 2011, where he worked on supramolecular self-healing materials and initiated the concept of vitrimers under the supervision of Prof. L. Leibler and Dr F. Tournilhac. He subsequently moved to UC Santa Barbara as a postdoctoral researcher with Profs C. J. Hawker, E. J. Kramer and G. H. Fredrickson, working on block copolymer self-assembly. He has been a CNRS research scientist since 2015, in the CP2M laboratory at the University Lyon 1. His research interests encompass all polymer materials in which reversible chemistry can modulate the structure, dynamics or physical properties. He was awarded the bronze medal of CNRS in 2021 and the prize of the French Polymer Group in Chemistry in 2022. |
 Franck D'Agosto | Franck D'Agosto completed his PhD in Polymer Chemistry at the joined unit between CNRS and bioMérieux (University of Lyon, France) before working at the University of Sydney (Australia) as a postdoctoral fellow in the Key Center for Polymer Colloids. He has been recruited at the French National Center for Scientific Research (CNRS) in 2002, and he has been appointed a research director at the Catalysis, Polymerization, Processes and Materials (CP2M) laboratory (Villeurbanne, France) in 2014. His research interests focus on the control of polymer architectures using different polymerization chemistries – such as catalytic and controlled free radical polymerizations – either performed in solution or in dispersed media. He was awarded the bronze medal of CNRS in 2006 and the joint prize of the French Chemical Society-French Polymer Group in 2010. |
 Christophe Boisson | Christophe Boisson received his PhD in Organic Chemistry from the University Paris-Saclay in 1996. The same year, he became a research associate at the French National Center for Scientific Research (CNRS). In 2008, he was appointed the CNRS research director at the Catalysis, Polymerization, Processes and Materials (CP2M) laboratory (Villeurbanne, France). Since 2019, he has been the director of the joint laboratory ChemistLab between the CNRS and the Michelin company. His research interests are focused on homogeneous and heterogeneous catalysts for the polymerization of olefins and conjugated dienes. In 2022, he was awarded the Prize Aymé-Poirson by the French Academy of Sciences. |
1. Introduction
Block copolymers (BCPs) represent a specific class of copolymers in which the individual monomers are not distributed randomly or alternately within the chain. Instead, they are grouped together as discrete blocks along the polymer.1 These materials received particular attention when the distinct blocks led to phase separation into various nanoscaled structures (5–200 nm). The corresponding morphologies, and in particular the connectivity of the various phases, drive for the most part how the specific properties of each phase are combined at the macroscopic level. BCPs have found a wide range of industrial applications as bulk materials or additives in thermoplastics, including compatibilizers for polymer blends, thermoplastic elastomer (TPE) materials, energy and data storage systems, coatings and adhesives.2,3 Controlled polymerization techniques are critical to obtain well-defined BCPs, and while linear block copolymers are the most extensively studied class,4 a variety of molecular architectures can also be obtained.5–7 Over the years, the increasing scope and reliability of numerical predictions such as self-consistent field theory for the self-assembly of BCPs have also contributed towards expanding the field of macromolecular engineering to functional or charged monomers for high performance lithographic applications requiring excellent pattern quality.8,9 Yet, combining inexpensive and available monomers into optimized block copolymer architectures for low-cost applications such as TPEs remains of critical interest to industry in the scope of developing sustainable alternatives to covalently crosslinked elastomers. Early strategies such as the development of styrene–butadiene–styrene (SBS) elastomers 70 years ago focused on triblock architectures featuring relatively incompatible segments and the strong segregation of glassy domains acting as physical crosslinks. While such materials display excellent properties at service temperature and are also amenable for post-polymerization modifications such as hydrogenation into styrene–ethylene butylene–styrene (SEBS) elastomers, they also demonstrate a high viscosity during processing as the phase separation persists in the melt. Single step processes using directly the raw olefins (ethylene or propylene) and relying on crystallization-induced phase separation are nowadays more desirable, yet they are much more difficult to achieve and drive significant academic interest.10–13 A major complexity is indeed to gain control over the stereoregularity of each segment while maintaining a well-defined macromolecular architecture. These olefin block copolymers (OBCs) typically do not present any phase separation in the melt, and feature low viscosities compatible with high throughput processing.The first single step strategy for OBCs was the use of living polymerization techniques. Living coordination–insertion polymerization enables polymer chains to grow in the absence of termination or chain transfer reaction while the metal center remains constantly coordinated at one end of the polymer chain until it is intentionally destroyed. Hence, block copolymers can be acquired by sequential addition of monomers or by varying reaction conditions.14,15 This method can produce very well defined OCBs with a versatile monomer composition.4 However, these well-controlled materials have not yet found commercial application due to high production costs as only one polymer chain is produced per metallic active site.16 Therefore, the development of catalytic systems capable of controlling the growth of the polymer chains while achieving a high atom economy is a main goal. In this case, the term “atom economy” refers to the fact that a catalyst molecule is consumed to form a polymer chain in a living system, whereas a catalytic system will only consume a fraction of it. Coordinative chain transfer (co)polymerization (CCT(co)P) is a recent methodology that allows the production of multiple polymer chains per metal complex. The catalytic feature of CCTP has triggered in this way a surge in academic research in recent years.17–19
CCT(co)P involves the use of a single transition metal (TM) catalyst and a chain transfer agent (CTA), an organometallic species (OS) such as MgR2, AlR3 or ZnR2.17,20,21 In this case, the growing macromolecular chain is reversibly transferred via transmetalation from the catalyst (active species) to the CTA (Scheme 1), which is generally considered as dormant species during polymerization. CCT(co)P is thus based on a degenerative transfer, i.e. a process involving a thermodynamically neutral dynamic equilibrium between propagating and dormant species.18 If transfer is faster than propagation, several chains per transition metal catalyst can grow in a controlled manner. This mechanism is indeed analogous to reversible addition fragmentation chain transfer (RAFT) in reversible deactivation radical polymerization (RDRP).22 Given that CTA is introduced in excess, the number of chains is governed by the concentration of CTA. In addition, as all the chains are connected to a metal center, chain-end functionalization and sequential block copolymerization are possible.
 | ||
| Scheme 1 Mechanism and principle of coordinative chain transfer (co)polymerization (CCT(co)P). TM is a transition metal and MGM is a main group metal. ktr is the transfer rate constant. | ||
In 2006, Arriola et al. at Dow Chemical Company extended the CCT(co)P to chain shuttling polymerization (CSP),23 in which two catalysts work together in the presence of a chain transfer agent – called in this case a chain shuttling agent (CSA) – and two monomers. The different reactivities of the monomers towards the two catalysts and the reversible transmetalation reactions can lead to multiblock copolymers that alternate two kinds of blocks, such as soft and hard crystalline blocks when TPEs are targeted (Scheme 2).19 However, the transfer rate must adequately commensurate with the propagation rate in order to allow sufficient polymer chain growth on each of the catalysts.
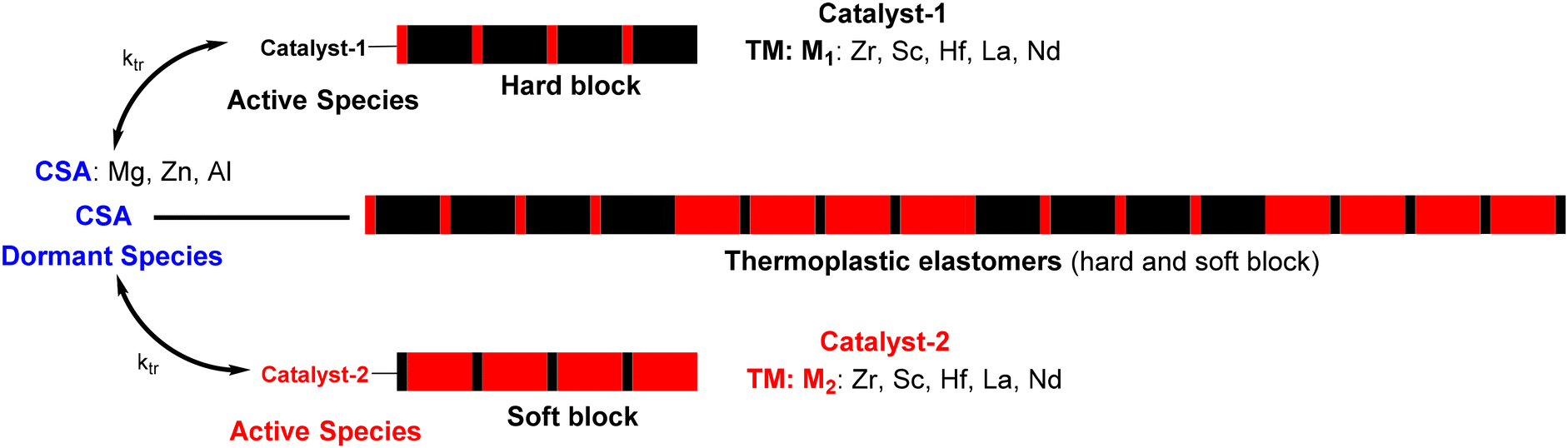 | ||
| Scheme 2 Chain shuttling copolymerization. Catalyst represents a transition metal center allowing propagation and CSA the chain shuttling agent. | ||
In addition to the fact that CCT(co)P offers a high degree of atom economy, efficient molar mass (Mn) control and the possibility to functionalize the final polymer, it is also possible to take advantage of coordination polymerization to control the stereospecificity of the polymerization of α-olefins, styrene and diene monomers (e.g. syndiospecific polymerization of propylene (P) and styrene, stereospecific trans-1,4 or cis-1,4 polymerization of butadiene and isoprene). Given that the polymer chains are connected to the TM or the metal of the CTA at the end of the polymerization, CCT(co)P can be efficiently combined with other controlled polymerization techniques such as anionic polymerization, RDRP and ring-opening polymerization (ROP), which are capable of producing highly controlled, low-dispersity block copolymers.19 Recently, Zinck and coworkers have reported in great detail the advancements made in CCT(co)P.18,19 However, little attention has been given to the mechanical properties of the resulting block copolymers. In the following, a comprehensive update of the synthesis methods for block copolymers based on olefins and conjugated dienes, including at least one CCTP or CSP step, will be provided in a first section. It will cover the various methodologies for the synthesis of BCPs from apolar monomers (olefins, styrene, and conjugated dienes) using CCT(co)P alone or in combination with other controlled polymerization methods, since the emergence of this field of research in the early 2000s. In a second section, the focus will be placed on the polyolefin thermoplastic elastomers (P-TPEs) obtained by this technique, to discuss the architecture, organization and mechanical properties of these materials.
2. CCT(co)P of olefins and 1, 3-dienes for the design of block and multiblock copolymers
This section will explore in a first part post-polymerization functionalization of polymers obtained by olefin CCTP to create reactive macroinitiators for other polymerization techniques and functional POs for coupling reactions. The second and third parts respectively examine sequential monomer addition methods and chain shuttling polymerization to form BCPs and MBCs. Finally, a last part is dedicated to BCP synthesis obtained using systems based on a polymerization switch from or to CCT(co)P conducted in situ. Tables (Tables 1–5) summarizing all the corresponding data are available at the end of each part.| Complex | CTA | Monomera | Conditions | FP (%F)b | M n (Đ) kg mol−1 | Ref. |
|---|---|---|---|---|---|---|
| a E = ethylene P = propylene, and B = butadiene. b Functional polymer (FP) and functionalization yield. c nd: not determined or not communicated by the authors. | ||||||
| 1 or 2 | MAO or TMA | E | P E = 1.3 atm, T = 50 °C, toluene | PE-OH (85) | 8.15 (1.70) | 24 |
| 3 | TIBA and DEZ | E | P E = 2 bar, T = 40 °C, toluene | PE-OH (88) | 2.23 (2.10) | 2 |
| 4 | DEZ | E | P E = 2 bar, T = 80 °C, toluene | PE-OH (nd) | ndc | 25 |
| 5 | ZnPh2 | P | P P = 5 psi, T = 0 °C, toluene | HOBn-aPP-BnOH (0.83) | 13.7 (1.11) | 26 |
| 6 | DEZ | E | P E = 4 bar, T = 40 °C, toluene | PE-OH (67) | 1.3 (nd) | 27 |
| 7 | BOMAG | E | P E = 3 bar, T = 80 °C, toluene | PE-SH (83) | 1.25 (1.40) | 28 and 29 |
| 8 | TEA | E | P E = 5 bar, | PE-OH (80) | 3.3 (1.90) | 30 |
| 9 | DEZ | E | P E = 1 atm, T = 25 °C, o-xylene | PE-Br (77) | 0.7 (nd) | 31 |
| 10 | Al(iBu)2H | BD | mB = 2 g, T = 50 °C, toluene | PB-b-PCL-Br (nd) | 21.2 (1.86) | 33 |
| 11 | TIBA | P | P p = 4 bar, T = 40 °C, toluene | iPP-Br (80) | 2.5 (1.99) | 34 |
| 7 | BOMAG | E | P E = 1.3 atm, T = 80 °C, toluene | PE-DD2 (40) | nd | 35 |
| 7 | BOMAG | E | P E = 3 bar, T = 80 °C, toluene | PE-NH-RAFT (84) | DPn = 41 | 36 |
| 12 | DEZ | E | P E = 1.3 MPa, T = 25 °C, toluene | PE-N3 (80) | M w = 2.1 (nd) | 38 |
| 7 | BOMAG | E | P E = 3 bar, T = 80 °C, toluene | PE-N3 (94) | nd | 37 |
| 7 | BOMAG | E | P E = 3 bar, T = 80 °C, toluene | PE-Ph (nd) | 0.85 (1.22) | 39 |
| 7 | BOMAG | E | P E = 3 bar, T = 80 °C, toluene | PE-Cp (72) | 1.4 (1.20) | 40 |
| 7 | BOMAG | E | P E = 3 bar, T = 80 °C, toluene | PE-SH (83) | 1.4 (1.40) | 41 |
| 9 | DEZ | E | P E = 0.4 MPa, T = 60 °C, toluene | PE-OH (63) | 0.72 (1.41) | 42 |
| 11 | TIBA and DEZ | P | P P = 3 bar, T = 40 °C, toluene | PP-OH (40) | 8 (2.20) | 43 |
| FP (%F)a | Secondary techniques | Secondary monomersb | Block copolymers | M n (Đ) kg mol−1 | Ref. |
|---|---|---|---|---|---|
| a Functional polymer (FP) and functionalization yield. nd: not determined or not communicated by the authors. b ε-CL = ε-caprolactone, PDL = pentadecalactone, EO = ethylene oxide, IB = isobutene, S = styrene, MMA = methylmethacrylate, n-BA = n-butyl acrylate, and tertBA = tert-butyl acrylate. c nd: not determined or not communicated by the authors. | |||||
| PE-OH (85) | ROP | ε-CL | PE-b-PCL | ndc | 24 |
| PE-OH (88) | ROP | ε-CL | PE-b-PCL | 51 (nd) | 2 |
| PDL | PE-b-PPDL | 64.58 (nd) | |||
| PE-OH (nd) | ROP | ε-CL | PE-b-PCL | 6.8 (nd) | 25 |
| HOBn-aPP-BnOH (83) | ROP | ε-CL | PCL-b-aPP-b-PCL | 23.5 (1.36) | 26 |
| PE-OH (67) | ROP | L-Lactide | PE-b-PLLA | 10.6 (nd) | 27 |
| PE-SH (83) | ROP | D,L-lactide | PE-b-PDLA | 3.8 (2.0) | 28 and 29 |
| PE-OH (80) | ROP | rac-Lactide | PE-b-PLA-1 | 4.5 (1.70) | 30 |
| PE-b-PLA-2 | 4.0 (1.60) | ||||
| PE-Br (77) | ATRP | n-BA | PE-b-PBA | 10.3 (1.16) | 31 |
| tertBA | PE-b-PtertBA | 10.4 (1.16) | |||
| PBD-b-PCL-Br | ATRP | ε-CL/S | PB-b-PCL-b-PS | 47.4 (1.87) | 33 |
| ε-CL /MMA | PB-b-PCL-b-PMMA | 60.3 (1.92) | |||
| iPP-Br | ARGET ATRP | S | iPP-b-PS | 8.2 (3.13) | 34 |
| PE-DD2 (40) | NMP | n-BA | PE-b-PBA | 67.9 (nd) | 35 |
| PE-NH-RAFT (84) | RAFT | n-BA | PE-b-PBA | nd | 36 |
| PE-N3 (80) | Cycloaddition | EO | PE-b-PEO | M w = 3.8 (1.21) | 38 |
| PE-N3 (94) | Cycloaddition | IB | PE-b-PIB-b-PE | nd | 37 |
| PE-Ph (nd) | Diels–Alder | S/MMA | PE-b-PS | 3.2 (1.08) | 39 |
| PE-b-PMMA | 1.8 (1.34) | ||||
| PE-Cp (72) | Hetero Diels–Alder | S | PE-b-PS | 2.5 (nd) | 40 |
| PE-SH (83) | Thia-Michael addition | EG | PE-b-PEO | 1.15 (1.40) | 41 |
| PE-OH (63) | Coupling | EG | PE-b-PEO | 0.9 (1.57) | 42 |
| PP-OH (40) | Transesterification | ε-CL | iPP-b-PCL | 14 (2.30) | 43 |
| Complex | CTA | Monomersa | Copolymers | M n (Đ) kg mol−1 | Ref. |
|---|---|---|---|---|---|
| a E = ethylene, ε-CL = caprolactone, P = propylene, Oct = 1-octene, Od = 1-octadecene, IP = isoprene, B = butadiene, S = styrene, CHO = cyclohexene oxide, TMC = trimethylene carbonate and nd: not determined or not communicated by the authors. | |||||
| 13 | BEM | E and ε-CL or MMA | PE-b-PCL | 2.3 (1.30) | 45 |
| PE-b-PMMA | 1.45 (1.20) | ||||
| 14 | Zn(Oct)2 | E and P | PE-b-PEP-b-PE | 83 (1.70) | 13 |
| 14 | TIBA | P and E | iPP-b-PEP-b-iPP | 198 (2.40) | 46 |
| 14 | TIBA | P and Oct or Od | iPP-b-(P-co-Oct) | Several | 47 |
| iPP-b-(P-co-Od) | |||||
| 14 | DEZ | E | HDPE-b-VLDPE | 44.5 (1.67) | 48 |
| 14 | DEZ | E | LLDPE-b-ULDPE | nd | 49 |
| 10 | Al(iBu)2H | IP and ε-CL | PIP-b-PCL | 7.2–11.8 (1.27–1.42) | 50 |
| 10 | Al(iBu)2H | IP and B and ε-CL | PB-b-PIP-b-PB | 9.83 (1.88) | 51 |
| PB-b-PIP-b-PCL | 9.54 (1.38) | ||||
| 10 | Mg(n-Bu)2 | B and ε-CL/L-lactide | PB-b-PCL | 18.1–37.1 (1.83–1.98) | 52 |
| PB-b-PLLA | |||||
| 15 | Al(iBu)2H | B and CHO/TMC | PB-b-PCHO | 8.25 (1.29) | 53 |
| PB-b-PTMC | 7.93 (1.78) | ||||
| 16 | BOMAG | E and B | EBR-b-PE | 5.2–19 (2.2–4.2) | 54 |
| 16 | PDMB | E and B | PE-b-EBR-b-PE | 71.2 (2.4) | 55 |
| (PE-b-EBR)n | 41.3 (2.3) | ||||
| 17 | BEM | S and IP | PS-b-trans-1,4-PB | 28.8 (1.20) | 56 |
| Complex | CSA | Ma | Multiblock copolymers | Ref. |
|---|---|---|---|---|
| a E = ethylene, Oct = 1-octene, C16 = hexadec-1-ene, N = norbornene, IP = isoprene, B = butadiene, and S = Styrene. | ||||
| 14, 18 | DEZ | E and Oct | E/Oct multiblock | 23 |
| 14, 19, 20, 21 | DEZ | E and Oct | E/Oct multiblock | 57 |
| 14, 18 | DEZ | E/C16 | E/C16 multiblock | 58 |
| 22, 23 | DEZ | E/N | E/N multiblock | 59 |
| 4, 24 | DEZ | E/N | E(PEHD)/N multiblock | 60 |
| 25, 26 | DEZ | E/N | E/N multiblock | 61 |
| 27, 29 | BEM, TIBA | IP | cis–trans-PIP multiblock | 62 |
| 29 | Mg(n-Bu)2 | B | cis–trans-PB multiblock | 63 |
| 30, 31, 32 | TIBA | S/IP | S(sPS)/IP multiblock | 64 |
| 33, 34 | BEM | S/IP | S/IP multiblock | 65 |
| Metal center | CTA | First monomer | Second monomer | Nature of switch | Block copolymers | M n (Đ) kg mol−1 | Ref. |
|---|---|---|---|---|---|---|---|
| E = ethylene, Oct = 1-octene, IP = isoprene, B = 1,3-butadiene, S = Styrene, Pent = 1-penetene, P = propylene, and H = 1-hexene. | |||||||
| 7, 35 | BEM | E | IP | CCTP to catalysis | PE-b-trans-1,4-PB | 40.7 (2.06) | 67 |
| 36 | Zn(benzyl)2 | E/Oct | S | CCTP to anionic | PO-b-PS | 97–193 (2.73–4.04) | 68 |
| 36 | Zn(1-hexyl)2 | E/P | S | CCTP to anionic | PEP-b-PS | 51–99 (1.26–1.39) | 69 |
| 36 | Zn(4-(isopropenyl)benzyl)2 | E/Oct or Pent | S | CCTP to anionic | PS-b-PO-b-PS | >120 (>2.40) | 70 |
| 14 | Et(Zn(CH2)6)αZnEt | E/P | S | CCTP to anionic | PS-b-PEP-b-PS | >120 (<1.64) | 71 |
| 14 | Zn(CH2CH2C6H4CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)2 CH2)2 |
E/P | S | CCTP to anionic | Triblocks and multiblocks | >94 (1.67–1.84) | 72 |
| 14 | Zn(CH2CH2CH2C6H4CHCH2)2 |
E/H | S | CCTP to anionic | PS-b-PEH-b-PS | 161 (1.80) | 66 |
| 16 | Macro-CTA | B and/or S | E or E/B | Anionic to CCTP | PB-b-EBR | 23.8 (1.13) | 73 |
| PB-b-PE | 14.2 (1.43) | ||||||
| PS-b-EBR | 44.5 (1.44) | ||||||
| SBR-b-EBR | 22.3 (1.24) | ||||||
| 16 | Macro-CTA | S | E/B then E | Anionic to CCTP | PS-b-EBR-b-PE | 61–154 (1.5–2.2) | 74 |
2.1 Synthesis of block copolymers from end-functional POs obtained by CCTP
CCTP is a polymerization method that produces metal-terminated POs that can be easily converted to functional POs. Subsequently, these end-functionalized POs can be used as precursors for the production of BCPs.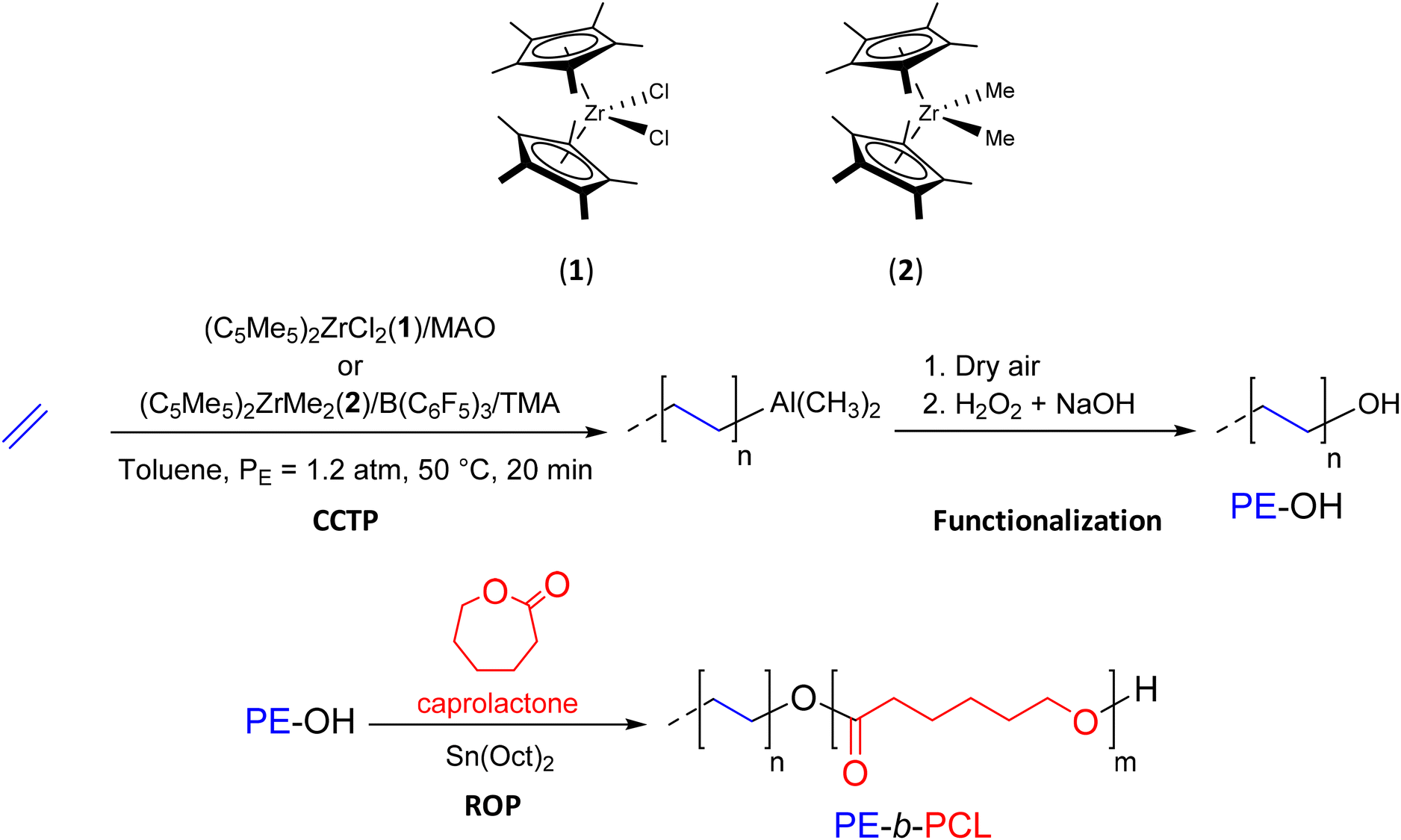 | ||
| Scheme 3 Synthesis of PE-OH and PE-b-PCL diblock copolymers by successive combination of CCTP and ROP.24 | ||
With the aim of reducing the interfacial tension in PO/polar polymer blends and improving their compatibility, Duchateau et al.2 have synthesized various compatibilizers based on PE and a polyester (Scheme 4). In a preliminary step, PE-OH was formed by using a guadinate titanium complex [Et2NC(NCy)2]TiCl3(3)/MAO (Cy: cyclohexyl) ([Al]/[Ti] = 2250) in the presence of triisobutylaluminum (TIBA) and a small amount of diethylzinc (DEZ) ([TIBA]/[Zn]/[Ti] = 400/100/1) to polymerize E. After exposure to synthetic dry air followed by acid treatment, the PE-OH macroinitiator was formed (Mn PE-OH = 2.23 kg mol−1, Đ = 2.10, 88%). Catalytic ROP polymerization of ε-caprolactone and ω-pentadecalactone (PDL) using a salen-aluminum complex ([Monomers]/[Al]/[PE-OH] = 1000/1/1) in the presence of PE-OH yielded the corresponding diblock copolymers, PE-b-PCL (Mn PE-b-PCL = 51 kg mol−1) and PE-b-PPDL (Mn PE-b-PPDL = 64.58 kg mol−1), respectively. However, despite the success of diblock copolymer synthesis, the authors only report the use of graft copolymers as compatibilizers for polymer blends.2 The latter were prepared via an ROP reaction using a PE macroinitiator randomly functionalized along the chain with a hydroxyl group.
 | ||
| Scheme 4 Synthesis of PE-OH and PE-b-PCL/PPDL diblock copolymers by the combination of CCTP and ROP.2 | ||
In a purely methodological approach, Ahmadi et al.25 wanted to develop an efficient synthetic route to make PE partially polar and more biocompatible. In this study, the synthesis of a PE-b-PCL diblock copolymer was reported using the same strategy as that used by Duchateau.2 PE-OH was obtained using a zirconium metallocene complex Et(Ind)2ZrCl2 (4) (Ind: indenyl) (Scheme 5) activated by MAO ([MAO]/[Zr] = 200) in the presence of DEZ ([Zn]/[Zr] = 500). After 30 min of reaction, this binary CCTP system leads to zinc-terminated PE chains, Zn(PE)2. The PE-OH is then formed in situ by purging the reactor with dry air and acid treatment. Finally, the hydroxy function was exploited to perform the ROP of ε-caprolactone in the presence of Sn(Oct)2 as a catalyst. Various diblock copolymers with different proportions of PCL were obtained. A reduction in the thickness of the PE crystal lamellae in the final copolymer, observed by X-ray diffraction, suggests the presence of PCL on the PE chain structure and the formation of the desired diblock copolymers. To evaluate the potential of these synthesized block copolymers as compatibilizers, 5 wt% PE-b-PCL (Mn PE-b-PCL = 6.8 kg mol−1) was added to a PE/PCL blend (80/20 wt/wt). A decrease in the PCL domain sizes observed by scanning electron microscopy (SEM) indicates the improved compatibility in the PE/PCL blend.
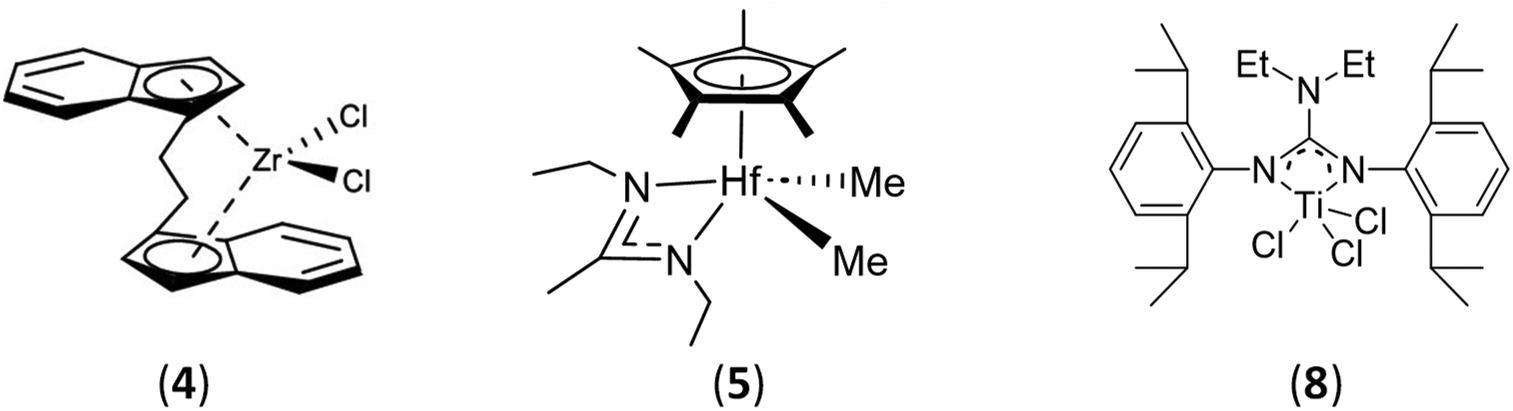 | ||
| Scheme 5 Metal complexes used for ethylene and propylene CCTP. | ||
With the goal of creating a new generation of POs that are more suitable for chemical or mechanical recycling or that function as compatibilizers in the processing of plastic waste, Sita et al.26 have reported an original strategy for the preparation of homotelechelic α,ω-bis(phenyl)polypropylene capable of initiating ROP of ε-caprolactone to provide PCL-based triblock copolymers (Scheme 6). To produce atactic polypropylene (aPP) functionalized with phenyl groups at both chain ends, the catalytic system based on a cyclopentadienyl amidinate hafnium complex (C5Me5)[(N,N)-κ2-N(Et)C(Me)N(Et)]Hf(CH3)2 (5) (Scheme 5)/[PhNH(CH3)2][B(C6F5)4] ([B]/[Hf] = 3.3) was combined with ZnPh2 used as the CTA ([Zn]/[Hf] = 40) in the polymerization of propylene. The Ph-aPP formed was functionalized with I2 and the resulting Ph-aPP-I underwent copper-catalyzed phenylation to form a Ph-aPP-Ph telechelic polymer. Friedel–Crafts acetylation of chain-end phenyl groups followed by reduction led to an α,ω-bis(benzylalcohol)-PP difunctional macroinitiator (HOBn-aPP-BnOH, Mn HOBn-aPP-BnOH = 13.7 kg mol−1, Đ = 1.11). In the end, after a ROP of ε-caprolactone in the presence of Sn(Oct)2, a PCL-b-aPP-b-PCL triblock copolymer with a Mn of 23.5 kg mol−1 and a Đ of 1.36 was obtained. Thermal analysis of the material showed a glass transition temperature (Tg) of −4 °C and a Tm of 54 °C, corresponding to the aPP and PCL blocks, respectively. Small-angle-X-ray scattering analysis of the material revealed a thermotropic behavior of the triblock copolymer, with scattering peaks indicating a lamellar phase below 100 °C and a hexagonal phase above 150 °C.
 | ||
| Scheme 6 Synthesis of telechelic aPP (HOBn-aPP-BnOH) and PCL-b-aPP-b-PCL triblock copolymers by the combination of CCTP and ROP.26 | ||
In this work, the authors have also reported the synthesis of the aPMP-b-PCL (PMP: poly-4-methyl-1-pentene) diblock copolymer (Scheme 7). The latter was obtained after ROP of ε-caprolactone using aPMP-BnOH as amacroinitiator.
 | ||
| Scheme 7 Synthesis of aPMP-BnOH and aPMP-b-PCL diblock copolymers by the combination of CCTP and ROP.26 | ||
The association of olefin coordination insertion polymerization with other polymerization techniques soon attracted the attention of researchers. In 2007, Dubois’ group reported the synthesis of a new BCP, PE-b-PLLA (PLLA: poly(L-lactide)), by combining CCTP of ethylene and ROP of L-lactide (Scheme 8).27 The key reaction intermediate, PE-OH, was prepared by ethylene polymerization with the catalytic system rac-dimethyl-silylen-bis(2-methyl-benz[e]indenyl)ZrCl2(6)/MAO ([Al]/[Zr] = 5000) in the presence of DEZ as the CTA ([Zn]/[Zr] = 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300). The reaction was carried out at 40 °C under 4 bar of ethylene for 30 min. Then, dry air was bubbled into the reaction medium for 5 h at 65 °C before an acidified methanol solution was added to give PE with 67% hydroxy function (Mn PE-OH = 1.3 kg mol−1). The diblock copolymer PE-b-PLLA (Mn PE-b–PLA = 10.6 kg mol−1) was obtained using PE-OH as a macroinitiator after the addition of an equimolar amount of Sn(Oct)2 and 231 equivalents of L-lactide. The presence of signals characteristic of CH2 of PE at the α position of PLLA in the 1H NMR spectra (PE-CH2-O-PLLA at 4.6 ppm) and the increase of the Mn observed after 60 min of reaction were good indications that the extension of PE-OH chains occurred.
300). The reaction was carried out at 40 °C under 4 bar of ethylene for 30 min. Then, dry air was bubbled into the reaction medium for 5 h at 65 °C before an acidified methanol solution was added to give PE with 67% hydroxy function (Mn PE-OH = 1.3 kg mol−1). The diblock copolymer PE-b-PLLA (Mn PE-b–PLA = 10.6 kg mol−1) was obtained using PE-OH as a macroinitiator after the addition of an equimolar amount of Sn(Oct)2 and 231 equivalents of L-lactide. The presence of signals characteristic of CH2 of PE at the α position of PLLA in the 1H NMR spectra (PE-CH2-O-PLLA at 4.6 ppm) and the increase of the Mn observed after 60 min of reaction were good indications that the extension of PE-OH chains occurred.
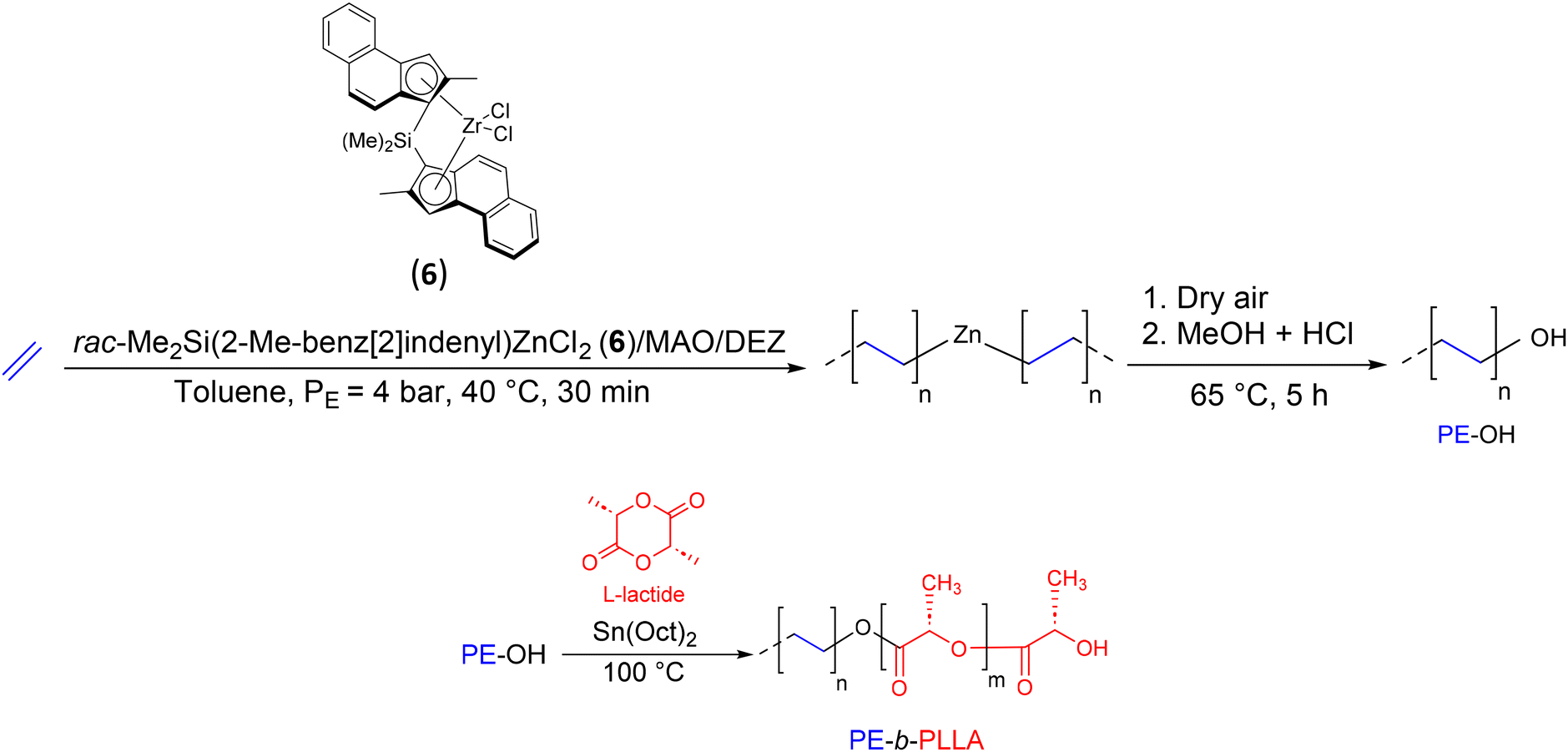 | ||
| Scheme 8 Synthesis of PE-OH and PE-b-PLLA diblock copolymers by the combination of CCTP and ROP.27 | ||
In contrast to previous studies that exploited the reactivity of the hydroxy terminus at the end of PE chains to synthesize BCPs, Boisson et al.28,29 introduced a new synthetic route to design thiol-terminated polyethylenes (PE-SH) (Scheme 9). This end was used as a macroinitiator for ring-opening of D,L-lactide and designing the PE-b-PDLLA (PDLLA: poly(D,L-lactide)) diblock copolymer. The catalytic system (C5Me5)2NdCl2Li(OEt2)2(7)/butyloctylmagnesium (BOMAG) with a ratio of nMg/nNd = 265 was used for the ethylene polymerization (T = 80 °C, P = 3 bar). The introduction of the thiol end to the PE chains was achieved by firstly reacting the Mg(PE)2 species with iodine to produce PE-I. The iodo end group was then reacted with potassium ethyl xanthate to form a dithiocarbonate-terminated PE (PE-S-C(S)-OEt). The subsequent reductive treatment of PE-S-C(S)-OEt with LiAlH4 gave PE-SH with 88% of thiol function (Mn PE-SH = 1.25 kg mol−1, Đ = 1.4). Lastly, the polymerization of D,L-lactide by ROP was initiated from PE-SH in the presence of 4-dimethylaminopyridine (DMAP) as a catalyst (nDMAP/nPE-SH = 4) to obtain the desired diblock copolymer after 24 h of reaction (Mn PE-b–PDLLA = 3.8 kg mol−1, Đ = 2.00). The evolution of the SEC chromatogram towards higher molar masses and the absence of the triplet corresponding to the methylene at the α position of the thiol (PE–CH2–SH) in the 1H NMR spectrum confirmed the efficient initiation of PE-SH chains and the formation of the corresponding diblock copolymer.
 | ||
| Scheme 9 Synthesis of PE-SH and PE-b-PDLLA diblock copolymers by the combination of CCTP and ROP.29 | ||
In 2012, Kempe et al. investigated the synthesis of PE-b-PLA block copolymers.30 Their study aimed to vary the molar mass and proportion of the PLA block to investigate its impact on the final morphology of the material. In contrast to other groups that mainly used metallocene complexes for the CCTP of ethylene, Kempe synthesized a monoguanidinate titanium complex (8) (Scheme 5). This complex was activated with ammonium borate ([R2N(CH3)H]+ [B(C6F5)4]−, R = C16H33-C18H37) ([Ti]/[B] = 1/1.1) to polymerize ethylene in the presence of triethylaluminum (TEA) as the CTA ([Al]/[Ti] = 25000). After 1 hour of reaction under 5 bar ethylene pressure, the Al–PE bond was oxidized to form a PE with 80% terminal hydroxy function (Mn PE-OH = 3.3 kg mol−1, Đ = 1.90). Subsequently, PE-b-PLA diblock copolymers were formed by extending the PE-OH block at 110 °C for 18 h in the presence of the rac-lactide monomer and Sn(Oct)2 catalyst ([PE-OH]/[Sn] = 57). Depending on the initial amount of monomer, two diblock copolymers with various PLA block Mn values were obtained, the first with a Mn of 4.5 kg mol−1 (Đ = 1.7) exhibiting a well-ordered lamellar structure and the second with a Mn of 4 kg mol−1 (Đ = 1.6) exhibiting a less ordered bicontinuous morphology. The Tm values of 130 °C and 127 °C for the PE and PLA blocks, respectively, were also reported by the authors.
N-2, 6-iPr2C6H3)2C5H3N}FeCl2 (9), previously activated with MAO, with the DEZ CTA (Fe = 5 μmol, [Fe]/[Al]/[Zn] = 1/100/500). After performing ethylene polymerization at 25 °C for 30 minutes, the resulting di-polyethylenylzinc (Zn(PE)2) was oxidized by exposure to dry air at 100 °C for 2 hours to form di-polyethyleneoxyzinc (PE–O–Zn–O–PE), and then a final treatment with a MeOH/HCl mixture yielded oligomeric PE with 77% of hydroxyl end-group. In a second step, the PE-OH was quantitatively transformed into α-bromoisobutyrate-terminated polyethylene to form the macroinitiator (PE-Br) to initiate ATRP. By reacting n-butyl acrylate (BA) in the presence of PE-Br and the CuBr/CuBr2/tris(2-(di(2-(n-butoxycarbonyl)ethyl)-amino)ethyl)amine mixture, a diblock copolymer PE-b-PBA was obtained after 8 hours with an n-butyl acrylate conversion of 72.8%. After removing the non-functionalized PE, the diblock copolymer was analyzed by SEC, the resulting chromatogram showed a monomodal shape, and the Mn measured was 10.1 kg mol−1 (Đ = 1.16). Under the same conditions, ATRP polymerization of tert-butyl acrylate (tertBA) was carried out for 6 hours to achieve a monomer conversion of 80.5% and form the diblock copolymer PE-b-PtertBA. The SEC chromatogram also showed a monomodal shape, and the Mn measured was 11.1 kg mol−1 (Đ = 1.16).
 | ||
| Scheme 10 Reaction pathway for the synthesis of PE-OH and diblock copolymers PE-b-PBA and PE-b-PtertBA by combining CCTP and ATRP.31 | ||
To design compatibilizers for incompatible binary polymer blends, Zhang et al.33 was inspired by Matyjaszewski's work to prepare BCP with a polybutadiene (PB) segment and various polar segments. Using a neodymium catalytic system, Nd(OiPr)3(10)/Al(iBu)2H/Me2SiCl2 ([Nd]/[Al]/[Cl] = 1/20/3), CCTP of butadiene was first performed followed by the ROP of ε-caprolactone initiated in situ to form a diblock copolymer terminated with a hydroxyl end group (Mn PB-b-PCL = 21.2 kg mol−1, Đ = 1.86) (Scheme 11). The hydroxyl group was subsequently reacted with 2-bromo-2-methylpropionyl bromide to form an α-bromoisobutyrate chain end, which can initiate ATRP of methyl methacrylate (MMA) and styrene (S). The targeted triblock copolymers PB-b-PCL-b-PMMA (Mn PBD-b-PCL-b-PMMA = 60.3 kg mol−1, Đ = 1.92) and PB-b-PCL-b-PS (Mn PBD-b-PCL-b-PS = 47.4 kg mol−1, Đ = 1.87) were successfully obtained after a third ATRP step using PBD-b-PCL-Br as a macroinitiator.
 | ||
| Scheme 11 Synthesis of PB-b-PCL-b-PMMA and PB-b-PCL-b-PS triblock copolymers by the combination of CCTP, ROP, and ATRP.33 | ||
Duchateau subsequently employed a similar strategy to synthesize compatibilizers for PPO/iPP polymer blends (PPO: poly(2, 6-dimethyl-1,4-phenylene oxide)) by combining CCTP with ATRP.34 The catalyst rac-Me2Si(2-Me-4-Ph-Ind)2ZrCl2 (11)/MAO was used alongside TIBA ([TIBA]/[Zr] = 625) and DEZ ([DEZ]/[Zr] = 156) as CTAs to form iPP-OH building blocks after a final oxidation step with dry synthetic air (Mn iPP–OH = 2.35 kg mol−1, Đ = 1.84) (Scheme 12). The deprotonation of the latter by triethylamine (NEt3) followed by the addition of α-bromoisobutyryl bromide in the presence of 4-dimethylaminopyridine (DMAP) led to the formation of an ATRP macroinitiator (iPP-Br, Mn iPP-Br = 2.5 kg mol−1, Đ = 1.99). Lastly, ATRP of styrene was initiated by iPP-Br to form the desired diblock copolymer iPP-b-PS (Mn iPP-b-PS = 8.2 kg mol−1, Đ = 3.13). The high Đ value is attributed to the presence of iPP in the final copolymer. This presence results from the low rate of functionalization of this latter with the α-bromoisobutyryl bromide group necessary to initiate the ARGET ATRP step.
 | ||
| Scheme 12 Synthesis of iPP-b-PS block copolymers by combining CCTP and ARGET ATRP.34 | ||
Boisson and D'Agosto were the first to report the combination of CCTP with nitroxide-mediated polymerization (NMP) for the synthesis of PE-b-PBA block copolymers.35 Initially, the 7/BOMAG catalytic system ([Nd]/[Mg] = 40) was employed for ethylene polymerization, before introducing a nitroxide of interest (DD2, Scheme 13), which reacts with Mg(PE)2 to produce a PE-DD2 intermediate. NMR analyses revealed a functionality rate of 40% and a number average polymerization degree (DPn) of 40. The nitroxide mediated radical polymerization of BA was subsequently carried out in bulk using PE-DD2 in the presence of a small amount of free nitroxide SG1 (N-(2-methyl-2-propyl)-N-(1-diethylphosphono-2,2-dimethylpropyl)-N-oxyl) at 120 °C. After 9 hours of reaction, the monomer conversion reached 51%, corresponding to a polymerization degree (DPn) of 522 for the PBA block and a number-average molar mass of 67.9 kg mol−1. The Mn evolution observed by SEC demonstrated the chain extension of the PE-DD2 intermediate and the formation of the desired PE-b-PBA block copolymer.
 | ||
| Scheme 13 Synthesis of PE-b-PBA block copolymers by combining CCTP and NMP.35 | ||
In continuation of these studies, this same group combined CCTP with RAFT to synthesize a PE-b-PBA diblock copolymer.36 Initially, Mg(PE)2 species was formed, as previously, using the catalytic system 7/BOMAG. Subsequently, through a series of functionalization steps including iodine and NaN3 addition, PE-N3 was obtained (further details in section 2.3.1)37 and further reduced with LiAlH4. The resulting PE-NH2 reacted with an activated ester containing a chain transfer agent for RAFT to form a PE-based macromolecular CTA for RAFT (PE-NH-RAFT). Finally, the targeted PE-b-PBA diblock copolymer was synthesized by conducting RAFT of BA at 85 °C in the presence of PE-NH-RAFT for 6 hours initiated by azobisisobutyronitrile (AIBN) ([PE-NH-RAFT]/[AIBN] = 5).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, to target a diblock copolymer, PE-b-PEO. The copolymer obtained showed a coupling efficiency of 65.4% and a Mw of 3.8 kg mol−1 (Đ = 1.21).
CH, to target a diblock copolymer, PE-b-PEO. The copolymer obtained showed a coupling efficiency of 65.4% and a Mw of 3.8 kg mol−1 (Đ = 1.21).
 | ||
| Scheme 14 Synthesis of PE-b-PEO diblock copolymers by combining CCTP and coupling reaction copper-catalyzed alkyne–azide cycloaddition.38 | ||
Boisson et al. explored the used of CuAAC reaction between azide and alkyne to potentially access a new family of thermoplastic elastomers, specifically PE-b-PIB-b-PE (PIB: poly(isobutene)) (Scheme 15).37 Initially, to obtain PE-N3 chains, the neodymium metallocene complex 7 was used with BOMAG ([Mg]/[Nd] = 40) for ethylene polymerization. A PE-I intermediate was obtained as previously described29 and then treated with NaN3 to obtain PE-N3 with 94% azide functionality (Mn PE-N3 = 2.25 kg mol−1, Đ = 1.20). The α,ω-alkyne end-functionalized PIB (CHC-PIB-CCH, Mn = 6.9 kg mol−1) was reacted via CuAAC with 2 equivalents of PE-N3 at 110 °C in the presence of 0.5 equivalents of CuBr and PMDTA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine). After 45 min of reaction, the product was analyzed using various characterization tools. The absence of the starting alkyne and azide signals and the presence of the triazole ring signal in the 1H NMR spectrum indicate the complete reaction between alkynes and azides. Additionally, the shift of the SEC chromatogram towards higher Mn compared to the starting polymers confirmed the formation of the target triblock copolymer. Diblock copolymers of PE-b-PIB have also been reported by the authors in this work.
 | ||
| Scheme 15 Synthetic strategy to obtain a PE-b-PIB-b-PE block copolymer by 1,3-dipolar cycloaddition between PE-N3 and CHC-PIB-CCH.37 | ||
Using essentially the same synthetic methodology, D'Agosto et al.39 designed and then functionalized the PE-N3 species with a photo-caged diene to form a highly photoreactive moiety upon irradiation with ultraviolet (UV) light, capable of reacting with double bonds in a Diels–Alder cycloaddition reaction to form PE-b-PS/PMMA diblock copolymers (Scheme 16). To achieve this, the authors first performed a CCTP of ethylene using the neodymium metallocene complex 7 combined with BOMAG. PE-N3 was then obtained as described above.37 Concurrently, a small photoreactive molecule with an alkyne end (Ph-alkyne) was synthesized through several steps to react with the azide-terminated compound (PE-N3) via a CuAAC reaction to form photoreactive polyethylene (PE-Ph) (Mn.PE-Ph = 1.2 kg mol−1, Đ = 1.25) (Scheme 16). Besides, PS and PMMA blocks containing a maleimide unit at their ends were synthesized by ATRP. Finally, two different diblock copolymers (PE-b-PS and PE-b-PMMA) were formed via a photoinduced Diels–Alder reaction by ligation of the o-methylbenzaldehyde-terminated PE-Ph with the maleimide-terminated PS or PMMA block (Scheme 16). The high temperature size exclusion chromatography (HT-SEC) traces of the block copolymers relative to the starting PE-Ph shifted to lower retention volumes, indicating a clear increase in Mn (Mn PE-b–PS = 3.2 kg mol−1, Đ = 1.08, Mn PE-b–PMMA = 1.8 kg mol−1, Đ = 1.34) and the successful formation of the block copolymers.
 | ||
| Scheme 16 Synthesis of PE-b-PS/PMMA diblock copolymers using the highly reactive o-methylbenzaldehyde diene species (PE-Ph(act)) through a Diels–Alder cycloaddition reaction with maleimides.39 | ||
The hetero-Diels–Alder (hDA) reaction involving a diene end-functionalized polymer and an electron-deficient dithioester has also been investigated by D'Agosto's group.40 The introduction of a cyclopentadienyl moiety at the end of a PE chain (PE-Cp) was demonstrated for the first time by reacting PE-I with bis(cyclopentadienyl)nickel(II) (4 eq.) in the presence of two equivalents of triphenylphosphine (Mn PE-Cp = 1.4 kg mol−1) (Scheme 17). Two out of the three possible isomers were obtained, resulting in an overall degree of functionalization of 72%. The PE-b-PS diblock copolymer was prepared by reacting an equimolar amount of a PS obtained by dithioester-mediated RAFT (PS-RAFT) with PE-Cp in the presence of trifluoroacetic acid (TFA) as a catalyst. The SEC chromatogram of the final compound showed a significant shift towards the higher Mn (Mn PS–b–PE = 2.5 kg mol−1) compared to the starting polymers. In contrast to the PE-b-PS diblock copolymer, the second targeted copolymer, PE-b-PIBA (PIBA: poly(isobornyl acrylate)), showed a retro Diels–Alder reaction at high temperature (120 °C).40
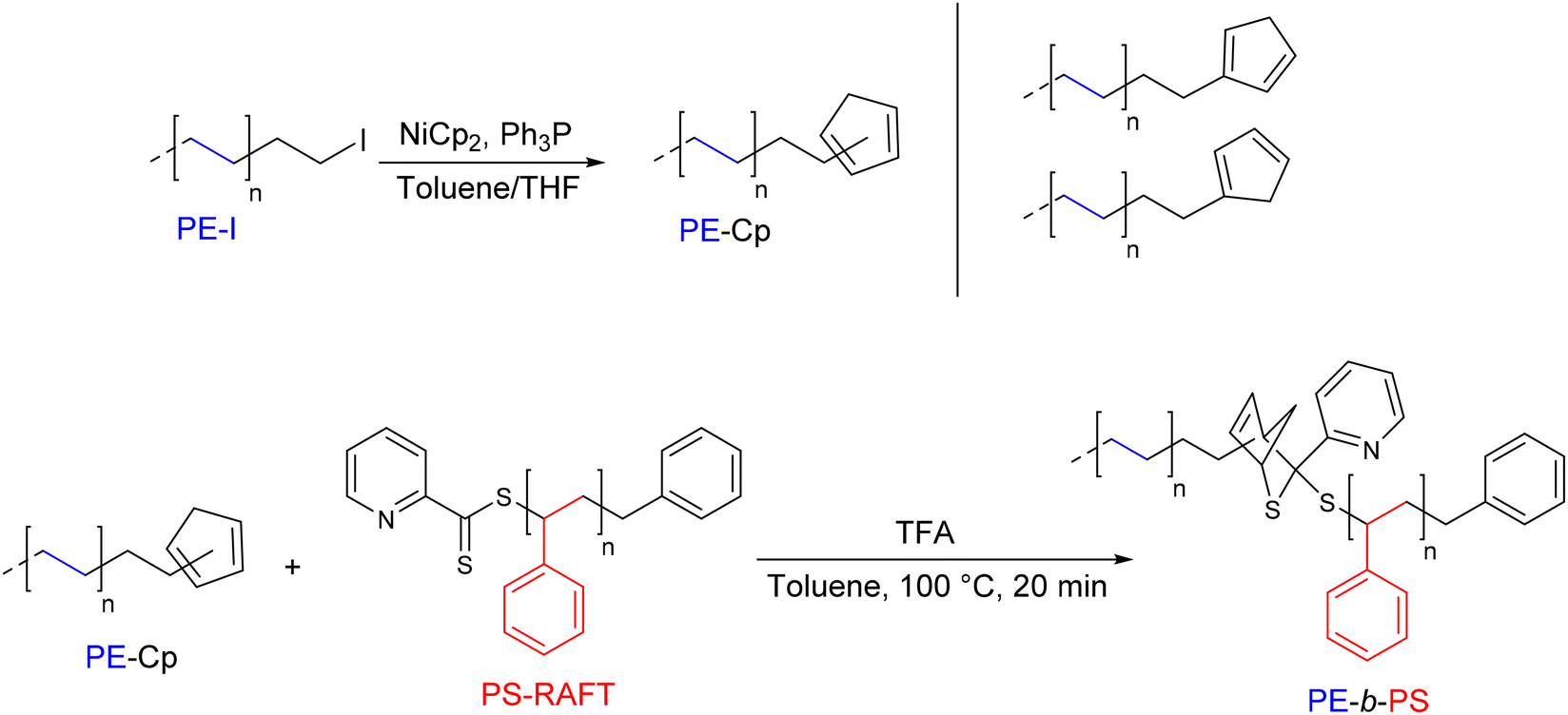 | ||
| Scheme 17 General synthetic strategy for the preparation of cyclopentadienyl end-functionalized PE (PE-Cp) and the formation of PE-b-PS via HDA.40 | ||
As mentioned above, Boisson et al. previously reported a thiol-terminated PE synthesis route to design PE-SH macroinitiators for the ROP of D,L-lactide.29 In continuation of this work, the authors aimed at further exploiting thiol nucleophilicity in thia-Michael addition reactions to synthesize PE-b-PEO diblock copolymers (Scheme 18).41 A commercially available polyethylene glycol with a terminal acrylate function (PEO-A) (5 eq.) was brought in contact with the PE-SH polymer (Mn PE-SH = 1.4 kg mol−1) (1 eq.) to form the amphiphilic diblock copolymer by the phosphine-mediated (PBu3) (0.5 eq.) thia-Michael reaction. The success and high efficiency of the reaction were demonstrated by the complete disappearance of the protons corresponding to the methylenes at α and β positions to the starting PE-SH in the 1H NMR spectrum of the recovered polymer.
 | ||
| Scheme 18 Thia-Michael addition reaction between a PEO-A macropolymer and PE-SH by CCTP.41 | ||
In order to design compatibilizing agents for LLDPE/PEO (LLDPE: linear low-density polyethylene) blends, Fan et al.42 reported the synthesis of PE-b-PEO diblock copolymers through a coupling reaction between PE-OH and isocyanate-terminated polyethylene glycol (PEO-NCO) (Scheme 19). In the initial approach, to produce PE-OH, the complex 9 was used in combination with dry ethylaluminoxane (DEAO). In the presence of DEZ, a CCTP system was efficiently implemented to produce zinc-bonded PE chains. After oxidation and hydrolysis of the resulting Zn(PE)2 intermediate, the final PE was obtained with a hydroxy functionality of 63% (Mn PE-OH = 0.72 kg mol−1, Đ = 1.41). PEO-NCO, previously prepared by a condensation reaction of monomethyl PEO with isophorone diisocyanate (IPDI) (Scheme 19), was then reacted with PE-OH for 24 hours at 120 °C in the presence of dibutyltin dilaurate (DBTDL) to form the desired diblock copolymer, PE-b-PEO (Mn PE-b–PEG = 0.9 kg mol−1, Đ = 1.57). Despite the low molar mass of the synthesized copolymer, the authors emphasized the industrial potential of such a copolymer as a compatibilizer for LLDPE/PEO blends.
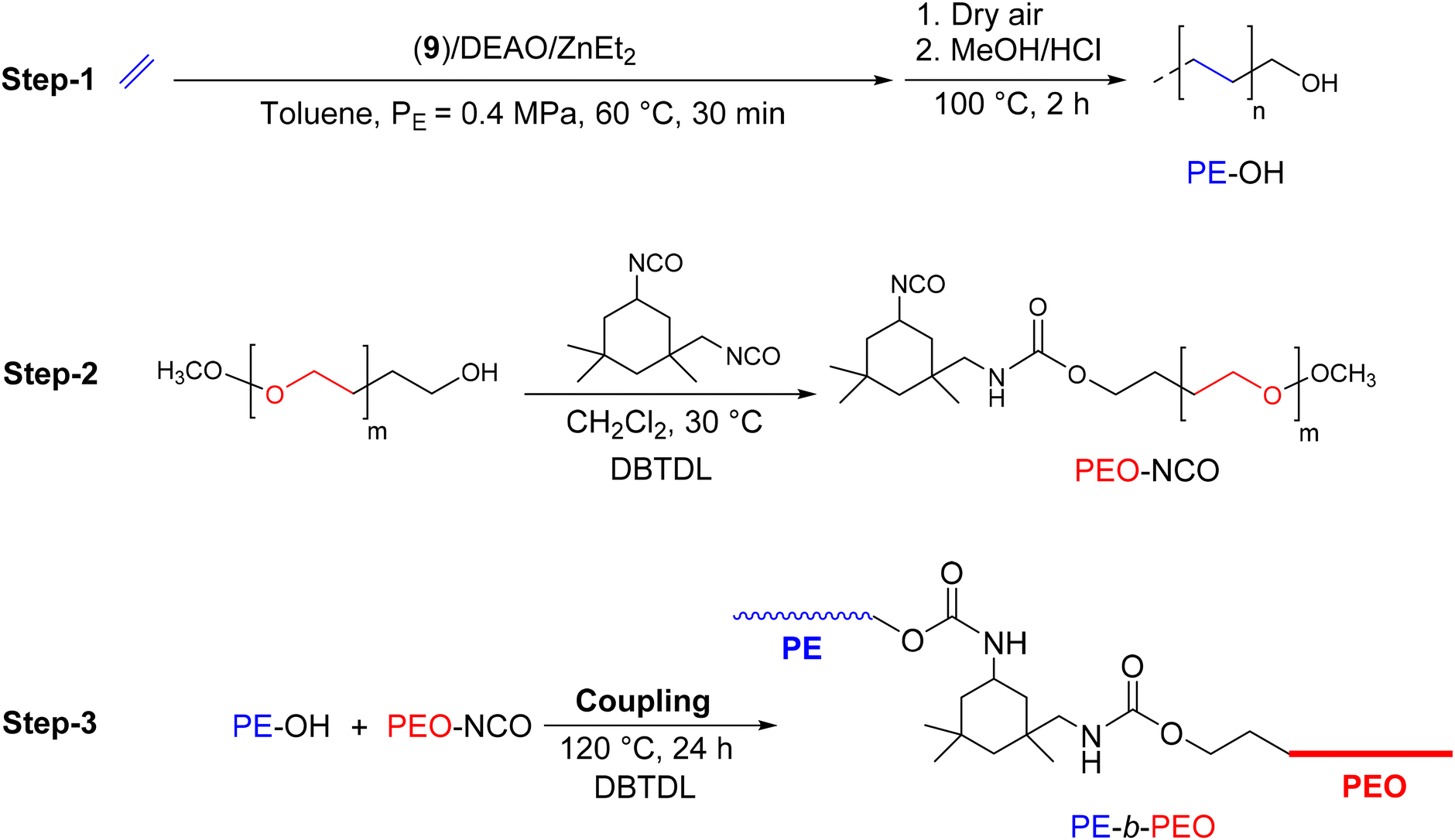 | ||
| Scheme 19 Synthesis of PE-b-PEO by a combination of CCTP and a coupling reaction.42 | ||
A similar strategy was used by Duchateau et al.43 to functionalize iPP chain ends with a hydroxy group capable of undergoing a transesterification reaction to form iPP-b-PCL (iPP: isotactic polypropylene) compatibilizers (Scheme 20). MAO-activated catalyst 11 was used in the presence of TIBA ([Al]/[Zr] = 1250) and a small amount of DEZ ([Zn]/[Zr] = 313) to prepare iPP connected to Al or Zn that were oxidized in situ followed by methanol acid treatment to produce iPP-OH. The oxidized iPP showed a Mn of 8 kg mol−1 (Đ = 2.2) and a iPP-OH content of 40%, and the remaining was non-functionalized iPP. In parallel, ε-caprolactone polymerization was carried out using Mg(BHT)2(THF)2 as a catalyst to obtain PCL blocks (Mn PCL = 38 kg mol−1, Đ = 2.1). By reacting iPP-OH and PCL in a twin-screw mini-extruder at 160 °C in the presence of Sn(Oct)2 ([iPP-OH]/[Sn] = 3) for 5 min, the diblock copolymer iPP-b-PCL (Mn iPP-b-PCL = 14 kg mol−1, Đ = 2.30) was obtained. The obtained Mn value was significantly lower than expected, possibly due to the presence of the non-functionalized propylene homopolymer in the final copolymer. Finally, the authors reported that the diblock copolymer was a poor compatibilizer for polypropylene/polycarbonate polymer blends.
 | ||
| Scheme 20 Synthesis of iPP-b-PCL by transesterification reaction between iPP-OH and polycaprolactone by reactive extrusion.43 | ||
Some researchers have also exploited CCTP products to form macro-initiators for RDRP. The combination of CCTP with techniques such as ATRP and NMP has enabled the formation of new block copolymers based on POs (or butadiene) and polyacrylate/polystyrene. Various coupling reactions, including cycloaddition, transesterification, hetero Diels–Alder, and thia-Michael reactions, have allowed for the coupling of the initially functionalized polyolefins produced by CCTP, resulting in the generation of block copolymers including novel apolar–polar copolymers.
However, the non-quantitative functionalization of olefinic end groups and sometimes the significant number of steps required to obtain the desired product remain major obstacles to the potential industrialization of these methods of block copolymer design. The approach allowing the formation of such copolymers, without the need for prior isolation of PO building blocks, would represent a significant breakthrough.
2.2 Block copolymer synthesis by varying the monomer feed during CCT(co)P
This work was further extended by Mortreux, who synthesized PE-b-PMMA and PE-b-PCL BCPs without requiring additional chain-end functionalization.45 In fact, the samarium metallocene chloride complexes (C5Me5)2SmCl2Li(OEt2)2(13) combined with an excess of n-butyl-ethyl-magnesium (BEM), provided active species for the CCTP of ethylene (Scheme 21). With a [Mg]/[Sm] ratio of 50 at 80 °C, a PE with Mn = 1.04 kg mol−1 was obtained after 30 min of reaction. Subsequently, MMA was added at −78 °C to extend the PE block and form the final low molar mass PE-b-PMMA diblock copolymer (Mn PE-b-PMMA = 1.45 kg mol−1, Đ = 1.20). The second PE-b-PCL diblock copolymer (Mn PE-b-PCL = 2.3 kg mol−1, Đ = 1.30) was obtained by simply changing the monomer feed from ethylene to ε-caprolactone at 80 °C without changing the polymerization conditions. However, unlike the PE-b-PMMA, which showed perfectly controlled chain extension and a monomodal distribution on the SEC chromatogram, PE-b-PCL exhibited a bimodal distribution. This was due to the higher reactivity of polymer chains bound to samarium towards lactone and lactide ROP compared to that of magnesium. The PCL chains grew essentially on the samarium leading to a loss of polymerization control.
 | ||
| Scheme 21 Synthesis of PE-b-PMMA and PE-b-PCL diblock copolymers by CCTP of ethylene followed by MMA polymerization on Mg and ROP on samarium.45 | ||
To develop a simple and practical method for producing ABA olefin block copolymers (OBCs), Lee et al.13 used CCT(co)P by sequentially feeding ethylene and ethylene/propylene mixed gas followed by a coupling reaction to produce PE-b-PEP-b-PE triblock copolymers (Scheme 22). The successive copolymerization of ethylene and ethylene/propylene with a pyridylamido hafnium complex (14), previously activated with [(C18H37)2MeNH]+[B(C6F5)4]−, was carried out in the presence of Zn(Oct)2 as the CTA and MMAO (modified methylaluminoxane) used as a scavenger ([Hf]/[B]/[Zn]/[Al] = 1/1/50/17) at 80 °C in methylcyclohexane. The treatment of the formed Zn-(PE-b-PEP)2 diblock copolymer intermediate (Mn PE-b-PEP = 56 kg mol−1, Đ = 2.10) with an excess of lauroyl peroxide led to C(sp3)–C(sp3) bond formation between the two polymeryl chains, yielding the targeted triblock copolymer (Mn PE-b-PEP-b-PE = 83 kg mol−1, Đ = 1.70) with a yield of 80%. With two semi-crystalline hard outer blocks and a soft middle block, this material showed TPE properties.
 | ||
| Scheme 22 Synthesis of PE-b-PEP diblock and PE-b-PEP-b-PE triblock copolymers through a combination of CCT(co)P of E and E/P, as well as coupling reactions using lauroyl peroxide in the case of the triblock copolymer.13 | ||
Using the well-known dimethyl(pyridylamido) hafnium complex 14, Pan and coworkers have reported a simple and rapid pathway to the synthesis of OBCs without additional coupling steps (Scheme 23).46 The pyridylamido hafnium 14/[Ph3C][B(C5F5)4] catalytic system in combination with a bulky aluminum CTA such as AliBu3 (TIBA) ([Hf]/[B]/[Al] = 1/2/100) was implemented in the polymerization of propylene for 2 min. An isotactic polypropylene (denoted as iPP2) was formed with a Mn of 57 kg mol−1 (Đ = 2). Through the addition of a mixture of ethylene/propylene 1:1 for 1, 3, 6 or 10 minutes, block copolymers with the same hard block iPP2 and different soft block PEP1–10 were produced. A clear increase in the Mn of the copolymers is observed when the reaction time was increased (from 57 for iPP2 to 94 kg mol−1 for iPP2-b-PEP10). All diblock copolymers (iPP2-b-PEP1–10) exhibited narrow, monomodal molar mass distributions. Dispersities increased only slightly (from 2.20 to 2.50) with increasing reaction time. These results clearly demonstrate that hard-b-soft diblock copolymers were successfully prepared by the two-step CCT(co)P reaction, featuring Tm and Tg around 155 °C and −32 °C, respectively. To obtain hard-b-soft-b-hard triblock copolymers, propylene must be fed for 6 minutes after diblock copolymer formation. Comparison of the Mn of the iPP2-b-PEP1 diblock copolymer (Mn iPP2-b-PEP1 = 70 kg mol−1, Đ = 2.20) with that of the iPP2-b-PEP1-b-iPP6 triblock copolymer (Mn iPP2-b-PEP1-b-iPP6 = 198 kg mol−1, Đ = 2.40) suggested that the triblock copolymer was obtained.46
 | ||
| Scheme 23 Synthesis of diblock iPP-b-PEP and triblock iPP-b-PEP-b-iPP copolymers through CCT(co)P of P and E/P.46 | ||
With a similar catalytic system, OBCs with hard semi-crystalline and soft amorphous blocks such as iPP and P(P/α-olefin) segment, respectively, were prepared (Scheme 24).47 The pyridylamido hafnium complex 14 activated with [Ph3C][B(C6F5)4] was used in the presence of TIBA as the CTA ([Hf]/[B]/[Al] = 1/2/150) to polymerize propylene in a first step. In a second step, propylene and 1-octene (Oct) (or 1-octadecene (Od)) copolymerization was performed to obtain diblock copolymers iPP-b-P(P-co-Oct) and iPP-b-P(P-co-Od). The HT-SEC chromatograms showed a significant increase in the Mn of the diblock copolymers compared to the iPP homopolymers. It was also found that under identical reaction conditions, 1-octene showed better incorporation into the soft block than 1-octadecene. Finally, the DSC thermograms of the copolymers showed high Tm and Tg well below those of iPP, suggesting that a promising new class of block copolymers has been established.47
 | ||
| Scheme 24 Synthesis of iPP-b-P(P-co-Oct/Od) diblock copolymers through CCTP of propylene followed by CCTcoP of P and 1-octene or 1-octadecene.47 | ||
By operating a CCTP system in a series of continuous stirred tank reactors (CSTRs), it was possible to form OBCs from a single catalyst through sequential monomer addition (Scheme 25).48 In a primary reactor (R-1) under ethylene feed in the presence of the pyridylamido complex 14 and DEZ ([Zn]/[Hf] = 380), the Dow Chemical Company teams successfully synthesized the high-density polyethylene (HDPE) block. In the secondary reactor (R-2), the very low-density polyethylene (VLDPE) block was subsequently formed after copolymerization of ethylene and 1-octene. The HDPE-b-VLDPE diblock copolymer obtained exhibited a Mn of 44.5 kg mol−1 and a Đ of 1.67. Analytical temperature rising elution fractionation analysis of the diblock copolymer sample shows a single peak at 93 °C, with no evidence of a shoulder at higher temperature that could indicate the absence of free HDPE.
 | ||
| Scheme 25 Cascade CSTRs for the preparation of HDPE-b-VLDPE diblock copolymers.48 | ||
Building up on these results, diblock copolymers based on ethylene and 1-octene with hard blocks of semi-crystalline linear low density polyethylene (LLDPE) and soft blocks of ultra-low density polyethylene (ULDPE) were targeted (Scheme 26).49 By employing the catalytic system based on complex 14 in combination with DEZ in a series of two CSTRs, self-assembled LLDPE-b-ULDPE diblock copolymers with well-ordered morphologies and photonic properties were reported.
 | ||
| Scheme 26 Cascade CSTRs for the preparation of LLDPE-b-ULDPE diblock copolymers.49 | ||
 | ||
| Scheme 27 Synthesis of cis-1,4-PIP-b-PCL diblock copolymers by CCTP of isoprene followed by ROP of ε-caprolactone on neodymium.50 | ||
Using the same traditional Ziegler–Natta system based on neodymium, various CTAs based on alkylaluminum and chloride donors were evaluated for their ability to produce PB by CCTP in a controlled manner.51 The ternary catalytic systems 10/Al(iBu)2H/Me2SiCl2 and 10/Al(iBu)2H/Al2Et3Cl3 exhibited medium catalytic efficiencies, yielding 7 and 9 polymer chains per Nd atom, respectively, in the presence of 20 eq. of CTA and 2 eq. of Cl. Butadiene was successfully polymerized with the 10/Al(iBu)2H/Me2SiCl2 catalytic system to produce a living Nd-polybutadienyl intermediate able to initiate a second block, yielding PB-b-PIP-b-PB and PB-b-PIP-b-PCL triblock copolymers (Scheme 28) with a Mn of 9.83 kg mol−1 (Đ = 1.88) and 9.54 kg mol−1 (Đ = 1.38), respectively. The significant displacement of the initial unimodal PB chromatogram towards higher molar masses after the formation of each block was evidence of the formation of the targeted copolymers.
 | ||
| Scheme 28 Synthesis of PB-b-PIP-b-PB and PB-b-PIP-b-PCL block copolymers by CCTP and ROP with the Nd(OiPr)3/Al(iBu)2H/Me2SiCl2 catalytic system, step 1: [BD]/[Nd] = 500, step 2: [IP]/[Nd] = 500, and step 3: [BD]/[Nd] = 500/[ε-CL]/[Nd] = 300.51 | ||
Unlike previous studies, Zhang et al.52 used a binary catalytic system 10/Mg(n-Bu)2 to form diblock copolymers based on PB having high trans-1,4 units (Scheme 29).52 The Nd complex 10 in contact with the cocatalyst and Mg(n-Bu)2 CTA ([Mg]/[Nd] = 4) provided an active species able to form a highly stereoregular (about 97%) trans-1,4-PB (Mn trans-1,4-PB = 9.28–20.8 kg mol−1, Đ = 1.65–1.80) with a transfer efficiency of up to 34%. The produced living PB was capable of initiating the ROP of ε-caprolactone/L-lactide to give trans-1,4-PB-b-PCL and trans-1,4-PB-b-PLLA (Mn = 18.1–37.1 kg mol−1, Đ = 1.83–1.98) copolymers in a controlled fashion. The formation of the diblock copolymers was monitored by SEC through the shift of the trans-1,4-PB chromatograms towards higher molar masses, and also by DSC, where new melting peaks were observed at 56 and 151 °C, corresponding to the PCL and PLLA blocks, respectively.
 | ||
| Scheme 29 Synthesis of trans-1,4-PB-b-PCL and trans-1,4-PB-b-PLLA diblock copolymers by CCTP of butadiene followed by ROP of ε-caprolactone/L-lactide.52 | ||
The in situ preparation of well-defined sequenced copolymers based on PB, such as PB-b-PCHO (CHO: cyclohexene oxide) and PB-b-PTMC (TMC: trimethylene carbonate), using a neodymium-based catalytic system Nd(vers)3(15)/AliBu2H/Me2SiCl2, has been reported (Scheme 30).53 After establishing the optimal polymerization conditions ([Al]/[Nd] = 20, [Cl]/[Nd] = 3), the first PB block with a high content of 1,4-units was efficiently synthesized via CCTP, and five polymer chains per Nd(vers)3 molecule were formed. Once the butadiene was completely consumed, a second polymerization with cyclohexene oxide was carried out to form the diblock copolymer PB-b-PCHO (Mn PB-b-PCHO = 8.25 kg mol−1, Đ = 1.29). Unimodal SEC curves, low dispersities, and the increased Mn compared to those of the first block indicated that the metal-capped polybutadienyl generated at the end of the first step initiated the polymerization of CHO. These same metal-polybutadienyl chains also initiated the ROP of TMC to form the corresponding PB-b-PTMC copolymer (Mn PB-b-PTMC = 7.93 kg mol−1, Đ = 1.78). Detailed analyses of the copolymers, particularly by 2D DOSY NMR, revealed that the products obtained are a mixture of diblock copolymers and PCHO and PTMC homopolymers primarily formed by AliBu2H that did not react in the first CCTP step.
 | ||
| Scheme 30 Synthesis of PB-b-PCHO and PB-b-PTMC diblock copolymers by CCTP of butadiene followed by ROP of cyclohexene oxide/trimethylene carbonate.53 | ||
With such a catalytic system, it is important to note that unlike previous studies where the authors had completely excluded the possibility of a ROP reaction on the CTA, the ring-opening of the cyclic ether and cyclic carbonate occurred on both the chains attached to Nd and those bound to the CTA. The CTAs that did not participate in the CCTP during the first stage also underwent ROP reactions to form PCHO and PTMC.
The possibility to control both the ethylene/butadiene copolymerization and the ethylene polymerization with the same catalytic system drove Boisson and coworkers to prepare new diblock copolymers by CCTP (Scheme 31)54 using the ansa-bis(fluorenyl) complex {Me2Si(C13H8)2Nd(BH4)2Li(THF)}2 (16) in combination with BOMAG ([Mg]/[Nd] = 40) as a CTA. The diblock copolymers incorporating a soft poly(ethylene-co-butadiene) segment, called ethylene butadiene rubber (EBR), and a hard PE can be produced by simply adjusting the different monomer feeds during polymerization. Since PISA (polymerization-induced self-assembly) is based on the use of a controlled polymerization for the growth of solvophobic segments from solvophilic chains, D'Agosto et al. investigated the formation of nano-objects by successive polymerization of E/B and E. Indeed EBR is fully soluble in the polymerization medium while PE is insoluble when the molar mass is high enough.
 | ||
| Scheme 31 Synthesis of diblock copolymers EBR-b-PE by CCT(co)P of ethylene/butadiene and ethylene.54 | ||
The ansa-bis(fluorenyl) complex 16 combined with a bimetallic CTA, namely pentanediyl-1,5-di(magnesium bromide) (PDMB) ([Mg]/[Nd] = 5–10), has allowed the synthesis of triblock and multiblock copolymers featuring highly semi-crystalline PE hard segments and amorphous EBR soft segments (Scheme 32).55 Triblock copolymers PE-b-EBR-b-PE were synthesized by sequential copolymerization of ethylene with butadiene followed by its chain extension in the presence of ethylene in a one-pot process. Subsequently, thanks to the precise control of the monomer feed and the pseudo-living nature of the polymerization, several feed switches from a mixture of ethylene/butadiene to pure ethylene led to a multiblock copolymer with Mn values of 10 and 3 kg mol−1 for the EBR and PE segments, respectively. A heptablock copolymer with a molar mass of 41.3 kg mol−1 and a dispersity of 2.3 was obtained.
 | ||
| Scheme 32 Synthesis of triblock copolymers PE-b-EBR-b-PE by CCT(co)P of ethylene/butadiene and ethylene using PDMB as a bimetallic CTA.55 | ||
 | ||
| Scheme 33 Synthesis of sPS-b-trans-1,4-PIP diblock copolymers by CCTP of styrene and isoprene.56 | ||
By incorporating innovative chain transfer agents like the bimetallic PDMB in the CCT(co)P process, it is possible to create triblock and multiblock macromolecular architectures with high efficiency and with a minimum number of steps. Substituting the α-olefins, commonly used to disrupt the crystallinity of PE blocks, with a diene monomer has proven to be an interesting alternative, allowing for the generation of OBCs with reactive double bonds in the main chain of the amorphous block. Successive polymerization of diene and cyclic monomers (lactone, lactide, cyclic ether and cyclic carbonate) has been shown to be a particularly promising approach for synthesizing apolar–polar block copolymers. Furthermore, the production of BCPs based on isoprene and styrene via CCTP opens up new perspectives for synthesizing TPEs based on styrenic blocks. These advancements offer unprecedented opportunities for BCP design. However, poor management of monomer feed variation during synthesis stages could quickly prove detrimental to achieving the desired polymers. Hence, finding a catalytic system enabling the formation of block copolymers without changing the feed is appealing.
2.3 Multiblock copolymers synthesized by chain shuttling polymerization (CSP)
Chain shuttling polymerization (CSP) is a process in which two catalysts work together in the presence of a chain shuttling agent (CSA) and two monomers. The different reactivities of the monomers vs. the two catalysts and the reversible transmetalation reactions give rise to original multiblock copolymers that alternate two kinds of blocks. | ||
| Scheme 34 Description of the likely chain shuttling mechanism in a single reactor, dual-catalyst approach ((14)/(18)/DEZ).23 | ||
Alternative chain shuttling catalysts with varying α-olefin selectivities were sought to enable the production of new OBCs. Zirconium and hafnium catalysts featuring imine–amine ligands (19–21) were investigated in combination with 14 as an alternative to bis(phenoxyimine) zirconium complexes.57 For the same monomer feed ([1-octene]/[ethylene] constant), the nature of the catalyst allows the hard block to be tuned to produce OBC with controlled Tm, crystallinity (ΔHf) and solubility (Scheme 35). All catalyst combinations led to polymers with characteristics indicating block architecture. Indeed, a single elution peak on TREF (temperature rising elution fractionation) analysis and Đ = 2 were obtained.
 | ||
| Scheme 35 Synthesis of multiblock copolymers from ethylene and 1-octene using (14)/(19–21)/DEZ catalytic systems. The thickness of the blue lines corresponds to the crystallinity rate of the block: the thicker the line, the more crystalline and richer in ethylene units the segment.57 | ||
A high-throughput experimentation approach was used by Cipullo, Auriemma et al.58 to report the synthesis of a special class of P-TPEs. Ethylene/hexadec-1-ene statistical MBCs were synthesized for the first time by CSP employing the mixed catalytic system (14, 18 and DEZ as the CSA). They are characterized by alternation of crystalline (hard) and amorphous (soft) blocks consisting of random copolymers of ethylene and hexadec-1-ene with low (0.5 mol%) and high (22 mol%) hexadec-1-ene content, respectively. The MBCs had a Mw greater than 100 kg mol−1 and hard block content of 20, 32 and 49 wt% when [14]/[18] = 4, 2 and 1, respectively. On the other hand, except for the fact that the soluble fraction decreases with the decrease of the soft block within the MBCs, the crystallization elution fractionation curves showed a crystalline peak with two maxima (at ≈95–100 and at ≈103–107 °C), which clearly indicates the presence of different chain populations characterized by blocks of different molar masses.
To further expand the properties of cyclic olefin copolymers (COCs), recent research efforts have been devoted to developing a synthesis strategy to modify poly(E-co-N) (N: norbornene) random copolymers.59 Exploitation of the chain shuttling strategy using the two ansa-zirconocene complexes, specifically isopropylidene(η5-cyclopentadienyl)(η5-indenyl)zirconium dichloride (22) and isopropylidene(η5-3-methylcyclopentadienyl)(η5-fluorenyl)zirconium dichloride (23) and DEZ as CSA, allowed the synthesis of multiblock copolymers based on E and N with monomodal molar mass distributions (Đ = 2) (Scheme 36). Complexes 22 and 23 feature high and low norbornene incorporation, respectively. By increasing the amount of CSA (2.8–11 mmol), the molar masses of the copolymers prepared with both catalytic systems decrease (44–14 kg mol−1). This indicates that DEZ actually acts as a CSA ([Zn]/[Zr] = 16).
 | ||
| Scheme 36 Synthesis of multiblock copolymers from ethylene and norbornene using (22), (23)/[PhNH(Me)2][B(C6F5)4]/DEZ catalytic systems.59 | ||
These positive results inspired Li and coworkers to construct new MBCs containing both PE (HDPE in this study) and COC segments, which are expected to exhibit unique and unprecedented performance.60 Two structurally similar ansa-metallocene precatalysts, ethylenebis(1-η5-indenyl)zirconium dichloride (4) and methylenebis(3-tert-butyl-1-η5-indenyl)zirconium dichloride (24), were adopted (Scheme 37). These two catalysts showed comparable catalytic activities but different selectivities towards the copolymerization of ethylene and norbornene, yielding P(E-co-N) random copolymer segments and semi-crystalline PE segments, under the same reaction conditions ([24] = [4], [MAO]/[Zr] = 1000, 100 °C, PE = 4 atm). Complex 4 showed high norbornene incorporation, while complex 24, bearing a sterically hindered tert-butyl substituent on each indenyl ligand, was almost unable to incorporate norbornene (incorporation < 1 mol%). In the presence of DEZ as a CSA ([Zn]/[Zr] = 5–10), polymeryl exchange between the two active zirconium centers occurred and multiblock copolymers containing both soft COC segments and hard linear PE segments were obtained. The average norbornene incorporation and Mn of the copolymer, which govern final performance parameters, were easily adjusted by the DEZ dosage, norbornene/ethylene ratio and/or reaction time. The monomodal distribution of molar masses and the presence of a Tg and a Tm, corresponding to soft and hard blocks, respectively, is an indication of the formation of multiblocks by CSP.
 | ||
| Scheme 37 Synthesis of multiblocks through CSP of ethylene and norbornene using (4), (24)/MAO/DEZ catalytic systems.60 | ||
Very recently, cyclic olefin block copolymers (COBCs) have also been prepared combining two bis(phenoxy-imine) titanium complexes (Scheme 38) in the presence of ZnEt2 as a chain shuttling agent.61 The catalytic system generated blocks with high and low norbornene contents corresponding to hard and soft blocks, respectively. The COBCs show properties ranging from plastic to elastomer, depending on norbornene incorporation.
 | ||
| Scheme 38 Bis(phenoxy-imine) complexes implemented for the synthesis of COBCs.61 | ||
 | ||
| Scheme 39 Synthesis of cis,trans-stereomultiblock PIP copolymers through CSP of isoprene using (27),(28)/BEM/TIBA catalytic systems.62 | ||
In a second contribution on conjugated diene polymerization, a ternary neodymium sulfonate catalytic system Nd(CF3SO3)3·H2O.3TBP (29)/Mg(n-Bu)2/MMAO (TBP: tributyl phosphate) able to form cis-1,4-PB and trans-1,4-PB multiblock copolymers through CSP was reported by Li (Scheme 40).63 The stereospecificity of the PB copolymers was regulated in this case by [MMAO]/[Nd], [Mg]/[Nd], and the delayed feeding time of MMAO. The polymerization of butadiene with the catalytic systems 29/Mg(n-Bu)2 and 29/MMAO resulted in PB with high trans-1,4 (95.3%) (Mn = 7.9 kg mol−1, Đ = 1.34) and cis-1,4 (90.8%) (Mn = 57.5 kg mol−1, Đ = 3.78) contents, respectively. The combination of the two systems in a chain shuttling process using [MMAO]/[Nd] > 80, [Mg]/[Nd] = 6 and the delayed feeding time of MMAO > 15 min yielded a stereoblock of trans-1,4-PB semicrystalline and cis-1,4-PB amorphous segments. The unimodal molar mass distributions and low dispersities (Đ = 1.87–2.20, Mn < 14 kg mol−1) confirm the CSP reaction process. The observation by DSC of two endothermic peaks (40 and 60 °C), indicating a polymorphic structure of the copolymer, and the absence of the crystalline peak in WAXD (wide-angle X-ray diffraction) shows that the degree of crystallinity is low and/or the crystalline domains are very small.
 | ||
| Scheme 40 Synthesis of cis,trans-stereomultiblock PB through CSP of butadiene using (29)/Mg(n-Bu2)/MMAO catalytic systems.63 | ||
 | ||
| Scheme 41 Chain-shuttling copolymerization of styrene and isoprene using two different scandium catalysts, 30 + 31/TIBA//[CPh3][B(C6F5)4] and 30 + 32/TIBA/[CPh3][B(C6F5)4].64 | ||
To extend the potential of CSP to the synthesis of a new class of thermoplastic elastomers, Zinck et al.65 have reported a simple synthetic approach that provides direct access to an unprecedented fully amorphous multiblock microstructure of soft and hard random copolymers. The lanthanum half-sandwich complex [(C5Me5)La(BH4)2(THF)2] (33) and the ansa-neodymocene [(C5H4CMe)2Nd(BH4)2]2Mg(THF)3 (34) were used in combination with BEM as the CSA ([Mg]/[33 + 34] = 1) in the presence of isoprene and styrene to design multiblock copolymers (poly(isoprene-co-styrene)). They display sequences rich in isoprene (trans-1,4-PIP) and sequences rich in styrene, respectively (Scheme 42). The monomodal molar mass distribution of the resulting MBC (Mn = 62.3 kg mol−1, Đ = 2.1) and the decrease of the Mn with increasing amounts of BEM ([Mg]/[33 + 34] = 1 to 10) showed that the reversible transmetalation of the growing polymer chains with BEM occurred efficiently during the CSP step. It should be noted, however, that in the presence of excess BEM, transmetalation does not allow sufficient chain growth on each of the catalysts to achieve the multiblock microstructure. The objective is only reached if one equivalent of BEM is used. The microstructure consists of poly(isoprene-co-styrene) blocks, richer in one of the two monomers leading to soft and hard blocks. Indeed, the resulting polymers were characterized by two Tg values, one at −15 °C attributed to the Tg of the isoprene-rich blocks and one at 41 °C attributed to the styrene-rich blocks.
 | ||
| Scheme 42 Chain shuttling copolymerizations of isoprene and styrene using 33 and 34 in combination with BEM.65 | ||
The remarkable atom efficiency of the CSP process of ethylene and 1-octene that leads to multiblock copolymers allows the formation of approximately 260 chains per catalyst molecule, along with the superior elastomeric qualities of the final product. It has paved the way for the synthesis of high-performance and cost-effective products.
Currently, the annual production of SBS is approximately 2 million tons, whereas that of their hydrogenated derivatives, SEBS, is limited to only 300000 tons.66 Despite their evident benefits (e.g. higher thermal and chemical stability), the high cost of SEBS restricts their utilization across various applications. The quest for a synthetic strategy offering significant atom economy and capable of producing copolymers with structures and compositions similar to SEBS, without necessitating additional expensive steps such as hydrogenation, is of importance for expanding the applications of these TPE materials. The switch from one polymerization technique to another one to combine blocks of different nature with such hard polystyrene and soft polyolefin blocks might thus be an attractive strategy. The following sections aim at providing the different strategies employed along these lines.
2.4 Polymerization switch (catalytic/catalytic or anionic/catalytic) for block copolymer synthesis
 | ||
| Scheme 43 Synthesis of PE-b-trans-1,4-PIP by switching from ethylene CCTP to isoprene coordination catalysis.67 | ||
 | ||
| Scheme 44 General description of the preparation of PO-b-PS diblock copolymers by the switch method from E/Oct CCTcoP to anionic polymerization of styrene.68 | ||
With the aim of synthesizing various PO (PEP) based block copolymers, the CCTcoP of ethylene and propylene was conducted with the pyridylamido hafnium complex 14 and Zn(1-hexyl)2 CTA([Zn]/[Hf] = 75).69 The Zn(PEP)2 species generated in this initial step was then brought in contact with the anionic polymerization preinitiator, pentylallyl-Li·(PMDTA), to form the active initiator complex [Zn(PEP)2(pentylallyl)]−[Li·(PMDTA)]+(Scheme 45). The PS chains were then efficiently developed by simple addition of the styrene monomer at elevated temperature to prevent precipitation of the polymers generated. The formation of the new PEP-b-PS was monitored by SEC. As anticipated, the final chromatogram shifted towards higher molar masses compared to that of the starting PO after the addition of styrene. However, it was found that 29 wt% of the starting styrene was used to form PS chains.
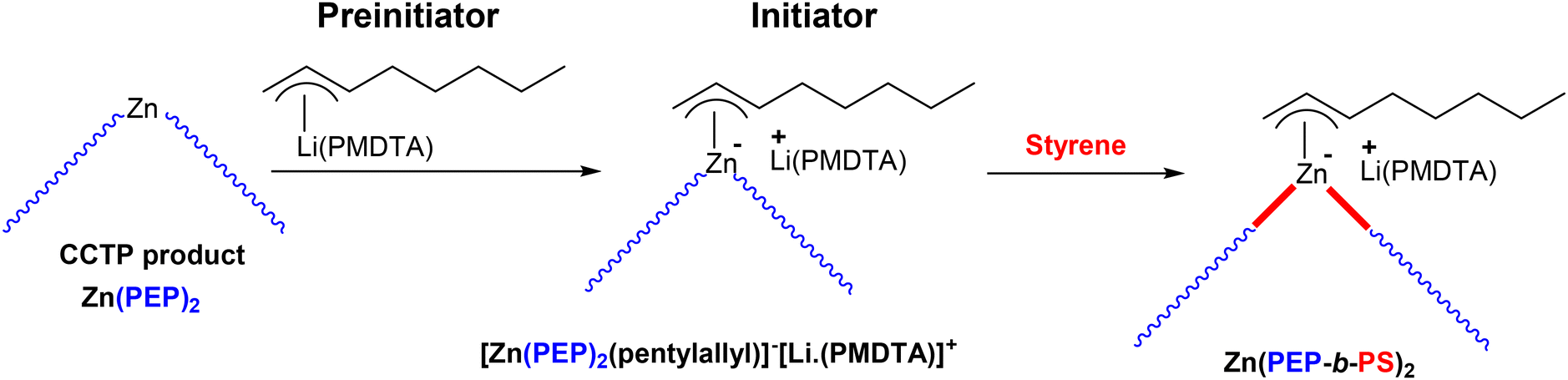 | ||
| Scheme 45 Preparation of PEP-b-PS diblock copolymers by switching from CCTcoP of ethylene and propylene to anionic polymerization of styrene. The diblock copolymer is obtained only after the reaction of a protic compound with the Zn(PEP-b-PS)2 species.69 | ||
Following his work, Lee et al. prepared PS-b-PO-b-PS triblock copolymers that mimic the chain structure of SEBS.70 In a first step, a dialkylzinc compound bearing an α-methylstyrene moiety (Zn[4-(isopropenyl)benzyl]2) was used in combination with the previous catalytic system 36 ([Al]/[36] = 200) during CCTcoP of ethylene and 1-octene or ethylene and 1-pentene (Scheme 46). The switch to anionic polymerization was then carried out by adding n-BuLi(TMEDA)2 ([Li] > [Zn] + [Al in MMAO]) and styrene monomer to achieve PS chain growth from Zn-PO sites and α-methylstyrene moieties. Depending on the experimental conditions, diblock or multiblock copolymers were generated. The formation of block copolymers was evident on comparing the molar mass of the samples before and after anionic polymerization. However, the formation of a significant amount of PS homopolymer initiated by n-BuLi(TMEDA)2 and undefined triblock copolymer was unavoidable during the process.
 | ||
| Scheme 46 Method for preparation of P(E-co-P)-b-PS and P(E-co-Oct)-b-PS diblock copolymers by switching from CCTcoP of ethylene and propylene/1-octene to anionic polymerization of styrene by using n-BuLi(TMEDA)2 as a preinitiator.70 | ||
In order to design well-defined triblock copolymers, a new multinuclear zinc species, Et[Zn(CH2)6]αZnEt, was prepared by successive addition of BH3 and Et2Zn to 1,5-hexadiene (Scheme 47a).71 Using this compound as a CTA, PEP chains were biaxially grown from –(CH2)6– units in Et[Zn(CH2)6]3ZnEt via a CCTcoP using the pyridylamido hafnium complex 14 previously activated with a borate [(C18H37)MeNH]+[B(C6F5)4]− ([Hf]/[B]/[Zn] = 1/1/37–50). With this catalytic system, undesired β-H elimination was suppressed. PS blocks were subsequently grown from these chains by the introduction of the anionic preinitiator Me3SiCH2Li-(PMDTA) ([Li]/[Zn] = 0.73 or 0.80) and styrene ([styrene]/[Zn] = 500) in MCH at 100–110 °C to form the targeted PS-b-PEP-b-PS triblock copolymers (Scheme 47b). The production of free PS (15 to 22% of the initial styrene mass) and diblock copolymers PEP-b-PS was not prevented. Nevertheless, the SEC curves of the obtained polymer chains shifted to higher molar masses after the anionic polymerization of styrene, and the observed Mn values (39 to 56 kg mol−1) were approximately twice those of the PS homopolymer (20–23 kg mol−1), in accordance with the assumption that the PS chains are generated from both chain ends of the PEP.
 | ||
| Scheme 47 (a) Synthesis of the dialkylzinc species. (b) Preparation of PS-b-PEP-b-PS triblock copolymers by switching from E/P CCTcoP to anionic polymerization of styrene by using Me3SiCH2Li(PMDTA) as a preinitiator.71 | ||
Similarly, a novel dialkylzinc species containing vinylbenzene moieties was synthesized via alkyl exchange between Et2Zn and the hydroboration product of divinylbenzene (Scheme 48a).72 From this species, PEP chains were grown via CCTcoP using the pyridylamido hafnium complex 14 ([Zn]/[Hf] = 38). Subsequently, PS chains were attached to both ends of the PEP chains by the introduction of styrene in addition to the anionic polymerization preinitiator Me3SiCH2Li·(PMDTA) ([Li]/[Zn] = 1, [St]/[Zn] = 500). The molar mass distributions obtained in this study (Đ = 1.67 to 1.84) are broader than those obtained in previous studies using Et-[Zn-(CH2)6]3-Zn-Et as a CTA (Đ = 1.37 to 1.64). These data suggest that the block copolymers generated are not simple PS-b-PEP-b-PS triblocks. Multiblock copolymers (e.g., PS-b-PEP-b-PS-b-PEP-b-PS) can be generated when the chain-growing styryl anion connected to PO chains attacks the styrene moieties at the ends of other PO chains (Scheme 48b).
 | ||
| Scheme 48 (a) Synthesis of the dialkylzinc species containing vinylbenzene moieties. (b) Species targeted and obtained after the combination of E/P CCTcoP and anionic polymerization of styrene using Me3SiCH2Li(pmdeta) as the preinitiator.72 | ||
One of the main issues encountered by Lee and his colleagues in the course of their research was the preparation of high-purity diorganozinc compounds with vinylbenzene moieties at the ends.66 In this contribution, the authors synthesized Zn(CH2CH2CH2C6H4CHCH2)2 from 4-vinylbenzyl chloride, a commercially available and inexpensive product (Scheme 49a).66 Triblock copolymer PS-b-P(E-co-1-hexene)-b-PS (SEHS) was then prepared by first performing a copolymerization (CCTcoP) of ethylene and 1-hexene (H) with the pyridylamido hafnium complex 14 ([Zn]/[Hf] = 300). This was followed by a one-pot polymerization of styrene using the anionic preinitiator pentylallyl-Li-(PMDTA) (Scheme 49b). The Mw value was substantially increased from 90 kg mol−1 to 161 kg mol−1 and the molar mass distribution was slightly narrowed (from 1.90 to 1.80) after the anionic polymerization step. DSC analyses revealed the Tg for PEH and PS chains at −55 °C and 99 °C, respectively, along with broad and weak Tm at 35 °C. By adjusting the contents of ethylene, 1-hexene, and styrene, and the Mn of block copolymers, TPE with stress–strain curves similar to those of commercial-grade SEBS (Kraton G1651) was obtained despite the presence of approximately 7 wt% of PS in the final SEHS.
 | ||
| Scheme 49 (a) Synthesis of Zn(CH2CH2CH2C6H4CHCH2)2 dialkylzinc species. (b) Preparation of PS-b-P(E-co-1-hexene)-b-PS triblock copolymers by switching from E/1-hexene CCTcoP to anionic polymerization of styrene by using pentylallyl-Li(PMDTA) as a preinitiator.66 | ||
 | ||
| Scheme 50 General description of the preparation of diblock copolymers based on successive anionic polymerization and coordinative chain transfer (co)polymerization (CCT(co)P).73 | ||
By varying the monomer feed composition to prepare block copolymers, the same team produced a series of PS-b-EBR-b-PE triblock copolymers.74 These original block copolymers combine glassy and a semi-crystalline hard block, respectively. Their structure and mechanical properties are highlighted in section 3.2.
Although there is currently only one example of copolymers obtained by the switch from anionic polymerization to CCT(co)P, this process has nonetheless demonstrated its robustness through a better-controlled anionic polymerization reaction, characterized by low dispersities and a close match between theoretical and experimental molar masses. This strategy, which allows for anionic polymerization under mild conditions, opens a very promising pathway for the design of new block copolymers, particularly triblock copolymers with elastomeric properties.
After reviewing the state-of-the-art of different techniques for synthesizing block copolymers with varied compositions, microstructures, and architectures, the following section will focus exclusively on block copolymers with elastomeric properties. A brief overview of the definition, history, architecture, and morphology of TPEs will be provided before discussing the mechanical performance of TPEs resulting from CCTcoP and CSP.
3. Mechanical properties of polyolefin thermoplastic elastomers (P-TPEs) based on block and multiblock copolymers
3.1. Introduction
Due to the recent global expansion of the TPE market that reached $16 billion in 2022 and is projected to reach $25.5 billion in 2028 according to estimates, many companies are producing these materials, including BASF, Arkema, Kraton, Dow Chemical Company, Huntsman, Covestro, and many others.75 The global TPE market includes several types of products, including styrenic block copolymers (SBC), thermoplastic polyurethanes (TPU), thermoplastic olefins (TPO), thermoplastic vulcanizates (TPV), thermoplastic copolyesters (COPE), and polyether block amides (PEBA). In this contribution, after an overview of TPEs, particularly their structure, architecture, and morphology, we will focus on describing the mechanical properties of linear P-TPEs. The investigation of grafted copolymers is out of the scope of this review. | ||
| Fig. 1 (a) Simplified structure of a TPE, consisting of soft rubbery segments and hard glassy or crystalline segments. (b) Typical dynamic mechanical analysis (DMA) curves for thermoplastic elastomers. Reproduced from ref. 77 with permission from American Chemical Society, copyright 1992. | ||
The discovery of living anionic polymerization by Szwarc in 1956 was a key step in providing a new fundamental concept for BCP synthesis by sequential monomer addition.80 In 1966, researchers at Shell Chemical Company synthesized and characterized ABA-type block copolymers based on polystyrene hard segments (block A) and polyisoprene soft segments (Block B).81 This discovery was of considerable academic and industrial importance, since triblock copolymers with an elastic midblock component were revealed as an attractive alternative for chemically crosslinked rubber materials. These TPEs, better known as Kraton, were first used in the formulation of leisure shoes and as a bitumen modifier, while their hydrogenated version, “Kraton G”, with improved oxidation, UV and heat stability, has found applications in medical devices, coatings, automotive components and adhesives.82
The toolbox of synthetic polymer chemistry has been considerably improved since then by advances in living and controlled polymerization techniques, including cationic polymerization,83 anionic polymerization,84 ring-opening polymerization,85 RDRP,86 and coordination polymerization.87 Well-defined polymers with various macromolecular architectures and functional groups have been prepared by these techniques for application in the TPE field.88–92 Most copolymers used as TPEs are linear ABA and ABC triblock copolymers, and multiblock copolymers (AB)n, with A and C serving as rigid blocks and B as elastic blocks.
 | ||
| Fig. 2 (a) Typical stress–strain curve observed in a TPE material. (b) Standard tests for elastic recovery conducted on TPEs. | ||
The elastic recovery of the specimen is evaluated through cyclic tensile tests (Fig. 2b). In this test, the sample is subjected to loading and unloading cycles at significantly high deformations (customarily about 300% strain in order to reach the vicinity of the strain hardening region). From the stress–strain curve, the elastic recovery (ER) can be calculated using the formula ER = 100 × (εa − εr)/εa, where εa is the applied deformation and εr is the residual deformation in the zero-load cycle after the applied deformation. Compression set experiments are typically carried out by applying 10 to 25% compression at different temperatures over 24 h. When comparing TPEs to thermosetting vulcanized rubbers, this test is undeniably the most rigorous, and most TPEs hardly reach values below 30% at 70 °C while best performance vulcanized rubbers (e.g. FFKM) reach values below 10% at temperatures above 150 °C.
While TPE is highly tunable, and commercial product lines feature grades with a very large range of properties, achieving the combination of high tensile strength, great extensibility, and excellent elastic recovery is more difficult, and combining the latter with a low melt viscosity enabling injection or extrusion processes at high throughputs is a true challenge.
 | ||
| Fig. 3 (a) Schematic illustration of possible chain conformations for ABA and ABC triblock copolymers. Reproduced from ref. 5 with permission from Elsevier, copyright 2019. (b) Visualization of possible conformations for symmetrical ABA triblock copolymers. Reproduced from ref. 97 with permission from Elsevier, copyright 2022. | ||
In contrast, AB diblock copolymers and BAB triblock copolymers, with the rubbery blocks at extremities, do not exhibit the characteristic behavior of TPEs, and fail in particular at showing high elastic recovery at large strains.
 | ||
| Fig. 4 χN versus fPI diagram for PI-b-PS diblock copolymers. Open and filled circles represent the order–order (OOT) and order–disorder (ODT) transitions, respectively. The dash–dot curve is the mean field prediction for the ODT. Solid curves have been drawn to delineate the different phases observed but might not correspond to precise phase boundaries. Reproduced from ref. 99 with permission from American Chemical Society, copyright 1995. | ||
Morphologies featuring isolated rigid domains such as spheres are strongly favored, as they ensure that macroscopic strains are mainly distributed locally in the continuous rubbery phase. Such morphologies promote desirable features such as low Young's moduli, high elastic recovery at intermediate deformations, and strain hardening at high deformations due to the anchoring effect of rigid domains. More extended and continuous rigid domains, such as cylinders, bicontinuous structures or lamellae, lead to mechanical reinforcement and higher tensile strengths and Young's moduli, at the expense of lower extensibility and elastic recovery.97 The presence of a yield point at low strains typically corresponds to irreversible rupture at interfaces between hard and soft domains, or partial fragmentation of elongated hard domains. Correspondingly, a significant stress softening is observed after the first cycle, which may lead in the subsequent cycles to very good elastic recoveries.
This feature also illustrates the importance of the processing during the manufacture of parts with TPEs: these materials have been intrinsically designed to enable fast processing such as injection molding or blow molding, and the resulting nanoscale morphologies might be far from those obtained after extended annealing using solvents or thermal processes. As a result, phase diagrams might be rather seen as guides for the design of the macromolecular structure rather than limitations to follow scrupulously.
The rules guiding the self-assembly in amorphous, strongly separated BCPs become much more complex in copolymers containing one or more crystallizable blocks, and in this phase separation is driven by crystallization.100 Complex processes can occur when two distinct phase separation mechanisms are simultaneously present (block incompatibility and crystallization): the final structure is influenced both by the segregation strength and the sequence of phase transformations occurring during the cooling from the melt.101–103 Distinct morphologies can be obtained when the crystallization remains confined within preformed microdomains, breaks out of the microdomains, or alters their shape by a templating process.104
The effective relationships between the macromolecular structures of the BCPs, their morphology, and the mechanical properties at large strains (e.g. strain hardening and elastic recovery) are highly complex. They can also depend on the processing, might be heavily modified by carefully selected or fortuitous formulations (e.g. the presence of a small fraction of diblock copolymers with the triblock copolymer), and are still largely studied experimentally with a case-by-case approach.
A few general rules can however be relevant: the minimum anchoring length for amorphous rigid segments in triblocks is expected to be approximatively their entanglement molar mass, and the middle rubbery block should be well entangled (Mn,rubbery > 3 Me-rubbery). In comparison, multiblock segmented copolymers can tolerate shorter rigid segments as the rubber elasticity is due to multiple anchoring sites. A high polydispersity in the BCPs is not necessarily disastrous as long as rigid segments are properly anchored in well-segregated domains, and if the presence of elastically inactive chains (e.g. diblocks and unentangled chains) is kept under control.
3.2. Thermoplastic elastomers resulting from CCT(co)P and CSP
In addition to the large availability and low cost of olefinic monomers, their main advantage as TPEs is derived from the high crystallinity of well-controlled segments such as iPP or linear PE and their superior chemical resistance (they are practically not soluble in any known solvent at room temperature). The low entanglement molecular weight of common polyolefins is also highly beneficial in yielding materials with high rubber moduli or to accommodate BCPs with relatively low molar mass (Mn < 150 kg mol−1). While the service temperature range is primarily limited by the melting temperature of the crystalline blocks, the whole melting range is also very important. In TPEs featuring less defined crystalline segments or poor crystallization-induced phase separation, the elastic recovery at large strains might be strongly degraded at even 30–60 °C below the Tm.In this section, we review more specifically the mechanical properties of TPEs synthesized using CCT(co)P and CSP. The nomenclature of the block copolymers discussed is kept the same as in the original publications in order to facilitate their identification in the figures. However, additional explanations will be provided when necessary for clarity.
As shown in the first part of this review, significant advancements in the design of OBCs were achieved in 2006, when Arriola et al.23 reported the synthesis of MBC composed of ethylene and 1-octene via the chain shuttling polymerization mechanism (section 2.3.1). These materials exhibited an alternation of semi-crystalline hard blocks (low 1-octene content) and amorphous soft blocks (high 1-octene content). Optimized formulations revealed tensile strengths ranging from 3 to 13 MPa and elongations at break between 480 and 1540%.105 The temperature gap between the Tg (−64 to −54 °C) and Tm (≈120 °C) made these TPEs suitable for applications covering a wide temperature range.23 Recently, through in-depth studies, Auriemma et al.58 provided additional information on the structure–property relationship of these commercial elastomers.106 While exact determination of segment lengths still remains a challenge, it was demonstrated that for comparable overall molar masses and compositions, an architecture featuring a large number of short segments (e.g. about 14) was preferred over a low number of long segments (e.g. about 4) (Fig. 5a and b). This example illustrates one of the many requisites that must be met for TPEs obtained by CSP: in addition, the hard segments must be highly crystalline and with sufficient length to induce phase separation.
 | ||
| Fig. 5 Illustration of multiblock copolymers with identical chain molar masses but varying mechanical properties. (a) Chains containing a low number of high molar mass hard–soft diblock units (loose elastomer network). (b) Chains containing a high number of low molar masses hard–soft diblock units (tight elastomer network). Adapted from ref. 106 with permission from Elsevier, copyright 2018. | ||
Since then, many research groups have been interested in this CSP methodology, involving chain transfer. In recent years, several successful syntheses of MBC composed of olefinic, styrenic, or diene monomers have been reported (see section 2.3). However, few of these materials, if any, have proven to be promising candidates for applications as TPE materials. One promising example from researchers at the University of Naples was recently reported with a multiblock copolymer based on ethylene and hexadec-1-ene (see section 2.3.1).58 This copolymer consists of an alternation of semi-crystalline hard blocks, with a low content of n-hexadec-1-ene units, and amorphous soft blocks, with high content of hexadec-1-ene units. Mechanical property analysis at 25 °C of a sample containing 20 wt% of hard blocks revealed a soft TPE behavior (Fig. 6a), with no pronounced yielding, good elastic recovery, and a deformation at break equal to 1600% for a low tensile strength (σb = 3.1 MPa).
 | ||
| Fig. 6 (a) Stress–strain curves for statistical multiblock copolymers based on ethylene and hexadec-1-ene containing 20 wt% of hard block obtained at 25 °C and −15 °C. (b) Simplified model for the crystallization behavior of the MBC samples stretched at high deformations at 25 °C and then cooled to −15 °C. The model does not account for the increase in the degree of deformation of the main chain crystals occurring at low temperatures. Adapted from ref. 58 with permission from American Chemical Society, copyright 2024. | ||
At −15 °C, the mechanical behavior of this sample undergoes considerable changes with a notable increase in Young's modulus reaching 59 MPa and a tensile strength of 8.7 MPa. This remarkable reinforcement was attributed to the crystallization of the long side chain of the hexadec-1-ene unit below 2 °C.
While CSP produces MBC from a starting monomer feed using a single polymerization process, the CCT(co)P method appears easier to implement in polyolefin TPEs as it involves only one catalyst and one CTA. The production of MBC, however, relies on successive changes in monomer feed or combination with other polymerization techniques.
One of the earliest examples of TPE obtained by CCTP was documented by Lee. PE-b-PEP-b-PE triblock copolymers were obtained by coupling PE-b-PEP diblocks at the extremities of the PEP block (see section 2.2.1).13 The transition from a diblock copolymer to a triblock copolymer resulted in a significant increase in tensile strength and elongation at break (entry 4 vs. entry 5 in Fig. 7a). As the fraction of the PE block increased from 15 to 25 wt% within the triblock copolymer (Fig. 7a, entry 4, 7 and 8), the elongation at break decreased from 1260 to 1100 and 820% while the tensile strength remained almost constant (5.2–5.4 MPa). In the cyclic tensile test with up to 100% deformation, the triblock copolymer (labeled entry 4 in Fig. 7a) exhibited an irreversible deformation of 38% during the first cycle (Fig. 7b), followed by relatively good recoveries and no rupture during the subsequent nine cycles. In contrast, the diblock copolymer showed increased brittleness and failed the cyclic tensile test even at low elongation. The presence of a clear yield point in the tensile tests, as well as a relatively modest elastic recovery performance typically indicated that PE domains are not sufficiently dispersed, and that the mechanical strength of these domains is not sufficient, probably due to an insufficient crystallinity.
 | ||
| Fig. 7 (a) Comparison of the stress–strain curves of PE-b-PEP-b-PE triblock copolymers (labeled entries 4, 7 and 8) with that of the PE-b-PEP diblock copolymer (labeled entry 5). (b) Cyclic tensile tests of the triblock copolymer PE-b-PEP-b-PE (labeled entry 4 in a). Adapted from ref. 13 with permission from American Chemical Society, copyright 2018. | ||
Triblock and multiblock copolymers containing highly semi-crystalline PE hard segments and soft EBR segments were obtained via the development of a bimetallic CTA (see Scheme 32 and section 2.2.2).55 These block copolymers are organized into lamellar structures induced by the crystallization of the disordered melt (Fig. 8).
 | ||
| Fig. 8 PeakForce AFM images (modulus) of PE3k-b-EBR30k-b-PE3k (left) and PE6k-b-EBR60k-b-PE6k (right). Reproduced from ref. 55 with permission from John Wiley and Sons, copyright 2023. | ||
The triblock copolymer PE3k-b-EBR30k-b-PE3k (Fig. 9a), composed of low Mn PE segments, showed limited strength (σmax = 1.5 MPa) and low elongation (εb = 580%) which were attributed to the relatively easy fragmentation or chain pullout from crystalline lamellae composed of a small number of Mn PE blocks (Fig. 9a). In contrast, the triblock PE6k-b-EBR60k-b-PE6k with longer PE segments exhibited significantly higher strength, with a slight yield point at low deformations and a pronounced strain hardening occurring beyond 600% elongation (Fig. 9a), similar to those of chemically crosslinked elastomers. The ultimate elongation and strength are 1450% and 4.4 MPa, respectively, while the elastic recovery is above 85% after 9 cycles. We note, as discussed above, that the non-recoverable deformation essentially originates from the first cycle. These data also illustrate clearly some features that must be considered when analyzing cycling testing, as the low molar mass copolymer PE3k-b-EBR30k-b-PE3k presented higher elastic recoveries while showing clearly much poorer tensile properties than PE6k-b-EBR60k-b-PE6k. These contradictory results are due to the combination of a much lower initial yield stress for PE3k-b-EBR30k-b-PE3k and the fact that 300% deformation in these samples is not yet sufficient to truly probe elasticity, as the strain hardening region rather starts above 600% deformation.
 | ||
| Fig. 9 (a) Graph showing the results of tensile tests performed on PE-b-EBR diblock, PE-b-EBR-b-PE triblock, and multiblock copolymers. (b) Elastic recovery following repetitive 300% tensile cycles of the block copolymers at room temperature. Adapted from ref. 55 with permission from John Wiley and Sons, copyright 2023. | ||
Finally, the mechanical property analysis of the multiblock (labeled 11d in Fig. 9a) composed of seven alternating PE3k and EBR10k segments showed considerably improved strength (σb = 3.2 MPa) compared to the PE3k-b-EBR30k-b-PE3k triblock copolymer, but with almost identical elongation at break (εb = 650%).
Very recently, Li et al. reported the synthesis of OBCs comprising semi-crystalline iPP segments and soft segments containing propylene and α-olefin (see section 2.2.1).47 Uniaxial tensile tests were conducted to examine the impact of the Mn of the hard and soft blocks, as well as the composition of the soft blocks, on the mechanical properties of the OBCs. In this case the copolymer samples were named Px-(P/Mn)y, where the subscripts x and y denote the polymerization time (min) for the propylene homopolymerization (synthesis of the first block) and the copolymerization (synthesis of the second block), respectively; P denotes propylene; M denotes the comonomer (O = 1-octene, Od = 1-octadecene, and E = ethylene) in the copolymerization; and n denotes the average comonomer content (mol%) in copolymers.
For a comparable α-olefin content, the P2-(P/O17)15 diblock copolymer in Fig. 10a exhibits significantly higher tensile strength and elongation at break than the random copolymer (P/O21)15. For the elastomeric OBCs with similar block molar masses but different α-olefin contents, the tensile strength decreased with increasing α-olefin content. For example, the tensile strength decreased from 25 MPa for P2-(P/O7)15 to 14 MPa for P2-(P/O17)15 when the 1-octene content increased from 7.1 to 16.8 mol% (Fig. 10a). According to the authors, these differences are attributed to variations in the crystallinity levels within the materials. To further investigate the elastomeric properties of OBCs with high soft block fractions, step cycle tensile tests were conducted to examine the elastic recovery of these OBCs (Fig. 10b). The block copolymers P2-(P/O7)15 and P2-(P/Od6)15 with similar Mn and α-olefin contents but different branch lengths exhibited similar ER values of 57.5% and 54.6%, respectively. For a similar Mn, the ER increased from 57.5% for P2-(P/O7)15 to 83.8% for P2-(P/O17)15, when the α-olefin content increased from 7.1 to 16.8 mol%, and the difference is due to better entanglement of chains containing more α-olefins in the soft block. These results suggest that α-olefin content is the main factor affecting the ER of OBCs with similar Mn, while the nature of α-olefin has a lower impact on ER.
 | ||
| Fig. 10 (a) Graph showing tensile test results for (P/O) homopolymers and P(P/O) and (P/Od) diblock copolymers.(b) Strain recovery of (P/O) homopolymers and representative P(P/O) and (P/Od) diblock copolymers as a function of applied stresses. Reproduced from ref. 47 with permission from American Chemical Society, copyright 2024. | ||
Furthermore, the tensile performance of OBCs containing poly(propylene-co-ethylene) copolymer segments as soft blocks was tested for comparison. The results showed a lower elongation at break presumably due to reduced chain entanglement (Fig. 11).
 | ||
| Fig. 11 Graph showing tensile test results for P-(P/E), P-(P/O) and P-(P/Od) diblock copolymers. Reproduced from ref. 47 with permission from American Chemical Society, copyright 2024. | ||
The limitations of current catalytic systems to the polymerization of specific olefins and α-olefins have motivated researchers to develop new synthetic strategies to access new classes of high-performance TPEs, particularly those incorporating styrenic groups capable of imparting rigidity and resistance to the material. As shown previously, the combination of a CTA bearing a vinylbenzene end group and an anionic polymerization preinitiator, pentylallyl-Li-(PMDTA), enabled the synthesis of PS-b-PEH-b-PS (SEHS) triblock copolymers,66 structurally similar to commercially available SEBS TPEs (see section 2.4.2).
The first resulting SEHS with a Mw of 146 kg mol−1 and a composition of ethylene, 1-hexene, and styrene at 45, 30, and 25 wt%, respectively (Table 6, and Fig. 12a, entry 1) was compared to a commercial SEBS (Kraton G1651) (Table 6, and Fig. 12a, entry 8) with an equivalent molar mass (Mw = 139 kg mol−1). Tensile tests showed similar tensile strength and elongation at break for the two samples, with σb = 22 MPa and εb = 1700% for SEHS compared to σb = 29 MPa and εb = 2000% for the SEBS. Increasing the 1-hexene content from 30 to 37 wt% resulted in an increase in elongation at break for the SEHS from 1700% to 2500% (Table 6, and Fig. 12a, entries 1 and 2), but the ultimate tensile strength decreased from 22 to 18 MPa. Varying the styrene content from 25 to 33 wt% increased the ultimate tensile strength from 22 to 35 MPa (Table 6, and Fig. 12a, entry 3), whereas the elongation at break decreased from 1700% to 1300%. SEHS with stress–strain characteristics comparable to commercial-grade SEBS (Table 6, and Fig. 12a, entry 7) was analyzed by TEM. The images (Fig. 12b) showed a well-ordered lamellar structure for the SEBS, while the SEHS exhibited a disordered spherical structure. This morphological difference was attributed to the dispersity differences between the two samples. Finally, as a comparative measure, blends of PP/SEHS and PP/SEBS were formulated to assess their efficacy as toughening agents for commercial PP (CB5230). It was notably observed that substituting SEBS with the prepared SEHS led to a noticeable enhancement in impact strength, while the tensile properties were maintained at a similar level.66
 | ||
| Fig. 12 (a) Stress–strain curves for of PS-b-PEH-b-PS (SEHS) and commercial-grade SEBS (Kraton G1651). (b) TEM images of prepared SEHS (entry 7) and commercial-grade SEBS (Kraton G1651). Reproduced from ref. 66 with permission from American Chemical Society, copyright 2020. | ||
| Entry | Ethylene contenta (wt%) | Hexene contenta (wt%) | Styrene contenta (wt%) | σ b (MPa) | ε b (%) |
|---|---|---|---|---|---|
| a Values determined by 1H NMR. b Data obtained from stress–strain curves. c wt% of 1-butene in a commercial SEBS copolymer determined by 1H NMR. | |||||
| 1 | 44.9 | 30.4 | 25.0 | 22 | 1700 |
| 2 | 41.4 | 36.6 | 22.1 | 18 | 2500 |
| 3 | 43.6 | 23.7 | 32.6 | 35 | 1300 |
| 4 | 37.9 | 28.5 | 33.6 | 7 | 1700 |
| 5 | 37.7 | 31.0 | 31.3 | 12 | 1800 |
| 6 | 41.1 | 33.2 | 25.7 | 20 | 2200 |
| 7 | 43.6 | 31.9 | 24.5 | 23 | 1900 |
| 8 | 43.4 | 24.8c | 31.8 | 29 | 2000 |
As we have discussed above, one major limitation of such styrenic symmetric triblocks is the high viscosity due to strong phase separation between the PS and olefinic domains occurring even in the melt. To overcome such limitations, CCT(co)P could be combined with anionic polymerization to yield asymmetric triblock copolymers. In a recent illustration, Boisson et al. showed that switching from the anionic polymerization of styrene to a sequenced CCT(co)P of ethylene/butadiene and then ethylene enabled obtaining PS-EBR-PE triblock copolymers featuring a central rubbery segment, and a glassy PS segment and a highly crystalline PE segment at the extremities (see section 2.4.3).74 This particular structure enabled a dual phase separation that combines strong segregation between PS and olefin domains in the melt, and crystallization-induced phase separation within the olefin domains at service temperatures (Fig. 13).
 | ||
| Fig. 13 Peak-force AFM adhesion images of asymmetric triblocks; from left to right: PS6k-b-EBR60k-b-PE6k, PS10k-b-EBR60k-b-PE6k and PS20k-b-EBR60k-b-PE6k. Reproduced from ref. 74 with permission from John Wiley and Sons, copyright 2025. | ||
In practice, this strategy offered an excellent compromise between moderate viscosities in the melt, as these TPEs rather behave effectively as phase-separated diblocks and not triblocks, and very good mechanical properties in service as the anchoring of rubbery chains occurs in two distinct rigid domains (one glassy and one crystalline). Comparison of the mechanical properties (Fig. 14) with those of purely semicrystalline TPE such as PE-EBR-PE (Fig. 9) shows significantly improved tensile strengths and elastic recovery (σb > 6 MPa and recovery > 95% after 9 cycles for the sample PS10k-b-EBR60k-b-PE6k).
 | ||
| Fig. 14 (a) Tensile testing of PS-EBR-PE triblock copolymers. (b) Elastic recovery after cyclic extension of the samples with up to 300% elongation. Reproduced from ref. 74 with permission from John Wiley and Sons, copyright 2025. | ||
3.3. Conclusion on the investigation of the mechanical properties of BCP obtained by CCTP and CSP
In recent years, numerous catalytic complexes have been employed to synthesize block or multiblock copolymers with precision and efficiency via CCTP or CSP (Table 7). Olefinic multiblock copolymers obtained through CSP, comprising semi-crystalline hard ethylene-rich blocks and soft amorphous α-olefin-rich blocks, have swiftly found industrial applications owing to their high deformation resistance and elongation capacity exceeding 1500%. Subsequently, the utilization of CCTP has facilitated the design of diblock and triblock copolymers consisting of semi-crystalline polyolefin hard segments and amorphous soft segments based on ethylene and dienes or α-olefins. The stress–strain curves of these copolymers have exhibited breaking strengths of up to 25 MPa for an elongation of 1100%, directly rivaling commercial TPEs. The switch method enabling the transition from CCTP to anionic polymerization has proven to be an effective route for producing olefin-based SBCs. Their tensile strength, reaching 35 MPa with a minimal elongation of 1300%, positions them as serious contenders to replace current SEBS obtained through anionic polymerization and post-polymerization hydrogenation. Advances in synthetic chemistry are crucial for the future development of TPEs. The preparation of polar, biodegradable, temperature-resistant, and environmentally resistant TPEs, using readily available monomers and polymerization methods such as CCTP, with high atom economy, could lead to high-performance materials with significant industrialization potential.| Polymerization method | Copolymer | σ b (MPa) | ε b (%) | E (MPa) | Ref. |
|---|---|---|---|---|---|
| Chain shuttling | Multiblock E/Oct | 3–13 | 450–1540 | — | 23 |
| Chain shuttling | Multiblock E/Oct | 25 °C:3.1 |
1600–800 | 2.4 | 58 |
| −15 °C:9 |
110 | ||||
| CCTP | PE-b-PEP-b-PE | 5.2–5.4 | 820–1260 | nd | 13 |
| CCTP | PE-b-EBR-b-PE and E/EBR multiblock | 1.5– > 4.4 | 580–1450 | 7.6–16.8 | 55 |
| CCTP | (P/O), P(P/O), P(P/Od), and P(P/E) | 4.9–25 | 990–1200 | 1.5–23.8 | 47 |
| CCTP and anionic polymerization | PS-b-EBR-b-PE | 3–9 | >1400 | 6–22 | 74 |
| CCTP and anionic polymerization | PS-b-PEH-b-PS | 7–35 | 1300–2500 | nd | 66 |
| Anionic polymerization | SEBS (Kraton G1651) | 29 | 2000 | nd | 66 |
4. Summary and outlook
The use of CCTP for olefin polymerization has emerged as a straightforward and economical approach to produce POs under mild conditions. The metal–polymer bonds generated during synthesis have been found to be easily functionalizable, allowing for the formation of species with hydroxyl, iodo, or thiol terminal groups. The reaction of these hydroxyl- and thiol-terminated POs with lactones or lactides facilitates the ring-opening polymerization of these monomers, thereby enabling the formation of diblock copolymers composed of apolar PO-based segments and polar polyester-based segments. Some researchers have also exploited CCTP products to form macro-initiators for RDRP. The combination of CCTP with techniques such as ATRP and NMP has enabled the formation of new block copolymers based on POs (or conjugated dienes) and polyacrylate or polystyrene. Various coupling reactions, including cycloaddition, transesterification, hetero-Diels–Alder, and Thia-Michael reactions, have been employed to couple initially functionalized polyolefins produced by CCTP, which has allowed the generation of a wider range of apolar–polar block copolymers. However, the non-quantitative functionalization of the chain end, along with the sometimes significant number of steps required to obtain the desired product, remain major obstacles to the potential industrialization of these block copolymer design methods.The successive polymerization of ethylene, dienes, or cyclic monomers (lactones, lactides, cyclic ethers, and cyclic carbonates) without prior end-group functionalization of the polymers derived from CCTP has demonstrated a particularly promising approach for the synthesis of block copolymers. The sole variation of monomer concentration in the reaction medium during polymerization enabled the synthesis of several well-defined block copolymers, suitable for high-performance applications such as TPEs (PE-b-EBR-b-PE and PE-b-PEP-b-PE).
The combination of two active sites (metal center) along with a reversible chain storage site (chain shuttling agent) during chain shuttling polymerization enabled the tailored synthesis of multiblock copolymers. By maintaining a constant monomer concentration in the reaction medium and selectively adjusting each catalyst's ability to stereospecifically insert a monomer or copolymerize an olefin, 1,3-dienes, and styrene, multiblock copolymers were efficiently produced via the chain shuttling mechanism. Recently, the CSP concept has also been extended to cyclic ester polymerization, leading to innovative polylactide-based block copolymers.107
The limitations of current catalytic systems for the polymerization of olefins and α-olefins have prompted researchers to develop new synthetic strategies to access new classes of high-performance TPEs, particularly those incorporating styrenic groups capable of conferring rigidity and strength to materials. The combination of a CTA bearing a vinylbenzene terminal group and an anionic polymerization pre-initiator, pentylallyl-Li(PMDTA), has enabled the synthesis of PS-b-PEH-b-PS (SEHS) triblock copolymers with structure and mechanical properties similar to commercially available SEBS TPEs. However, further optimization of this synthetic process is crucial to better control anionic polymerization and prevent side reactions such as the undesired formation of homopolymers. Moreover, the high temperature (110 °C) required to form the PS blocks by anionic polymerization constitutes a major drawback for optimal control of the reaction.
The switch from anionic polymerization to CCTP has demonstrated high robustness through a better-controlled anionic polymerization reaction. This is characterized by low dispersities and a close match between theoretical and experimental molar masses. This strategy, which allows for anionic polymerization under mild conditions (40 °C), opens a very promising pathway for the design of new block copolymers, particularly triblock copolymers with elastomeric properties. For instance, it would be possible to design with this methodology P-TPEs that incorporate glassy blocks with high Tg, thereby broadening the application range of these materials.
There is no doubt that CCTP will continue to develop over the coming years, opening the way to new macromolecular architectures. The possibilities are numerous. They concern not only the combination of catalysts in CSP, but also the combination of different polymerization chemistries, which can be carried out successively or in a single step. A major challenge concerns the design of TPEs with a higher service temperature, particularly at high temperatures, which requires the introduction of high Tg or Tm hard blocks.
Author contributions
Conceptualization, CB; writing—original draft preparation, SA; and writing—review and editing, SA, FDA, DM and CB. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
The authors thank the Centre National de la Recherche Scientifique (CNRS) for funding under the interdisciplinary program ‘80 Prime’, and the Manufacture Française des Pneumatiques Michelin for support.References
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS PubMed.
- S. Rutkowski, A. Zych, M. Przybysz, M. Bouyahyi, P. Sowinski, R. Koevoets, J. Haponiuk, R. Graf, M. R. Hansen, L. Jasinska-Walc and R. Duchateau, Macromolecules, 2017, 50, 107–122 CrossRef CAS.
- H. Feng, X. Lu, W. Wang, N.-G. Kang and J. W. Mays, Polymers, 2017, 9, 494 Search PubMed.
- H. Dau, G. R. Jones, E. Tsogtgerel, D. Nguyen, A. Keyes, Y.-S. Liu, H. Rauf, E. Ordonez, V. Puchelle, H. Basbug Alhan, C. Zhao and E. Harth, Chem. Rev., 2022, 122, 14471–14553 Search PubMed.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N.-G. Kang, K. Hong and J. W. Mays, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- C. M. Bates and F. S. Bates, Macromolecules, 2017, 50, 3–22 Search PubMed.
- K. T. Delaney and G. H. Fredrickson, J. Phys. Chem. B, 2016, 120, 7615–7634 CrossRef CAS.
- H. Wang, S. Li, J. Zeng and T. Zhang, J. Chem. Phys., 2025, 162, 104105 Search PubMed.
- N. Hu, C.-K. Mai, G. H. Fredrickson and G. C. Bazan, Chem. Commun., 2016, 52, 2237–2240 Search PubMed.
- S. Nojiri, S. Kimata, K. Ikeda, T. Senda, A. W. Bosman, J. W. Peeters and H. M. Janssen, Macromolecules, 2017, 50, 5687–5694 Search PubMed.
- H. Ohtaki, F. Deplace, G. D. Vo, A. M. LaPointe, F. Shimizu, T. Sugano, E. J. Kramer, G. H. Fredrickson and G. W. Coates, Macromolecules, 2015, 48, 7489–7494 CrossRef CAS.
- S. D. Kim, T. J. Kim, S. J. Kwon, T. H. Kim, J. W. Baek, H. S. Park, H. J. Lee and B. Y. Lee, Macromolecules, 2018, 51, 4821–4828 CrossRef.
- A. Sakuma, M.-S. Weiser and T. Fujita, Polym. J., 2007, 39, 193–207 CrossRef.
- G. W. Coates, P. D. Hustad and S. Reinartz, Angew. Chem., Int. Ed., 2002, 41, 2236 CrossRef.
- Y. Kang, H. Wang, X. Li, F. Meng, H. Liu, Y. Xiao, Z. Jiang, H. Gao, C. Liu, F. Wang, L. Pan and Y. Li, Macromolecules, 2024, 57, 4208–4219 CrossRef.
- R. Kempe, Chem. – Eur. J., 2007, 13, 2764–2773 CrossRef PubMed.
- A. Valente, A. Mortreux, M. Visseaux and P. Zinck, Chem. Rev., 2013, 113, 3836–3857 CrossRef.
- R. Mundil, C. Bravo, N. Merle and P. Zinck, Chem. Rev., 2024, 124, 210–244 CrossRef.
- V. C. Gibson, Science, 2006, 312, 703–704 CrossRef.
- L. R. Sita, Angew. Chem., Int. Ed., 2009, 48, 2464–2472 CrossRef.
- F. D'Agosto and C. Boisson, Aust. J. Chem., 2010, 63, 1155–1158 CrossRef.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef.
- C. J. Han, M. S. Lee, D.-J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923–8925 CrossRef.
- F. Jamali, D. Hassanian-Moghaddam, S. Ahmadjo, S. M. M. Mortazavi, Y. Maddah and M. Ahmadi, Eur. Polym. J., 2022, 169, 111142 CrossRef.
- A. A. Burkey, D. M. Fischbach, C. M. Wentz, K. L. Beers and L. R. Sita, ACS Macro Lett., 2022, 11, 402–409 CrossRef.
- J. O. Ring, R. Thomann, R. Mülhaupt, J. Raquez, P. Degée and P. Dubois, Macromol. Chem. Phys., 2007, 208, 896–902 CrossRef.
- J. Mazzolini, I. Mokthari, R. Briquel, O. Boyron, F. Delolme, V. Monteil, D. Bertin, D. Gigmes, F. D'Agosto and C. Boisson, Macromolecules, 2010, 43, 7495–7503 CrossRef.
- C. Lefay, D. Glé, M. Rollet, J. Mazzolini, D. Bertin, S. Viel, C. Schmid, C. Boisson, F. D'Agosto, D. Gigmes and C. Barner-Kowollik, J. Polym. Sci., Part A:Polym. Chem., 2011, 49, 803–813 CrossRef.
- S. K. T. Pillai, W. P. Kretschmer, M. Trebbin, S. Förster and R. Kempe, Chem. – Eur. J., 2012, 18, 13974–13978 CrossRef.
- H. Kaneyoshi, Y. Inoue and K. Matyjaszewski, Macromolecules, 2005, 38, 5425–5435 CrossRef.
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. Van Meurs, J. Am. Chem. Soc., 2004, 126, 10701–10712 CrossRef PubMed.
- F. Wang, B. Dong, H. Liu, J. Guo, W. Zheng, C. Zhang, L. Zhao, C. Bai, Y. Hu and X. Zhang, Macromol. Chem. Phys., 2015, 216, 321–328 CrossRef.
- L. Jasinska-Walc, M. Bouyahyi, P. Lorenc, A. L. Heeneman, R. Duchateau, A. Różański and K. V. Bernaerts, Polymer, 2018, 147, 121–132 CrossRef.
- R. Godoy Lopez, C. Boisson, F. D'Agosto, R. Spitz, F. Boisson, D. Gigmes and D. Bertin, J. Polym. Sci., Part A:Polym. Chem., 2007, 45, 2705–2718 CrossRef.
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311–9313 CrossRef.
- E. Espinosa, B. Charleux, F. D'Agosto, C. Boisson, R. Tripathy, R. Faust and C. Soulié-Ziakovic, Macromolecules, 2013, 46, 3417–3424 CrossRef.
- T. Li, W. J. Wang, R. Liu, W. H. Liang, G. F. Zhao, Z. Li, Q. Wu and F. M. Zhu, Macromolecules, 2009, 42, 3804–3810 CrossRef.
- J. T. Offenloch, S. Norsic, H. Mutlu, M. Taam, O. Boyron, C. Boisson, F. D'Agosto and C. Barner-Kowollik, Polym. Chem., 2018, 9, 3633–3637 RSC.
- E. Espinosa, M. Glassner, C. Boisson, C. Barner-Kowollik and F. D'Agosto, Macromol. Rapid Commun., 2011, 32, 1447–1453 CrossRef.
- J. Mazzolini, O. Boyron, V. Monteil, F. D'Agosto, C. Boisson, G. C. Sanders, J. P. A. Heuts, R. Duchateau, D. Gigmes and D. Bertin, Polym. Chem., 2012, 3, 2383 RSC.
- P. Li, Z. Fu and Z. Fan, J. Appl. Polym. Sci., 2015, 132, 42236 CrossRef.
- A. Ginzburg, V. Ramakrishnan, L. Rongo, A. Rozanski, M. Bouyahyi, L. Jasinska-Walc and R. Duchateau, Rheol. Acta, 2020, 59, 601–619 CrossRef.
- H. Yasuda, M. Furo, H. Yamamoto, A. Nakamura, S. Miyake and N. Kibino, Macromolecules, 1992, 25, 5115–5116 CrossRef.
- T. Chenal, X. Olonde, J.-F. Pelletier, K. Bujadoux and A. Mortreux, Polymer, 2007, 48, 1844–1856 CrossRef.
- H. Gao, X. Lu, S. Chen, B. Du, X. Yin, Y. Kang, K. Zhang, C. Liu, L. Pan, B. Wang, Z. Ma and Y. Li, Macromolecules, 2022, 55, 5038–5048 CrossRef.
- Y. Kang, H. Wang, X. Li, F. Meng, H. Liu, Y. Xiao, Z. Jiang, H. Gao, C. Liu, F. Wang, L. Pan and Y. Li, Macromolecules, 2024, 57, 4208–4219 CrossRef.
- P. D. Hustad, R. L. Kuhlman, D. J. Arriola, E. M. Carnahan and T. T. Wenzel, Macromolecules, 2007, 40, 7061–7064 CrossRef.
- P. D. Hustad, G. R. Marchand, E. I. Garcia-Meitin, P. L. Roberts and J. D. Weinhold, Macromolecules, 2009, 42, 3788–3794 CrossRef.
- F. Wang, C. Zhang, Y. Hu, X. Jia, C. Bai and X. Zhang, Polymer, 2012, 53, 6027–6032 CrossRef.
- F. Wang, H. Liu, W. Zheng, J. Guo, C. Zhang, L. Zhao, H. Zhang, Y. Hu, C. Bai and X. Zhang, Polymer, 2013, 54, 6716–6724 CrossRef.
- W. Zheng, N. Yan, Y. Zhu, W. Zhao, C. Zhang, H. Zhang, C. Bai, Y. Hu and X. Zhang, Polym. Chem., 2015, 6, 6088–6095 RSC.
- M. Zhang, L. Liu, R. Cong, J. Dong, G. Wu, F. Wang, H. Liu and X. Zhang, Eur. Polym. J., 2021, 148, 110355 CrossRef.
- N. Baulu, M. Langlais, P. Dugas, J. Thuilliez, F. Jean-Baptiste-dit-Dominique, M. Lansalot, C. Boisson and F. D'Agosto, Chem. – Eur. J., 2022, 28, e202202089 CrossRef PubMed.
- M. Langlais, N. Baulu, S. Dronet, C. Dire, F. Jean-Baptiste-dit-Dominique, D. Albertini, F. D'Agosto, D. Montarnal and C. Boisson, Angew. Chem., Int. Ed., 2023, 62, e202310437 CrossRef PubMed.
- P. Zinck, A. Valente, A. Mortreux and M. Visseaux, Polymer, 2007, 48, 4609–4614 CrossRef.
- R. L. Kuhlman and J. Klosin, Macromolecules, 2010, 43, 7903–7904 Search PubMed.
- A. Vittoria, G. Urciuoli, S. Costanzo, V. Ianniello, R. Cipullo, F. D. Cannavacciuolo, O. R. De Ballesteros, V. Busico, N. Grizzuti and F. Auriemma, Macromolecules, 2024, 57, 1532–1545 Search PubMed.
- I. Tritto, L. Boggioni, G. Scalcione, D. Sidari and N. G. Galotto, J. Organomet. Chem., 2015, 798, 367–374 CrossRef.
- H. Gao, S. Chen, B. Du, Z. Dai, X. Lu, K. Zhang, L. Pan, Y. Li and Y. Li, Polym. Chem., 2022, 13, 245–257 RSC.
- N. Qiu, Z. Sun, F. Yu, K. Wang, C. Long, Z. Dong, Y. Li, K. Cao and Z.-R. Chen, Macromolecules, 2024, 57, 5729–5738 CrossRef.
- Y. Phuphuak, F. Bonnet, G. Stoclet, M. Bria and P. Zinck, Chem. Commun., 2017, 53, 5330–5333 Search PubMed.
- Q. Dai, X. Zhang, Y. Hu, J. He, C. Shi, Y. Li and C. Bai, Macromolecules, 2017, 50, 7887–7894 CrossRef.
- L. Pan, K. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 12012–12015 CrossRef.
- A. Valente, G. Stoclet, F. Bonnet, A. Mortreux, M. Visseaux and P. Zinck, Angew. Chem., Int. Ed., 2014, 53, 4638–4641 Search PubMed.
- J. C. Lee, K. L. Park, S. M. Bae, H. J. Lee, J. W. Baek, J. Lee, S. Sa, E. J. Shin, K. S. Lee and B. Y. Lee, Macromolecules, 2020, 53, 7274–7284 CrossRef.
- T. Chenal and M. Visseaux, Macromolecules, 2012, 45, 5718–5727 CrossRef.
- J. Y. Jeon, S. H. Park, D. H. Kim, S. S. Park, G. H. Park and B. Y. Lee, J. Polym. Sci., Part A:Polym. Chem., 2016, 54, 3110–3118 CrossRef.
- T. J. Kim, J. W. Baek, S. H. Moon, H. J. Lee, K. L. Park, S. M. Bae, J. C. Lee, P. C. Lee and B. Y. Lee, Polymers, 2020, 12, 537 CrossRef PubMed.
- D. H. Kim, S. S. Park, S. H. Park, J. Y. Jeon, H. B. Kim and B. Y. Lee, RSC Adv., 2017, 7, 5948–5956 RSC.
- S. S. Park, C. S. Kim, S. D. Kim, S. J. Kwon, H. M. Lee, T. H. Kim, J. Y. Jeon and B. Y. Lee, Macromolecules, 2017, 50, 6606–6616 CrossRef CAS.
- C. Kim, S. Park, S. Kim, S. Kwon, J. Baek and B. Lee, Polymers, 2017, 9, 481 CrossRef.
- N. Baulu, M. Langlais, R. Ngo, J. Thuilliez, F. Jean-Baptiste-dit-Dominique, F. D'Agosto and C. Boisson, Angew. Chem., Int. Ed., 2022, 61, e202204249 CrossRef CAS.
- S. Alioui, M. Langlais, R. Ngo, K. Habhab, S. Dronet, F. Jean-Baptiste-dit-Dominique, D. Albertini, F. D'Agosto, C. Boisson and D. Montarnal, Angew. Chem., Int. Ed., 2025, 64, e202420946 CrossRef CAS PubMed.
- Exactitude Consultancy, https://exactitudeconsultancy.com/fr/rapports/1584/march%C3%A9-des-%C3%A9lastom%C3%A8res-thermoplastiques/, (accessed May 24, 2024).
- G. Zanchin and G. Leone, Prog. Polym. Sci., 2021, 113, 101342 CrossRef CAS.
- R. B. Seymour and G. B. Kauffman, J. Chem. Educ., 1992, 69, 967 CrossRef CAS.
- M. Sun, Y. Xiao, K. Liu, X. Yang, P. Liu, S. Jie, J. Hu, S. Shi, Q. Wang, K. H. Lim, Z. Liu, B. Li and W. Wang, Can. J. Chem. Eng., 2023, 101, 4886–4906 CrossRef CAS.
- J. G. Drobny, Handbook of thermoplastic elastomers, 2nd edn., 2014 Search PubMed.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- H. Geoffrey and M. Ralph, Shell Oil Company, US3265765, 1966.
- Kraton Corporation, https://kraton.com/about/.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS.
- A. Hirao, R. Goseki and T. Ishizone, Macromolecules, 2014, 47, 1883–1905 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- G. W. Coates, P. D. Hustad and S. Reinartz, Angew. Chem., Int. Ed., 2002, 41, 2236 CrossRef CAS.
- A. Hotta, E. Cochran, J. Ruokolainen, V. Khanna, G. H. Fredrickson, E. J. Kramer, Y.-W. Shin, F. Shimizu, A. E. Cherian, P. D. Hustad, J. M. Rose and G. W. Coates, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15327–15332 CrossRef CAS.
- K. E. Crawford and L. R. Sita, ACS Macro Lett., 2015, 4, 921–925 CrossRef CAS.
- M. B. Harney, Y. Zhang and L. R. Sita, Angew.Chem., Int.Ed., 2006, 45, 2400–2404 CrossRef CAS.
- Adhesives & Sealants Industry, https://www.adhesivesmag.com/articles/91909-self-assembling-acrylic-block-copolymers-for-enhanced-adhesives-properties, (accessed May 20, 2024).
- H. Wang, W. Lu, W. Wang, P. N. Shah, K. Misichronis, N. Kang and J. W. Mays, Macromol. Chem. Phys., 2018, 219, 1700254 Search PubMed.
- G. Holden, E. T. Bishop and N. R. Legge, J. Polym. Sci., 1969, 26, 37–57 Search PubMed.
- H. Watanabe, Macromolecules, 1995, 28, 5006–5011 CrossRef CAS.
- K. R. Albanese, J. R. Blankenship, T. Quah, A. Zhang, K. T. Delaney, G. H. Fredrickson, C. M. Bates and C. J. Hawker, ACS Polym. Au, 2023, 3, 376–382 CrossRef CAS.
- K. R. Albanese, J. R. Blankenship, T. Quah, A. Zhang, K. T. Delaney, G. H. Fredrickson, C. M. Bates and C. J. Hawker, ACS Polym. Au, 2023, 3, 376–382 CrossRef CAS.
- M. Steube, T. Johann, R. D. Barent, A. H. E. Müller and H. Frey, Prog. Polym. Sci., 2022, 124, 101488 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- A. K. Khandpur, S. Foerster, F. S. Bates, I. W. Hamley, A. J. Ryan, W. Bras, K. Almdal and K. Mortensen, Macromolecules, 1995, 28, 8796–8806 CrossRef CAS.
- C. De Rosa, A. Malafronte, R. Di Girolamo, F. Auriemma, M. Scoti, O. Ruiz De Ballesteros and G. W. Coates, Macromolecules, 2020, 53, 10234–10244 CrossRef CAS.
- Y.-L. Loo, R. A. Register and A. J. Ryan, Macromolecules, 2002, 35, 2365–2374 CrossRef CAS.
- S. B. Myers and R. A. Register, Macromolecules, 2009, 42, 6665–6670 CrossRef CAS.
- A. B. Burns and R. A. Register, Macromolecules, 2016, 49, 269–279 CrossRef CAS.
- C. De Rosa, R. Di Girolamo, A. Malafronte, M. Scoti, G. Talarico, F. Auriemma and O. Ruiz De Ballesteros, Polymer, 2020, 196, 122423 CrossRef CAS.
- INFUSETM Olefin Block Copolymers Product Selection Guide, https://www.dow.com/en-us/document-viewer.html?randomVar=2768608821355614010&docPath=/content/dam/dcc/documents/788/788-08201-01-infuse-olefin-block-copolymers-selection-guide.pdf, (accessed May 23, 2025).
- F. Auriemma, C. De Rosa, M. Scoti, R. Di Girolamo, A. Malafronte, G. Talarico and E. Carnahan, Polymer, 2018, 154, 298–304 CrossRef CAS.
- J. Meimoun, C. Sutapin, G. Stoclet, A. Favrelle, P. Roussel, M. Bria, S. Chirachanchai, F. Bonnet and P. Zinck, J. Am. Chem. Soc., 2021, 143, 21206–21210 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |