DOI:
10.1039/C6RA15395J
(Paper)
RSC Adv., 2016,
6, 77752-77759
Green synthesis and characterization of zinc oxide nanoparticles using carboxylic curdlan and their interaction with bovine serum albumin
Received
14th June 2016
, Accepted 12th July 2016
First published on 9th August 2016
Abstract
Carboxylic curdlan (Cc), as a versatile β-1,3-polyglucuronic acid derivative, was used as both reducing and capping agents for the green synthesis of zinc oxide nanoparticles (ZnO NPs). The as-prepared Cc–ZnO NPs had the hexagonal wurtzite structure with maximum absorption at 370 nm assigned to the intrinsic band-gap absorption. Scanning and transmission electron microscopy (SEM and TEM) images showed that the Cc–ZnO NPs were spherical and agglomerated particles with an average diameter of 58 ± 6 nm. The interaction between Cc–ZnO NPs and bovine serum albumin (BSA) was evaluated using various spectroscopic techniques. The results indicated that the binding between Cc–ZnO NPs and BSA molecules occurred due to static quenching, leading to the formation of a ground state complex. Synchronous fluorescence spectra indicated changes in the microenvironment close to the tryptophan residues; circular dichroism spectra revealed conformational changes in the secondary structure of BSA, which were attributable to electrostatic interactions during the binding process.
Introduction
Zinc oxide nanoparticles (ZnO NPs) are unique semiconductor nanomaterials and have attracted much attention for their potential applications in electronics, optics, optoelectronics and the biomedical fields.1,2 Up to now, a variety of methods have been reported for the synthesis of ZnO NPs such as sol–gel solvothermal, direct precipitation and hydrothermal etc.1 However, most of these approaches require tedious processes, expensive substrates, sophisticated equipment and rigorous experimental circumstances. Furthermore, chemical methods lead to the presence of some toxic chemicals adsorbed on the surface that may have adverse effects in medical applications.3 Therefore, further development of simpler and greener methods for ZnO NPs is needed to widen their range of applications in pharmacy, pharmacology and biochemistry.
With increasing concerns over environmental pollution, more and more efforts are being put in developing green synthesis strategies for the preparation of ZnO NPs using non-toxic, eco-friendly and renewable materials, such as those originated from biological sources including plants,4,5 bacteria,6 fungus,7 and microalgae.8 Green technology (biosynthesis) can avoid inert gases, high pressure, high temperature, laser radiation, toxic chemicals and so on, when compared with those conventional methods, and is playing a vital role in the synthesis of ZnO NPs.9 Polysaccharides are favorable for food and biomedical applications because of their desirable biocompatibility and biodegradability as well as a broad spectrum of functional and bioactive properties.10 Several natural and modified polysaccharides such as starch and its carboxymethyl derivatives,11,12 chitosan,13 and gum tragacanth (GT)14 have been used as both chelating and stabilizing agents for the green synthesis of ZnO NPs due to the presence of hydroxyl, carboxyl and amino groups in the polysaccharide molecules. Compared with uncapped ones, the polysaccharide-capped ZnO NPs are more favorable owing to their more prominent surface functionalization and hydrophilicity.15 Besides oxides, biomolecules/polysaccharides have also been frequently developed to prepare other functional and versatile nanomaterials, such as noble metal nanomaterials,16–18 and semiconductor quantum dots.19 Very recently our group has successfully used carboxylic curdlan (Cc) as a reducing and stabilizing agent for the facile and green synthesis of metal nanoparticles in an aqueous solution.20,21 This study was to apply Cc for the green synthesis of ZnO NPs and to evaluate the interactions of ZnO NPs with a model protein bovine serum albumin (BSA).
Nanoparticles in an aqueous protein solution can be coated by a layer of proteins, resulting in the formation of a protein 'corona' that surrounds the nanoparticles and shields their original surface properties.22,23 Therefore, the protein-nanoparticle interaction in a physiological environment is of great importance for biomedical applications of nanoparticles in the emerging areas of nanomedicine and nanotoxicity. Of particular interest is the interaction between nanoparticles and proteins in the plasma. Serum albumin is a water-soluble protein that responsible for binding and transporting a wide range of endogenous and exogenous compounds in the bloodstream to their target organs.24 Bovine serum albumin is most frequently used as model protein due to its well-characterized properties and its structural homology to human serum albumin.25 BSA has also been used in previous studies on the protein interactions with metal oxide nanoparticles such as ZnO,15,26–28 Al2O3,29 and magnetic iron oxides.30 Particularly, Chakraborti et al. reported that the interaction between PEI-functionalized ZnO NPs and BSA was mainly attributed to electrostatic forces, instead of hydrophobic forces for the uncapped ZnO NPs.15 Additionally, Fan et al. demonstrated that the interaction between TEMPO-SNPs and BSA was mainly attributed to electrostatic forces.31 However, to the best of our knowledge, there is no reported study on the interaction between polysaccharide-functionalized ZnO NPs and BSA.
In the present study, a simple and eco-friendly approach was developed for the synthesis of ZnO NPs in an aqueous medium by using Cc as both reducing and stabilizing agents. The structural characteristics of the as-prepared Cc–ZnO NPs were elucidated through electron microscopy and spectroscopy analyses. Furthermore, the interaction of the Cc–ZnO NPs with BSA was investigated.
Materials and methods
Materials and chemicals
Carboxylic curdlan (Cc) bearing the β-1,3-polyglucuronic acid structure was prepared by using 4-acetamido-TEMPO-mediated oxidation, whose details could be found in our previous work.32 The carboxylate content and molecular weight (MW) of Cc were 4.87 mmol g−1 and 1.2 × 105 Da, respectively. Zinc acetate dihydrate [Zn(Ac)2·2H2O, ≥98%], and bovine serum albumin (BSA, MW 6.6 × 104 Da) were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). All other chemicals and solvents were of laboratory grade and used without further purification.
Green synthesis of zinc oxide nanoparticles (ZnO NPs)
0.2 g of Zn(Ac)2·2H2O and 0.2 g of Cc were respectively dissolved in 40 mL of deionized water under constant stirring at room temperature for 30 min. After that, the zinc acetate solution was added to an equal volume of Cc aqueous solution, and the resultant mixture was maintained at 70 °C under continuous stirring in a water bath for 6 h. A pale white solid product was collected through centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min after thorough washing with deionized water and then freeze-dried. The resultant dried precursor was crushed into powder and stored in an airtight container for further analysis.
000 rpm for 20 min after thorough washing with deionized water and then freeze-dried. The resultant dried precursor was crushed into powder and stored in an airtight container for further analysis.
Characterization of Cc–ZnO NPs
The UV-visible absorption spectrum of the Cc–ZnO NPs solution was measured by a Lambda 35 spectrophotometer (Perkin Elmer, USA) in the wavelength range of 200–700 nm with an interval of 1.0 nm. The particle size and morphology of the Cc–ZnO NPs were observed using transmission electron microscopy (TEM) (Tecnai 12, Philips, 120 kV). For TEM analysis, the sample was prepared by placing a drop of the Cc–ZnO NPs solution on a carbon coated copper grid (300 meshes), followed by drying at room temperature (20 °C) for 30 min. The shape, size and microstructures of the sample were characterized by using scanning electron microscopy (SEM, S4800, Hitachi Ltd., Japan) at acceleration voltages of 5.0 kV. The elemental composition of the nanoparticles was determined by energy dispersive X-ray spectroscopy (EDX, Noran, Thermo Fisher Scientific, USA) attached to the SEM (S4800, Hitachi Ltd., Japan). The crystalline size and structural property of the Cc–ZnO NPs were determined by X-ray diffraction (XRD, D8-Advance, Bruker, Co., Germany). The XRD patterns were recorded with Cu Kα radiation (λ = 0.1541 nm) at 40 kV and 40 mA in the 2θ ranging from 20° to 90° with a step speed of 4° min−1. The Cc and freeze-dried Cc–ZnO NPs were used for Fourier-transform infrared (FT-IR) analysis. The FT-IR analysis was performed on a Nexus 670 FT-IR spectrometer (Thermo Nicolet Co., USA) in the wavenumber range of 400–4000 cm−1 with KBr pellets and referenced against air.
Interaction of BSA with Cc–ZnO NPs
A stock solution of BSA (1.5 × 10−5 M) was prepared in advance by dissolving 0.1 g of BSA in 100 mL of deionized water and kept in a refrigerator before use. For the interactions between BSA and Cc–ZnO NPs, 0.3 mL of BSA stock solution was added to 2.7 mL of the as-prepared Cc–ZnO NPs aqueous solution with different concentrations. The mixture was then homogenized and incubated at 25 °C in a water bath for 1 h under continuous stirring. After the incubation, the mixture was kept at 4 °C before measurement.
Characterization of BSA and Cc–ZnO NPs interactant
All fluorescence measurements were recorded on a Cary Eclipse fluorescence spectrophotometer (Varian Co., USA) at ambient temperature. For fluorescence quenching, the excitation wavelength was set at 295 nm and emission was measured in the range of 300–470 nm and with excitation and emission slit widths fixed at 5 nm each. For synchronous fluorescence spectra, the initial excitation wavelength was set at 200 nm and scanned up to 500 nm with a scanning speed of 200 nm min−1. The difference between excitation and emission wavelength (Δλ) was set at 15 nm for tyrosine residue and at 60 nm for tryptophan residue. Excitation and emission slit width was set at 5.0 nm. The UV-vis absorption spectra were recorded using a Lambda 35 spectrophotometer (Perkin Elmer, USA) at room temperature in the wavelength range of 200–500 nm. Circular dichroism (CD) spectra were recorded using a J-815 CD spectrometer (JASCO, Japan) at room temperature in the wavelength range of 200–250 nm. The measurements were taken in the far-UV region in a quartz cuvette with a path length of 1 cm, and band width of 5 nm.
Results and discussion
Green synthesis and characterization of Cc–ZnO NPs
As demonstrated in our previous studies,20,21 carboxylic curdlan (Cc) derived from 4-acetamido-TEMPO-mediated oxidation was a negatively charged molecule bearing a number of carboxyl (COO–) groups. The carboxyl groups may be responsible for chelating and reducing zinc ions, and to form zinc oxide nanoparticles (ZnO NPs) in an aqueous medium.
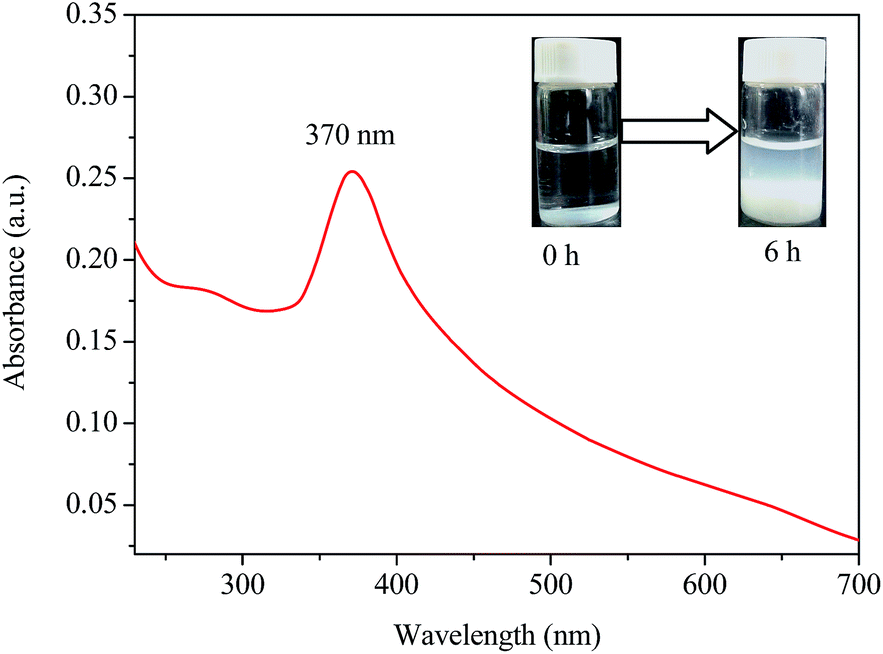
UV-vis analysis. UV-vis spectrometry is a simple and effective technique for detection of formation, shape and stability of nanoparticles in an aqueous solution.33 The UV-vis spectrum of the as-prepared ZnO NPs by Cc and zinc acetate aqueous solutions is shown in Fig. 1. A characteristic absorption peak was clearly observed at 370 nm which could be attributed to the intrinsic band-gap absorption of ZnO because of the electron transitions from valence band to the conduction band (O2p → Zn3d).12 The band gap energy of the Cc–ZnO NPs can be estimated according to the formula E = hc/λ, where h (6.626 × 10−34 J s) is plank's constant, c (3 × 108 m s−1) is the velocity of light and λ (370 nm) is the wavelength. The band gap energy of the Cc–ZnO NPs was calculated as 3.3 eV, which was in good agreement with some reported studies.4,34 The characteristic absorption peak of the Cc–ZnO NPs at wavelength of 370 nm was slightly blue-shifted relative to the absorption maximum of 377 nm for the bulk ZnO NPs. Similar change in the absorption peak location has been reported by Elumalai et al.35 Additionally, the relatively sharp absorption peak at 370 nm further suggested that the Cc–ZnO NPs was in the nano-size range with a narrow particle size distribution. As can be seen in the inset image in Fig. 1, the mixture solution of Cc and zinc acetate was initially clear and colorless. After incubation at 70 °C for 6 h, a pale white precipitation appeared at the bottom of the mixture reaction, indicating the formation of the ZnO NPs.
 |
| | Fig. 1 UV-vis absorption spectrum of Cc–ZnO NPs synthesized by Cc (0.5%, w/v) and zinc acetate (0.5%, w/v) at 70 °C for 6 h (inset images: reaction mixtures at different durations). | |
XRD and EDX analyses. Fig. 2A shows the XRD pattern of the Cc–ZnO NPs prepared with 0.5% (w/v) Cc and 0.5% (w/v) zinc acetate solution at 70 °C for 6 h. The diffraction peaks at 2θ corresponding to 31.7, 34.5, 36.3, 47.5, 56.5, 62.8, 66.2, 67.9, 69.0, 72.6 and 76.9 are attributed to the crystal planes of (100), (002), (101), (102), (110), (103), (200), (112), (201), (004) and (202) of Cc–ZnO NPs, respectively, and they were in line with characteristics of the hexagonal wurtzite structure (JCPDS card no. 36-1451, The International Centre for Diffraction Data ver. 2002). The strong and narrow peaks indicates the good crystalline nature of Cc–ZnO NPs. Among them, the peak at 101 with a relatively high intensity suggests anisotropic growth as a preferred orientation for the Cc–ZnO NPs.35 Moreover, no other diffraction peaks could be detected in the XRD spectrum of the Cc–ZnO NPs, indicating that the precursor synthesized by Cc and zinc acetate has been completely decomposed and no other crystalline impurities have been found. The average crystal size (D) of the Cc–ZnO NPs can be calculated from the Scherrer's formula D = 0.90λ/(Bcosθ),36 where D is the crystal size, λ the X-ray wavelength (0.1541 nm), B the full width at half maximum of the (101) peak in radians and θ the Bragg's angle in radians. The average particle size of the Cc–ZnO NPs was found to be about 56 nm.
 |
| | Fig. 2 (A) XRD pattern of Cc–ZnO NPs, and (B) EDX spectrum of Cc–ZnO NPs. | |
Fig. 2B presents the results from EDX analysis of the Cc–ZnO NPs. Strong signals at 0.5 keV, 1.1 keV, and 8.6 keV in the EDX spectrum are assigned to O Kα, Zn Lα, and Zn Kα, respectively.37 The atomic percent values for Zn and O were 0.48 and 0.17, respectively. These results suggested that the as-prepared Cc–ZnO NPs were essentially free from impurities. The presence of C (atomic percent 0.22) element at 0.3 keV in the EDX spectrum should be attributed to the Cc content in the nanocomposite. In addition, other peaks were observed at 2.2 keV and 9.7 keV for Au element attributed to the gold sputtering.7 Therefore, the XRD and EDX analyses confirmed the composition and crystallinity of the as-prepared Cc–ZnO NPs.
SEM and TEM images. As shown by the SEM micrograph (Fig. 3A), the as-prepared Cc–ZnO NPs were spherical in shape and uniformly distributed. The TEM image (Fig. 3B) of the Cc–ZnO NPs also showed spherical particles in the nano-size range, which were surrounded by a thin Cc layer. These morphological characteristics were very similar to those shown in a previous report.7 The microscopic analysis showed that the particle size of the Cc–ZnO NPs was distributed in a range of 50–70 nm with an average diameter of 58 ± 6 nm, which was similar to the crystal size obtained from XRD analysis. The larger size of the ZnO nanoparticles prepared with Cc might be due to agglomerations observed in Fig. 3B, which was probably attributable to the high surface energy of ZnO NPs and to densification of the NPs.38,39 Similar phenomenon has also been observed in previous studies.5,8,35
 |
| | Fig. 3 (A) SEM image of Cc–ZnO NPs, and (B) TEM image of Cc–ZnO NPs. | |
FT-IR analysis. In the present study, FT-IR measurement was used to explore and confirm the possible interaction ZnO NPs with Cc, and the results are shown in Fig. 4. The Cc spectrum shows that the absorption peaks at 1610 and 1416 cm−1 can be ascribed to the asymmetrical and symmetrical COO– stretching vibrations, respectively.21 In the FT-IR spectrum of Cc–ZnO NPs, the peaks at about 3394, 2989, 1610, 1416, 1151, and 1075 cm−1 are attributed to O–H, C–H, COO–, and C–O–H, respectively, which are agreement with the characteristic peaks of the polysaccharides. This result indicates that the skeleton structure of Cc remains unchanged after the formation of ZnO NPs. More importantly, the peaks observe in the region between 600 and 400 cm−1 have been allotted to the metal–oxygen, and the characteristic peak at 420 cm−1 can be ascribed to the stretching vibrations of ZnO.40
 |
| | Fig. 4 FT-IR spectra of Cc and Cc–ZnO NPs. | |
Interaction of Cc–ZnO NPs with BSA
The introduction of nanoparticles in biological systems is prone to induce corona formation with proteins, which affects the surface properties of nanoparticles and their functions.23 We have previously investigated the interaction of silver and gold nanoparticles with BSA and found that the BSA molecules were adsorbed on the surface of the nanoparticles, without significant changes in the secondary structure.20,21 Here, the interaction between the as-prepared Cc–ZnO NPs and BSA at room temperature was investigated by using various spectroscopic techniques as follows.
Fluorescence quenching of BSA by Cc–ZnO NPs. Fig. 5 shows the fluorescence spectra of the BSA before and after the addition of Cc–ZnO NPs in different concentrations. The maximum fluorescence emission wavelength of the BSA in water was 342 nm, and the fluorescence intensity gradually decreased with increasing Cc–ZnO NPs concentration. The fluorescence peak was slightly blue-shifted by about 8 nm with increasing concentration of Cc–ZnO NPs. The change in the fluorescence characteristics of BSA suggests the formation of a certain complex through the binding of Cc–ZnO NPs and BSA. Similar fluorescence quenching have also been reported from several previous studies on the interactions between ZnO NPs and BSA.15,26–28 Chakraborti et al. reported that the binding site on the surface of the ZnO-PEI was in close proximity to the Trp 134 residues but not the Trp 212 residues of BSA, because the Trp 212 residues laid in a hydrophobic cavity.15 Therefore, it can be concluded that the Trp 134 residues were mainly responsible for the fluorescence quenching of BSA by Cc–ZnO NPs with negligible contribution from Trp 212 residues.
 |
| | Fig. 5 Fluorescence spectra of BSA (1.5 × 10−6 M) in the absence and presence of Cc–ZnO NPs at different concentrations. Cc–Zn NPs concentrations were (a) 0 M, (b) 3.0 × 10−6 M, (c) 6.0 × 10−6 M, (d) 9.0 × 10−6 M, (e) 1.2 × 10−5 M, and (f) 1.5 × 10−5 M, respectively. | |
In general, fluorescence quenching takes place by two mechanisms, dynamic and static, depending on the nature of interaction between quencher and BSA.41 Dynamic quenching usually occurs when quencher molecules have sufficient energy to collide with the fluorophore of BSA at excited state, to bring it to the ground state. On the other hand, static quenching results from the formation of non-fluorescent ground state complex between quencher and fluorophore. The fluorescence quenching data were determined by using the Stern–Volmer equation (eqn (1)).42
| | |
F0/F = 1 + Ksv[Q] = 1 + Kqτ0[Q]
| (1) |
where
F0 and
F are the fluorescence intensities of BSA in the absence and presence of quencher, respectively,
Ksv the Stern–Volmer constant,
Kq the biomolecular quenching rate constant, [Q] the concentration of quencher, and
τ0 the average lifetime of BSA, 10
−8 s.
43 A linear Stern–Volmer plot between
F0/
F against the concentration of Cc–ZnO NPs was obtained according to
eqn (1), and the result was presented in
Fig. 6A. The quenching rate constant (
Kq) for the complexation could be calculated from the slope and the result was 3.3 × 10
12 M
−1 s
−1, which was higher than the maximum diffusion collision rate constant for various biopolymers (2.0 × 10
10 M
−1 s
−1),
43 indicating a static quenching mechanism. Bhogale
et al. also suggested that the interaction between BSA and ZnO NPs was a static quenching process.
26
 |
| | Fig. 6 (A) Stern–Volmer plotting of BSA (1.5 × 10−6 M) with Cc–ZnO NPs at different concentrations. (B) The plot of lg[(F0 − F)/F] versus lg[Cc–ZnO NPs]. | |
For static quenching, the binding constant (K) and the number of binding sites (n) between Cc–ZnO NPs and BSA were determined with the following equation (eqn (2)).41
| | |
lg[(F0 − F)/F] = nlg[Q] + lgK
| (2) |
where
K is the binding constant of Cc–ZnO NPs with BSA, [Q] the concentration of Cc–ZnO NPs, and
n the number of binding sites.
Fig. 6B shows the linear plot of lg[(
F0 −
F)/
F] against lg[Cc–ZnO NPs] according to
eqn (2). From the slope and intercept, the binding constant (
K) and the number of binding sites (
n) were obtained as 5.5 × 10
4 M
−1 and 0.81, respectively. The value of
n for the interaction of BSA with Cc–ZnO NPs was close to 1, indicating that there was only one binding site available in BSA for Cc–ZnO NPs, similar to those reported previously.
15,27 However, Bhogale
et al. found that the value of
n was greater than 1, which was due probably to the formation of an unstable complex of BSA with ZnO NPs.
26 The value of
K obtained in this work was almost equal to the ZnO-PEI's (6.4 × 10
4 M
−1) reported by Chakraborti
et al.15 Several other reports on colloidal ZnO–BSA binding also presented a binding constant of ∼10
4 M
−1.
27,44 It is generally considered that both electrostatic and hydrophobic forces are responsible for the binding in the interaction process.
15 The hydrophobic forces play an important role in the binding between uncapped ZnO NPs and BSA. On the contrary, the Cc-capped ZnO NPs, as well as the reported ZnO-PEI,
15 have a negative charged layer on the surface to become more hydrophilic due to the presence of hydroxyl and carboxyl groups of Cc, contributing to electrostatic interactions. Therefore, the Cc-functionalized ZnO NPs could bind with BSA by electrostatic interactions at the surface close to the Trp 134 residues which is located on the surface of the Subdomain Ib, but not Trp212 residues which is located near the vicinity of the hydrophobic cavity (Sudlow's site I).
Synchronous fluorescence spectroscopy. Synchronous fluorescence spectroscopy, which is a simple and useful method, has been widely used to study the molecular environment in the vicinity of the fluorophore functional group and evaluate the conformational changes of the protein.45 The difference between excitation wavelength and emission wavelength (Δλ = λemission − λexcitation) is an useful parameter for investigating the synchronous fluorescence characteristics of BSA. Δλ values of 15 nm and 60 nm are suitable for characterizing the environment changes for the tyrosine (Tyr) or tryptophan (Trp) residues, respectively. Therefore, synchronous fluorescence spectroscopy was used in the present work to determine the specific binding mode of Cc–ZnO NPs with BSA in the interaction process.Fig. 7 shows the synchronous fluorescence spectra of BSA with various concentrations of Cc–ZnO NPs recorded at Δλ = 15 nm and Δλ = 60 nm, respectively. When Δλ was kept at 15 nm (Fig. 7A), there was a gradual decrease in the emission intensity of the peak at about 288 nm with increasing Cc–ZnO NPs concentration, but no changes in wavelength. At the same time, the addition of Cc–ZnO NPs also resulted in a decrease in the intensity of the maximum peak at 280 nm and a blue-shift of about 3 nm in the peak wavelength at Δλ = 60 nm (Fig. 7B). The results indicated that the Cc–ZnO NPs had an obvious effect on the microenvironment of Trp residues in BSA but not on the microenvironment close to Tyr residues in BSA, which was in good agreement with the results of fluorescence quenching.
 |
| | Fig. 7 (A) Synchronous fluorescence spectra of BSA (1.5 × 10−6 M) at Δλ = 15 nm in the absence and presence of Cc–ZnO NPs at different concentrations. (B) Synchronous fluorescence spectra of BSA (1.5 × 10−6 M) at Δλ = 60 nm in the absence and presence of Cc–ZnO NPs at different concentrations. Cc–ZnO NPs concentrations were (a) 0 M, (b) 3.0 × 10−6 M, (c) 6.0 × 10−6 M, (d) 9.0 × 10−6 M, (e) 1.2 × 10−5 M, and (f) 1.5 × 10−5 M, respectively. | |
It has been shown previously the blue shift in the emission maxima of Trp residue can be mainly ascribed to the increase in hydrophobicity around the Trp residue after the addition of nanoparticles.41,46 Bhogale et al. reported that both the Tyr and Trp residues in BSA were perturbed in the presence of ZnO NPs, leading to an increase in the hydrophobicity of both residues.26 Kathiravan et al. also showed that the colloidal ZnO NPs affected only the Tyr residues in BSA.27 However, the results from this work were different from those in the previous reports, which may be due to the difference in the preparation of ZnO NPs. As mentioned previously, the ZnO NPs reported in this study were synthesized and functionalized by the hydrophilic Cc molecules so that the Cc–ZnO NPs were more hydrophilic. As reported by Chakraborti et al.,15 the PEI-functionalized ZnO NPs that were more hydrophilic favored the interaction with BSA on its surface via the surface-exposed Trp 134 by electrostatic interactions but not at the hydrophobic Sudlow's site I (Trp 212). This was similar to that found for the citrate-stabilized AuNPs–BSA interaction,47 which supported the general belief that the association of BSA to the citrate-stabilized AuNPs could take place by an electrostatic attraction mechanism. Casals et al. also reached the same conclusion according to the observation that AuNPs became negatively charged due to the formation of a protein corona.22 Based on the above discussion, it can be concluded that the microenvironment close to the Trp 134 residue was perturbed in the presence of the Cc–ZnO NPs and the binding between Cc–ZnO NPs and BSA was through electrostatic interactions, as illustrated in Fig. 8.
 |
| | Fig. 8 A possible mechanism of the as-prepared Cc–ZnO NPs approaching BSA. | |
Absorption studies of BSA–Cc–ZnO NPs interaction. Fig. 9A shows the UV-vis absorption spectra of BSA with and without Cc–ZnO NPs. The maximum absorption peak (λmax) of BSA at 280 nm increased gradually with the increase of the Cc–ZnO NPs concentration. However, there was no absorption band observed at around 280 nm for the Cc–ZnO NPs (Fig. 1). This result clearly indicates that the interaction between BSA and Cc–ZnO NPs has undergone ground state complex formation. Kathiravan et al. reported a similar increase in the intensity of the absorbance peak at 280 nm of BSA, which was attributed to the interaction of colloidal ZnO NPs with BSA by static quenching mechanism.27
 |
| | Fig. 9 (A) Absorption spectra of BSA (1.5 × 10−6 M) in the absence and presence of Cc–ZnO NPs at different concentrations. Cc–ZnO NPs concentrations were (a) 0 M, (b) 3.0 × 10−6 M, (c) 6.0 × 10−6 M, (d) 9.0 × 10−6 M, (e) 1.2 × 10−5 M, and (f) 1.5 × 10−5 M, respectively. (B) CD spectra of BSA in the absence and presence of Cc–ZnO NPs. The concentrations of BSA and Cc–ZnO NPs were 1.5 × 10−6 and 1.5 × 10−5 M, respectively. | |
Circular dichroism (CD) spectroscopy. CD is a powerful and sensitive technique for detecting conformational changes in protein arising from interaction with nanoparticles.48 Fig. 9B illustrates the far-UV CD spectra of BSA both in the absence and in the presence of Cc–ZnO NPs as recorded in the wavelength range of 200–250 nm. It is generally recognized that BSA possesses two strong negative bands at 208 nm and 222 nm, which are characteristic of the transition of π–π* and n–π* of the α-helical structure of BSA molecule.49 Fig. 8B shows a decrease in intensity of the two bands which represent the α-helical contents in the presence of Cc–ZnO NPs, though the shape and position of the two bands remained almost unchanged. This result indicates that changes in CD signals after the addition of Cc–ZnO NPs were attributed to the local conformational changes in BSA due to the interaction, and the essential structural features of BSA remained intact.
Conclusions
A simple and green approach has been developed to synthesize ZnO NPs by using carboxylic curdlan (Cc) as both reducing and stabilizing agents. The as-prepared Cc–ZnO NPs had the hexagonal wurtzite structure with an absorption maximum at 370 nm. The Cc–ZnO NPs were spherical and uniformly distributed with an average particle size of 58 ± 6 nm. The as-prepared Cc–ZnO NPs could form complex with BSA in an aqueous medium, resulting in slight conformational changes in BSA. The as-prepared Cc–ZnO NPs could be further explored as a biocompatible nanomaterial for food and biomedical applications.
Acknowledgements
This work was supported financially by the Natural Science Foundations of Jiangsu Province (BK20140542, BK20130511), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and Jiangsu Overseas Research & Training Program for University Prominent Young & Middle-aged Teachers and Presidents.
References
- A. Kołodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833 CrossRef.
- M. H. Huang, S. Mao, H. Feick, H. Yan, Y. Wu, H. Kind, E. Weber, R. Russo and P. Yang, Science, 2001, 292, 1897 CrossRef CAS PubMed.
- D. Jain, H. K. Daima, S. Kachhwaha and S. L. Kothari, Dig. J. Nanomater. Biostruct., 2009, 4, 557 Search PubMed.
- K. Elumalai and S. Velmurugan, Appl. Surf. Sci., 2015, 345, 329 CrossRef CAS.
- P. C. Nagajyothi, S. J. Cha, I. J. Yang, T. V. M. Sreekanth, K. J. Kim and H. M. Shin, J. Photochem. Photobiol., B, 2015, 146, 10 CrossRef CAS PubMed.
- C. Jayaseelan, A. A. Rahuman, A. V. Kirthi, S. Marimuthu, T. Santhoshkumar, A. Bagavan, K. Gaurav, L. Karthik and K. V. B. Rao, Spectrochim. Acta, Part A, 2012, 90, 78 CrossRef CAS PubMed.
- N. Jain, A. Bhargava, C. Jagadish, K. Tarafdar, K. Sunil and S. Panwar, Appl. Microbiol. Biotechnol., 2013, 97, 859 CrossRef CAS PubMed.
- S. Azizi, M. B. Ahmad, F. Namvar and R. Mohamad, Mater. Lett., 2014, 116, 275 CrossRef CAS.
- M. Thamima and S. Karuppuchamy, Adv. Sci., Eng. Med., 2015, 7, 18 CrossRef CAS.
- S. W. Cui, Food carbohydrate: chemistry, physical properties, and applications, Taylor & Francis, Boca Raton, FL, 2005 Search PubMed.
- S. T. Lin, M. Thirumavalavan, T. Y. Jiang and J. F. Lee, Carbohydr. Polym., 2014, 105, 1 CrossRef CAS PubMed.
- A. K. Zak, W. H. A. Majid, M. R. Mahmoudian, M. Darroudi and R. Yousefi, Adv. Powder Technol., 2013, 24, 618 CrossRef.
- M. Khajeh and A. R. Golzary, Spectrochim. Acta, Part A, 2014, 131, 189 CrossRef CAS PubMed.
- M. Darroudi, Z. Sabouri, R. K. Oskuee, A. K. Zak, H. Kargar and M. H. N. A. Hamid, Ceram. Int., 2013, 39, 9195 CrossRef CAS.
- S. Chakraborti, P. Joshi, D. Chakravarty, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2012, 28, 11142 CrossRef CAS PubMed.
- X. R. Song, N. Goswari, H. H. Yang and J. Xie, Analyst, 2016, 141, 3126 RSC.
- N. Goswari, K. Zheng and J. Xie, Nanoscale, 2014, 6, 13328 RSC.
- J. Huang, L. Lin, D. Sun, H. Chen, D. Yang and Q. Li, Chem. Soc. Rev., 2015, 44, 6330 RSC.
- J. Zhou, Y. Yang and C. Zhang, Chem. Rev., 2015, 115, 11669 CrossRef CAS PubMed.
- J. K. Yan, P. F. Cai, X. Q. Cao, T. T. Fan and H. L. Ma, J. Inorg. Organomet. Polym., 2013, 23, 1383 CrossRef CAS.
- J. K. Yan, J. L. Liu, Y. J. Sun, S. Tang, Z. Y. Mo and Y. S. Liu, Carbohydr. Polym., 2015, 117, 771 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. J. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623 CrossRef CAS PubMed.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40 CrossRef CAS.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780 RSC.
- U. K. Hansen, Pharmacol. Rev., 1981, 33, 17 Search PubMed.
- A. Bhogale, N. Patel, P. Sarpotdar, J. Mariam, P. M. Dongre, A. Miotello and D. C. Kothari, Colloids Surf., B, 2013, 102, 257 CrossRef CAS PubMed.
- A. Kathiravan, G. Paramaguru and R. Renganathan, J. Mol. Struct., 2009, 934, 129 CrossRef CAS.
- R. Žūkienė and V. Snitka, Colloids Surf., B, 2015, 135, 316 CrossRef PubMed.
- A. Rajeshwari, S. Pakrashi, S. Dalai, M. V. Iswarya, N. Chandrasekaran and A. Mukherjee, J. Lumin., 2014, 145, 859 CrossRef CAS.
- Q. Yang, J. Liang and H. Han, J. Phys. Chem. B, 2009, 113, 10454 CrossRef CAS PubMed.
- H. Fan, N. Ji, M. Zhao, L. Xiong and Q. Sun, Int. J. Biol. Macromol., 2015, 78, 333 CrossRef CAS PubMed.
- J. K. Yan, H. L. Ma, P. F. Cai, Q. Zhang, N. Z. Hu, X. B. Feng and J. Y. Wu, Food Chem., 2014, 143, 530 CrossRef CAS PubMed.
- W. R. Rajesh, R. L. Jaya, S. K. Niranjan, D. M. Vijay and B. K. Sahebrao, Curr. Nanosci., 2009, 5, 117 CrossRef.
- P. Rajiv, S. Rajeshwari and R. Venckatesh, Spectrochim. Acta, Part A, 2013, 112, 384 CrossRef CAS PubMed.
- K. Elumalai, S. Velmurugan, S. Ravi, V. Kathiravan and S. Ashokkumar, Spectrochim. Acta, Part A, 2015, 143, 158 CrossRef CAS PubMed.
- B. D. Cullity, Elements of X-ray Diffraction, Addison-Wesley Publishing Company, Inc., London, 1978 Search PubMed.
- H. Bahadur, A. K. Srivastava, R. K. Sharma and S. Chandra, Nanoscale Res. Lett., 2008, 2, 469 CrossRef.
- B. Alessio, D. Maximilian, L. N. Pierandrea and B. Piero, J. Nanopart. Res., 2008, 10, 679 CrossRef.
- J. H. Ryu, C. S. Lim and K. H. Auh, J. Korean Ceram. Soc., 2002, 39, 321 CrossRef CAS.
- K. Elumalai, S. Velmurugan, S. Ravi, V. Kathiravan and S. Ashokkumar, Spectrochim. Acta, Part A, 2015, 136, 1052 CrossRef CAS PubMed.
- J. Mariam, P. M. Dongre and D. C. Kothari, J. Fluoresc., 2011, 21, 2193 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- T. G. Dewey, Biophysical biochemical aspects of fluorescence spectroscopy, in Trends in Biochemical Sciences, Plenum Press, New York, 1991 Search PubMed.
- M. Bardhan, G. Mandal and T. Ganguly, J. Appl. Phys., 2009, 106, 034701 CrossRef.
- G. Z. Chen, X. Z. Huang, J. G. Xu, Z. B. Wang and Z. Z. Zhang, Method of fluorescent analysis, Science Press, Beijing, 2nd edn, 1990, p. 123 Search PubMed.
- A. Bhogale, N. Patel, J. Mariam, P. M. Dongre, A. Miotello and D. C. Kothari, Colloids Surf., B, 2014, 113, 276 CrossRef CAS PubMed.
- S. Dominguez-Medina, S. McDonough, P. Swanglap, C. F. Landes and S. Link, Langmuir, 2012, 28, 9131 CrossRef CAS PubMed.
- S. Laera, G. Ceccone, F. Rossi, D. Gilliland, R. Hussain, G. Siligardi and L. Calzolai, Nano Lett., 2011, 11, 4480 CrossRef CAS PubMed.
- C. H. Yu, A. Al-Saadi, S. J. Shih, L. Qiu, K. Y. Tam and S. C. Tsang, J. Phys. Chem. C, 2009, 113, 537 CAS.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.