
Design of bio-molecular interfaces using liquid crystals demonstrating endotoxin interactions with bacterial cell wall components†
Dibyendu Das,
Sumyra Sidiq and
Santanu Kumar Pal*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector-81, Knowledge City, Manauli-140306, India. E-mail: skpal@iisermohali.ac.in
First published on 30th July 2015
Abstract
Interaction of different bacterial cell membrane components such as, peptidoglycan (PG) and lipoteichoic acid (LTA) with bacterial endotoxin (LPS) shows diverse consequences on the toxicity of Gram negative bacteria in mammalian hosts, implying the huge importance of studying this interaction for clinical understanding associated with Gram negative bacterial infections. In this advance, herein, we report a liquid crystal (LC) based simple, robust experimental design for rapid and precise recognition of the interaction of LPS with PG and LTA. The optical appearance of nematic 4-cyano-4′-pentylbiphenyl (5CB) LCs changed from dark to bright (consistent with an ordering transition of the LCs) in contact with an aqueous solution of PG and LTA on LPS–laden aqueous–LC interfaces. The ordering transition demonstrates the strong interaction between PG and LTA with LPS at these interfaces. Our experiment also revealed that the interaction of PG and LTA towards LPS is highly specific. In addition, PG and LTA shows different binding affinity towards LPS and response of the LC is found to vary significantly from one to another which is conveniently quantified by measurement of the light intensity transmitted through the LC under crossed polars. Langmuir Blodgett (LB) and polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS) measurements provide further insight on LPS laden aqueous–LC interfaces. Finally, we have also quantified the different binding affinity of PG and LTA towards LPS by measuring the optical retardance of the LC at aqueous–LC interfaces. Overall, the results presented in this paper offer a promising approach to study and quantify the interactions between different bacterial cell membrane components with LPS at aqueous–LC interfaces.
Introduction
In the last two decades, the incidence of Gram negative bacterial infections has progressively increased.1–4 Currently, among one third of all microorganism mediated infections are caused by Gram negative bacteria and it is expected that these bacterial contagions will continue to rise and predominate in the years to come. Consequently, with increasing concern over Gram negative bacterial infection at present, the substantial medicinal efforts to combat microorganism infections are mainly focused on Gram negative bacteria.5–7 From the light of vast pathophysiological studies, it has been well approved that the cell membrane components of Gram negative bacteria play as principal mediators in inducing Gram negative bacterial infections in mammalian hosts.8–14 Lipopolysaccharide (LPS), also known as bacterial endotoxin, is a major constituent of outer cell membrane of Gram negative bacteria. This amphipathic macromolecule is well recognized as important contributing factors to the pathogenesis of Gram negative bacterial infection. LPS consists of varying length of hydrophilic polysaccharide chains, covalently attached to hydrophobic lipid A moiety which is considered as most active toxic agent during Gram negative bacterial infection leading to high fever, septic shock, multiorgan failure syndrome and even death.8–10 More recently, several clinical and pathophysiological studies on induced endotoxic behavior of LPS in mammalian cells have revealed and documented very interesting and crucial fact that these severe biological activities caused by LPS are largely intervened by the coexistence of different other bacterial (Gram negative as well as Gram positive) cell membrane components.11 In this regard, peptidoglycan (PG) and lipoteichoic acid (LTA), two active outer cell membrane components of bacteria, have drawn widespread interest in clinically investigating their influence over the consequence of endotoxicity induced by LPS in mammalian hosts.15–22PG is a glycan polymer containing long sugar chains of two alternating sugar derivatives, N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) which are highly cross-linked through peptide bridges forming 3D mesh like layer. In both Gram negative and Gram positive bacteria PG plays an important role in maintaining structural integrity of bacterial cell membrane, as well as counteracting the osmotic pressure of the cytoplasm. Unlike PG, LTA is only found in the cell wall of Gram positive bacteria. It is a linear polymer of phosphodiester linked glycerol phosphate and polysaccharide attached to diacylglycerol chains. The main function of LTA is to provide structural rigidity in bacterial cell wall and also regulate the function of auto wall enzymes.
The interaction of LPS with these bacterial membrane constituents has been found to result in different effects on LPS endotoxicity, varying from reduction to enhancement. In this context, Thiemermann and his coworkers have reported synergistic behavior of PG towards LPS in mammalian hosts.18 They have shown that co administration of PG and LPS from pathogenic Gram negative (E. coli) bacteria synergies to cause multiple organ dysfunctions in rat.18 The presence of PG dramatically amplifies the lethal toxicity of LPS in mammalian host.15–18
In contrast to this agonist behavior of PG towards endotoxicity of LPS, LTA exhibits antagonistic influence on the activity of LPS by lowering its lethal toxicity in mammalian cell.20–22 Hailman and his coworkers reported that LTA prevents LPS induced release of TNF from monocytes into blood serum via CD14 dependent pathway, ensuing in many fold decrease in LPS toxicity.21 For example, Sugawara et al. have reported that LTA of Gram-positive cocci in the oral cavity may inhibit the action of LPS from periodontopathic Gram-negative bacteria resulting in the inhibition of the initiation of periodontal disease and therefore, LTA can be considered as useful agent for suppressing LPS induced periodontal diseases.20 Overall these entire clinical investigations on synergism between bacterial endotoxin and different bacterial cell membrane components (PG and LTA) clearly demonstrate its practical and pathophysiological significance from the aspect of better understanding in Gram negative bacterial infection in mammalian hosts.
Interleukine-1 (IL-1),23 toll like receptor (TLR) expression,24 tumor necrosis factor (TNF),25 are major biological assays available to study the interaction of endotoxin with cell membrane components. Although all of these methods provide several essential information regarding these interactions, but lack of sensitivity, high cost, complex instrumentation and many other drawbacks limit their practical applications. In this advance, herein, we report a simple, robust liquid crystal (LC) based system for rapid and precise detection of the interaction of LPS with different cell membrane components (PG, LTA).
In recent years, the liquid crystal-based sensor has become an innovative and promising tool for transducing and amplifying biomolecular interactions with high sensitivity and spatial resolution at the aqueous/LC interface. Due to its rapid orientational response on chemically functionalized surface, the nematic LC has served as an attractive medium to report chemical/biochemical events occurring at the interfacial biological membranes.26–38 So, far fluid biometric membrane systems supported on LCs have been coupled to the screening of specific protein binding event,28,29,31–35 DNA hybridization,39–42 monitoring enzymatic reaction,30,36,43–45 and different pathogenic toxic detections.46–49 For example, Abbott and his coworkers reported the specific interaction of immunoglobulin (IgG) antibody to the surface immobilized antigen resulting in a change in optical response of LCs.50 K.-L. Yang, et al. designed pH sensitive liquid crystal sensor for monitoring enzymatic activities of penicillinase at aqueous–LC interface.36 Wu et al. designed LC biosensor based on target triggering DNA dendrimers for the detection of p53 mutation gene.51 D. Liu, et al. demonstrated the interaction between chitosan, a biopolymer, and lipid membrane at aqueous–LC interface.52 More recently, C.-H. Jang et al. reported LC based detection of coagulating protease thrombin coupled to interactions between a polyelectrolyte and a phospholipid monolayer.53 To the best of our knowledge, as yet, there is no report regarding the study of interaction of LPS with bacterial cell membrane components, PG and LTA, on LC based sensing platform.
The study reported in this paper is mainly two fold. First, we sought to determine if it is possible to report the interaction of bacterial endotoxin (LPS) with PG and LTA at aqueous–LC interface through surface-driven ordering transition in LC. Second, we sought to demonstrate if it is possible to quantify these interactions that would explore the use of the LC as novel quantitative analytical tool to report bimolecular interaction at aqueous–LC interfaces.
The approach that we report in this paper revolves around the formation of LPS laden aqueous–LC interface. Past reports established that the hydrophobic interaction between alkyl chain of lipid A moiety of LPS interact with 5CB leads to homeotropic orientation of LCs at aqueous/LC interface.29,31 With this idea keeping in mind, we hypothesized that strong, specific interactions of LPS with cell membrane components, PG and LTA, may attribute to the disruption of LPS resulting in the orientational ordering transition of the LC from homeotropic to tilted state which can be easily visualized under polarized optical microscopy (Scheme 1). Overall, the results of the study served as a promising tool to the design of responsive LC-based system that can report LPS–PG/LTA interactions at aqueous/LC interfaces.
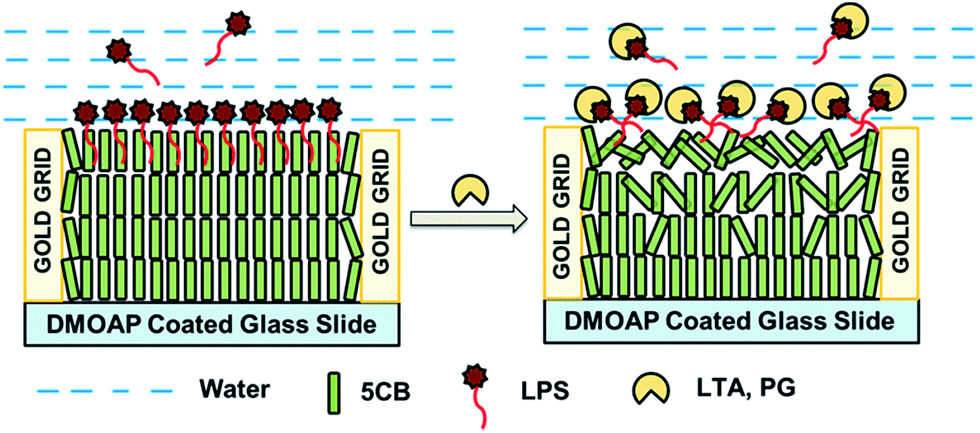 | ||
| Scheme 1 Schematic representations of the anchoring transitions of 5CB hosted in TEM gold grid supported on DMOAP-coated glass slide at LPS laden 5CB/aqueous interface before (A) and after (B) being exposed to different biomolecules. | ||
Experimental section
Materials and methods
Lipopolysaccharides (from E. coli 0111:B4), peptidoglycan (from Micrococcus luteus), lipoteichoic acid (from Staphylococcus aureus), 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DOPG), Tris buffered saline (pH 7.4) and N,N-dimethyl-N-octadecyl-3-aminopropyltrimethoxysilyl chloride (DMOAP), FITC conjugated lipopolysaccharides (from E. coli 0111:B4) were purchased from Sigma-Aldrich (St. Louis, MO). 1,2-Didodecanoyl-sn-glycerol-3-phosphocholine (DLPC) and lysophosphatidic acid (LPA) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Sulfuric acid and hydrogen peroxide (30% w/v) were purchased from Merck. Ethanol was obtained from Jebsen & Jenssen GmbH and Co., Germany (Sd. fine-chem limited). The 5CB LC was obtained from Merck. Deionization of a distilled water source was performed using a Milli-Q-system (Millipore, bedford, MA). Fischer's Finest Premium Grade glass microscopic slides and cover glass were obtained from Fischer Scientific (Pittsburgh, PA). Gold specimen grids (20 μm thickness, 50 μm wide bars, 283 μm grid spacing) were obtained from Electron Microscopy Sciences (Fort Washington, PA).Cleaning of glass substrates
Glass microscope slides were cleaned according to published procedures using ʻpiranhaʼ solution [70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 (% v/v) H2SO4:H2O2 (30%)], as described in detail elsewhere.31 Briefly, the glass slides were immersed in a piranha bath at 100 °C for at least 1 h and then rinsed in running deionized (DI) water for 5–10 min. Finally, the slides were rinsed sequentially in ethanol and then dried under a stream of nitrogen. The clean slides were stored in an oven at 100 °C for overnight. All other glassware was cleaned prior to use.
:30 (% v/v) H2SO4:H2O2 (30%)], as described in detail elsewhere.31 Briefly, the glass slides were immersed in a piranha bath at 100 °C for at least 1 h and then rinsed in running deionized (DI) water for 5–10 min. Finally, the slides were rinsed sequentially in ethanol and then dried under a stream of nitrogen. The clean slides were stored in an oven at 100 °C for overnight. All other glassware was cleaned prior to use.
Treatment of glass microscope slides with DMOAP
The cleaned glass slides were dipped into 0.1% (v/v) DMOAP solution in DI water for 5 min at room temperature and were then rinsed with DI water to remove unreacted DMOAP from the surface. The DMOAP coated glass slides were dried under a stream of nitrogen gas and kept in oven at 100 °C for 3 h to allow crosslinking of DMOAP.Preparation of optical cells
The DMOAP coated glass slides were then cut into squares for supporting LC. Then, a gold grid was placed on the slide, and approximately 0.3 μL of 5CB was dispensed onto the grid. Excessive LC was removed by using a capillary tube.Formation of LPS laden LC films
LCs laden with a LPS were prepared following procedures published in previous literature.31 Powdered LPS (endotoxin) was dissolved in Milli-Q water at room temperature to obtain the required concentration. The resulting solutions were then sonicated for 5 min and vortexed for 10 min at room temperature. The LPS vesicle size was found to be 174.01 ± 2.88 nm (Fig. S1, see ESI†). The LPS laden 5CB interface was formed by contacting the gold grid impregnated with 5CB to the LPS solution in the optical cell for a period of 2 h. The LPS laden interface was washed twice with Tris buffer (pH 7.4) prior to use.Preparation of vesicles
Vesicles were prepared according to the published procedures.32 Briefly, the lipids were dissolved in chloroform (0.5 mL) and dispensed into round bottomed flask. Prior to re-suspension, the chloroform was evaporated from the flask under vacuum for at least 2 h until it formed a thin film along the inner walls of the flask. The lipid film formed in the flask was then placed under a stream of nitrogen for 30 min. The dried lipid was then hydrated in the aqueous solution (DI water) for at least 1 h and vortexed for 1 min. This results in the cloudy solution indicative of large multilamellar vesicles. Subsequent sonication of lipid suspension using a probe ultrasonicator (1 × 15 min at 25 W) resulted in a clear solution.Optical characterization of LC films in aqueous solutions
The grid containing the LC was immersed in Tris buffer (10 mM, pH 7.4). The optical appearance was observed by using a polarizing optical microscope (Nikon ECLIPSE LV100POL, Japan) in the transmission mode. Each image was captured with a Q-imaging digital camera mounted on the microscope with an exposure time of 40 ms.Tilt angle measurements
The optical retardance of LCs was measured using tilting compensator (type 2357 K, equipped with a calcite compensator plate, Leitz, Germany). The retardance values reported in this paper are the average obtained within four squares of the gold specimen grid used to host the LCs. For a thin film of nematic LCs with strong homeotropic anchoring (θ1 = 0°) at the DMOAP-treated glass interface and a tilt of angle of θ2 away from the surface normal at the aqueous–LC interface, the tilt of LCs across the film varies linearly with position so as to minimize the elastic energy of the LC film (assuming splay and bend elastic constants of the LCs to be equal). This result permits the establishment of a relationship between optical retardance (Δr) of the film of LCs and the tilt of the director at the aqueous–LC interface (θs), namely
 | (1) |
Epifluorescence imaging of aqueous–5CB interface
Fluorescence imaging was performed with Zeiss (Observer. A1) fluorescence microscope. The samples were viewed using a fluorescence filter cube with a 480 nm excitation filter and a 534 nm emission filter. Images were obtained with Axio cam camera.Fluorimetric measurement
All fluorescence intensity measurements were performed using a SHIMADZU RF-5301PC SPECTROFLUROPHOTOMETER (Shimadzu Corp., Kyoto, Japan). With an excitation wavelength 490 nm (0.5 nm excitation slit) and an emission wavelength range of 500–534 nm (5 nm emission slit) for the detection of FITC fluorescence.Preparation of LPS monolayer
Surface pressure (π)–area per molecule (Am) isotherm experiments were carried out in an Langmuir Blodgett (LB) trough. LPS monolayers were prepared at the air–water interface. The surface pressure was measured using the standard Wilhelmy plate technique in a trough (MINITROUGH, KSV, Finland) enclosed in a Plexiglas box to reduce surface contamination. The subphase of the trough was filled with 1 M NaCl. Using a microsyringe, 50 μL of lipid (LPS and LPS doped with 0.2% FITC–LPS) solution (1 mg mL−1) in chloroform:methanol (3:1) was carefully spread onto the aqueous subphase. After spreading, the film was left for 20 min, allowing the solvent to evaporate. The π–Am isotherms were obtained by symmetric compression of the barriers with a constant compression rate of 10 mm min−1. Surface pressure and trough area were recorded simultaneously using Nima software. Based upon the volume deposited, the average molecular weight, and concentration of solution, the average area per molecule was calculated. All the measurements were performed at a room temperature of 25.0 ± 1 °C. Once deposited and transferred onto the 5CB confined in gold grid supported on DMOAP coated glass slide supported on gold films at a surface pressure of 56 mN m−1, these supported LPS monolayers were kept under vacuum.
Polarization modulation infrared reflection absorption spectroscopy
The gold films with thicknesses of ≥2000 Å were deposited onto micropillars (array of nickel (Ni) micropillars electroplated on a glass substrate fabricated as described in detail elsewhere54) mounted on rotating planetaries (no preferred direction or angle of incidence) by using thermal evaporator (Excel Instruments, India). These gold coated micropillars were dipped into 0.1% (v/v) DMOAP solution in DI water for 5 min at room temperature and were then rinsed with DI water to remove unreacted DMOAP from the surface. The DMOAP coated micropillars were dried under a stream of nitrogen gas and kept in oven at 100 °C. Then, 5CB was dispersed onto the micropillars. Deposited LPS onto a 5CB immobilized on DMOAP supported on micro-pillars coated with a uniformly deposited film of gold (2000 Å) were examined by using PM-IRRAS. A Bruker PMA 50 connected to the external beam port of a Bruker Tensor 27 FT-IR spectrometer was used for PM-IRRAS measurements. The deposited LPS on a gold sample was mounted on an attachment for PM-IRRAS measurements within the PMA 50 compartment. After reflection of the polarized light incident on the substrate at an angle of incidence of 82° from the surface normal, the IR beam was focused on a liquid nitrogen-cooled photovoltaic MCT detector in the PMA 50 cabinet. A photoelastic modulator (Hinds, PEM 90) was used to modulate the polarization of the light at a frequency of 50 kHz. Demodulation was performed with a lock-in-amplifier (Standford Research Systems, SR830 DSP). Before measurements, the spectrometer was allowed for a complete purge with nitrogen for at least 30 min. Each spectrum is the sum of 100 individual spectra collected at a resolution of 4 cm−1 with photoelastic modulator (ZnSe, 42 kHz, AR-coated) set to 1600 cm−1. Data was collected as differential reflectance (ΔR/R)/absorbance versus wavenumbers.Dynamic light scattering
(DLS) Hydrodynamic size (diameter) measurements of bacterial cell wall components and LPS were performed on a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Southborough, Massachusetts). The DLS instrument was operated under the following conditions: temperature: 25 °C, detector angle: 90° incident laser wavelength: 632 nm, and laser power: 4 mW. Samples were prepared in Tris buffer (pH 7.4) followed by equilibration typically 5 minutes.Zeta potential measurement
A Malvern Zetasizer Nano ZS instrument (Malvern Instruments, Southborough, Massachusetts) was used to measure zeta potential at 25 °C for bacterial cell wall components. Samples prepared for the DLS measurements were loaded into a pre-rinsed polystyrene cuvette for the zeta potential measurements. An applied voltage of 100 V was used for the measurement. A minimum of three measurements were made per sample.Results and discussions
In order to investigate the interaction of LPS with different cell membrane components, we first studied the interaction of LPS with LC at aqueous–LC interface. Fig. 1A represents the optical response of 5CB, confined within a gold grid supported on DMOAP coated glass slides, dipped into an aqueous solution of Tris buffer (pH 7.4). The bright and colorful optical appearance reflects the planar orientation of LCs due to its interaction with water at aqueous/LC interface. | ||
| Fig. 1 Polarized optical micrographs of 5CB films confined in TEM gold grid supported on DMOAP coated glass slides: (A) immersed in Tris buffer of pH 7.4 (10 mM), (B) incubated with LPS of 0.1 mg mL−1 for 2 h to form stable LPS laden aqueous/LC interface. Scale bar = 40 μm. | ||
Here it is noteworthy that, the orientation of LCs at DMOAP coated glass surface still remains homeotropic due to the interaction between alkyl chain of 5CB and DMOAP leading to an orientation of the nematic molecules perpendicular to the surface. Next, we observed a fast change in optical appearance of 5CB from bright to dark (Fig. 1B) when 0.1 mg mL−1 aqueous LPS solution in Tris buffer (pH 7.4) was introduced on LC interface, as expected. This observation clearly indicates that the homeotropic orientation of the LC mediated by LPS is a consequence of strong hydrophobic interaction of lipid chains of the LPS with the mesogens of the LCs.29–31 Notably, the bright rim observed around the grid circumference in Fig. 1B is due to direct interaction of peripheral 5CB with gold surface.
Prior to study the interactions between LPS with cell membrane components at aqueous–LC interfaces, we verified that the surface used in our study was decorated with LPS. For this, first we exploited Langmuir film balance technique55,56 to preorganize monolayer of LPS molecules at the air–water interface at well-defined densities, followed by transferring this LPS monolayer in a vertical dip fashion to LC–water interfaces stabilized within gold grids supported on DMOAP coated glass slide. Before attempting transfer of LPS from the air–water interface, we verified that the surface pressure (π) verses area (Am) isotherms at the air–water interfaces. Fig. S2 (see ESI†) shows the representative π–Am isotherm of LPS at the air–water interface. Collapse of the film was observed at surface pressures in the range of 56 mN m−1. Next, we quantitatively transfer LPS/LPS doped with 0.2% FITC–LPS monolayers from the air–water interface to the LC–aqueous interface at different surface pressures. The preparation of LPS monolayers via Langmuir transfer from the air–water interface was performed at a surface pressure of 0, 30 and 52 mN m−1. Fig. 2 shows the polarized optical micrographs and the respective epifluorescence micrographs of LPS monolayers formed via Langmuir transfer from the air–water interface at different surface pressures. The lower density of LPS monolayers at the LC interface gave rise to planar orientation of the LC (Fig. 2A and B) while the higher area density (higher surface pressure) LPS film transferred gave rise to homeotropic LC orientation as shown in Fig. 2C. Interestingly, the corresponding epifluorescence measurements (Fig. 2D–F) shows the increase in fluorescence intensity indicate the quantitative transfer of LPS from the aqueous–air interface onto the aqueous–LC interface. Fig. 2G represents the linear increase of fluorescence intensity of LPS monolayers formed via Langmuir transfer from the air–water interface at increasing areal density. These results confirmed that LPS–LC interactions lead to the ordering transition of the LC at aqueous interface.
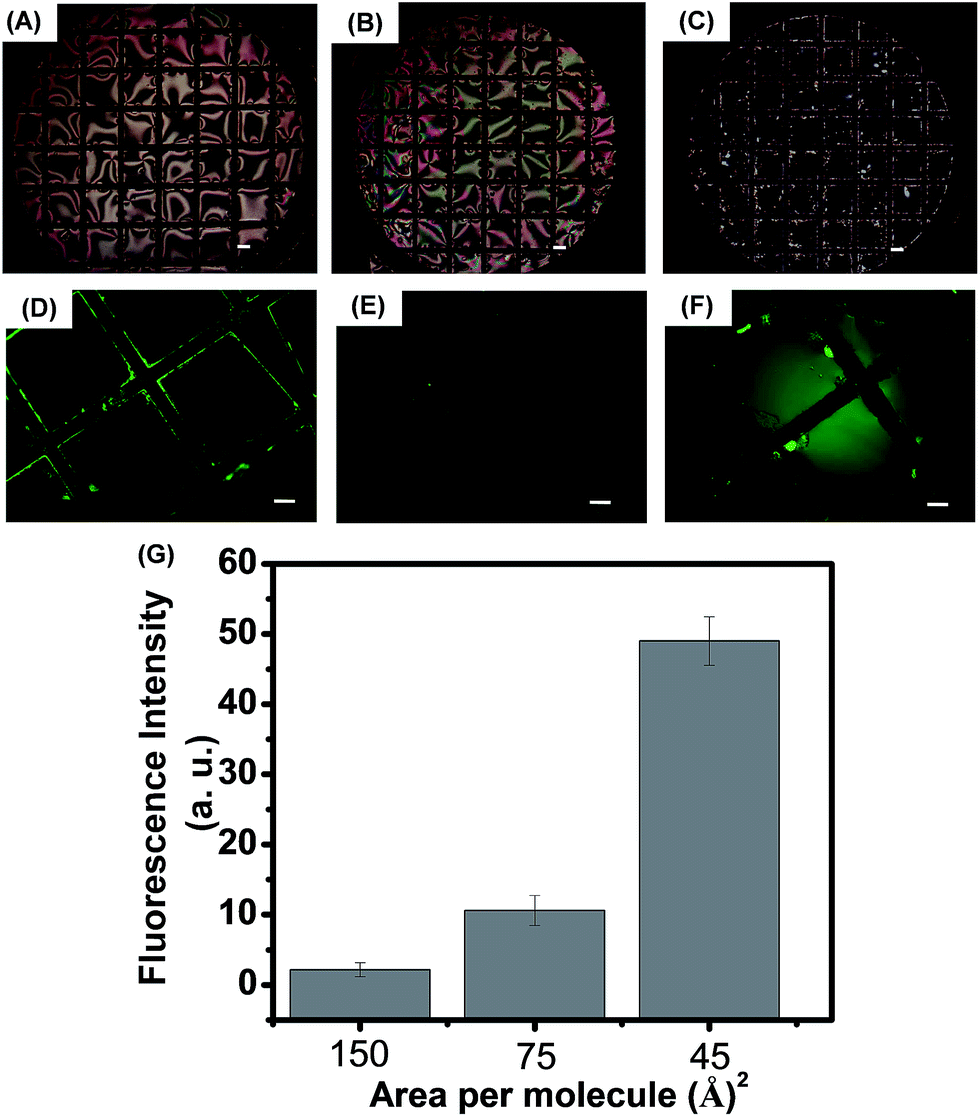 | ||
| Fig. 2 Polarized light micrographs of 0.5% (mol) FITC–LPS/LPS monolayers after transfer to the LC–water interface at surface pressure of (A) 0, (B) 30 and (C) 52 mN m−1. (D)–(F) Corresponding fluorescence images of films at the LC–water interface. Scale bar = 40 μm. (G) Represents fluorescence intensity measured for a 0.5% (mol) FITC–LPS doped Langmuir monolayer of LPS at different areal density, measured at 25 °C. | ||
We further characterized the adsorption of LPS on 5CB aqueous interface using PM-IRRAS measurements. PM-IRRAS is used to evaluate the structural features of organic films of thickness less than 200 nm.57 First, we functionalized gold coated micro-pillars (2–3 μm) with DMOAP. The IR spectra using polarization modulation of DMOAP coated surface shows the characteristic peaks of Si–C (1265 cm−1), Si–O (1177 cm−1, 1110 cm−1), C–O (1043 cm−1) and CH3 and CH2 stretching (2960 cm−1, 2924 cm−1, 2853 cm−1) as shown in Fig. 3A. Next we poured the 5CB into the DMOAP coated micro-pillars. Fig. 3B shows the strong absorption bands of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N (2218 cm−1) along with aromatic C–H stretching (3144 cm−1, 3030 cm−1), aliphatic CH2 and CH3 stretching (2924 cm−1, 2852 cm−1, 2801 cm−1) and C–H bending (1474 cm−1). Next we incubated 5CB confined in DMOAP coated micro-pillars with an aqueous solution of 0.1 mg mL−1 LPS for 6 h and kept this LPS adsorbed on 5CB film under vacuum for complete drying. In PM-IRRAS spectra of this LPS (Fig. 3C) absorbed 5CB films, we observed amide carbonyl stretching (1631 cm−1), broad O–H stretching (3441 cm−1), amide N–H stretching (3239 cm−1), sharp amide N–H bending (1556 cm−1), symmetric and anti-symmetric stretching of phosphate (1138 cm−1, 1298 cm−1), CH2 and CH3 stretching (2983 cm−1, 2919 cm−1, 2801 cm−1) and C–H bending (1461 cm−1). Overall these peaks strongly support presence of LPS58 over 5CB film. In addition to this, in this spectrum we also observed aromatic C–H stretching (3108 cm−1) and CN stretching (2205 cm−1) correspond to the presence of 5CB film.
N (2218 cm−1) along with aromatic C–H stretching (3144 cm−1, 3030 cm−1), aliphatic CH2 and CH3 stretching (2924 cm−1, 2852 cm−1, 2801 cm−1) and C–H bending (1474 cm−1). Next we incubated 5CB confined in DMOAP coated micro-pillars with an aqueous solution of 0.1 mg mL−1 LPS for 6 h and kept this LPS adsorbed on 5CB film under vacuum for complete drying. In PM-IRRAS spectra of this LPS (Fig. 3C) absorbed 5CB films, we observed amide carbonyl stretching (1631 cm−1), broad O–H stretching (3441 cm−1), amide N–H stretching (3239 cm−1), sharp amide N–H bending (1556 cm−1), symmetric and anti-symmetric stretching of phosphate (1138 cm−1, 1298 cm−1), CH2 and CH3 stretching (2983 cm−1, 2919 cm−1, 2801 cm−1) and C–H bending (1461 cm−1). Overall these peaks strongly support presence of LPS58 over 5CB film. In addition to this, in this spectrum we also observed aromatic C–H stretching (3108 cm−1) and CN stretching (2205 cm−1) correspond to the presence of 5CB film.
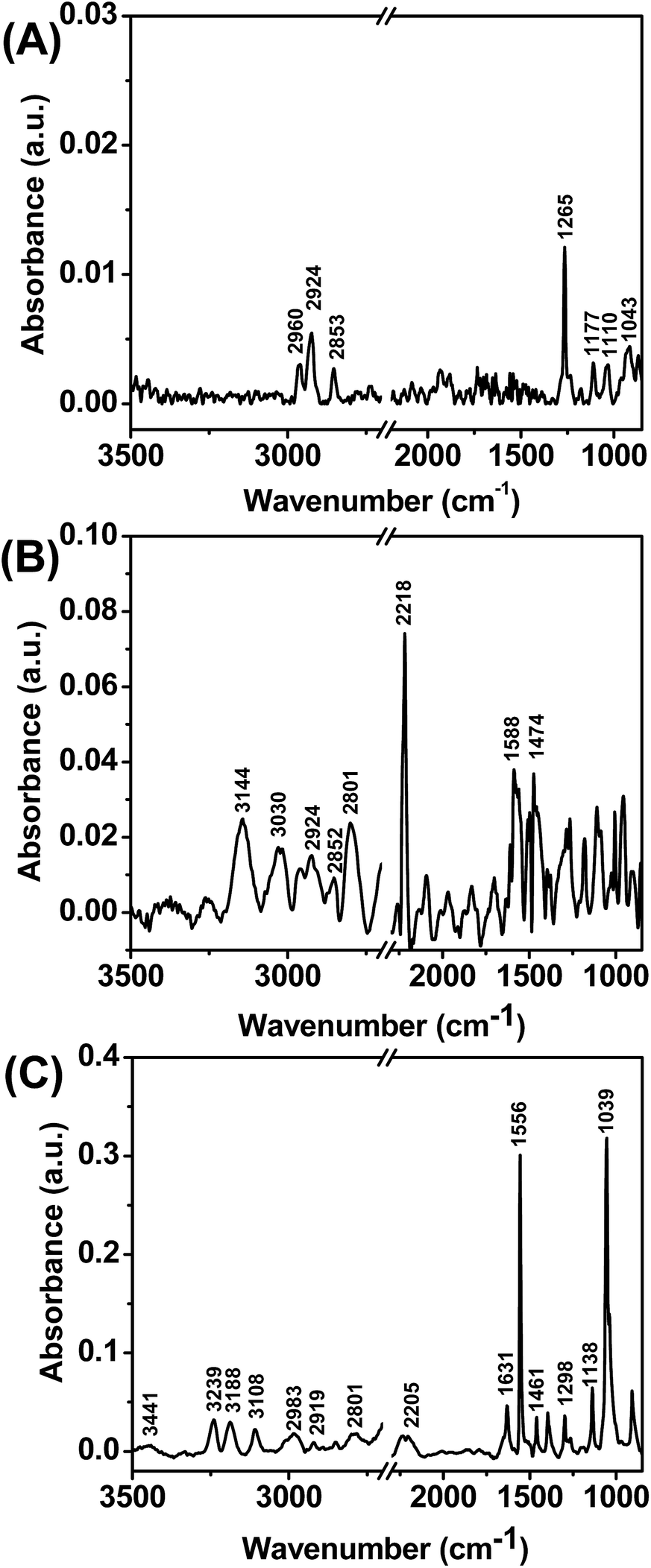 | ||
| Fig. 3 PM-IRRAS spectra generated from (A) DMOAP, (B) 5CB and LPS supported on micro-pillars coated with a uniformly deposited film of gold. | ||
To study the interactions of LPS with cell membrane components, we first incubated this optical cell containing LPS solution for 2 h. Next, we exchanged the LPS solution with Tris buffer (pH 7.4) three times to remove excess free LPS from bulk solution and then exposed this LPS laden aqueous/5CB interface in contact with different cell membrane components. In our first experiment, we added 0.1 mg mL−1 solution of PG in Tris buffer (pH 7.4) onto the LPS laden aqueous–LC interface. We observed an immediate change in optical response of the LC from dark to bright indicating an ordering transition of LCs from homeotropic to tilted state (Fig. 4A and B). This ordering transformation of LC was construed due to strong interaction between PG and LPS, which, in turn, disturb the ordered arrangement of LPS at aqueous–LC interface leading to the tilted orientation of LC molecules.
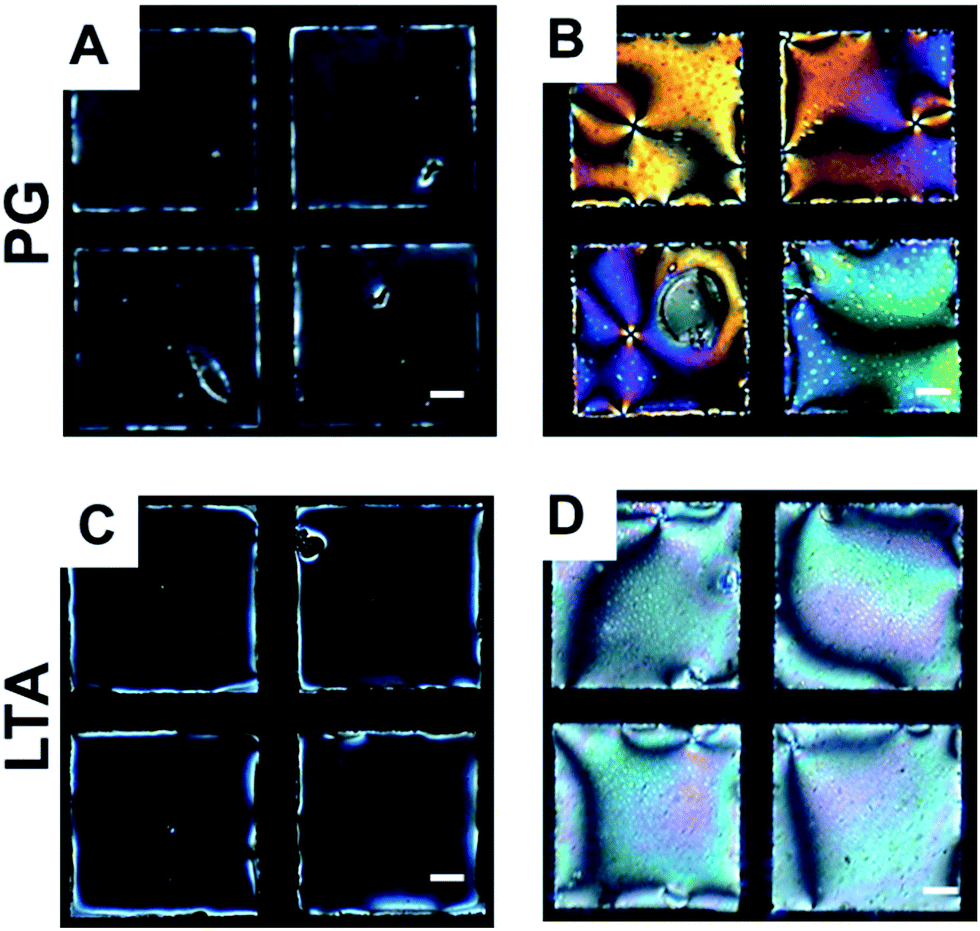 | ||
| Fig. 4 Optical images (crossed polars) of 5CB hosted in gold grids supported on DMOAP-coated glass slides in contact with (A and C) LPS for 2 h of incubation. (B and D) after introducing aqueous PG and LTA solution onto LPS laden aqueous/LC interface respectively. Scale bar = 40 μm. | ||
Next, we sought to investigate if the interaction of LTA with LPS interface could lead an ordering transition in the LC. For this, we exposed 0.1 mg mL−1 of aqueous LTA solution in Tris buffer (pH 7.4) onto the LPS laden aqueous–LC interface. We observed a rapid change in optical appearance of the LC from dark to bright (Fig. 4C and D) consistent with an ordering transition of 5CB from homeotropic to planar/tilted state. We interpreted the orientational ordering transition of the LCs as a result of hydrophobic interaction of LTA with LPS decorated aqueous–LC interfaces.
To confirm, whether electrostatic interactions play any role in determining ordering transitions of LCs at these interfaces, we performed several control experiments in different pH conditions other than the physiological pH. Interestingly, in all cases the dynamic ordering transitions towards a planar ordering in different pH conditions other than the physiological pH was found to be same (images not shown). These observations, as a whole, suggest that the interactions between LPS with LTA and PG are not driven by electrostatically. This study demonstrate that specific and non-specific interactions of different cell membrane components (PG and LTA) with endotoxin leading to changes in the optical appearance of LCs can be attributed to changes in the ordering of LPS molecules at the interface through strong interactions and thus, provide facile approach to study these interactions at aqueous–LC interfaces.
Next, to provide further insight into the above proposition that strong interaction of LPS with PG and LTA is responsible for the rapid ordering transitions of the LCs at aqueous–LC interface we performed several control experiments. First, we sought to investigate whether direct interaction of PG and LTA with interfacial 5CB molecules could able to alter the orientation of LCs in the absence of LPS membrane at the interface. To validate this, we added 0.2 mg mL−1 of aqueous solution of PG, LTA in Tris buffer (pH 7.4) directly onto LPS free aqueous/LC interface. We found that the optical appearance of LCs remained bright (even after 12 h of incubation or more) indicating a planar/tilted orientation of 5CB molecules at aqueous–LC interface (Fig. 5). This observation clearly demonstrates that there are no direct interactions present between interfacial 5CB molecules and the cell membrane components which could perturb the orientation of LCs at these interfaces.
 | ||
| Fig. 5 Polarized optical images of 5CB contained in gold grids supported on DMOAP-treated glass slides and placed into contact with (A and C) aqueous Tris buffer (pH 7.4) and (B and D) in contact with an aqueous solution of 0.2 mg mL−1 of PG and LTA respectively. Scale bar = 40 μm. | ||
Second, we focused to carry out investigations on the specificity of the interactions of LPS with PG and LTA, respectively. For this, we replaced LPS with other three different phospholipids. We chose zwitterionic DLPC and negatively charged LPA and DOPG which can form self-assembled at aqueous–LC interfaces and orient the LCs homeotropically. This study was motivated by two goals. First, we wanted to see whether these lipids (in addition to LPS) could able to interact with PG and LTA resulting in an orientational ordering transition of the LCs from homeotropic to tilted/planar at these interfaces. Second, we sought to find out if any favorable electrostatic interactions between these phospholipids (charged) and PG, LTA are responsible for the LC ordering at those interfaces. Interestingly, we observed that the optical appearance of LCs coupled to DLPC, LPA and DOPG decorated interfacial membrane remained dark (Fig. 6) after addition of 0.2 mg mL−1 aqueous solution of PG, LTA in Tris buffer (pH 7.4) even after 6 h or more incubation. This observation strongly suggests that the interaction of PG and LTA coupled to these phospholipids decorated interfaces is not strong enough, if present, to disrupt the ordering of LC–aqueous interfaces. In addition, we have measured the zeta potential of LPS, PG and LTA solutions (Tris buffer 20 mM, pH 7.4) as shown in Table S1 (see ESI†). The zeta potential was found to be negative in all cases. This also led us to conclude that the interaction of these cell membrane components (PG and LTA) with LPS is highly specific and not driven by electrostatically but could be through hydrophobic interactions.
 | ||
| Fig. 6 Polarized optical micrographs of LC aqueous interface laden with (A) DOPG (C) DLPC and (E) LPA. (B, D and F) represents the optical response of 5CB decorated with DOPG, DLPC and LPA after adding biomolecules respectively. Scale bar = 40 μm. | ||
To provide further insight into the mode of interaction of PG and LTA with LPS at aqueous–LC interface, we performed another control experiment using aqueous starch solution. This investigation is guided by the proposition from a recent report where Vagenende et al. have shown the self-assembly of LPS on allantoin crystals is initiated through hydrogen-bond attachment of hydrophilic LPS regions with amide-groups of allantoin.59 Therefore, the principle motive behind performing this control experiment with starch was to find out whether hydrogen bonding plays any role in the interaction of these biomolecules (PG and LTA) with LPS self-assembled at aqueous–LC interface. Starch is a polysaccharide based macromolecule. PG and LTA also contain sugar units, having several hydroxyl functionalities, similar to starch. Therefore, we hypothesized that, like PG and LTA, if starch also could able to induce an ordering transition of LPS laden interfacial 5CB, we would able to confirm that the hydrogen bonding between the polysaccharide moieties of these biomolecules (PG, LTA, starch) and LPS is mainly responsible to induce ordering transition of LPS decorated interfacial 5CB. But when we carried out this experiment, we found that the optical appearance of LPS laden 5CB interface remained dark over 2 hours of observation period after exposing 500 μg mL−1 aqueous starch solution onto 5CB–aqueous interface (Fig. S3, see ESI†). This result strongly suggests the absence of any interaction between LPS and starch at 5CB interface and hydrogen bonding between LPS and starch does not play any role in inducing ordering transition of interfacial 5CB. In addition to that, from this experiment, we also confirmed that the interaction of these cell membrane components (PG and LTA) with LPS is highly specific and not driven by hydrogen bonding but dominated mostly through hydrophobic interactions.
The above experiments demonstrate that the strong interaction of LPS with PG and LTA results in rapid ordering transition of the LCs from homeotropic to tilted/planar and these interactions are proven to be highly specific towards LPS at those interfaces. As the consequence of these interactions of cell membrane components with LPS in mammalian hosts is highly divergent towards the endotoxic behavior of LPS, therefore, in addition to, studying the interaction of PG, LTA with LPS at these interfaces, it is very important to determine the sensitivity of the LC based system to the realization of a novel biosensor for detection of such biomolecular interactions. With this idea keeping in mind, we thought to determine limit of detection (LOD) and response time of the LC based system to study biomolecular interactions. For this, we compared the dynamic response of the LCs at different concentrations of PG and LTA onto LPS decorated aqueous/LC interface. We first optimized the concentration of LPS (i.e. minimum concentration required) which is required to align the LCs homeotropically at aqueous–LC interfaces. After exposing LPS of different concentrations onto aqueous–LC interface followed by removal of excess LPS from the solution, we found that 60 μg mL−1 is the optimum concentration at which LPS orients the LCs homeotropically and results a uniform dark optical image under crossed polars (Fig. S4, see ESI†). Second, we varied the concentration of PG and LTA onto LPS laden (at a concentration of 60 μg mL−1) aqueous–LC interfaces to determine the sensitivity of the LC based system (see below for details).
Fig. 7A and B exhibits the optical appearance of the LCs after addition of 60 μg mL−1 PG, LTA solutions (for 1 h of incubation) onto LPS laden aqueous/LC interface. We observed a rapid change in the optical appearance of LCs from dark to bright within 5 minutes-a little longer time span compare to the concentration (0.1 mg mL−1) of PG/LTA used previously. After careful observation we revealed that the birefringence colors of 5CB (20 μm thick film) in presence of PG onto LPS laden LC interface was distinct from that of LTA, which clearly indicates the different tilted states of 5CB molecules at aqueous–LC interfaces. On further varying the concentrations of PG and LTA to successive lower concentrations values, we observed an increase in the time span of the change in the optical appearance of the 5CB (from completely dark to bright) (Fig. S5 and S6, see ESI†).
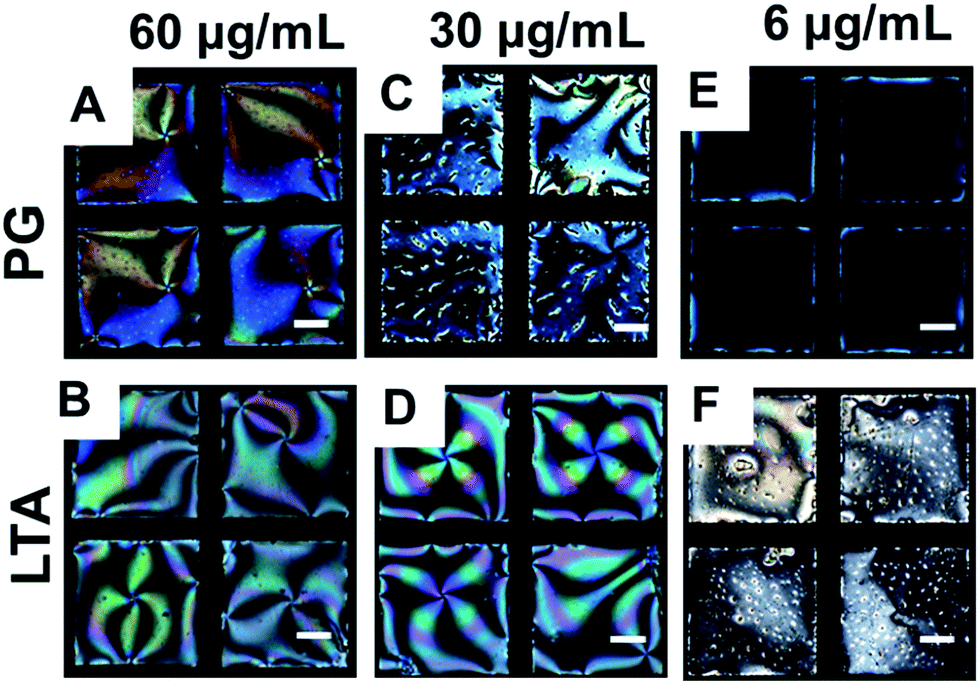 | ||
| Fig. 7 Optical micrographs of 5CB on exposing LPS laden aqueous LC interface for 60 min incubation with 60 μg mL−1, 30 μg mL−1 and 6 μg mL−1 aqueous solutions of (A, C and E) PG and (B, D and F) LTA respectively. Scale bar = 40 μm. | ||
We also observed a faster change in the optical appearance of the LC in presence of LTA in comparison to that of PG. Careful inspection of the LC ordering at different concentrations of PG and LTA, we found that 5CB exhibits only few bright spots (for 1 h of incubation, see Fig. 7C) at a concentration of 30 μg mL−1 of PG, whereas, LTA at the similar concentration (same 1 h of incubation) induced bright appearance of the LCs (Fig. 7D). Further lowering the concentration to 28 μg mL−1 or below, we found that in presence of PG, the optical appearance of 5CB remained completely dark even after 2 h of incubation (Fig. 7E and S5, see ESI†). Therefore, we confirmed that the concentration of 30 μg mL−1 as LOD for PG in our LC based sensing system. In contrast, we observed that 20 μg mL−1 of LTA could able to alter the optical appearance of the LC from dark to bright (Fig. S6, see ESI†) within 1 h of incubation period suggesting higher sensitivity of LTA in comparison to PG. Therefore, we further decreased the concentration of LTA to determine the LOD value. Consequently, we found that 6 μg mL−1 is the LOD for LTA (Fig. 7F) which could induce an ordering transition of the LC at LPS–laden aqueous–LC interfaces. Further decreasing the concentration (<6 μg mL−1), we observed that LC retained its dark optical view, in presence of LTA even after 2 h of incubation period or more (Fig. S6†). The LOD values found as invariant at different pH (pH 2, pH 9) (images not shown) suggesting these binding events are independent of any electrostatic interaction and mainly driven by hydrophobic forces.
In a consequence, to report the change in the optical appearance of the LC with varying concentrations of PG and LTA, we measured the average gray scale intensity and quantified the optical response as a function of varying concentration of PG and LTA. Interestingly, we observed a continuous decrease in gray scale intensity with decreasing in concentration on addition of both PG and LTA (Fig. 8). It is noteworthy; however, the measured gray scale intensity obtained for LTA on LPS–laden aqueous–5CB interfaces is higher than that of PG in all concentration range measured. This experiments demonstrate that LTA exhibits stronger binding affinity towards LPS with respect to PG which is also well-supported with their LOD values (6 μg mL−1 for LTA, whereas 30 μg mL−1 for PG).
 | ||
| Fig. 8 Represent the average gray scale intensity of optical images of 5CB films as a function of varying concentrations of PG and LTA on LPS decorated aqueous/5CB interface. | ||
In order to further validate our observation that the changes in the organization of the adsorbed LPS at the aqueous–LC interfaces underlie the different orientational behaviors of 5CB in presence of PG and LTA, we measured the tilt angle of LCs during LPS–PG/LTA binding event at those interfaces. According to our hypothesis, the extent of disorderness of LPS membrane at aqueous–LC interface solely depends on the strength of binding of these cell membrane components (PG and LTA) with LPS which, in turn, lead to different tilt of LC molecules. Therefore, we thought that by measuring tilt angle at aqueous–LC interfaces (during LPS–PG/LTA binding events) it is possible to quantify the LC ordering at those interfaces. For this experiment, first, we chose 30 μg mL−1 of PG and LTA which is the minimum concentration required to show the change in the ordering transition of LC from homeotropic to tilted through interfacial binding with LPS–laden aqueous interface. Next, we calculated tilt angle of 5CB from the measured values of optical retardance using previously reported procedures (see Experimental section for details).37 Fig. 9 shows the dynamic change in the tilt angle of 5CB coupled to PG and LTA onto LPS decorated aqueous–LC interface, respectively. We observed the (for 3.5 h of incubation or more) maximum tilt angle of 5CB obtained for PG–LPS and LTA–LPS were 26.76° ± 1.7 and 38.6° ± 2.5, respectively, at these interfaces. During the experiment, it may be pointed out that the tilt of 5CB is highest (38.6° ± 2.5) within 80 min in case of LTA–LPS binding event and the value remained constant in rest of the observation period. In contrast, the highest tilt angle (26.76° ± 1.7) of 5CB observed after 180 min in case of PG–LPS binding event. Consequently, these observations clearly indicate different binding affinity of LTA and PG towards LPS–laden aqueous–LC interface. We have mentioned earlier that stronger binding event could disrupt the LPS arrangement to a greater extent which, in turn, will lead to greater change in the tilt angle of 5CB at aqueous interfaces. Therefore, on this basis from tilt angle measurement we can say that LTA shows greater binding affinity towards LPS compared to PG. These experiments also reveal a quantitative approach to report the interaction of bacterial endotoxin with cell membrane components (PG and LTA) at aqueous–LC interface.
 | ||
| Fig. 9 The tilt angle of 5CB decorated with LPS at aqueous/LC interface on exposure of 30 μg mL−1 aqueous solution of PG and LTA, respectively. | ||
Prior to conclusion, another key observation we would like to report is that specific binding of PG/LTA to LPS at aqueous–LC interface triggers a continuous orientational ordering transition (continuous change in the tilt) in the LC. It is observed that contact of an aqueous solution of LPS vesicles with the interface of a micrometer-thick film of 5CB formed a spontaneous arrangement of LPS on the interface of the LC, resulting in a discontinuous ordering transition in which the LC changes from parallel to the perpendicular orientation. Such lipid induced discontinuous ordering switch of interfacial LCs is well supported by past studies31 where spontaneous adsorption of lipid, DLPC, on LC–aqueous interface has been reported to give rise to a fast ordering transition in LCs mediated by discontinuous alteration of the LC alignment in molecular level at those interfaces. In contrast, the binding of PG/LTA with LPS at interface led to a continuous ordering transition of LCs from homeotropic to tilted orientation. The continuous process can be well understood by observing a gradual decrease of tilt of 5CB during PG/LTA–LPS binding event at the interface. In addition, we can also illustrate this phenomenon from previous studies that have reported the heterogeneous interfaces comprised of nanoscopic patches cause homeotropic or planar anchoring of LCs and give rise to micrometer-scale tilting of the LC. The pattern of local surface-imposed orientations of the LC becomes homogeneous in the bulk of the LC in order to minimize the elastic energy of the LC.60–62 Previously, Abbott and his coworkers reported the continuous LC ordering transitions induced by binding of vesicles to protein-decorated LC interfaces are consistent with an inhomogeneous LC interface comprised of nano-domains of proteins and phospholipids.32 In our study similar continuous transition of interfacial LCs decorated with LPS has been observed in presence of PG/LTA at aqueous/LC interface. Overall, our work reported in this paper hold two key features. First, we demonstrated specific binding of these cell membrane components on LPS laden aqueous/LC interface for the first time. Second, these binding exhibited ordering transitions of the LCs from homeotropic to tilted state in a continuous manner.
Conclusions
In summary, we have developed a subtle and robust LC-based sensing platform to study quantitatively the interaction of LPS with bacterial cell membrane components. We have characterized the LPS laden aqueous/5CB interface using Langmuir–Blodgett technique and PM-IRRAS measurement. We also measured hydrodynamic diameter of all the cell membrane components. Strong interaction between the cell membrane components (PG, LTA) and LPS induces the orientational ordering transition of LCs at aqueous–LC interfaces through changes in the optical appearance of the LCs. These binding events remain invariant as a function of pH and therefore, suggest that the ordering of the LC is independent of electrostatic interactions. We demonstrated that these interactions of PG and LTA are highly specific towards LPS in response to different lipids. The detection limit of our LC based system towards these biomolecular interactions was found 6 μg mL−1 and 30 μg mL−1 for LTA–LPS and PG–LPS interactions, respectively, indicating high sensitivity of our system towards these biomolecular binding events at aqueous/LC interface. From average gray scale intensity measurement, we affirmed that binding affinity of LTA towards LPS is higher compared to PG. Finally, we have shown the quantitative approach of studying different binding affinities of PG and LTA towards LPS in light of tilt angle measurement. Overall, the results presented in this paper suggest that LCs offer the basis of a novel analytical tool for fundamental studies of bacterial cell membrane components and endotoxin at interface and, specifically, they offer methods to quantify specific binding of PG and LTA on LPS decorated interface.Acknowledgements
This work was carried out with the financial support from IISER Mohali and Department of Atomic Energy (DAE-BRNS) bearing sanction No. 2012/20/34/7/BRNS. SKP is thankful to IISER Mohali for SAXS/WAXS facility. D. Das and S. Sidiq acknowledge the receipt of a graduate fellowship from IISER Mohali and UGC, respectively.Notes and references
- C. G. Giske, D. L. Monnet, O. Cars and Y. Carmeli, Antimicrob. Agents Chemother., 2008, 52, 813–821 CrossRef CAS PubMed.
- L. S. Engel, Emerg. Med., 2009, 41, 18–27 Search PubMed.
- G. D. Wright, Nat. Rev. Microbiol., 2007, 5, 175–186 CrossRef CAS PubMed.
- V. Turhan, M. Mutluoglu, A. Acar, M. Hatipoglu, Y. Onem, G. Uzun, H. Ay, H. Oncul and L. Gorenek, J. Infect. Dev. Countries, 2013, 7, 707–712 Search PubMed.
- B. Beutler and E. T. Rietschel, Nat. Rev. Immunol., 2003, 3, 169–176 CrossRef CAS PubMed.
- V. L. Tesh and D. C. Morrison, J. Immunol., 1988, 141, 3523–3531 CAS.
- R. I. Roth and W. Kaca, Biomater., Artif. Cells, Immobilization Biotechnol., 1994, 22, 387–398 CAS.
- C. Galanos and M. A. Freudenberg, Mediators Inflammation, 1993, 2, S11–S16 CrossRef CAS PubMed.
- L. Chu, T. E. Bramanti, J. L. Ebersole and S. C. Holt, Infect. Immun., 1991, 59, 1932–1940 CAS.
- S. M. Opal, P. J. Scannon, J. L. Vincent, M. White, S. F. Carroll, J. E. Palardy, N. A. Parejo, J. P. Pribble and H. Lemke, J. Infect. Dis., 1999, 180, 1584–1589 CrossRef CAS PubMed.
- A. E. Myhre, A. O. Aasen, C. Thiemermann and J. E. Wang, Shock, 2006, 25, 227–235 CrossRef CAS PubMed.
- R. Daziarski, J. Biol. Chem., 1991, 266, 4719–4725 Search PubMed.
- C. Natanson, R. L. Danner, R. J. Ellin, J. M. Hosseini, K. W. Peart, S. M. Banks, T. J. Macvitte, R. I. Walker and J. E. Parrillo, J. Clin. Invest., 1989, 83, 243–251 CrossRef CAS PubMed.
- K. Takayama, Z. Z. Din, P. Mukerjee, P. H. Cooke and T. N. Kirkland, J. Biol. Chem., 1990, 265, 14023–14029 CAS.
- J. E. Wang, P. F. Jorgensen, E. A. Ellingsen, M. Almiof, C. Theimermann, S. J. Foster, A. O. Aasen and R. Solberg, Shock, 2001, 16, 178–182 CrossRef CAS.
- H. Takada and C. Galanos, Infect. Immun., 1987, 55, 409–413 CAS.
- M. Parant, F. Parant, M. A. Vinit, C. Jupin, Y. Noso and L. Shedid, J. Leukocyte Biol., 1990, 47, 164–169 CAS.
- G. M. Wary, S. J. Foster, C. J. Hinds and C. A. Theimermann, Shock, 2001, 15, 135–142 CrossRef.
- M. A. Wolfert, T. F. Murray, G. J. Boons and J. N. Moore, J. Biol. Chem., 2002, 277, 39179–39186 CrossRef CAS PubMed.
- S. Sugawara, R. Arakaki, H. Rikiishi and H. Takada, Infect. Immun., 1999, 67, 1623–1632 CAS.
- T. Kusunoki, E. Hailman, T. S.-C. Juan, H. S. Lichenstein and S. D. Wright, J. Exp. Med., 1995, 182, 1673–1682 CrossRef CAS.
- Oral microbiology and immunology, ed. R. J. Nisengard and M. G. Newman, The W.B. Saunders Co., Philadelphia, Pa., 2nd edn, 1994, pp. 360–384 Search PubMed.
- S. N. Lichtman, J. J. Wang, H. Schwab and J. J. Lemasters, Hepatology, 1994, 19, 1013–1022 CrossRef CAS PubMed.
- J. S. Hadley, J. E. Wang, S. J. Foster, C. Theimermann and C. J. Hinds, Infect. Immun., 2005, 73, 7613–7619 CrossRef CAS PubMed.
- E. Mattsson, L. Verhage, J. Rollof, A. Fleer and H. V. Dijk, FEMS Immunol. Med. Microbiol., 1993, 7, 281–287 CAS.
- D. Hartono, W. J. Qin, K.-L. Yang and L.-Y. Lanry Yung, Biomaterials, 2009, 30, 843–849 CrossRef CAS PubMed.
- D. Hartono, C.-Y. Xue, K.-L. Yang and L.-Y. Lanry Yung, Adv. Funct. Mater., 2009, 19, 3574–3579 CrossRef CAS PubMed.
- J.-S. Park and N. L. Abbott, Adv. Mater., 2008, 20, 1185–1190 CrossRef CAS PubMed.
- A. Agarwal, S. Sidiq, S. Setia, E. Bukusoglu, J. J. de Pablo, S. K. Pal and N. L. Abbott, Small, 2013, 9, 2785–2792 CrossRef CAS PubMed.
- Q.-Z. Hu and C. H. Jang, ACS Appl. Mater. Interfaces, 2012, 4, 1791–1795 CAS.
- J. M. Brake, M. K. Daschner, Y.-Y. Luk and N. L. Abbott, Science, 2003, 302, 2094–2097 CrossRef CAS PubMed.
- L. N. Tan, V. J. Orler and N. L. Abbott, Langmuir, 2012, 28, 6364–6376 CrossRef CAS PubMed.
- S. Sidiq, D. Das and S. K. Pal, RSC Adv., 2014, 4, 18889–18893 RSC.
- S. J. Woltman, G. D. Jay and G. P. Crawford, Nat. Mater., 2007, 6, 929–938 CrossRef CAS PubMed.
- Y.-Y. Luk, M. L. Tingey, K. A. Dickson, R. T. Raines and N. L. Abbott, J. Am. Chem. Soc., 2004, 126, 9024–9032 CrossRef CAS PubMed.
- X. Bi, D. Hartono and K.-L. Yang, Adv. Funct. Mater., 2009, 19, 3760–3765 CrossRef CAS PubMed.
- D. Das, S. Sidiq and S. K. Pal, ChemPhysChem, 2015, 16, 753–760 CrossRef CAS PubMed.
- S. Sidiq, I. Verma and S. K. Pal, Langmuir, 2015, 31, 4741–4751 CrossRef CAS PubMed.
- A. D. Price and D. K. Schwartz, J. Am. Chem. Soc., 2008, 130, 8188–8194 CrossRef CAS PubMed.
- S. Yang, C. Wu, H. Tan, Y. Wu, S. Liao, Z. Yu, G. Shen and R. Yu, Anal. Chem., 2013, 85, 14–18 CrossRef CAS PubMed.
- H. Tan, S. Y. Yang, G. L. Shen, R. Q. Yu and Z. Y. Wu, Angew. Chem., Int. Ed., 2010, 49, 8608–8611 CrossRef CAS PubMed.
- S. Y. Yang, Y. M. Liu, H. Tan, C. Wu, Z. Wu, G. L. Shen and R. Yu, Chem. Commun., 2012, 48, 2861–2863 RSC.
- C.-H. Chen and K.-L. Yang, Sens. Actuators, B, 2013, 181, 368–374 CrossRef CAS PubMed.
- Q. Z. Hu and C. H. Jang, J. Biotechnol., 2012, 157, 223–227 CrossRef CAS PubMed.
- Q. Z. Hu and C. H. Jang, Colloids Surf., B, 2011, 88, 622–626 CrossRef CAS PubMed.
- I.-H. Lin, M.-V. Meli and N. L. Abbott, J. Colloid Interface Sci., 2009, 336, 90–99 CrossRef CAS PubMed.
- L. A. T. Espinoza, K. R. Schumann, Y.-Y. Luk, B. A. Israel and N. L. Abbott, Langmuir, 2004, 20, 2375–2385 CrossRef.
- L.-L. Cheng, Y.-Y. Luk, C. J. Murphy, B. A. Israel and N. L. Abbott, Biomaterials, 2005, 26, 7173–7182 CrossRef CAS PubMed.
- C.-H. Jang, L.-L. Cheng, C. W. Olsen and N. L. Abbott, Nano Lett., 2006, 6, 1053–1058 CrossRef CAS PubMed.
- J. J. Skaife and N. L. Abbott, Langmuir, 2001, 17, 5595–5604 CrossRef CAS.
- H. Tan, X. Li, S. Liao, R. Yu and Z. Wu, Biosens. Bioelectron., 2014, 62, 84–89 CrossRef CAS PubMed.
- D. Liu, Q. Z. Hu and C. H. Jang, Colloids Surf., B, 2013, 108, 142–146 CrossRef CAS PubMed.
- M. Zhang and C.-H. Jang, Anal. Biochem., 2014, 455, 13–19 CrossRef CAS PubMed.
- D. Cheng, S. S. Sridharamurthy, J. T. Hunter, J.-S. Park, N. L. Abbott and H. Jiang, J. Microelectromech. Syst., 2009, 18, 973–981 CrossRef CAS.
- M. V. Meli, I. H. Lin and N. L. Abbott, J. Am. Chem. Soc., 2008, 130, 4326–4333 CrossRef CAS PubMed.
- I. H. Lin, D. S. Miller, P. J. Bertics, C. J. Murphy, J. J. de Pablo and N. L. Abbott, Science, 2011, 332, 1297–1300 CrossRef CAS PubMed.
- B. L. Frey, R. M. Corn and S. C. Weibel, Polarization–ModulationApproaches to Reflection–Absorbtion Spectroscopy, in Handbook of Vibrational Spectroscopy, ed. J. Chalmers and P. R. Griffins, John Wiley& Sons, Chichester, U.K., 2001, vol. 2, p. 1042 Search PubMed.
- K. Bradenburg, Biophys. J., 1993, 64, 1225–1231 Search PubMed.
- V. Vagenende, T.-J. Ching, R.-J. Chua, Q. Z. Jiang and P. Gagnon, Colloids Surf., B, 2014, 120, 8–14 CrossRef CAS PubMed.
- K. Zhang, N. Liu, R. Twieg, B. Auman and P. Bos, Liq. Cryst., 2008, 35, 1191–1197 CrossRef CAS PubMed.
- H. S. Kwok, Y. W. Li and F. S. Yeung, Mol. Cryst. Liq. Cryst., 2009, 507, 26–40 CrossRef CAS PubMed.
- F. S. Yeung, J. Y. Ho, Y. W. Li, F. C. Xie, O. K. Tsui, P. Sheng and H. S. Kwok, Appl. Phys. Lett., 2006, 88, 051910 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the size and zeta potential measurements of biomolecules using dynamic light scattering, surface pressure–area isotherm of LPS using LB technique and optical response of the LCs in presence of LPS and other biomolecules at LPS–laden aqueous–LC interfaces at different concentration. See DOI: 10.1039/c5ra09640e |
| This journal is © The Royal Society of Chemistry 2015 |