
Pd and Pd–CuO nanoparticles in hollow silicalite-1 single crystals for enhancing selectivity and activity for the Suzuki–Miyaura reaction†
Chengyi Daiab,
Xinmin Lia,
Anfeng Zhangab,
Chun Liu*a,
Chunshan Songabc and
Xinwen Guo*ab
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: cliu@dlut.edu.cn; Tel: +86-411-84986182
bPSU-DUT Joint Center for Energy Research, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: guoxw@dlut.edu.cn; Fax: +86-411-84986134; Tel: +86-411-84986133
cEMS Energy Institute, PSU-DUT Joint Center for Energy Research and Department of Energy & Mineral Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA
First published on 27th April 2015
Abstract
Pd and Pd–CuO nanoparticles were successfully encapsulated in hollow silicalite-1 single crystals by tetrapropylammonium hydroxide (TPAOH) hydrothermal treatment with an “impregnation-dissolution-recrystallization” process. The size and number of particles in the hollow zeolite depended mainly on the nature of the metal. For palladium, the palladium nanoparticles easily aggregated into larger particles in the hydrothermal process, which displays excellent substrate selectivity for the meta- and para-substituted aryl bromides in the Suzuki–Miyaura reaction. For Pd–CuO binary metals (oxide), introducing copper oxide prevents aggregation of palladium, which shows about 3 times higher activity than encapsulated single Pd catalyst for the above reaction. The strategy using a hollow zeolite crystal as a support is a more reliable method for preparing multi-metallic (oxide) catalysts with well-dispersed nanoparticles.
1 Introduction
The palladium-catalyzed Suzuki–Miyaura reaction of aryl halides with arylboronic acids is one of the most important and powerful tools to form biaryls and has been widely used in the synthesis of natural products, pharmaceuticals and advanced functional materials.1 The popularity of this reaction has been further enhanced by the increasing availability of organoboron reagents, the low toxicity of the inorganic by-products, and the tolerance of a broad range of functional groups.2 Because of the great significance of this reaction, it was recognized with the 2010 Nobel Prize in Chemistry. The generic palladium-catalyzed Suzuki–Miyaura reaction employs phosphine ligands, which are often water- and/or air-sensitive.3 Although some modified ligands could work in water, and even in air, the tedious multi-step synthesis and high cost of these ligands restrict their applications.4 In recent years, there has been an increasing interest in developing efficient and green processes for the nano-palladium catalyzed Suzuki–Miyaura reaction in the absence of a ligand.5 A variety of supports, such as polymers,6 metal–organic frameworks (MOFs),7 organic–inorganic composites,8 silica9 and zeolites,10 are used to support Pd nanoparticles. However, obtaining the target product with high selectivity and activity remains difficult.In the past years, hollow zeolites have attracted considerable interests due to their well-defined hollow structure, thermal/mechanical stability, and shape selectivity.11 However, there is none report on hollow silicalite-1 single crystals as containers of catalysts for the Suzuki–Miyaura reaction. Herein, using “impregnation-dissolution-recrystallization” method, Pd and Pd–CuO nanoparticles located on the surface of solid silicalite-1 were successfully encapsulated in the regular void of the hollow crystal. To the best of our knowledge, this is the first report on the encapsulation of palladium nanoparticles in hollow silicalite-1 single crystals. This material exhibits excellent catalytic selectivity for the meta- and para-substituted aryl bromides in the Suzuki–Miyaura reaction under ligand-free and environmentally friendly conditions. In addition, we report, also for the first time, Pd–CuO binary metal (oxide) particles displaying outstanding metal nanoparticle dispersion in the hollow silicalite-1 single crystals. The binary metal (oxide) particles show approximately 3 times higher activity than encapsulated single Pd catalyst for the above reaction.
2 Experimental
2.1 Preparation of silicalite-1 zeolite
Silicalite-1 was synthesized from the clear solution method. Typically, 15.4 mL of tetraethyl orthosilicate (TEOS) was mixed with a certain amount of TPAOH solution. The molar composition of the synthesis mixture was 1 TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.27 TPAOH:37 H2O. After being stirred for 3 h at 35 °C, the resultant solution was heated at 80 °C in order to remove the ethanol generated during the hydrolysis of TEOS and then water was added to maintain constant volume. After crystallization at 170 °C for 3 days, the product was recovered by centrifugation and dried overnight at 100 °C. Finally, the template was removed by calcination in static air at 540 °C for 6 h.
:0.27 TPAOH:37 H2O. After being stirred for 3 h at 35 °C, the resultant solution was heated at 80 °C in order to remove the ethanol generated during the hydrolysis of TEOS and then water was added to maintain constant volume. After crystallization at 170 °C for 3 days, the product was recovered by centrifugation and dried overnight at 100 °C. Finally, the template was removed by calcination in static air at 540 °C for 6 h.
2.2 Preparation of Pd@Hol S-1 and Pd–Cu@Hol S-1
Pd/S-1 was synthesized by the incipient-wetness impregnation method. In brief, the calcined silicalite-1 was impregnated with an aqueous solution of H2PdCl4, after drying overnight at 100 °C; the product was calcined in static air at 500 °C for 4 h. The Pd loading on the Pd/S-1 was 0.97 wt%, as measured by inductively coupled plasma (ICP) mass spectrometry.The as-prepared Pd/S-1 was treated with 0.3 M TPAOH (20 mL of solution per gram of zeolite) at 170 °C for 72 h, after dried overnight at 100 °C and calcined in static air at 500 °C for 4 h, the Pd@Hol S-1 was obtained. The Pd loading on the Pd@Hol S-1 was 1.3 wt%, as measured by ICP mass spectrometry.
Pd–CuO binary metals (oxide) encapsulated in the hollow S-1 were prepared by the similar method with single metal, which used H2PdCl4 and CuCl2 as Pd and Cu source, respectively, and co-impregnation method to synthesize Pd–CuO/S-1. The as-prepared Pd–CuO/S-1 was treated with 0.3 M TPAOH (20 mL of solution per gram of zeolite) at 170 °C for 72 h, after drying overnight at 100 °C and calcining in static air at 500 °C for 4 h, the Pd–CuO@Hol S-1 was obtained. The Pd and Cu loading on the Pd–CuO@Hol S-1 was 1.2 and 0.82 wt%, respectively, as measured by ICP mass spectrometry.
2.3 Characterization
Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku Smartlab diffractometer using a nickel-filtered CuKα X-ray source at a scanning rate of 0.02° over the range between 5° and 80°.Transmission electron microscopy (TEM) images were taken on a Tecnai G2 20 S-twin instrument (FEI Company) with an acceleration voltage of 200 kV. The samples for TEM analysis were prepared by dipping the carbon-coated copper grids into ethanol solutions of the samples and drying at ambient condition.
Ar isotherms were measured in a Quantachrome autosorb-iQ2 gas adsorption analyzer at 87 K. Prior to the measurement, the samples were degassed in vacuum at 300 °C for 10 h. The Brunauer–Emmett–Teller (BET) method was applied to calculate the total surface area (SBET), while the t-plot method was used to discriminate between micro- and meso-porosity. In the t-plot, the reported mesopore surface area (Smeso) consists of contributions from the outer surface of the particles as well as mesopores and macropores.
Scanning electron microscopy (SEM) images were obtained on a Hitachi S-5500 instrument with an acceleration voltage of 3 kV. Some samples were sputtered with a thin film of gold.
X-ray photoelectron spectroscopy (XPS) was conducted on an ESCALAB 250 (Thermo VG Corporation) using Mg Kα radiation (1253.6 eV, 15 kV, 10 mA, 150 W). The recorded spectra were fitted by a least square procedure to a product of Gaussian–Lorentzian functions. The concentration of each element was calculated from the area of the corresponding peak.
The elemental analysis of catalysts was carried out on a Perkin Elmer OPTIMA 2000DV ICP Optical Emission Spectrometer.
2.4 Catalytic tests
All commercially available reagents (from Acros, Aldrich, Fluka) were used without further purification. All reactions were carried out in air. NMR spectra were recorded on a Brucker AdvanceII 400 spectrometer using TMS as an internal standard (400 MHz for 1HNMR). All products were isolated by short chromatography on a silica gel (200–300 mesh) column using petroleum ether (60–90 °C). Compounds described in the literature were characterized by 1H NMR spectra and compared to reported data.A mixture of aryl bromides (0.25 mmol), phenylboronic acid (0.375 mmol), K2CO3 (0.5 mmol), catalysts (1.6 mol%), and EtOH/H2O (1 mL/1 mL) was stirred at 80 °C in air for the indicated time. The reaction mixture was added to brine (15 mL) and extracted with ethyl acetate (4 × 10 mL). The solvent was concentrated under vacuum, and the product was isolated by short column chromatography on silica gel.
3 Results and discussion
Fig. 1a shows a typical TEM image of the prepared Pd/S-1, which clearly exhibits that palladium nanoparticles are located on the crystal surface. After TPAOH treatment, a large regular hollow void was created in the interior of the silicalite-1 and the thickness of the shell was about 20 nm. The zeolite nanocubes were still single-crystals and the palladium particles with 11.1 nm in average particle size were encapsulated in the large cavity (Fig. 1b). Fig. 1c and d are images of this sample with high resolution and the zeolite lattice fringes align clearly parallel to each other, confirming the nanocubes are still single-crystals. From Fig. 1d, it can be seen that the lattice fringes are not aligned parallel to each other, confirming the Pd particle is agglomerated from smaller clusters. The lattice spaces are measured to be 0.194 and 0.225 nm, which matches the values of the d-spacing of Pd (200) and Pd (111), respectively (JCPDS: 46-1043). The energy-dispersive X-ray spectrometry (EDX) elemental analysis along the line shown in Fig. 1e shows the core rich in Pd and the shell rich in Si and O, thereby confirming that palladium is mainly located in the core of hollow silicalite-1 (Fig. 1f).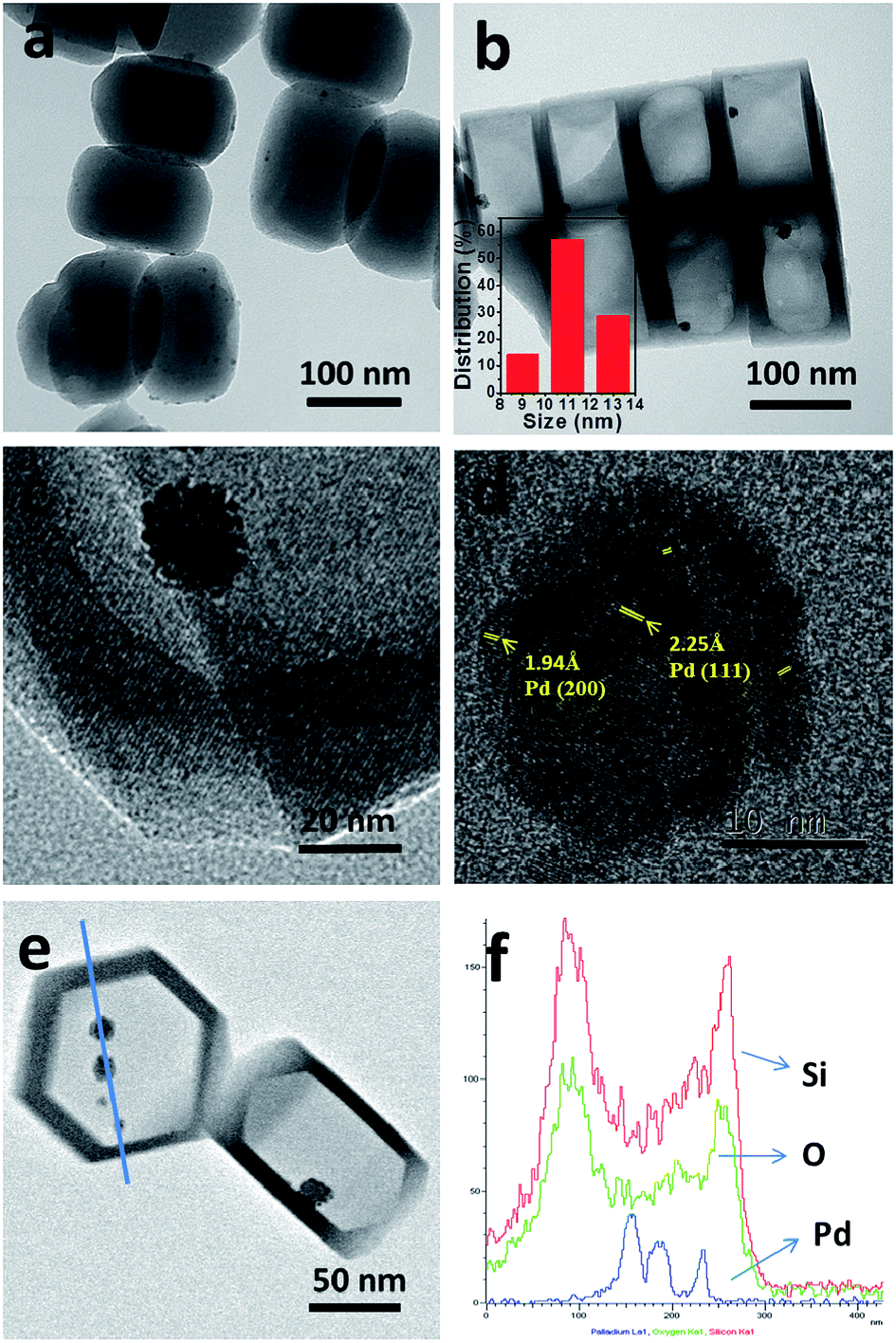 | ||
| Fig. 1 TEM images of prepared Pd/S-1 (a) and Pd@Hol S-1 (b–d); (e) STEM image of Pd@Hol S-1; and (f) EDX analysis along the line shown in (e). | ||
The Pd@Hol S-1 formation process is shown in Fig. 2. During TPAOH treatment, the silicate oligomers are leached from interior of the crystal and recrystallize at the surface of silicalite-1 crystal. In this process, the palladium particles move from surface of the crystal to the interior of cavity, just like the process of cell phagocytosis (Fig. 2 step I and step II). During the encapsulation process, the palladium nanoparticles aggregated into larger particles (Fig. 2 step III, Fig. 1b). The SEM images of Pd/S-1 and Pd@Hol S-1 show that, after alkaline treatment, the sizes of nanocubes are larger than that of the parent samples, and their shells were not damaged (Fig. 3).
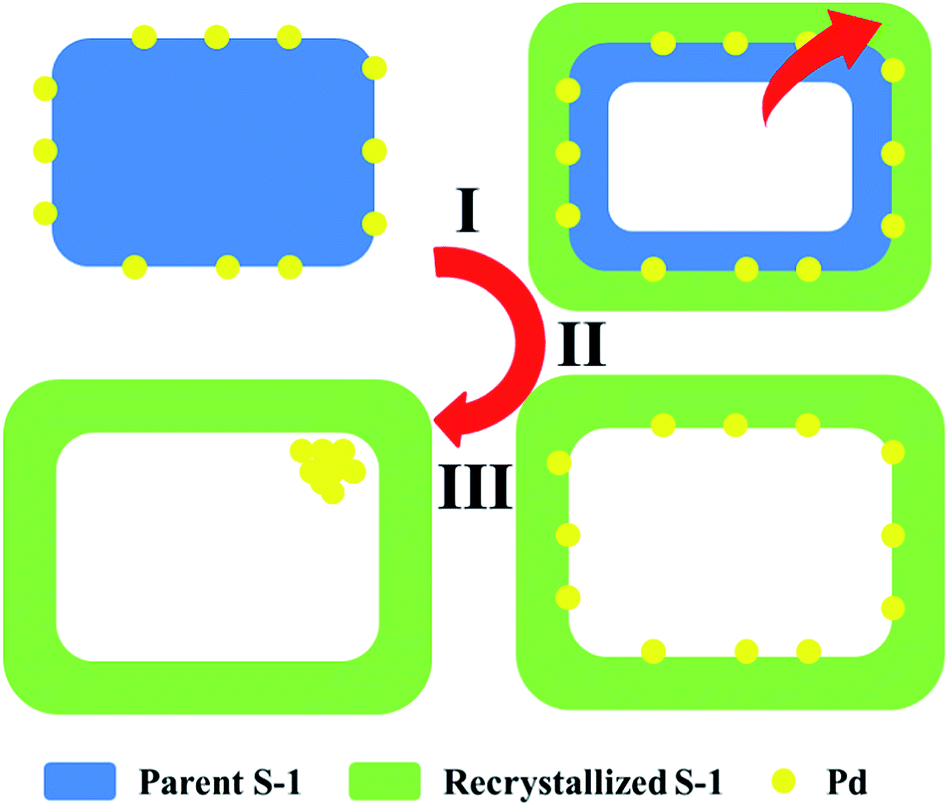 | ||
| Fig. 2 Pd@Hol S-1 formation process. | ||
 | ||
| Fig. 3 SEM images of prepared Pd/S-1 (a) and Pd@Hol S-1 (b). | ||
The Ar adsorption–desorption isotherms of Pd@Hol S-1 show the presence of a H2 hysteresis loop with an abrupt step around p/p0 = 0.45 in the desorption branch (Fig. 4 inset). This shows that during the TPAOH treatment, most of the hollow structure of the zeolite is preserved, which is consistent with the TEM and SEM results. The micropore size distributions derived from the argon adsorption isotherms indeed confirm that the original micropore size is not affected during the alkaline treatment (Fig. 4). X-ray photoelectron spectroscopy (XPS) reveals that Pd/S-1 exhibits two energy bands at 341.0 and 335.7 eV, which are values for the Pd 3d3/2 and 3d5/2 electrons of metallic Pd0, while the peaks around 342.4 and 337.1 eV correspond to Pd2+ species (Fig. 5). Compared to the Pd/S-1 sample, the Pd0 content of Pd@Hol S-1 was increased from 24.9% to 41.3%.
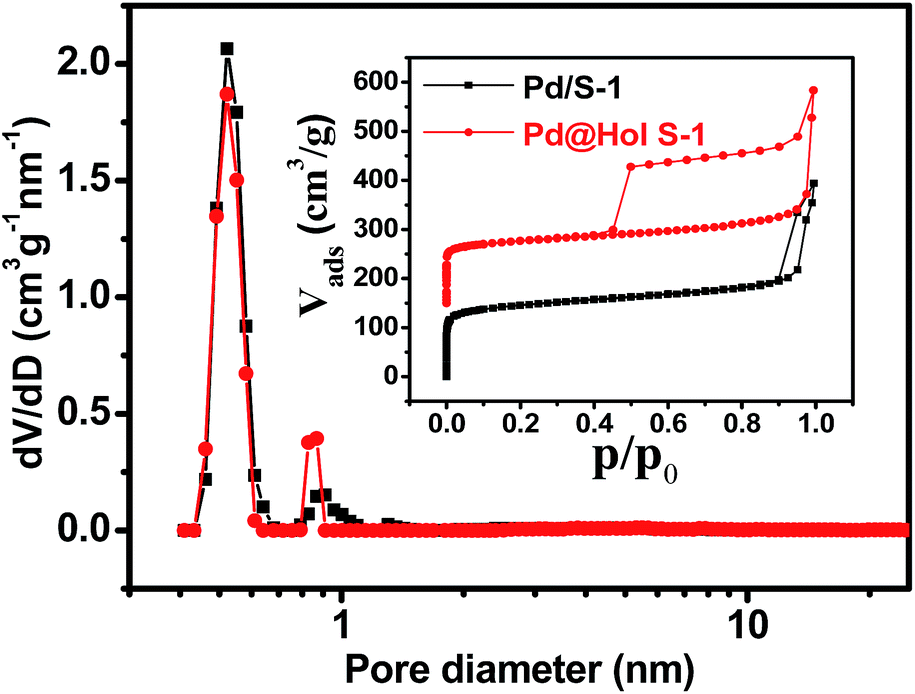 | ||
| Fig. 4 Ar adsorption and desorption isotherms at −186 °C (inset) and pore size distributions of Pd/S-1 and Pd@Hol S-1. The isotherms of Pd@Hol S-1 have been shifted upward by 150 cm3 g−1. The pore size distributions were determined by non-local density functional theory (NLDFT). | ||
 | ||
| Fig. 5 Pd 3d XPS spectra for Pd/S-1 and Pd@Hol S-1. | ||
In situ XRD and TEM were used to investigate the thermal stability of Pd@Hol S-1 (Fig. 6 and 7). The diffraction peaks were almost unchanged when the temperature rose to 800 °C, suggesting that the MFI framework structure of hollow S-1 was not destroyed and the Pd nanoparticles were not sintered during heating. Fig. 7 shows TEM images of Pd/S-1 and Pd@Hol S-1 after calcination in static air at 800 °C for 30 min. For Pd/S-1, palladium particles grow significantly compared to the sample after calcination in static air at 500 °C for 4 h (Fig. 1a). For Pd@Hol S-1, the particle sizes of palladium were almost unchanged compared to that obtained at 500 °C (Fig. 1b), because the Pd nanoparticles in the cavity hardly transport through the microporous walls of the silicalite-1 crystals.
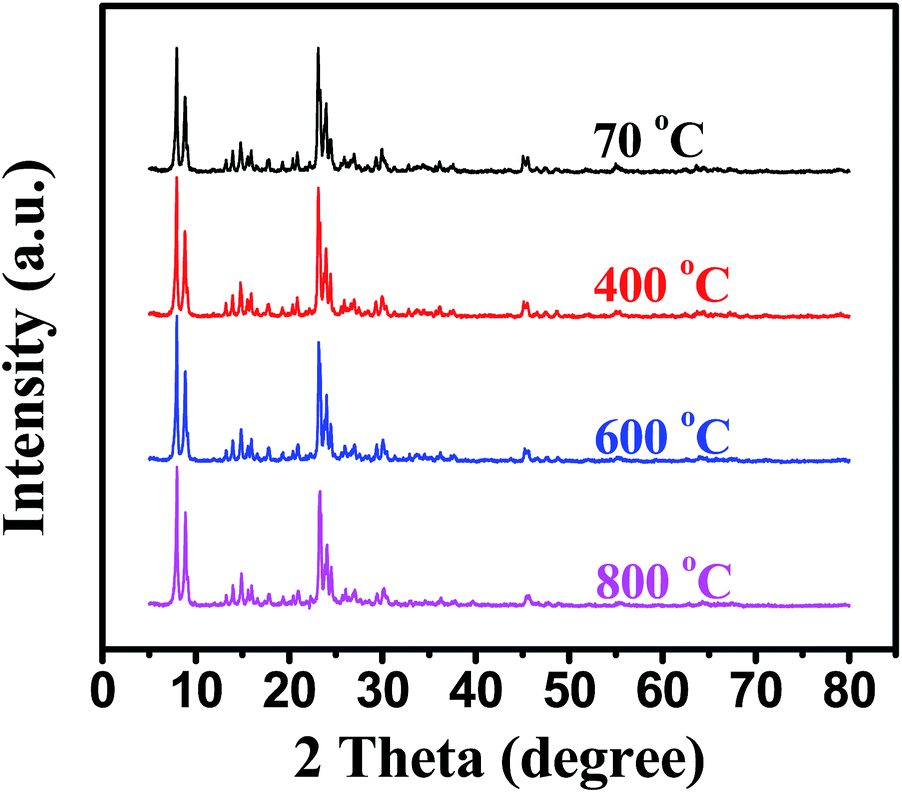 | ||
| Fig. 6 In site XRD patterns of Pd@Hol S-1. | ||
 | ||
| Fig. 7 TEM images of Pd/S-1 (a) and Pd@Hol S-1 (b) after calcinate in static air at 800 °C for 30 min. | ||
To explore the catalytic activity of Pd@Hol S-1, the Suzuki–Miyaura reactions were performed between various aryl bromides and phenylboronic acid using Pd@Hol S-1 at 80 °C in 50% aqueous ethanol. As shown in Table 1, 4-substituted aryl bromides bearing either electron-donating or electron-withdrawing groups, such as CN, NO2, CHO and OCH3, provided the corresponding products in excellent yields (Table 1, entries 1, 3–5), and negligible loss of activity is detected even after reaction of 5 runs (Fig. S2†). In addition, 3-bromonitrobenzene also exhibited high reactivity and afforded a 97% yield in 2 h (Table 1, entry 6). Interestingly, ortho-substituted aryl bromides did not undergo the Suzuki–Miyaura reaction under the same conditions (Table 1, entries 7, 11, 13 and 15). However, moderate to excellent yields were obtained for the same ortho-substituted aryl bromides when using Pd/S-1 (Table 1, entry 8), Pd(OAc)2 (Table 1, entry 10) or Pd/C (Table 1, entries 9, 12, 14, 16) as the catalyst. For example, no product was observed using Pd@Hol S-1 as the catalyst in the Suzuki–Miyaura reaction of 2-bromonitrobenzene with phenylboronic acid (Table 1 entry 7). Contrarily, 32% and 49% yields were observed in 2 h using Pd/S-1 and Pd/C, respectively (Table 1 entries 8 and 9). These results demonstrate that the Pd@Hol S-1 catalyst displays excellent substrate selectivity for the meta- and para-substituted aryl bromides for the Suzuki–Miyaura reaction.
|
||||
|---|---|---|---|---|
| Entry | Ar–Br | Product | Time (h) | Yieldb (%) |
| a Reaction conditions: aryl bromide (0.25 mmol), phenylboronic acid (0.375 mmol), Pd@Hol S-1 (1.6 mol%), K2CO3 (0.5 mmol), EtOH/H2O (1 mL/1 mL), under air.b Isolated yields.c Pd/S-1 (2 mol%).d 5% dry Pd/C (1 mol%).e Pd(OAc)2 (1 mol%).f Pd–CuO@Hol S-1 (2 mol%). | ||||
| 1 |  |
 |
1.5 | 96 |
| 2 | 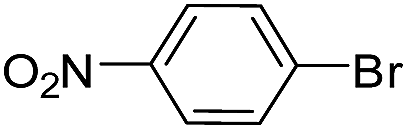 |
 |
1.0 | 96c |
| 3 |  |
 |
1.0 | 95 |
| 4 |  |
 |
2.0 | 96 |
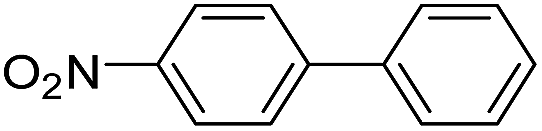
| 5 |  |
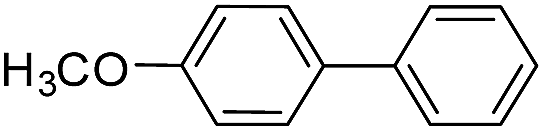 |
4.0 | 93 |
| 6 |  |
 |
2.0 | 97 |
| 7 |  |
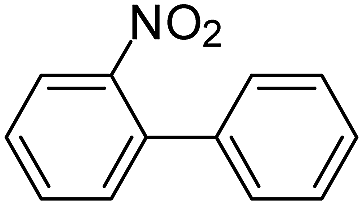 |
2.0 | 0 |
| 8 |  |
 |
2.0 | 32c |
| 9 |  |
 |
2.0 | 49d |
| 10 |  |
 |
2.0 | 93e |
| 11 |  |
 |
2.0 | 0 |
| 12 | 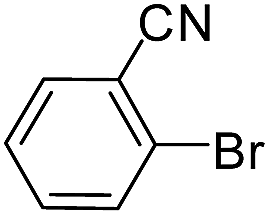 |
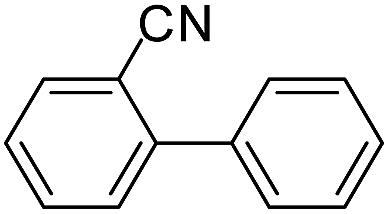 |
0.5 | 96d |
| 13 | 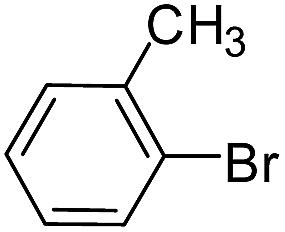 |
 |
2.0 | 0 |
| 14 | 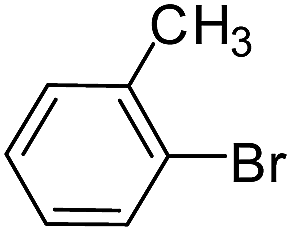 |
 |
0.5 | 96d |
| 15 |  |
 |
2.0 | 0 |
| 16 |  |
 |
0.5 | 94d |
| 17 |  |
 |
0.5 | 96f |

In addition, the Pd–CuO@Hol S-1 was used in the coupling reaction of 4-nitrobromobenzene. As expected, phenylboronic acid resulted in a 96% isolated yield in 30 min (Table 1, entry 17), which demonstrated about 3 times higher activity than Pd@Hol S-1 (Table 1, entry 1). Fig. 8 shows TEM images and metal particle size distributions of prepared Pd–CuO@Hol S-1. Two populations of metal species can be clearly seen in the hollow crystals (Fig. 8a). According to EDX analysis, the larger particle with an average of 10.7 nm (Fig. 8c) consisted mainly of palladium, and the smaller particles with an average of 2.7 nm (Fig. 8b) contained both copper and palladium. The outstanding dispersion of Pd–CuO binary metal (oxide) nanoparticles in the hollow silicalite-1 single crystals enhances the bimetallic promotion and improves the activity of the catalyst.
 | ||
| Fig. 8 TEM image (a) and metal particle size distributions (b and c) of prepared Pd–CuO@Hol S-1. | ||
4 Conclusions
The palladium-encapsulated silicalite-1 micro capsular catalyst (Pd@Hol S-1) has been successfully synthesized. Characterization results show that the zeolite nanocubes are single-crystals with very high thermal stability and the Pd particles were not sintered during heating. The Pd@Hol S-1 catalyst displays excellent substrate selectivity in the ligand-free Suzuki–Miyaura reaction of meta- and para-substituted aryl bromides. In addition, Pd–CuO binary metal (oxide) sample displayed outstanding metal nanoparticle dispersion in the hollow silicalite-1 single crystals, which resulted in approximately 3 times higher activity than encapsulated single Pd catalyst for the above reaction. The strategy using hollow zeolite crystal as a support is a more reliable method for preparing multi-metallic (oxide) catalysts with well-dispersed nanoparticles.Author contribute
C. Y. Dai and X. M. Li contributed equally.Acknowledgements
C. Liu and X. Li thank the financial support from the National Natural Science Foundation of China (21276043).Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed; (c) C. Liu and W. Yang, Chem. Commun., 2009, 41, 6267 RSC; (d) Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed.
- (a) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, John Wiley & Sons, 2012 Search PubMed; (b) A. Suzuki, J. Organomet. Chem., 2002, 653, 83 CrossRef CAS; (c) G. A. Molander, B. Canturk and L. E. Kennedy, J. Org. Chem., 2009, 74, 973 CrossRef CAS PubMed.
- (a) N. Miyaura, T. Yanagi and A. Suzuki, Synth. Commun., 1981, 11, 513 CrossRef CAS PubMed; (b) L. Wu, Z. Li, F. Zhang, Y. He and Q. Fan, Adv. Synth. Catal., 2008, 350, 846 CrossRef CAS PubMed; (c) K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 7023 CrossRef PubMed; (d) J. P. Wolfe, R. A. Singer, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1999, 121, 9550 CrossRef CAS; (e) I. D. Kostas, F. J. Andreadaki, D. Kovala-Demertzi, C. Prentjas and M. A. Demertzis, Tetrahedron Lett., 2005, 46, 1967 CrossRef CAS PubMed.
- (a) J. Shi, J. Bertoia, K. R. Czerwinski and C. Bae, Green Chem., 2009, 11, 1576 RSC; (b) P. Conelly-Espinosa and D. Morales-Morales, Inorg. Chim. Acta, 2010, 363, 1311 CrossRef CAS PubMed; (c) A. N. Marziale, S. H. Faul, T. Reiner, S. Schneider and J. Eppinger, Green Chem., 2010, 12, 35 RSC; (d) S. M. Islam, P. Mondal, A. S. Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067 CrossRef CAS PubMed.
- (a) W. Han, C. Liu and Z. L. Jin, Org. Lett., 2007, 9, 2005 Search PubMed; (b) W. Han, C. Liu and Z. L. Jin, Adv. Synth. Catal., 2008, 350, 501 CrossRef CAS PubMed; (c) P. Li, P. P. Huang, F. F. Wei, Y. B. Sun, C. Y. Caoand and W. G. Song, J. Mater. Chem. A, 2014, 2, 12739 RSC.
- M. Chtchigrovsky, Y. Lin, K. Ouchaou, M. Chaumontet, M. Robitzer, F. Quignard and F. Taran, Chem. Mater., 2012, 24, 1505 CrossRef CAS.
- B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054 CrossRef CAS PubMed.
- J. Liu, H. Q. Yang, F. Kleitz, Z. G. Chen, T. Yang, E. Strounina, G. Q. Lu and S. Z. Qiao, Adv. Funct. Mater., 2012, 22, 591 CrossRef CAS PubMed.
- (a) Z. Chen, Z. M. Cui, F. Niu, L. Jiang and W. G. Song, Chem. Commun., 2010, 46, 6524 RSC; (b) J. Goebl and Y. Yin, ChemCatChem, 2013, 5, 1287 CrossRef CAS PubMed.
- Z. Guan, J. Hu, Y. Gu, H. Zhang, G. Li and T. Li, Green Chem., 2012, 14, 1964 RSC.
- (a) J. Shi, X. Li, Q. R. Wang, Y. H. Zhang and Y. Tang, J. Catal., 2012, 291, 87 CrossRef CAS PubMed; (b) N. Ren, Y. H. Yang, Y. H. Zhang, Q. R. Wang and Y. Tang, J. Catal., 2007, 246, 215 CrossRef CAS PubMed; (c) S. W. Li, A. Tuel, D. Laprune, F. Meunier and D. Farrusseng, Chem. Mater., 2015, 27, 276 CrossRef CAS; (d) C. Y. Dai, A. F. Zhang, L. L. Li, K. K. Hou, F. S. Ding, J. Li, D. Y. Mu, C. S. Song, M. Liu and X. W. Guo, Chem. Mater., 2013, 25, 4197 CrossRef CAS; (e) C. Y. Dai, A. F. Zhang, J. J. Li, K. K. Hou, M. Liu, C. S. Song and X. W. Guo, Chem. Commun., 2014, 50, 4846 RSC; (f) D. Fodor, L. Pacosova, F. Krumeich and J. A. van Bokhoven, Chem. Commun., 2014, 50, 76 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR data of products. See DOI: 10.1039/c5ra05952f |
| This journal is © The Royal Society of Chemistry 2015 |