
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled synthesis of copper sulfide-based catalysts for electrochemical reduction of CO2 to formic acid and beyond: a review
Anirban Mukherjee
 a,
Maryam Abdinejad
*b,
Susanta Sinha Mahapatra
c and
Bidhan Chandra Ruidas
*a
a,
Maryam Abdinejad
*b,
Susanta Sinha Mahapatra
c and
Bidhan Chandra Ruidas
*a
aDepartment of Chemical Engineering, Birla Institute of Technology, Mesra, Ranchi 835215, India. E-mail: bidhanruidas@gmail.com
bDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
cDepartment of Chemistry, Birla Institute of Technology, Mesra, Ranchi 835215, India
First published on 3rd September 2024
Abstract
Converting carbon dioxide (CO2) into value-added chemicals is considered as a promising strategy to mitigate climate change. Among the various CO2 reduction techniques, electrochemical CO2 reduction (ECO2R) using renewable energy sources holds significant potential. Consequently, the design and development of electrocatalysts capable of offering both high performance and cost-effectiveness hold the potential to expedite reaction kinetics and facilitate widespread industrial adoption. In recent years, abundant copper sulfide (Cu/S)-based nanomaterials among various metal–chalcogenides have attracted extensive research interest due to their semiconductivity and low toxicity, enabling them to be used in a wide range of applications in the ECO2R field. This review highlights the progress in engineered Cu/S-based nanomaterials for ECO2R reactions and elaborates on the correlations between engineering strategies, catalytic activity, and reaction pathways. This paper also summarises the controllable synthesis methods for fabricating various state-of-the-art Cu/S-based structures and outlines their possible implementation as electrocatalysts for CO2 reduction. Finally, challenges and prospects are presented for the future development and practical applications of Cu/S-based catalysts for ECO2R to value-added chemicals.
 Anirban Mukherjee | Mr Anirban Mukherjee completed his Master of Engineering at Jadavpur University, Kolkata. Currently, he is pursuing his PhD as an institute research fellow at the Birla Institute of Technology, Mesra (BIT Mesra), Ranchi 835215, India. His research mainly focuses on synthesizing and characterizing high-performance electrocatalysts for the electrochemical reduction of CO2 into value-added products. |
 Maryam Abdinejad | Dr Maryam Abdinejad completed her PhD at the University of Toronto, followed by a postdoc at the Delft University of Technology. In 2022 she continued her research as an associate postdoc researcher at the Massachusetts University of Technology (MIT). Her research mainly focuses on the rational design and development of novel high-performance homogeneous and heterogeneous catalysts for the capture and electrochemical reduction of CO2 to value-added materials. |
 Susanta Sinha Mahapatra | Dr Susanta Sinha Mahapatra is an Assistant professor at the Chemistry Department at the Birla Institute of Technology, Mesra, Ranchi, India. He received his MSc in Chemistry and MTech in Energy Science & Technology from Visva-Bharati, Santiniketan, India, and Jadavpur University, Kolkata, India, respectively, and his PhD from Indian Institute Engineering Science & Technology, Shibpur, India. His research mainly focuses on direct alcohol fuel cells, electrochemical supercapacitors, electrochemical CO2 reduction and hydrogen evolution reactions. |
 Bidhan Chandra Ruidas | Dr Bidhan Chandra Ruidas completed his PhD in Chemical Engineering at the Indian Institute of Technology Kharagpur, India. Currently, he is working as an Assistant Professor in the Department of Chemical Engineering at the Birla Institute of Technology, Mesra Ranchi, India. His research mainly focuses on enhanced oil recovery and electrochemical reduction of CO2 to value-added chemicals. |
1. Introduction
Since industrialization, the utilization of non-renewable energy sources, which encompass fossil fuels, i.e., coal, crude oil, and natural gas, has tremendously increased.1 Fossil fuels, as non-renewable energy sources, have two disadvantages: (1) limited supplies cannot meet increasing demands for energy, resulting in a severe energy crisis and (2) the consumption of fossil fuels for energy generation releases a substantial amount of CO2 into the earth's atmosphere, contributing to global ecological issues such as global warming, global sea level rise, land degradation, and many more.1–3 To achieve a sustainable future, reducing CO2 concentrations in the atmosphere is crucial.4 In this context, the scientific community has made numerous efforts to achieve this goal by using the CO2 capture, utilization, and storage (CCUS) approach, as depicted in Fig. 1.5,6 The conversion of CO2 into C1 and highly dense C2 products through chemical routes has been considered a promising technology for reutilizing CO2.7,8 It can be achieved by several methods, such as photochemical,9 electrochemical,10,11 photo-electrochemical,12 and biochemical methods.13 Among these methods, electrochemical CO2 reduction (ECO2R) has gained a lot of attention owing to its (a) controllable process parameters (i.e., potential and temperature), (b) feasibility with the reaction environment (e.g., organic and aqueous electrolytes), and (c) ability to scale up.14,15 Additionally, ECO2R can be conducted using renewable energy resources such as solar and wind as power sources, allowing for renewable energy storage and redistribution.16 Therefore, ECO2R has emerged as a significant research area with industrial prospects, and, in the recent past, much progress has been made in this prospering domain.17,18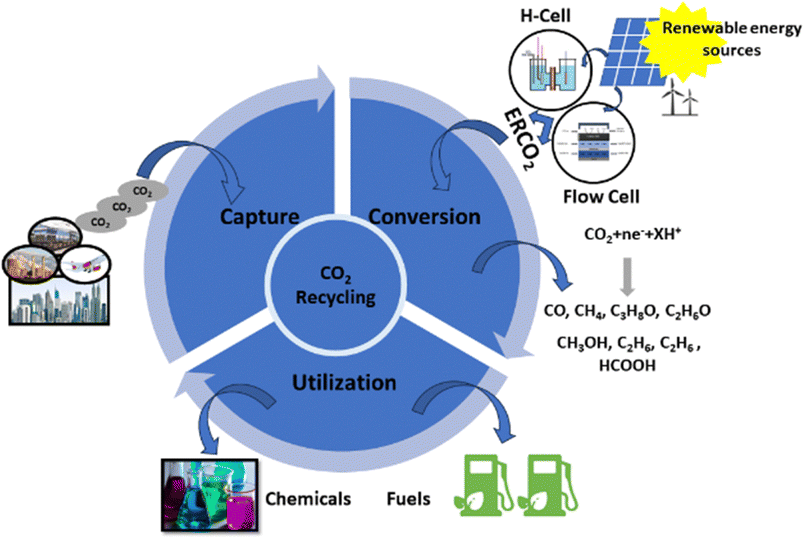 | ||
| Fig. 1 A graphical illustration of CO2 capture, utilization, and storage (CCUS). | ||
As reported in the scholarly literature and with the progress of research outcomes, the ECO2R catalysts are classified into metal and metal-free catalysts based on their design, synthesis, and product selectivity.19 Nonmetal electrocatalysts are mostly made of carbon-based nanoparticles.20 On the other hand, metal electrocatalysts include bare metals and their hybrid complexes, such as metal alloys,19,21,22 metal oxides,23,24 metal sulfides,25 and metal complexes.26,27 The activity and selectivity for the ECO2R vary with different metal catalysts owing to variations in the activity and adsorption strength of the intermediates.28 Thus, metal catalysts are further classified into four categories based on their selectivity for final products: (a) Cd, Hg, Tl, Sn, In, Bi, and Pb produce the HCOO− intermediate, resulting in formate as their primary product;29–34 (b) Au, Ag, Zn, and Pd produce CO as their main reduction product because of the weak CO adsorption capacity;35–42 (c) Ni, Fe, Si, V, and Pt produce H2 owing to the low HER potential;43–47 and (d) Cu produces up to sixteen carbon-based nanomaterials, comprising several highly energy-dense multi-carbon products, under different conditions.48,49 Hori et al.50 conducted ECO2R in 0.5 M KHCO3 at 5 mA cm−2 for an hour on various polycrystalline metal electrodes. The outcomes showed that each metal electrode needs a significant overpotential to reach a particular current density (in this case, 5 mA cm−2). Au requires the lowest overpotential (−0.6 VRHE) to obtain CO (87%), but Pb needs the highest overpotential (−1.1 VRHE) to produce formate (97%).50
Although significant progress has been made, some challenges still exist with the industrial aspects of ECO2R,51,52 such as (1) high overpotential for ECO2R as CO2 is a linear molecule that is thermodynamically stable and chemically inert;53 (2) sluggish kinetics of CO2 electroreduction because the complex proton-coupled electron transfer (PCET) steps and kinetics of electron transfer were slowed down by the insolubility of CO2 in the aqueous medium;54 (3) low exchange current densities; a majority of CO2 electrocatalysts reported thus far operate at less than 20 mA cm−2; however, this is significantly less than those of the commercial electrolyzers, which generally operate at over 70% efficiency at current densities exceeding 200 mA cm−2;55 (4) unsatisfactory selectivity, suggesting expensive separation procedures;56 (5) catalyst deactivation in less than 100 h, limiting practical application and industrialization of technology;57 and (6) competitive hydrogen evolution reaction (HER); the HER surpasses the ECO2R due to its favourable energy conditions at higher overpotentials, leading to a higher FEH2 (faradaic efficiency) than FE for other ECO2R products.58 Recently, much research has concentrated on designing and synthesizing innovative, cost-effective, and robust electrocatalysts that can counter these bottlenecks and reduce CO2 at high rates with minimal overpotential.57,58
As pointed out in an earlier paragraph, Cu can electrochemically reduce CO2 to highly energy-dense C2+ products, which has gained massive attention.59 However, the critical path of the C–C coupling process required for the C2+ product formation introduces a high activation energy barrier, resulting in low activity and poor selectivity.60,61 In this context, synthesizing Cu-based electrocatalysts is crucial for overcoming the energy barrier in ECO2R. Researchers showed that the CO2 electroreduction performance (i.e., activity and selectivity) of Cu catalysts is affected by multiple factors. For example, in their work, Hori et al.62,63 showed that Cu with several crystal facets could yield various ECO2R products. The Cu(111) surface produces methane as a primary product, while C2 products such as ethylene and ethanol are formed over Cu(100) surfaces. Reske and colleagues64 also showed that faradaic efficiency and current density of Cu nanoparticles were significant compared to bulk Cu, while Chen et al.65 demonstrated that the single-atom Cu showed better ECO2R performance than other reported Cu-based materials, i.e., bulk metal Cu.
Moreover, bimetallic Cu-based electrocatalysts have shown promise in enhancing copper's selectivity and overall catalytic performance. For example, Burdyny's group58 could electrochemically reduce CO2 to formate as the primary product using bimetallic Cu–Pd with a faradaic efficiency of 93% and a current density of 150 mA cm−2 at a cell potential of −2.9 VRHE using a zero-gap flow cell, also known as the membrane electrode assembly cell. They successfully demonstrated how the engineering design of an electrochemical cell, coupled with catalyst structure, achieves the highest overall reported energy efficiency (EE) for formate production at 47%. These results show the benefit of using non-post-transition metals as primary catalysts for formate production. Another study shows that catalyst morphology is also essential: Chorkendorff et al.66 found that metallic Cu with varying surface roughness exhibits distinct selective ECO2R product formation. They discovered that the Cu nanoparticle-coated electrocatalysts have improved hydrocarbon selectivity compared to non-coated electrocatalysts. Motivated by the aforementioned work, several research groups synthesized different morphologies, such as nanowires,67,68 films,69 microcubes,70,71 core–shell,72,73 and Cu-based bimetallic58 catalysts and studied their catalytic activity towards ECO2R. They showed that the catalytic activity of Cu-based catalysts towards ECO2R can be remarkably enhanced by tailoring the catalysts' structure and morphology.
In recent years, Cu/S-based nanomaterials have gained significant attention as electrocatalysts due to their p-type semiconducting, earth-abundance and nontoxic characteristics. Various phases of copper sulfide are reported in the literature, namely chalcocite (Cu2S), djurleite (Cu1.97S), digenite (Cu1.80S), anilite (Cu1.75S), geerite (Cu1.60S), spionkopite (Cu1.40S), yarrowite (Cu1.12S) and covellite (Cu1.00S) as copper-rich systems, while villamanite (CuS2) as a sulfur-rich system and covellite (CuS) as a 1/1 system, usually denoted as Cu2−xS having minimal values of x.74–77 Based on the packing of sulfur atoms in the lattice, the aforementioned crystal structures have been grouped into three categories, as illustrated in Fig. 2, specifically cubic close packing (anilite and digenite), close hexagonal packing (djurleite and chalcocite), and a combination of close hexagonal packing and covalent bonding of the sulfur atoms (covelline).77 But for the remaining forms, i.e., yarrowite, spionkopite, and geerite, crystal structures remain unknown. It is to be noted that copper sulfides' electrical conductivity depends on their phases, decreasing from copper-deficient to copper-rich.78,79 For example, it has been observed that at 1.63 K, the naturally occurring covellite phase of CuS exhibits exceptional electrical conductivity.80 Therefore, owing to their unique versatile properties, copper sulfides are fascinating nanomaterials for various applications, i.e., optoelectronic devices,81 photocatalysis,82 photovoltaic cells,83 sensors,84 battery electrodes,85 and biomedical applications.86 Thus, numerous studies have focused on the engineering strategies of these materials for modifying their properties, including the electronic structure modulation for ECO2R. Notably, developing ECO2R catalytic systems that can overcome bottlenecks is becoming an essential topic with the increasing use of this electrocatalytic technology. According to the reported literature, a variety of physical87 and chemical75 methods, such as hydrothermal and solvothermal,88,89 ball-milling,90 electrodeposition,91 microwave irradiation,92 thermolysis,93 and template-assisted94,95 approaches, have been widely used for constructing different nano-dimensional (i.e., zero-dimensional, one-dimensional, two-dimensional, and three-dimensional) Cu/S-based nanomaterials. So far, several synthetic processes have yielded various shapes of Cu/S-based nanostructures, such as nanoparticles, nanoplates, hollow spheres, nanorods, nanowires, nanotubes, nanosheets, etc. Therefore, the significant connections within several nanomaterials and their distinct properties have prompted research into the controlled fabrication of valuable electrocatalysts with altered nanostructures.
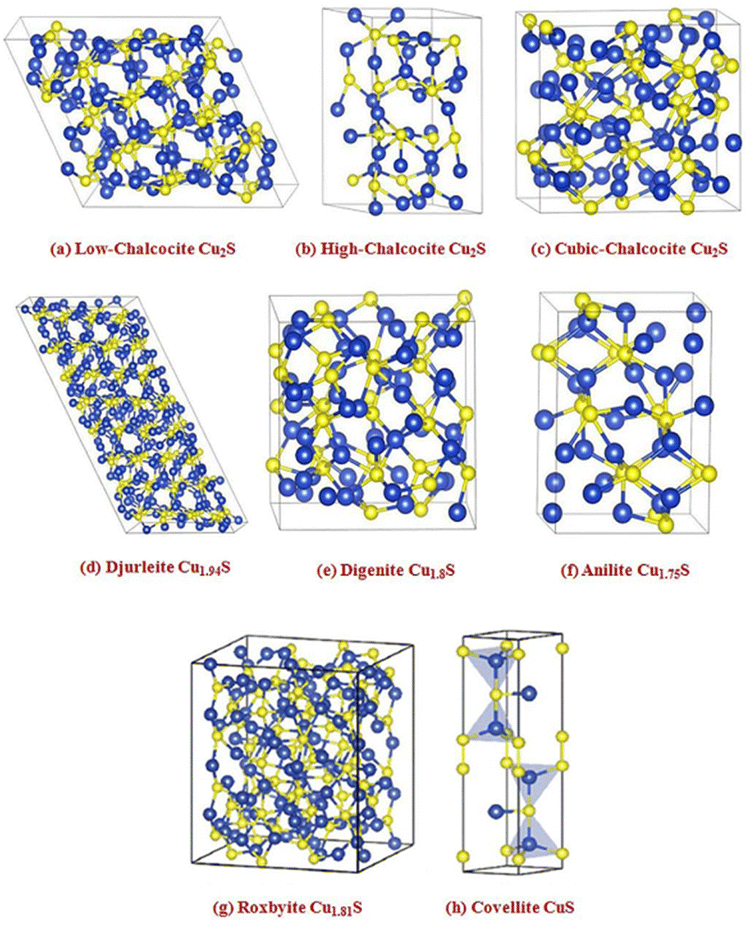 | ||
| Fig. 2 Crystal structures of some representative copper sulfide phases. Blue spheres – copper atoms and yellow spheres – sulfur atoms, surrounded by black lines showing each unit cell boundary ((a)–(f) reproduced with permission from ref. 95, copyright American Institute of Physics 2012 and (g) and (h) reproduced with permission from ref. 77, copyright RSC 2019). | ||
Until recently, many outstanding review articles on Cu/S-based nanomaterials for several energy applications have been published,74,79,96 including the preparation, characterization methods, and the effects of modification strategies on catalytic activity. However, an in-depth and systematic investigation of synthesis strategies of Cu/S-based electrocatalysts and electron modulation effects on ECO2R is lacking and is desired. This review summarizes the most recent advancements in Cu/S-based catalysts for ECO2R reactions and outlines the connection between catalytic efficiency and several engineering approaches. The primary section discusses various modification strategies for Cu/S-based nanomaterials. After this, the effects of these strategies on electron modulation will be summarized. This fundamental understanding can help with the design of high-efficiency ECO2R catalysts. Finally, this article highlights the research challenges and future prospects of Cu/S-based nanomaterials for ECO2R.
2. Characteristics for improving the ECO2R performance of Cu/S-based catalysts
This section discusses the essential features of Cu/S-based catalysts to understand their fundamental characteristics, which are mentioned below in each subsection.2.1 Exposed facets for improving electrocatalytic performance
As reported by many literature studies, tuning the exposed facets of nano-electrocatalysts may alter atomic rearrangements, reaction intermediate affinity, and surface energy, influencing the electrochemical activity.97,98 Inspired by this, a research group presented a simple and effective technique for limiting product distribution using sulfur-modified Cu2O electrocatalysts.99 A wet chemical technique was used to synthesize distinct morphologies of Cu2O effectively with varied exposed facets (Fig. 3a). Surprisingly, they observed that the faradaic efficiency of formate on sulfur-modified Cu2O electrocatalysts was significantly sensitive to Cu2O crystal facets, as shown in Fig. 3b–i, with selectivity in the order of Cu2O(100) > Cu2O(100)/(111) > Cu2O(111). Among all the sulfur-modified Cu2O electrocatalysts prepared, the optimized S3-Cu2O-70 demonstrated a FE of 90% at −0.9 VRHE and an extended stability of more than 80 h in an H-type cell. Furthermore, a flow-associated cell system achieved a jformate of 260 ± 16 mA cm−2, outperforming most formate-producing Cu-based electrocatalysts. Sulfur can improve water activation for synthesizing unique H2 species and reduce the activation energy of *OCHO intermediate formation on the surface of sulfur-modified Cu2O, boosting formate selectivity in ECO2R, according to experimental data and density functional theory (DFT) calculations. Similarly, He et al.100 investigated the role of rich high-index facets of polycrystalline Cu (Cu-s) nanoparticles successfully derived from Cu2−xS nanocrystals, as illustrated in Fig. 3j. They observed that the formation of high-index facets during surface reconstruction is beneficial for providing surface active sites for C–C coupling, thus boosting C2H4 generation. The Cu–S nanocatalysts exhibited high catalytic performance with an FE of 68.6% (a jC2H4 of 40.8 mA cm−2) for C2H4 because of vicinal facet formation during surface engineering. Furthermore, in situ studies demonstrated that Cu–S electrocatalysts use the *COCHO intermediate route for producing C2H4 via ECO2R. Similarly, Dou et al.101 investigated the facet-dependent selectivity and activity of CuS nanosheet arrays on brass mesh for ECO2R prepared via a facile and green chemical bath deposition approach (Fig. 3k). Meanwhile, connecting CuS with BM increased overall performance (j = 75 mA cm−2 at −0.7 VRHE and FEHCOO− = 67.8 ± 1%) for ECO2R. Instead, PTFE-coated CuS/BM achieved more enhanced CO2 conversion to generate HCOOH/HCOO− (FE = 70.2 ± 1% at 0.7 VRHE) than CuS/BM. Their investigations reveal that the reconstruction of CuS/BM resulted in a uniform nanowire framework with abundant active surfaces during the ECO2R, considerably increasing catalytic reactivity. They integrated DFT research with experimental findings. The work attributes the high selectivity for HCOO− production to the reconstructed formation of the Cu(111)/CuS(102) facets throughout the electrolysis process. According to the theoretical investigation, S under the CuO layer reduces the binding energies of HCOO* and *COOH on Cu(111)/CuS(102) compared to the Cu(111) plane, allowing the development of HCOOH or HCOO*.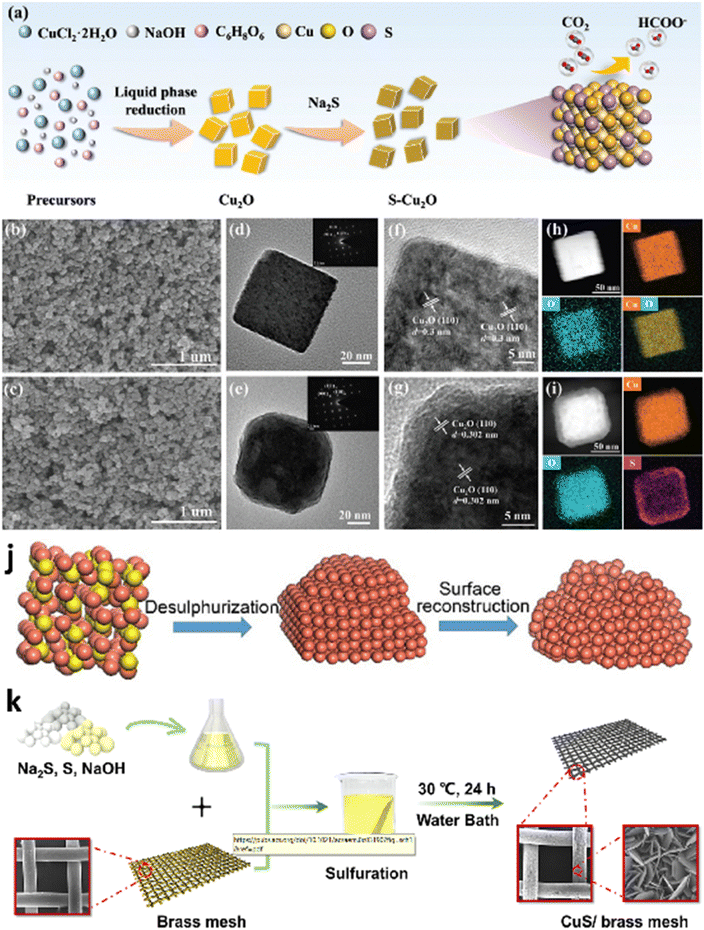 | ||
| Fig. 3 Synthesis and morphological characterization. (a) Schematic illustration of the electrocatalyst preparation process. SEM images of (b) S0-Cu2O-70 and (c) S3-Cu2O-70. TEM images of (d) S0-Cu2O-70 and (e) S3-Cu2O-70 (the inset shows the selected area electron diffraction (SAED) patterns). HRTEM images of (f) S0-Cu2O-70 and (g) S3-Cu2O-70. The corresponding elemental mappings of (h) S0-Cu2O-70 and (i) S3-Cu2O-70 (reproduced with permission from ref. 99, copyright Wiley-VCH GmbH 2023). (j) Diagrammatic representation of the conversion from Cu2−xS to Cu-s (reproduced with permission from ref. 100, copyright Springer Nature 2021). (k) Diagrammatic representation of the CuS NS arrays/BM synthesis method (reproduced with permission from ref. 101, copyright ACS 2021). | ||
2.2 Edge engineering for improving electrocatalytic performance
Recent research has established that the exposed edges of transition metal chalcogenides are more catalytically active than planar surfaces in electrochemical reactions.102,103 The more exposed edges on the electrode/electrolyte interfaces can improve the electrochemical performance owing to more active sites. Owing to the sufficiently exposed-edge planes, the hierarchical hollow CuS microcubes (MCs) exhibited remarkable electrocatalytic properties for CO2 reduction with a FECO of 32.7% at a lower onset potential of −0.2 VRHE. Shao et al.104 reported a novel engineered surface and morphology-enhanced exposed edge sites, offering significantly higher electrocatalytic activities and selectivity toward ECO2R. They demonstrated CO2 electro-reduction with lower overpotential for hollow CuS microcubes (MCs). According to the morphological characterization, it has been shown that as-synthesized h-CuS MCs lead to a higher density of defective edge exposed sites, essential for enhanced conversion activity and selectivity.2.3 Porosity effect for improving electrocatalytic performance
Owing to their high specific surface areas, nanoporous structures significantly accelerate surface reactions and facilitate mass transfer by enhancing contact between the electrolyte and the active site.105 The adsorption and transformation of intermediates, the release and diffusion of gas products, and the lowering of electron transfer resistance are all significantly more accessible by the active sites of the nanoporous structures.106,107 Inspired by this, Li et al.108 designed and synthesized hollow-ordered porous copper sulfide cuboctahedra (HOP CuS-CO) with regulated shell thicknesses, as shown in Fig. 4a–h, to selectively produce formate from ECO2R. The hollow shells of HOP CuS-CO possess uniformly distributed and interconnected pores. With these advantages and benefits of porous cages, the HOP CuS-CO catalyst exhibited an exceptional FEformate of 70.3% with stability of up to 26 h at a potential of −1.1 VRHE. In situ Raman spectroscopy studies showed that the HCOO* intermediate adsorption energy is favourable on the surfaces of HOP CuS-CO by a spatial confinement effect, resulting in highly effective ECO2R for formate generation. The above studies provided insights into designing novel morphologies for more outstanding formate production using ECO2R. Recently, Yabuki et al.109 employed the thermal breakdown of a sulfur and copper–amine complex ink to create copper sulfide film electrodes. Furthermore, the XRD study confirmed that Cu1.8S and CuS nanoparticles were present in the film. The copper sulfide film possessed variations in the surface area caused by the film's micropores, resulting in ECO2R to CO, CH4, and C2H4, with a more significant percentage of C2H4 (C2 product) than a copper electrode.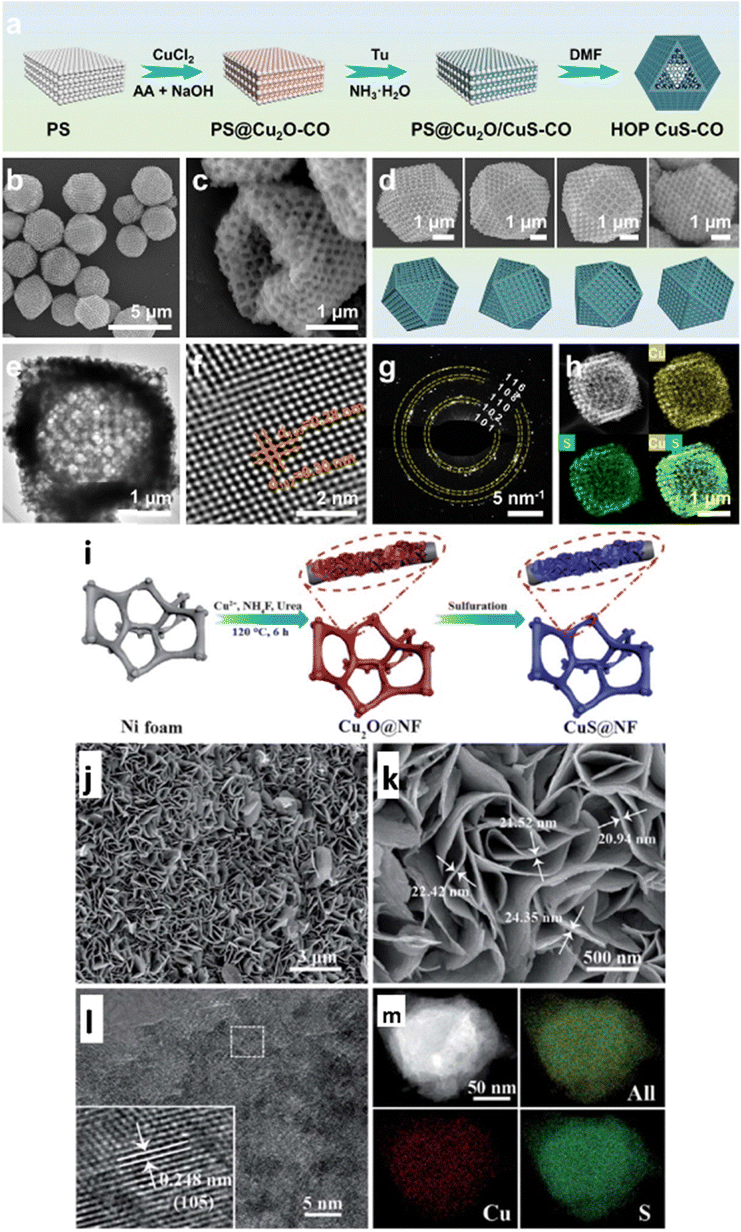 | ||
| Fig. 4 (a) Schematic illustration of synthesis of HOP CuS-CO. (b) SEM image of HOP CuS-CO. (c) SEM images of one individual HOP CuS-CO crystal projected from four different directions. (d) SEM image of partially broken HOP CuS-CO particles. (e) TEM and (f) HRTEM images and (g) SAED patterns of HOP CuS-CO. (h) HAADF-STEM image and the corresponding elemental maps of Cu (yellow) and S (green) (reproduced with permission from ref. 108, copyright Elsevier 2024). (i) Schematic illustration of synthesis of CuS@NF samples. (j and k) SEM images of CuS@NF samples with lower and higher magnification. (l) HR-TEM and (m) HAADF STEM images and corresponding EDS elemental mapping of CuS@NF samples. (reproduced with permission from ref. 110, copyright RSC 2017). | ||
Zhu et al.111 synthesized highly porous Cu2S-decorated copper foam (Cu-foam), an active CO2 reduction electrocatalyst, in an H-cell system. They used anodization followed by a heat treatment process to deposit Cu2S nanoarrays on Cu foam. Anodization was placed in the first phase in an electrocatalytic cell, employing platinum foil as the cathode and Cu-foam as the anode in an aqueous Na2S solution. After the anodization, the anodized Cu-foam was washed multiple times with distilled water, followed by thermal treatment. The 3D-shaped Cu2S/Cu-foam electrode produces much more HCOOH (FEco = 85% with j = 5.3 mA cm−2 at −2.0 VAg/AgCl) than the Cu-foam (FEco = 38.9% at −1.8 VAg/AgCl). Using a simple hydrothermal procedure, Zhao et al.110 efficiently fabricated economical, robust and highly porous CuS nanosheet arrays successfully decorated on a porous nickel foam support, as illustrated in Fig. 4i, for ECO2R activity. The CuS nanosheet is highly dense and is evenly dispersed over the highly porous Ni foam structure, forming a 3D organized foam CuS/NF, as shown in Fig. 4j–m. The thicknesses of the highly porous CuS/NF framework range from 20 to 25 nm. Their findings revealed that S concentration in the electrodes facilitates CO2 adsorption and speeds up the rate-limiting step by converting CO2 to CO2˙− intermediates. Next, the CO2˙− intermediate produces CH4 by PCET reaction. Thus, CuS@NF achieved an extraordinary faradaic efficiency of 73.5% at −1.1 VRHE for CH4 formation and was stable up to 60 h.
3. Emerging regulations for improving the ECO2R activity
3.1 Phase effect for improving electrocatalytic performance
This section discusses the ECO2R activity of catalysts, which is determined by the final stage of the active metal's conversion, not the initial phase.112–114 Notably, Cu/S-based materials typically act as pre-catalysts for ECO2R due to their conversion to oxide form during electrolysis. Due to their high oxidation potential, Cu/S-based materials could be almost totally/partially transformed into their oxide species.115,116 For example, Chen et al.117 observed that during the ECO2R, copper sulfide nanoflowers (Fig. 5a and b) undergo restructuring to metallic Cu (Fig. 5c), and S2− ions are released into the electrolyte and adsorbed on the surface of Cu catalysts. This phenomenon suppressed the formation of other hydrocarbon products, resulting in high selectivity towards HCOOH during ECO2R. Copper sulfide pre-catalysts with high S content may absorb more S2− ions on the Cu surface, resulting in higher FEHCOOH. To assess this hypothesis, they used copper foil as the electrode and added varying amounts of K2S to the KHCO3 electrolyte. Later, they discovered that the FEHCOOH of copper gradually increased with increasing K2S concentrations. In this context, Phillips et al.118 used in situ electro-reduction to investigate selective formate production over copper sulfide-derived copper surfaces (S-derived copper) (Fig. 5d–i). As evidenced by surface-enhanced infrared absorption (SEIRA) spectroscopy, the aforementioned electrocatalyst reduces H2 and CO formation while enhancing formate selectivity. The authors interpreted this increase in formate selectivity by describing a plausible reaction mechanism in which the active sites are occupied by COads, preventing the adsorbed hydrogen molecules (Hads) from combining and producing an H2 molecule. As a result, Hads could only generate H2 in solution by making bonds with protons via PCET. Theoretical simulations suggest that Hads might create HCOOH by reacting with a solution-phased-CO2 molecule rather than an H+-containing solution. As a result, Hads molecules adsorbed on S-derived Cu surfaces and generated HCOOH by interacting with solution-phase CO2 via PCET. | ||
| Fig. 5 (a) SEM image and (b) HR-TEM image of CuS nanoflowers before ECO2R; (c) SEM image of CuS nanoflowers after ECO2R (reproduced with permission from ref. 117, copyright Elsevier 2024). SEM image of catalyst surfaces (d)–(f) before and (g)–(i) after ECO2R. (d) and (g) Copper foil, (e) and (h) copper sulfide (CuS) electrodeposited for 30 min (CuS-1), and (f) and (i) CuS electrodeposited for 2 h (CuS-2). The CuS samples convert to SD-Cu after ECO2R (top scale bars – 250 nm and bottom scale bars – 10 μm for each panel, reproduced with permission from ref. 118, copyright ACS 2018). | ||
In a different work, through a straightforward two-step coupling procedure via a hydrothermal method followed by pyrolysis, as illustrated in Fig. 6a, Zhang et al.119 created a Cu1.81S catalyst supported by a multi-walled carbon nanotube (MWCNT). Due to the highly active sites of the uniformly dispersed Cu1.81S particles and the effective electron transport and active sites provided by the MWCNT, the Cu1.81S@MWCNT-600 composite catalyst (Fig. 6b–e) was able to achieve superior ECO2R performance with 30 h stability during continuous operation. Later, they reported that Cu1.81S@MWCNT-600 (Cu1.81S@MWCNT-600-OD) with oxide modification showed improved catalytic activity and had a high FEFormate of 82%. According to the authors, copper oxide, which changed the phase into a needle-shaped structure during ECO2R, provided more active sites and improved electrocatalytic activity (Fig. 6f–i). In another work, Oversteeg et al.120 investigated the role of phase engineering via Cu2−xs derived copper sulfide-supported carbon (CuS/C and Cu2S/C) (Fig. 6j, m and n) ECO2R catalysts synthesized using the liquid phase sulfidation of CuO/C nanoparticles (Fig. 6l and o). All the prepared phases are confirmed by XRD analysis, as shown in Fig. 6k. According to the electrochemical and in situ X-ray absorption (XAS) spectroscopy investigations, the metallic Cu reduction occurs in CuS@C and Cu2S/C nanoparticles during electrochemical CO2 reduction (Fig. 6p and q). Later, their observation revealed that CuS/C- and Cu2S/C-derived catalysts had higher selectivity towards creating formate at low current densities than the CuO/C-derived electrocatalyst. Surprisingly, the catalyst only needed less carbon surface coverage (<4%) for total formate selectivity to achieve the highest FE (12%). The efficiency of formate generation in ECO2R can be increased using sulfur-derived copper with carbon catalysts.
 | ||
| Fig. 6 (a) Schematic illustration of the preparation of Cu1.81S@MWCNT. (b) SEM and (c) TEM images of Cu1.81S@MWCNT-600. (d) HAADF-STEM image and the corresponding EDS maps revealing the uniform distribution of Cu (blue) and S (purple) in the Cu1.81S particles. (e) HRTEM image of Cu1.81S@MWCNT-600. SEM images of used Cu1.81S@MWCNT-600-OD after (f) 10 min, (g) 20 min, (h) 1 h, and (i) 20 h of ECO2R (reproduced with permission from ref. 119, copyright Elsevier 2020). (j) SEM images of the carbon paper substrate with Cu2S@C deposited on the carbon fibres by spraying. (k) XRD patterns of the bare GNP-500 carbon support (orange) and of CuO@C (black), CuS@C (red) and Cu2S@C (blue) nanoparticles on this carbon support. TEM images of (l) CuO@C, (m) CuS@C and (n) Cu2S@C nanoparticles before electrolysis; TEM images of the (o) CuO@C-, (p) CuS@C-, and (q) Cu2S@C-derived catalysts after 5 h of ECO2R (reproduced with permission from ref. 120, copyright Elsevier 2021). | ||
An interesting approach has been reported recently, in which they created a series of CuS catalysts using different precursors and examined the relationship between phase restructuring and ECO2R catalytic activity during electrolysis. First, Guo et al.121 employed hydrothermal synthesis to create several CuS-based catalysts using different sulfur precursors (i.e., TU = thiourea, STS = sodium thiosulfate, TAA = thioacetamide, and SS = sodium sulfide). Surprisingly, in terms of ECO2R activity, CuS-TU outperformed the other electrodes (i.e., CuS-STS, CuS-TAA, and CuS-SS) in CO2-saturated 0.1 M KHCO3 electrolytes, with an FECO of 72.67% (−0.51 VRHE) and a high CO selectivity. They hypothesized that the thiourea precursor's rapid S decomposition led to a higher concentration of dissolved S2− in the electrolyte, enabling quicker nucleation and restructuring of a nanoflower like CuS–thiourea electrocatalyst for more enhanced mass transfer kinetics and favourable ECO2R.
The phase engineering of a target nanocrystal (NC) can be systematically varied by carefully substituting metal cations in a prefabricated NC template using an emerging electrochemically driven cation exchange (ED-CE) approach. He and coworkers122 recently designed a Cu2S catalyst for ECO2R from the CoS2 template via the ED-CE approach (Fig. 7a). Employing the ion exchange process, Cu almost entirely replaces Co cations in CoSx, and the Cu/S atomic ratio is ∼2.4. The Cu2S catalyst produced by converting the predesigned template retains the initial morphology of CoS2 by preserving its high grain-boundary density, improving CO2 adsorption. Also, the electronic structure of the nearby Cu sites has changed due to electronegative S heteroatoms, creating positively charged Cu+ sites. The primary formate intermediate, *OCHO species, is formed when partially positive-charged Cu sites adsorb CO2 molecules with more electronegative oxygen. The as-prepared 3D-shaped Cu2S catalysts showed an FE of 87% with a current density of 19 mA cm−2 at −1.9 VRHE in a 0.1 M NaHCO3 medium for CO2 conversion to HCOOH. Li et al.123 utilized copper sulfide nanosheets as a template to develop Ag/Cu electrocatalysts through a straightforward ED-CE process (Fig. 7b and c). When the Ag+ concentration in the exchange solution increased, the crystal structure of Cu2−xS nanosheets with lateral dimensions of 100 nm and a thickness of 14 nm progressively changed from Cu7S4 to Ag2S (CA-nano-x, cation-exchanged nanosheets, where x indicates a higher concentration of Ag). The Ag/Cu mass ratio varied between 0.3 and 25. Hence, at an average overpotential (−0.2 VRHE), both C-nano-0 and CA-nano-x exhibited outstanding FEHCOOH. Surprisingly, when the Ag content increases, formate-producing C-nano-0 can generate C2+ products at −1.0 VRHE. This observation concludes that the nanosheets show shape distortion as the Ag content rises while maintaining their original morphology after the cation exchange process.
 | ||
| Fig. 7 (a) Schematic illustration of the experimental pathways and mechanisms for electrochemically driven cation exchange (ED-CE). Synthetic strategies for Ag/Cu sulfide catalysts (reproduced with permission from ref. 122, copyright Wiley-VCH 2020). (b) Cu sulfide nanosheets (C-nano-0, 100 nm lateral dimension, 14 nm thick) were obtained through colloidal synthesis with CuSCN in oleylamine (OAM). (c) Cu sulfides on Cu foil (C-foil-x) were obtained through electrooxidation in 1 M NaOH to produce an oxide layer of a few 10 s of microns thick, followed by sulfurization with 0.1 M Na2S. After cation exchange where Ag+ replaces the Cu+ in the Cu sulfides, Ag/Cu sulfide nanosheets (CA-nano-x) remain in nanosheet structure with some distortion in shape as the Ag/Cu mass ratio ranges from 0.3 to 25, while for C-foil-x, Ag nucleates at higher Ag concentration, which impedes the uniform distribution of Ag and Cu (reproduced with permission from ref. 123, copyright RSC 2021). | ||
In 2024, Goh et al.124 investigated phase-engineered sulfide-derived Cu–Sb electrodes for electrochemical CO2 conversion in a gas diffusion electrode (GDE) based cell. They synthesized several distinct Cu–Sb–S phases, skinnerite (SK; Cu3SbS3), tetrahedrite (TH; Cu12Sb4S13), and chalcostibite (CS; CuSbS2) using a heat-up colloidal nanoparticle route, as illustrated in Fig. 8a–f, and each showed a different selectivity for ECO2R with CO as the main product, which contrasts with the individual CuSx and SbSx control samples, which show a preference for the formate product. They also demonstrated that the different elemental compositions caused the different selectivity patterns when the parent phases were reduced using fundamental composition characterization after reduction. Interestingly, lower Cu concentrations reduce phase segregation into harmful S-doped Cu that converts CO2 to HCOO and H2, whereas higher sulfur concentrations disrupt crystallinity and promote CO formation. This outstanding performance is attributed to the tetrahedral Cu–Sb–S sample, which has the highest residual sulfur, with a COFE of about 80.5% at −1.0 VRHE and a jgeometrical of 37.6 mA cm−2. Post-electrocatalysis characterization combined with DFT calculations demonstrated that adding sulfur to Sb sites enhances *COOH binding compared to *CO, rupturing scaling relations and aiding in CO(g) formation afterwards.
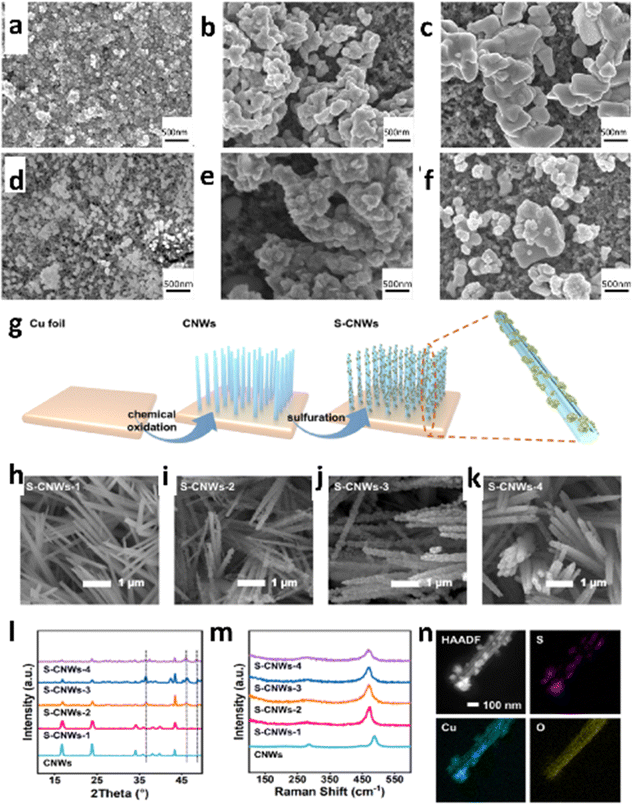 | ||
| Fig. 8 SEM characterization of the samples before reduction. (a)–(c) SEM images of the samples SK (a), TH (b) and CS (c) sprayed on carbon paper. SEM characterization of the samples after reduction. (d)–(f) SEM images of the samples SK (d), TH (e) and CS (f) sprayed on carbon paper (reproduced with permission from ref. 124, copyright RSC 2024). Morphological and structural characterization; (g) schematic illustration of S-CNW preparation, (h)–(k) SEM images of S-CNWs with different loadings of TAA at 75 °C for 0.75 h, (50, 75, 100, and 200 mg per 80 mL) for S-CNWs-1, S-CNWs-2, S-CNWs-3, and S-CNWs-4, respectively; (l) XRD patterns of CNWs and S-CNWs with different degrees of vulcanization, (m) Raman spectra of CNWs and S-CNWs with different degrees of vulcanization, (n) HAADF-STEM and the corresponding EDS images of S-CNWs (reproduced with permission from ref. 125, copyright ACS 2023). | ||
Similarly, in 2023, Mai et al.125 used a facile solvothermal technique to create several cuprous sulfide nanoparticle-modified copper hydroxide nanowire array (S-CNW) pre-catalysts for elucidating the reaction mechanism of ECO2R (Fig. 8g–n). They explored the effect of cuprous sulfide nanoparticle modification on formate generation during CO2 reduction and observed sulfur modification changes in the intermediate during CO2 reduction, leading to improved formate selectivity (60% of FEco with jHCOO− = 10 mA cm−2 at −0.58 VRHE). Therefore, the role of trace sulfur alteration of copper surfaces in selective formate production is investigated using DFT. According to the study, sulfur modification in copper, as compared to a pure copper surface, can accelerate the synthesis of *OCHO, a critical step along the formate pathway.
3.2 Size effect for improving electrocatalytic performance
Researchers discovered that the surface of tiny nanoparticles is rich in unsaturated atoms and defective sites compared to the bulk, exposing the number of active sites to improve catalytic activity.126 Reducing nanoparticles even further to nanoclusters (NCs), which are made up of a few hundred or fewer atoms and have an average size of about 2 nm, reveals intriguing electrochemical performance with significant quantum size effects.127 Shinagawa et al.128 used a wet chemistry approach to create carbon-supported CuS nanocatalysts by controlling the size. The experimental investigation demonstrated that during ECO2R the aforementioned nanometric-sized CuS was restructured and reformed to S-modified copper (Cu–S). As a result, at considerable overpotential (−0.8 V vs. RHE), Cu–S catalysts produced formate with an FE of >60% and negligible quantities of CO as a byproduct. They discovered that as the particle size dimension became 3 nm to 20 nm, there was a slight increase in HCOOH production, highlighting the catalytic size–activity relationship. Following this, in a similar article, it was demonstrated that solvothermally prepared submicron-sized CuS electrodes had better FEHCOOH (80%) for ECO2R to HCOOH than nanometric CuS (FEHCOOH > 60%), indicating the important link between the particle size and electrochemical activity. Later, Lim et al.129 studied the impact of size engineering by fabricating a size-controlled CuSx electrocatalyst in an aqueous medium using Cu foil dipped in industrial CO2 that contains H2S (Fig. 9a–c). As per their observations, the Cu foil and the sulfur interacted appropriately when the concentration of sulfur in the solution increased. The simultaneous interactions raised the average particle size and surface sulfur density of CuSx nanoparticles (NPs) to 133.2 ± 33.1 nm and 86.2 ± 3.3%, respectively, as shown in energy dispersive X-ray spectroscopy (EDX) images (Fig. 9c). When the sulfur percentage and sizes of the CuSx nanoparticles increased steadily, the FEFormate improved from 22.7 to 72.0% at −0.6 VRHE (Fig. 9d–g). Although the CuSx nanocatalysts had a lower current density, the 72-hour stability must be addressed for industrial CO2 conversion.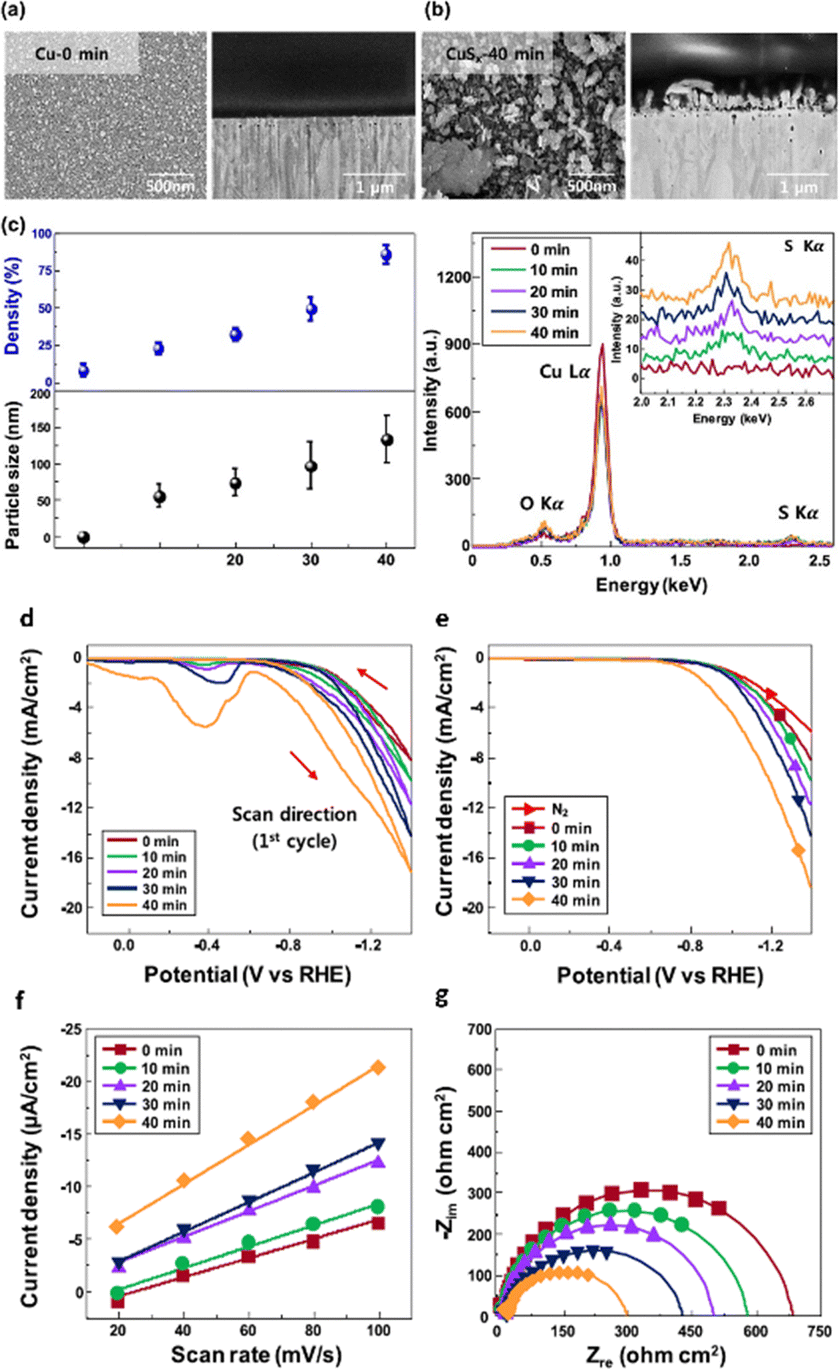 | ||
| Fig. 9 Top-view and cross-sectional SEM images of (a) Cu-0 min without sulfur species and (b) CuSx-40 min catalysts. (c) The average particle size and surface density of CuSx NPs were calculated using a computing-based image analyzer and EDX spectra of Cu and CuSx catalysts. (d) CV (1st scan) of CuSx catalysts with different amounts of sulfur species. (e) LSV curves in N2-purged and industrial CO2-purged 0.1 M KHCO3 electrolytes at a scan rate of 50 mV s−1. (f) Current density plots at various CV scan rates. The current densities were obtained from the double-layer charge/discharge curves at 0.40 VRHE in industrial CO2-purged 0.1 M KHCO3 electrolyte. (g) Nyquist impedance plots of catalysts in industrial CO2-purged 0.1 M KHCO3 electrolyte from 1 MHz to 0.1 Hz with 30 mV amplitude at −0.6 VRHE (reproduced with permission from ref. 129, copyright ACS 2020). | ||
3.3 Grain boundary effect for improving electrocatalytic performance
Grain boundaries (GBs) can be efficient active sites for catalytic processes because they provide an electrodynamically beneficial surface.130,131 Also, it has been shown that catalytically energized surfaces can be stabilized using grain boundaries (GBs).132,133 For example, Yang et al.134 used electrochemical reduction techniques to synthesize S-Cu2O/Cu hybrid catalysts derived from Cu7S4/Cu nanoflowers (Fig. 10a–g). They showed that GB surface defects in Cu2O/Cu interfaces were more energized than grain surfaces for ECO2R. They observed that Cu7S4/Cu is dynamically restructured in situ to provide an S-Cu2O/Cu hybrid catalyst for efficient ECO2R to formate with a FEHCOOH of up to 70% at −1.0 VRHE and a partial current density of 5 mA cm−2), thus outperforming Cu2O/Cu and Cu7S4. The authors attributed this performance to (i) thermodynamic and experimental investigations suggesting that the optimized adsorption of the HCOO* intermediate on the S-Cu2O/Cu surface is modified, and S-doping suppresses the H2 route (surface H), (ii) GBs at the Cu2O/Cu interfaces reduce the adsorption energy favoured by S-doping and increased formate efficiency by inhibiting the HER route and CO2-to-CO conversion. In another work, Wang et al.135 fabricated S-doped Cu2O derived from CuS 811 (consisting of CuS and CuSO4·3H2O) using the electrolysis method to study the relationship between the catalytic activity and GBs. This catalyst performed exceptionally well for formate production in ECO2R, reaching an optimal FE of 92% in an H-type cell and an excellent jformate of 321 mA cm−2 in a flow cell while retaining an FE of more than 80%. The authors explained this outstanding performance from two viewpoints. (1) Structural studies showed that CuSO4·3H2O inhibits CuS growth and vice versa, leading to lower grain sizes and more primary GBs (Fig. 10h–k). After electrolysis and electrochemical reduction, these grains break down into the smaller ones, resulting in more dense grain boundaries suitable for electrocatalysis (Fig. 10l and m). (2) The kinetics of S shedding during electrochemical ECO2R are comparatively slow because of the initial mixture of multi-crystalline phases; this leads to a higher S content and a relatively complete CuS crystalline phase in CuS 811 post ECO2R. They did DFT calculations and showed that retaining sulfur–sulfur bonds from covellite may reduce the binding energy, mainly by weakening the binding energies with several reaction intermediates, thus decreasing the energy barriers and facilitating the desorption steps and increasing formate generation activity. The catalytic impact of the GB was comparable to its dislocation strain field, establishing a way for a more comprehensive application of the GB effects in heterogeneous catalysts. More extensive research is required to understand the fundamental principle of GBs and apply it to developing advanced catalytic schemes for the ECO2R into valuable products. | ||
| Fig. 10 (a) Schematic illustration of the synthetic process, (b) SEM image, (c) TEM image, (d) HRTEM image, (e) EDS elemental mapping, and (g) XPS pattern of Cu7S4/Cu. (f) XRD patterns of Cu7S4/Cu and Cu9S5 (reproduced with permission from ref. 134, copyright Wiley-VCH 2024). Morphological and structural characterization of CuS 811 and CuS MKL before ECO2R by electron imaging. (h) and (i) Aberration-corrected TEM images of (h) CuS 811 and (i) CuS MKL. (j) and (k) HRTEM images of (j) CuS 811 and (k) CuS MKL. The crystal facets with heterogeneous species are highlighted in (j) and (k). Structural characterisation of CuS 811 and CuS MKL post ECO2R by electron imaging. (l) and (m) HRTEM images of (l) CuS 811 and (m) CuS MKL. Red dash-dotted lines highlight the boundaries of induced grains, and the grains with heterogeneous lattice fringes are marked in various colours (l) and (m). The marking colours are only used to distinguish every grain, and there is no correlation between the colour and phase (reproduced with permission from ref. 135, copyright Wiley-VCH 2024). | ||
3.4 Defect/vacancy creation effect for improving electrocatalytic performance
Defect engineering, such as vacancy creation, exposing edge sites, and heteroatom doping, is essential for electrocatalytic activities because defects are usually rich in active sites.136,137 Furthermore, defects can modulate the electronic properties of active sites, increasing the density of catalytic active sites. As a result, defect engineering has become a strategy mostly used to increase active sites and improve charge transfer ability to fine-tune electrocatalytic activity.138,139 Therefore, considering the connection between diverse defects and the specific catalytic properties of various materials is critical for developing advanced catalysts.140,141For example, it is difficult to obtain the n-propanol product during electrolysis because of the complicated C3 creation mechanism that requires the stabilization of *C2 intermediates and subsequent C1–C2 coupling. Zhuang and colleagues142 synthesized a bifunctional core–shell nanostructure (Fig. 11a) showing that adding sulfur atoms to the catalyst core and copper vacancies to the shell named core–shell vacancy engineered Cu (CSVE-Cu) results in excellent ECO2R activity towards n-propanol formation. The CSVE-Cu electrocatalyst exhibited satisfactory reduction performance by generating highly energy-dense C2+ alcohols (i.e., C3H7OH and CH3CH2OH) with a FE of 32% ± 1%. The alcohol-to-ethylene ratio increased sixfold compared to simple copper nanoparticles, indicating an alternative route for producing alcohols instead of alkenes. According to DFT modelling, the incorporated vacancy, as shown by morphology analysis from Fig. 11b–g, on a bifunctional core–shell catalyst raises the activation energy of the C2H4 route (1.148 eV). Still, it does not affect the CH3CH2OH path (0.427 eV). Peng et al.143 created a double-sulfur vacancy (DSV) engineering structure to achieve enhanced ECO2R performance. The mechanistic study showed that the DSV-engineered CuS(100) planes facilitated the stabilization of both CO* and OCCO*, a *C2 dimer which undergoes the subsequent interaction with a third *CO via CO–OCCO coupling (Fig. 11h–k). The DSV-engineered CuSx exhibited an improved FEn-PrOH of ∼15.4% −1.05 VRHE for n-propanol formation in 0.1 M KHCO3 medium in a H-cell set up, but in flow cells, jn-PrOH was increased to 9.9 mA cm−2. This study provides an appealing strategy for using the lithium electrochemical tuning method to create an array of novel frameworks with ion vacancies as active sites for electrochemical reactions.
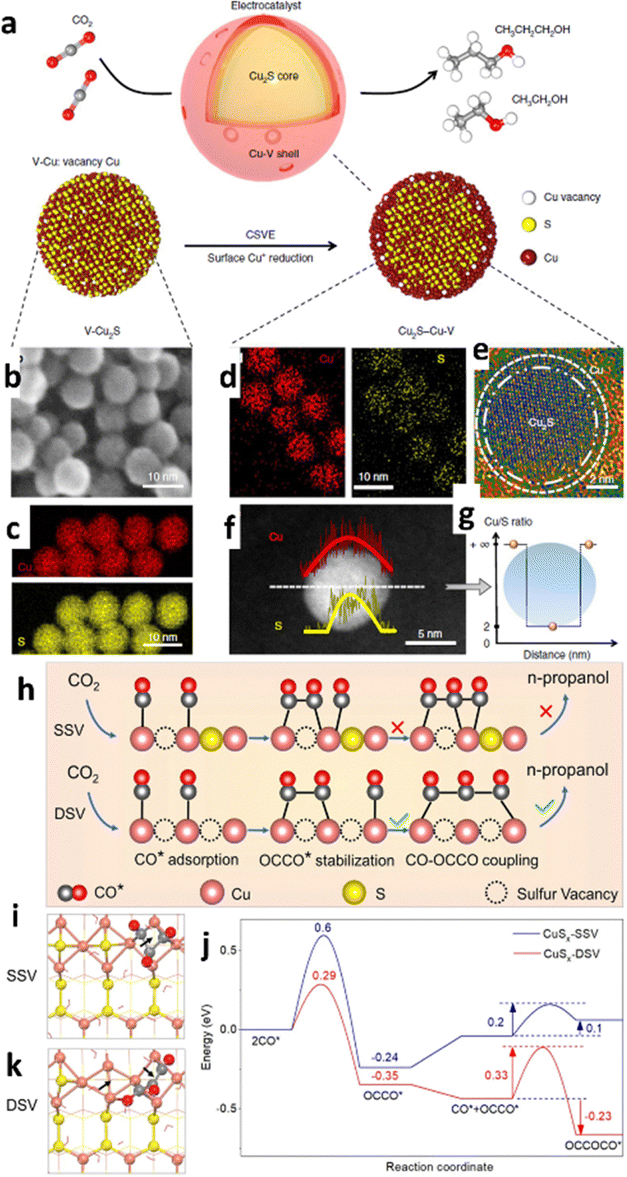 | ||
| Fig. 11 Catalyst design and structural characterization. (a) Schematic illustration of Cu2S–Cu–V CSVE electrocatalyst design for production of multi-carbon alcohols by CO2 reduction. (b) and (c) TEM (b) and EDS (c) mapping of the original V–Cu2S nanoparticles, showing the uniform size and the homogeneous distribution of Cu and S. (d)–(g) EDS mapping (d), high-resolution TEM (e), EDS line scan (f) and the ratio of Cu/S concentration (g) of the reduced CSVE nanocatalysts after electrochemical reduction, showing the removal of S from the nanoparticle surface. V–Cu indicates Cu with surface vacancies (reproduced with permission from ref. 142, copyright Nature 2018). (h) The mechanism of n-propanol formation on adjacent CuSx-DSV shows the dimerization of CO–CO followed by CO–OCCO coupling. (i) and (k) Top views of the optimized OCCOCO* intermediate configurations on the (100) surface of (i) CuSx-SSV and (k) CuSx-DSV. The arrows indicate the positions of sulfur vacancies. (j) The corresponding energy diagrams of CuSx-SSV (blue curve) and CuSx-DSV (red curve) at 0 V vs. RHE. The pink, yellow, grey, and red spheres and red wireframe in (h)–(k) represent copper, sulfur, carbon, and oxygen atoms and water molecules, respectively (reproduced with permission from ref. 143, copyright Nature 2021). | ||
3.5 Heterostructure effect for improving electrocatalytic performance
A heterostructure commonly comprises more than one component that accomplishes different roles in the electrocatalytic reactions.144 Modification of physical and chemical properties will enhance each component's combined advantages. As a result, the various components will produce a synergistic effect, contributing to ECO2R kinetics.145,146 Unlike single-component catalysts, the synergistic effects caused by heterogeneous interfaces can significantly improve catalytic activity.147 Inspired by this, recently, Tao et al.148 developed a novel Cu2S/SnO2@C nanocomposite by solvothermal heating, where SnO2@C is confined on the snowflake-like Cu2S surface and has a combined interfacial effect at the Cu2S sites (Fig. 12a–e). Specifically, the Cu2S snowflake improves the CO2 concentration near the surface, and carbon spheres increase the surface electron transport capacity. Afterwards, the as-synthesized Cu2S/SnO2@C nanocomposites are used as a working electrode to investigate ECO2R to HCOOH. They varied the loading amount to examine the impact of the SnO2@C content on the ECO2R. The prepared Cu2S/1%SnO2@C catalyst shows good selectivity and activity for ECO2R to form liquid HCOOH products, thus outperforming all the existing Cu-based electrocatalysts. The electrochemical study demonstrated a high ECSA, exceptional CO2 adsorption capacities and a fast electron transport rate on the surface of Cu2S/1%SnO2@C. Additionally, DFT calculations revealed the plausible reaction pathways for the enhanced HCOOH production by Cu2S/SnO2@C. Notably, through heterostructure construction, this work provides an easy method to fabricate effective Cu-based catalysts that can improve HCOOH selectivity and activity in the ECO2R process. | ||
| Fig. 12 SEM image (a) and TEM image (b) of Cu2S/SnO2@C. HRTEM images of Cu2S/SnO2@C (c) and SnO2@C (d). The HAADF-STEM image and the corresponding elemental mapping of Cu2S/SnO2@C (e) reveal the distribution of Cu (green), S (yellow), and Sn (rosy), respectively (for interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article; reproduced with permission from ref. 148, copyright Elsevier 2023). | ||
Another work used a redox process at ambient temperature to create a heterostructure of CuS and S-doped SnO2 (CuS/SnO2-S) for ECO2R, as shown in Scheme 1 in Fig. 13.149 Structure analysis methods revealed the structural regeneration phase that had occurred during the first electrolysis (Fig. 13a–i). The unique restructuring of the CuS/SnO2-S heterostructure to Cu/Sn/Cu6.26Sn5 nanowires reduces CO2 adsorption energy while increasing *H adsorption and reducing the competing HER. During ECO2R, at −0.8 VRHE, it achieves a formate conversion with a FE of 84.9% and a yield of 8860 μmol h−1 cm−2 in an H-cell at a jformate of 18.8 mA cm−2. This research focused on the structural development of CuSn sulfides from precursor materials' early state as well as the process of formate production.
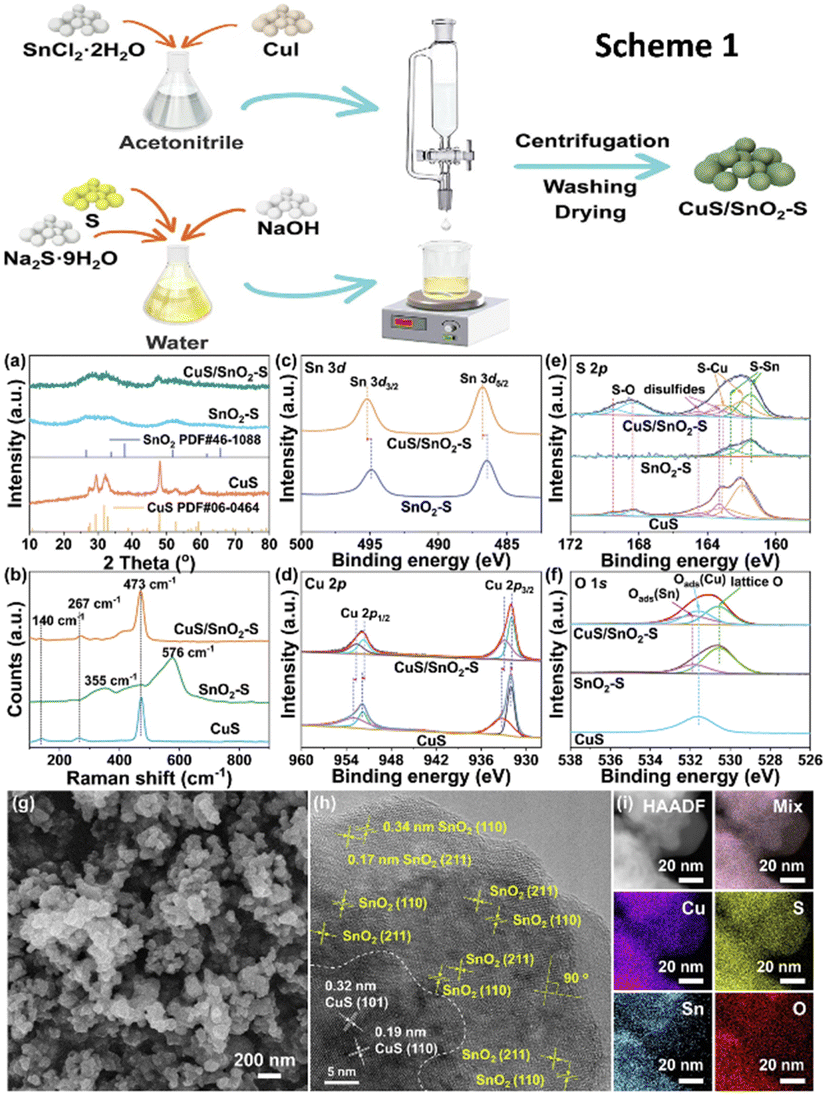 | ||
| Fig. 13 Scheme 1: the synthetic process of CuS/SnO2-S. (a) XRD patterns, (b) Raman spectra and (c)–(f) XPS analysis of Sn 3d, Cu 2p, S 2p and O 1s of CuS/SnO2-S RCu/Sn high, SnO2-S and CuS. (g) SEM images and (h) HRTEM and (i) HAADF image and EDS mapping of as-prepared CuS/SnO2-S RCu/Sn high (reproduced with permission from ref. 149, copyright Elsevier 2023). | ||
Wang et al.150 created an innovative framework of 0D/2D composites of SnO2 nanoparticles dispersed on CuS nanosheets (SnO2/CuS) for selective syngas generation (a CO/H2 ratio of 0.11–3.86), as illustrated in Scheme 1 in Fig. 14. The electrocatalytic system was highly efficient for syngas selectivity, with a faradaic efficiency of nearly 85%, a turnover frequency (TOF) of 96.12 h−1, and stability of 24 hours. They explained the increased catalytic activity based on two factors: (a) the most active sites are provided by the uniformly distributed ultrasmall SnO2 nanoparticles on ultrathin CuS nanosheets, enabling a faster electron transfer rate (Fig. 14a–c) and (b) the interfaces between SnO2 and CuS lower the activation energy of reaction intermediates, enhancing ECO2R performance to generate high-ratio tunable syngas. Both the SnO2(110) and CuS(001) facet surfaces, as shown in Fig. 14d and e, favoured HCOOH creation. In contrast, the SnO2/CuS contact considerably lowered the free energy of COOH* intermediate synthesis by 0.52 eV and encouraged CO formation.
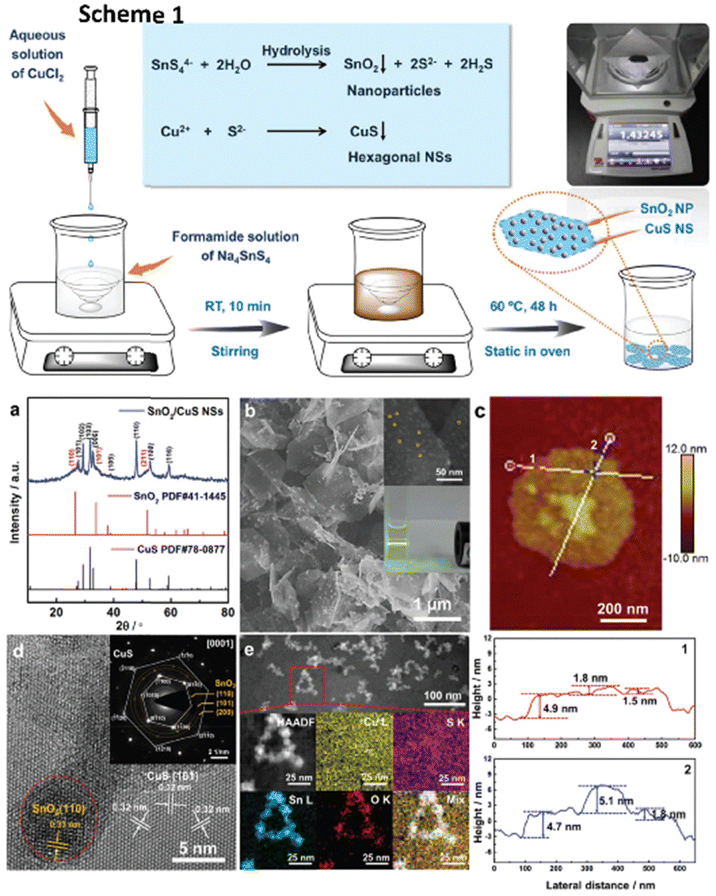 | ||
| Fig. 14 Scheme 1: schematic illustration and the proposed reaction mechanism for the one-pot scalable synthesis of hierarchical SnO2/CuS NSs. Structural characterization of SnO2/CuS NSs. (a) XRD patterns. (b) SEM and (inset) magnified SEM image of the surface and photograph of the dispersion of SnO2/CuS NSs in EtOH, demonstrating the Tyndall effect with a laser pointer. (c) AFM image and the corresponding height profiles of the as-obtained ultrathin SnO2/CuS NSs marked by lines in different colours. (d) HRTEM image and the corresponding SAED pattern (inset). (e) HAADF-STEM images and the corresponding elemental mapping images of SnO2/CuS NSs (reproduced with permission from ref. 150, copyright RSC 2020). | ||
In 2023, Liu and group151 reported the production of monoclinic-phase colloidal Cu2SnS3 nanoplates with precise surfaces (Fig. 15a–h). Their findings revealed that thiocyanate-capped plate-shaped Cu2SnS3 nanoparticles exhibit outstanding formate selectivity over a broad spectrum of potentials, observing a formate production with a FEmax of 92% with a jformate as high as 181 mA cm−2 in a GDA-based flow cell. Compared to prior studies of mono metal- and bimetal-based Cu– and Sn–sulfide nanoparticles that typically experienced phase separation or the creation of metal-based domains, Cu2SnS3 demonstrated outstanding structural robustness, as demonstrated by the concurrent retention of nanoplate morphology and the crystal phase during ECO2R. In situ and DFT studies, as shown in Fig. 15i–m, have demonstrated that the Sn site basal-planes are the multi-active sites for favourable HCOO* adsorption to produce formate by ECO2R. According to DFT studies, thiocyanates also inhibit Cu sites on the surface, and the Sn site's electronic structure modulation is observed, favouring the activation energy barrier of ECO2R to formate.
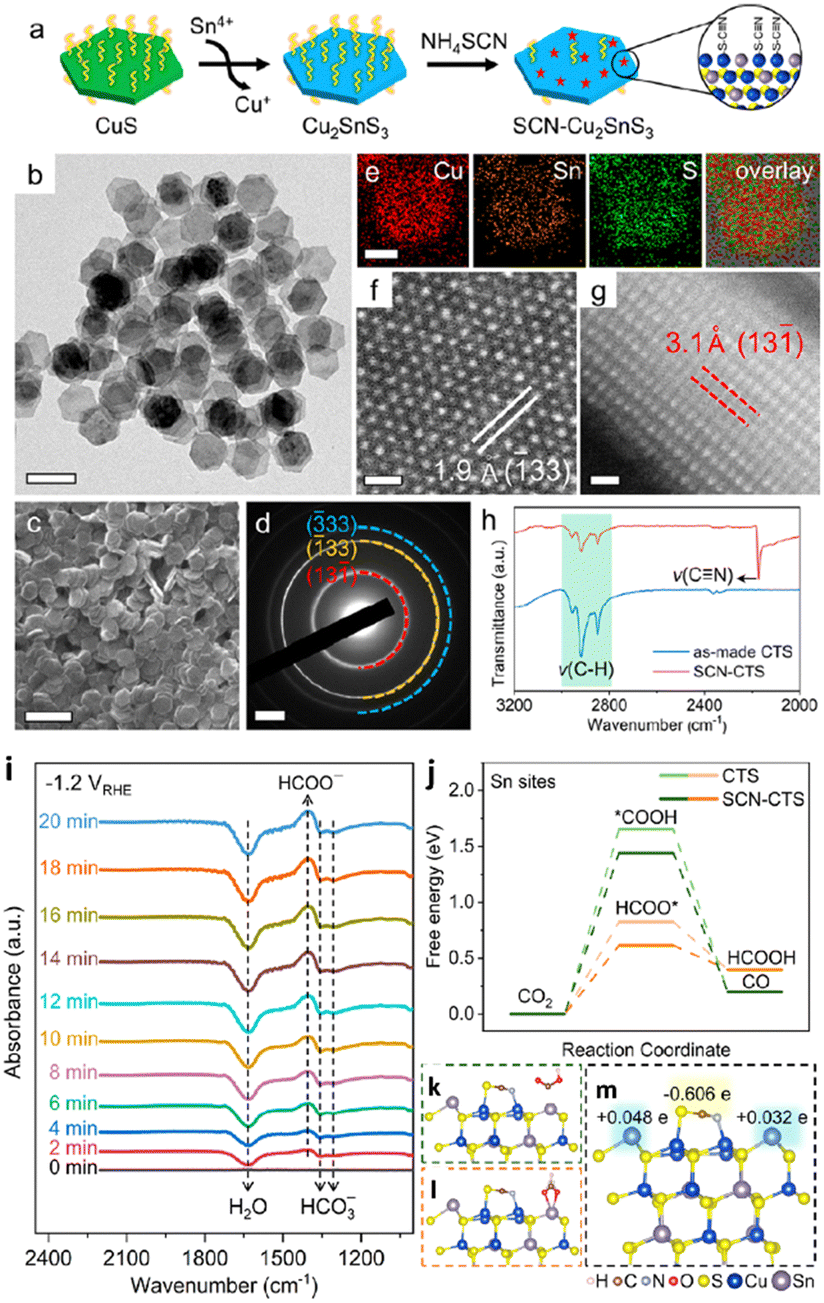 | ||
| Fig. 15 Synthesis and characterization of Cu2SnS3 nanoplates. (a) Synthetic scheme of Cu2SnS3 nanoplates and ligand exchange with NH4SCN. (b) TEM and (c) SEM images, (d) SAED pattern, (e) STEM-EDS elemental maps, (f) top-view and (g) side-view ACSTEM images of Cu2SnS3 nanoplates. (h) FTIR spectra of Cu2SnS3 nanoplates (basal plane edge length: 41.2 ± 2.4 nm) before and after ligand exchange with NH4SCN. Scale bars: (b) 100 nm, (c) 200 nm, (d) 2 nm−1, (e) 30 nm, (f) and (g) 0.5 nm. (i) In situ FTIR spectra recorded at different times for CTS-3 at −1.2 V vs. RHE. (j) Free energy diagrams of ECO2R into formate and CO on Cu2SnS3 nanoplates. (k)–(m) Optimized geometric structures of *COOH (j), HCOO* (k), and SCN-modified Cu2SnS3 (001) surface (m). Also shown in (m) are charge transfers obtained using Bader charge analysis where e is the elementary charge (reproduced with permission from ref. 151, copyright ACS 2023). | ||
Also, Xiong et al.152 used a homogenous mixing approach to create bimetallic CuInS2 hollow nanoparticles. According to their observations, the synergy between metal centres and hollow-shaped nanostructures accelerates the electron transfer kinetics. Consequently, the bimetallic catalyst had a FECO of 82.3% at −1.0 VRHE and a FEHCOOH of 72.8% at −0.7 VRHE. In situ studies showed faster conversion of CO2 to CO2˙− radicals as the rate-limiting step, and afterwards, electron redistribution happened at different potentials, leading to a product distribution shift (CO to HCOOH). Furthermore, Nyquist plots showed that hollow-like CuInS2 nanocomposites have a substantially greater interfacial charge-transfer rate during electrocatalysis than Cu2In because the interfacial charge-transfer impedance (Rct) of Cu2In is lower than that of CuInS2.
Graphdiyne (GDY) is a novel 2D all-carbon structure where alkyne bonds (sp-hybridized C) bind each benzene ring (sp2-hybridized C). GDY's particular sp-/sp2-hybridized architecture provides numerous distinctive and intriguing qualities that are exceptional to standard carbon materials, i.e., abundant carbon chemical bonds, massive conjugated pi structures, a favourable band gap, etc.153 More enticingly, the exceptionally variable distribution of surface charges and incomplete charge transfer between GDY and metal centres can offer more active sites, higher intrinsic activity, and efficient control of reaction intermediates' adsorption and desorption behaviour on functional site surfaces.153 Owing to these advantages, in 2023, Li and colleagues153 discovered a novel graphdiyne/copper sulfide (GDY/CuSx) heterostructured electrocatalyst (Fig. 16a) controlling in situ development of GDY over the surface of CuSx, as illustrated in Fig. 16b–x. The authors showed that the imperfect charge transfer between GDY and atomic Cu increased catalytic conductivity, providing additional active sites and enhancing the conversion performance. Thus, the heterostructure accomplished an FE of 70% and a jtotal of 65.6 mA cm−2 at −0.9 VRHE for ECO2R to formic acid.
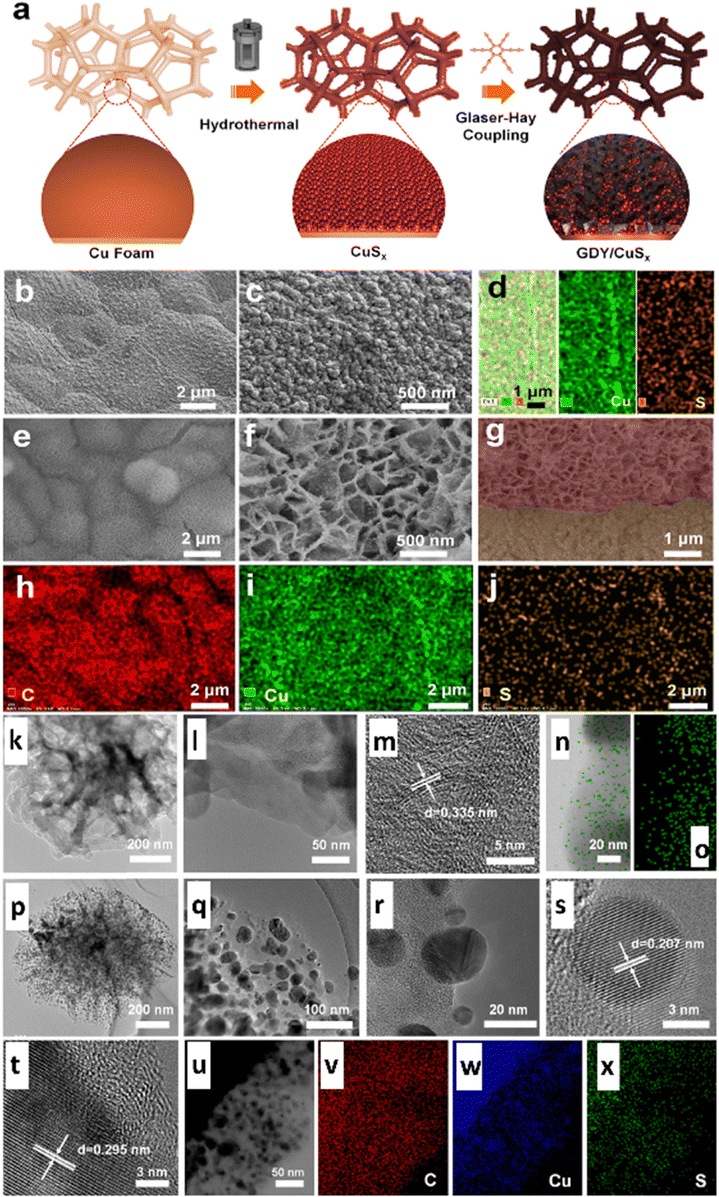 | ||
| Fig. 16 (a) Schematic representation of the synthesis route of GDY/CuSx; and (b) low- and (c) high-magnification SEM images of CuSx. (d) Energy dispersive spectroscopy mapping of CuSx. (e) Low- and (f) high-magnification SEM images of GDY/CuSx. (g) Cross-sectional SEM image of GDY/CuSx; and (h)–(j) energy dispersive spectroscopy mapping of GDY/CuSx. (k) TEM and (l) and (m) HRTEM images of the GDY nanosheet. (n) STEM image (left) and (o) the corresponding elemental mapping (right) of the elemental carbon of GDY. (p)–(r) TEM and (s) and (t) HRTEM images of GDY/CuSx, (u) STEM image and the corresponding elemental mapping of (v) C, (w) Cu, and (x) S elements of the GDY/CuSx electrode (reproduced with permission from ref. 153, copyright RSC 2023). | ||
Heterostructure engineering modulates interfacial charge distribution and promotes CO2 adsorption. Wen et al.154 used a local sulfur doping strategy (SHKUST-1) to appropriately develop an isolated Cu@S motif on the HKUST-1 pre-catalyst (Fig. 17a). The in situ reconstruction of S-HKUST-1 results in a Cu(S) array and active biphased copper/copper sulfide (Cu/CuxSy) interfaces (Fig. 17b), achieving highly selective ethylene (C2H4) formation in the H-cell with a FEmax of 60.0%. In a flow cell configuration, ECO2R occurred at a high current density of 400 mA cm−2 with a FEC2H4 of approximately 57% and FEC2 (FE of C2H4, C2H5OH and CH3COOH) of 88.4%. The S motif is stable before and after the ECO2R, as evidenced by the systematic characterization (Fig. 17c–h). The researchers explain S-HKUST-1's superior selectivity as follows: the approximate distance between Cu0 and Cuδ+ favours the *CO dimerization step at the interface of Cu/Cu2S. Also, a high S-concentrate electrocatalyst (i.e., Cu2S) showed a significant Cu–Cu distance that practically restricted *CO dimerization. Compared to a pure metallic Cu catalyst, the Cu/Cu2S interface decreased to *CO species binding energy at the surface and fastened *CO dimerization (Fig. 17i–m). Similarly, in 2023, Yu et al.155 constructed a Cu2S nanocrystal on Cu nanosheets (Cu–Cu2S), as shown in Fig. 18a, for ECO2R to C2H5OH. The author's design focuses on attaining three discrete features: (i) the nanocomposites produce a positive charge locally on Cu (Cuδ+) to offer multi-active sites during ECO2R; (ii) the evenly dispersed tiny Cu2S on Cu generate interfaces, as illustrated in Fig. 18b–g, between Cuδ+ and Cu in the zero-valence state Cu0; and (iii) the uneven and stepped Cu–Cu2S surface offers a spatially advantageous arrangement for C2H5OH production. Due to these structural features, a total FE of 90% for C2+ products (a FE of 6% for C2H5OH and a FE of 15% for C2H4) with a partial current density of 45 mA cm−2 at −1.2 VRHE was attained in a H-cell configuration. According to the in situ spectroscopy and DFT investigations, as illustrated in Fig. 18h–l, they showed that the three characteristics of Cu–Cu2S work together to (1) improve CO2 adsorption by enabling high electronic conductivity, (2) facilitate the adsorption of the *CO intermediate on the Cu–Cu2S surface, (3) lower the energy barrier for forming the *COCO intermediate, and (4) make the reaction path more thermodynamically viable for C2H5OH over C2H4. This study emphasizes the potential for commercializing alcohol and related product formation from CO2 by proposing an effective method and the underlying mechanism for the significant increase of ECO2R to C2H5OH conversion.
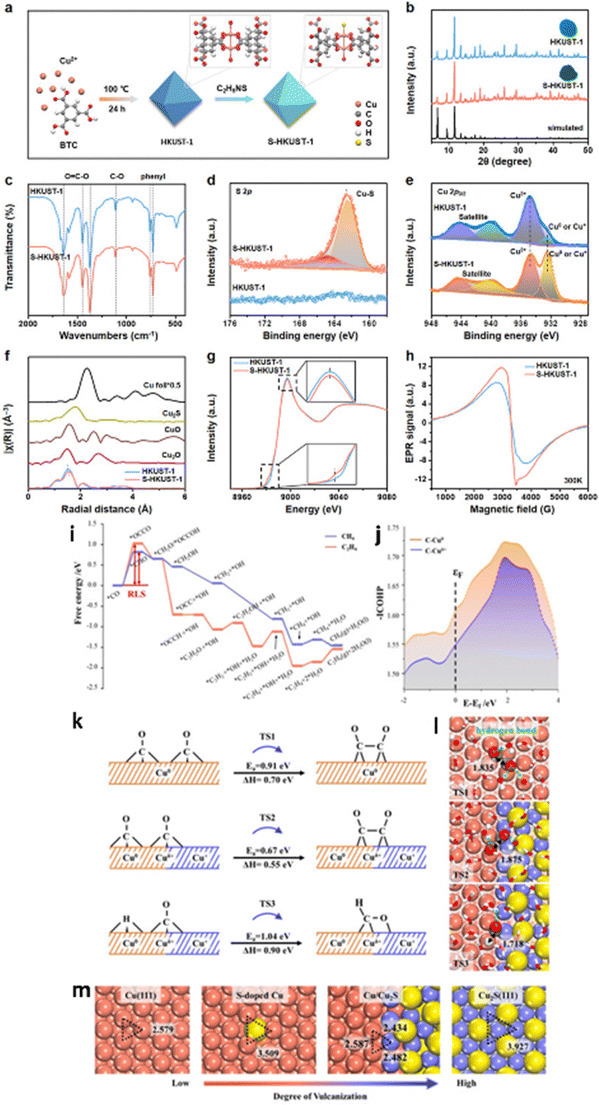 | ||
| Fig. 17 (a) Schematic diagram of the preparation of S-HKUST-1. (b) XRD patterns of the prepared S-HKUST-1 and HKUST-1 precatalysts are well indexed to the simulated HKUST-1. The insets are digital photos of S-HKUST-1 (dark green) and HKUST-1 (blue). (c) FTIR spectra of S-HKUST-1 and HKUST-1 precatalysts indicate negligible differences. The XRD and FTIR results prove that the long-range ordered structure is not destroyed after S incorporation. (d) and (e) High-resolution XPS spectra of S-HKUST-1 and HKUST-1 precatalysts in the (d) S 2p region, showing the characteristic Cu@S bonds in the S-HKUST-1 precatalyst and (e) Cu 2p region, showing the increased content of Cuδ+ or Cu0 species. (f) FT of the EXAFS spectra and (g) Cu K-edge XANES spectra of HKUST-1 and S-HKUST-1 precatalysts. The inset in (g) is the magnified image. The XAFS results in (f) and (g) prove the successful incorporation of local heteroatoms, which might be bonded to Cu atoms in MOFs. (h) EPR spectra of the samples measured at 300 K. (i) Calculated free energy profiles for ECO2R to CH4 and C2H4 over a pure Cu(111) surface, indicating that the initial *CO hydrogenation and dimerization steps determine the CH4 and C2H4 product distribution. (j) Integrated crystal orbital Hamilton population (−ICOHP) curves of Cu Cuδ+–CO and Cu0–CO bonds. (k) The reaction barriers together with enthalpies and (l) the corresponding transition state configurations for *CO dimerization and hydrogenation over Cu(111) and Cu/Cu2S surfaces, respectively. Yellow, red, grey, white, orange and blue balls refer to S, O, C, H, Cu0 and Cuδ+ atoms, respectively. (m) Surface configuration (top view) of Cu-based structures with different degrees of vulcanization. The distances between two neighbouring Cu atoms on different surfaces are given in b (reproduced with permission from ref. 154, copyright Wiley-VCH GmbH 2021). | ||
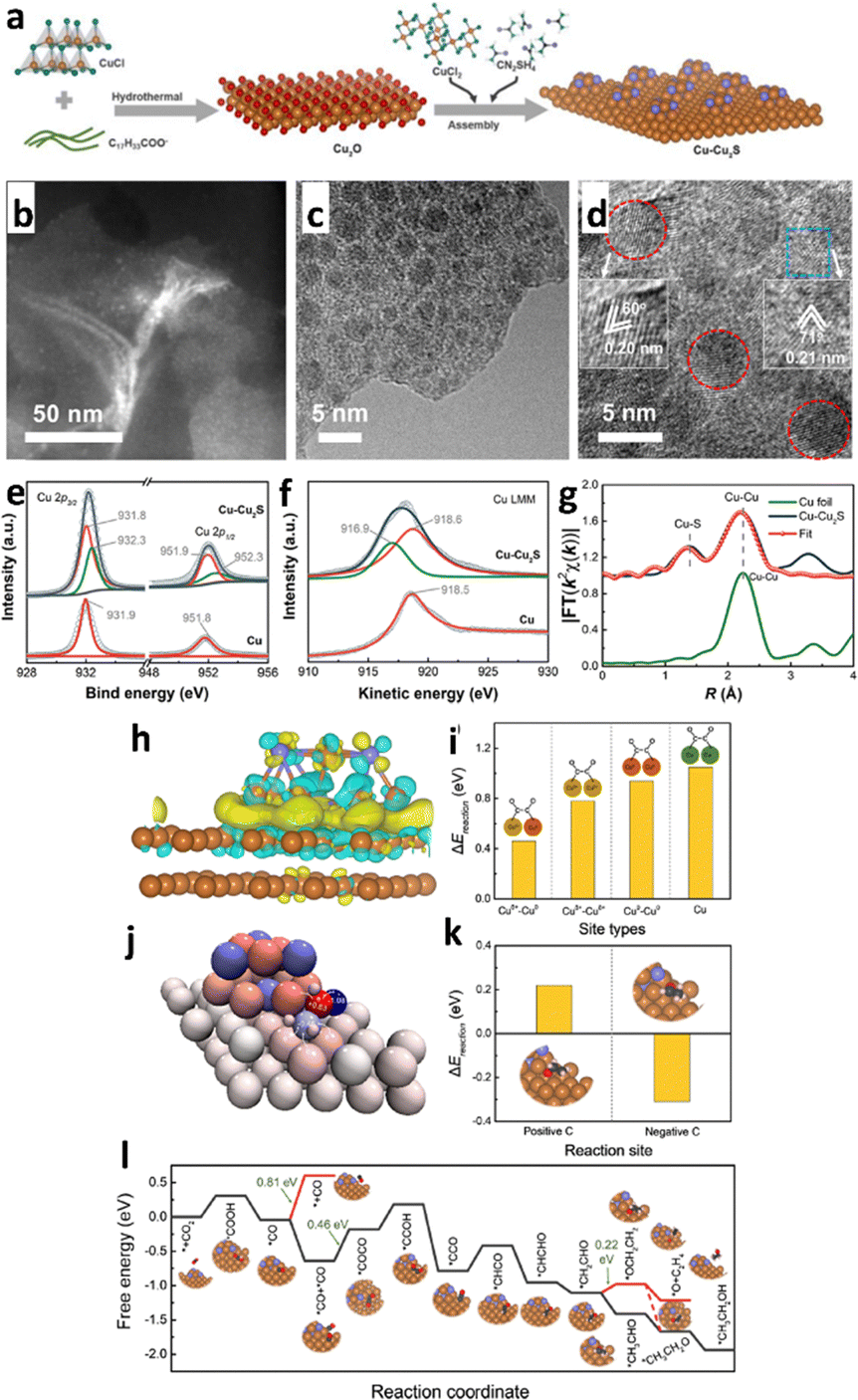 | ||
| Fig. 18 (a) Schematic illustration of the synthesis of Cu–Cu2S. Characterization of the Cu–Cu2S sample. (b) and (c) TEM, (d) high-resolution TEM (HRTEM), (e) high-resolution Cu 2p XP spectra, (f) Cu LMM Auger spectra, and (g) Cu K-edge K2-weighted χ(k) function of Fourier transform extended X-ray absorption fine structure (EXAFS) spectra of the sample, where pristine Cu and Cu foil are used as controls in panels (e)–(g), revealing that both Cu(0) and Cu(I) states are involved, respectively. Calculated ECO2R on Cu–Cu2S. (h) DFT-calculated charge density difference of Cu–Cu2S (the Cu and S atoms are in brown and violet, respectively; the yellow and cyan colours represent charge accumulation and depletion, respectively; the isosurface value is 0.002 e Å−3). (i) Energy barrier (ΔEreaction) for two *CO forming one *OCCO via C–C coupling at Cuδ+–Cu0, Cuδ+–Cuδ+, and Cu0–Cu0 sites of Cu–Cu2S and at the pristine Cu surface, where Cuδ+ and Cu0 of Cu–Cu2S and Cu of pristine are in brown, red, and green, respectively, showing that the energy barrier at the Cuδ+–Cu0 site of Cu–Cu2S is the lowest. (j) Bader charge of *CH2CHO adsorbed on Cu–Cu2S, where the colour ranging from blue to red indicates the negative to the positive charge of the atoms, respectively. (k) ΔEreaction for hydrogenation on the positively charged C and negatively charged C of *CH2CHO converting to CH2CH2O* and CH3CHO*, respectively. (l) Calculated free energy for each step of the reaction pathway converting CO2 to C2H5OH on Cu–Cu2S (reproduced with permission from ref. 155, copyright ACS 2023). | ||
Mosali et al.156 developed an array of CuxZnyS nanoparticles (Fig. 19a–l) with varying Cu:Zn compositions and that are electrochemically stabilized, resulting in SD-CuxZnyS catalysts. The catalysts showed composition-dependent product selectivity during ECO2R. Operating in an H-cell configuration, it was discovered that SD-CuxZnyS catalysts with a higher copper content exhibited a significantly higher FECH4 than the Zn-rich catalysts. In contrast, Zn-rich counterparts produced CO as the primary ECO2R product. The best composition of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Cu:Zn resulted in highly selective CH4 as the main product, with an FE of 76 ± 3% at −0.98 VRHE. The current density increased after switching to a flow cell arrangement for ECO2R in 1.0 M KHCO3 or 1.0 M KOH electrolyte. However, the selectivity changed to produce syngas, with molar ratios of 2:3 to 3:2 for the formation of H2/CO. This tendency holds irrespective of the applied potential, particularly in the 1.0 M KOH electrolyte. An ex situ investigation revealed a significant reduction in CuS in electrocatalysts with higher copper content. In addition, pre-reduction of CuZnS catalysts and time-dependent observations showed the importance of copper's higher oxidation state and the interaction of partially reduced CuS and ZnS present in the catalysts in attaining methane selectivity. These findings demonstrate that copper's oxidation state is crucial in determining selective product formation using copper-based catalysts. Likewise, the same research group created S-derived copper–cadmium (SD-CuxCdy) electrocatalysts earlier, where x and y denote the molar ratio of Cu/Cd to assist in generating vital intermediates.157 In 0.1 M KHCO3 solution, the SDCuCd2 catalyst observed 32% ethanol selectivity at a low overpotential of −0.89 VRHE. They exhibited selective ethanol production at lower overpotentials with the best-performing SD-CuCd2 electrocatalyst of Cu3Cd10alloy/Cu2S/CdS phased composites, as illustrated in Fig. 19m and n.
:1 Cu:Zn resulted in highly selective CH4 as the main product, with an FE of 76 ± 3% at −0.98 VRHE. The current density increased after switching to a flow cell arrangement for ECO2R in 1.0 M KHCO3 or 1.0 M KOH electrolyte. However, the selectivity changed to produce syngas, with molar ratios of 2:3 to 3:2 for the formation of H2/CO. This tendency holds irrespective of the applied potential, particularly in the 1.0 M KOH electrolyte. An ex situ investigation revealed a significant reduction in CuS in electrocatalysts with higher copper content. In addition, pre-reduction of CuZnS catalysts and time-dependent observations showed the importance of copper's higher oxidation state and the interaction of partially reduced CuS and ZnS present in the catalysts in attaining methane selectivity. These findings demonstrate that copper's oxidation state is crucial in determining selective product formation using copper-based catalysts. Likewise, the same research group created S-derived copper–cadmium (SD-CuxCdy) electrocatalysts earlier, where x and y denote the molar ratio of Cu/Cd to assist in generating vital intermediates.157 In 0.1 M KHCO3 solution, the SDCuCd2 catalyst observed 32% ethanol selectivity at a low overpotential of −0.89 VRHE. They exhibited selective ethanol production at lower overpotentials with the best-performing SD-CuCd2 electrocatalyst of Cu3Cd10alloy/Cu2S/CdS phased composites, as illustrated in Fig. 19m and n.
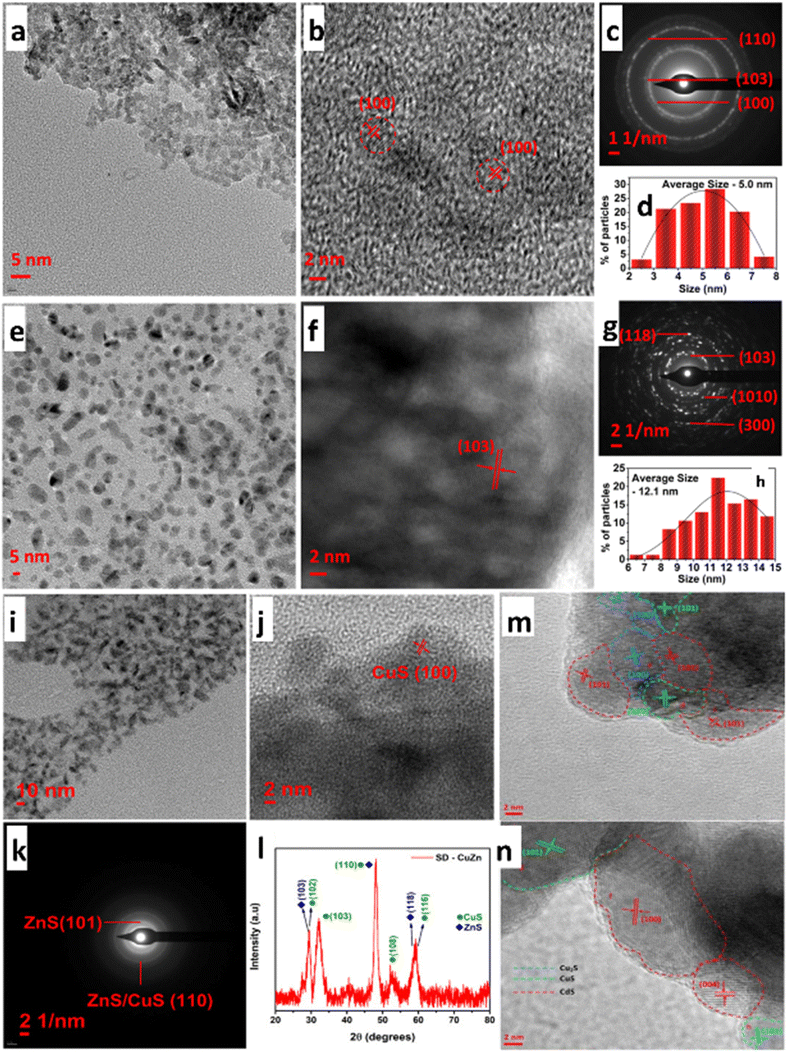 | ||
| Fig. 19 (a) and (e) TEM images, (b) and (f) HR-TEM images, (c) and (g) SAED pattern and (d) and (h) particle size distribution of (a)–(d) CuS and (e)–(h) ZnS nanoparticles. (i) TEM images, (j) HR-TEM images, (k) SAED pattern, and (l) XRD powder pattern of SD-CuZn nanoparticles obtained upon pre-reduction at −0.80 V vs. RHE in 0.10 M KHCO3 solution saturated with CO2 (blue diamonds (♦) represent the peaks corresponding to ZnS, and green circles (•) represent the peaks corresponding to CuS; reproduced with permission from ref. 156, copyright Elsevier 2024). HR-TEM images of (m) CuCd2S; (n) Cu2CdS showing boundary distributions of CdS, CuS and Cu2S (reproduced with permission from ref. 157, copyright Wiley-VCH GmbH 2023). | ||
Xi and colleagues158 recently developed and built CeO2/CuS (Fig. 20a–k) with a −0.5 VRHE overpotential for ECO2R to ethanol and a FEC2+ of 75% at −0.8 VRHE. During ECO2R, they investigated catalysts' structural development, interactions, and activity sources, as illustrated in transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images (Fig. 20b–d). The fast electron movement route provided by CeO2 regions in catalysts prevents electron aggregation around Cu+ sites, preserving Cu+ sites during ECO2R. The experimental in situ studies and DFT calculations reveal that altering CeO2 on CuS thermodynamically reduces the production energy for *COCHO compared to CuS nanoplates. The fast water molecule activation near CeO2 speeds up the synthesis of *COCHO. As a result, the C–C coupling is accelerated via the *CHO route, providing CeO2/CuS catalysts with exceptional electrocatalytic efficacy towards C2+ products. Wang et al.159 proved that heterointerfaces of Bi/CeO2/CuS nanohybrids (Fig. 20l–s) can be advantageously employed as a highly efficient and selective catalyst for ECO2R to produce formate over a wide potential range. The nanohybrid demonstrated excellent activity at −0.9 VRHE, with a formate efficiency of 88% and a current density of −17 mA cm−2. The morphological analysis showed no change in the structure after a one-hour reaction at −0.9 VRHE, and the FE of formate remained stable at that potential. The Bi/CeO2/CuS heterostructure substantially decreases the formation energy barrier of OCHO* intermediates because, during electrolysis, Ce4+ rapidly suffers Ce3+ reduction, forming a conductive network of Ce4+/Ce3+, resulting in high activity and selectivity of ECO2R to formate. Overall, the system increased electron mobilization, stabilized Cu+ species, and improved CO2 adsorption and activation over the catalytic surface. Furthermore, sulfur boosts the transformation of OCHO* to formate.
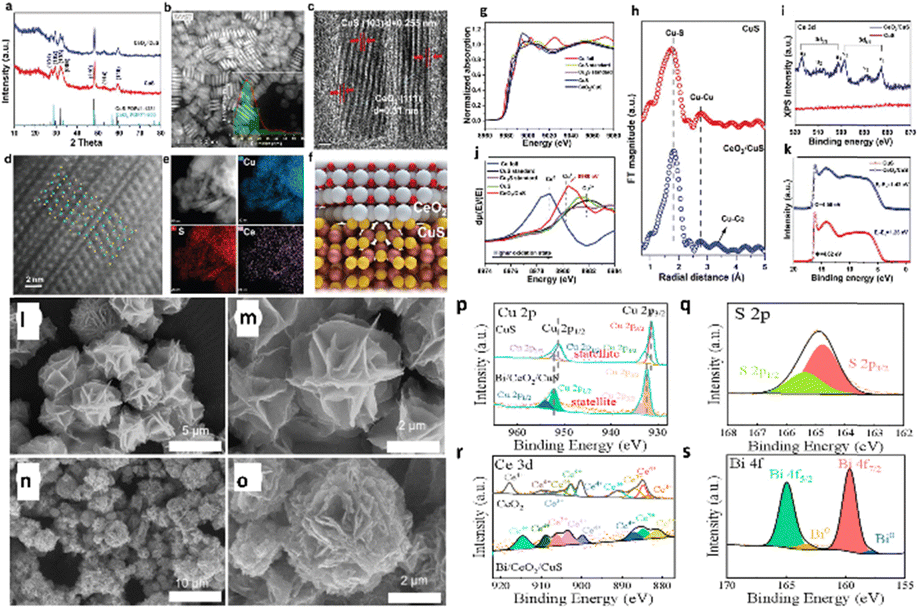 | ||
| Fig. 20 Crystal structures and composition of CuS and CeO2/CuS. (a) XRD patterns of CuS and CeO2/CuS. (b) TEM image of CeO2/CuS (inset: size distribution). (c) HRTEM image of CeO2/CuS. (d) The HAADF-STEM image of CeO2/CuS (inset: CuS 9 000 523. cif). (e) The HAADF-STEM image and corresponding EDS element mapping of CeO2/CuS. (f) Structural representation of CeO2/CuS, white: Ce atoms; red: O atoms; yellow: S atoms; gold: Cu atoms. Electronic structures of CuS and CeO2/CuS. (g)–(i) X-ray absorption spectra of CeO2/CuS, CuS catalyst, standard CuS, standard Cu2S, and Cu foil. (g) Normalized Cu K-edge XANES. (h) the first derivative μ(E)/dE. (i) Fourier-transformed k2χ(k) of CeO2/CuS (lower panel) and CuS (upper panel). (j) Ce 3d XPS spectra. (k) UPS spectra (reproduced with permission from ref. 158, copyright Wiley-VCH GmbH 2023). SEM images of (l) and (m) CuS and (n) and (o) Bi/CeO2/CuS. XPS patterns of Bi/CeO2/CuS: (p) Cu 2p, (q) S 2p, (r) Ce 3d, and (s) Bi 4f (reproduced with permission from ref. 159, copyright MDPI 2024). | ||
Han et al.160 employed a simple solution-phase method to synthesize a ternary metal–metal sulfide Bi–Cu2S heterostructure electrocatalyst (Fig. 21a and b). Due to the high synergistic and interfacial effects between Bi and Cu2S, the heterostructure showed a lower overpotential (240 mV) than Bi with an outstanding FEformate of >98% and 2.4 and 5.2 times higher partial current density than bare Cu2S and Bi at −1.0 VRHE. According to the theoretical study, the HCOOH generation was aided by stabilizing the *OCHO intermediates over *COOH and *H due to the higher electron transfer rate between the Bi and Cu2S interfaces (Fig. 21c–i).
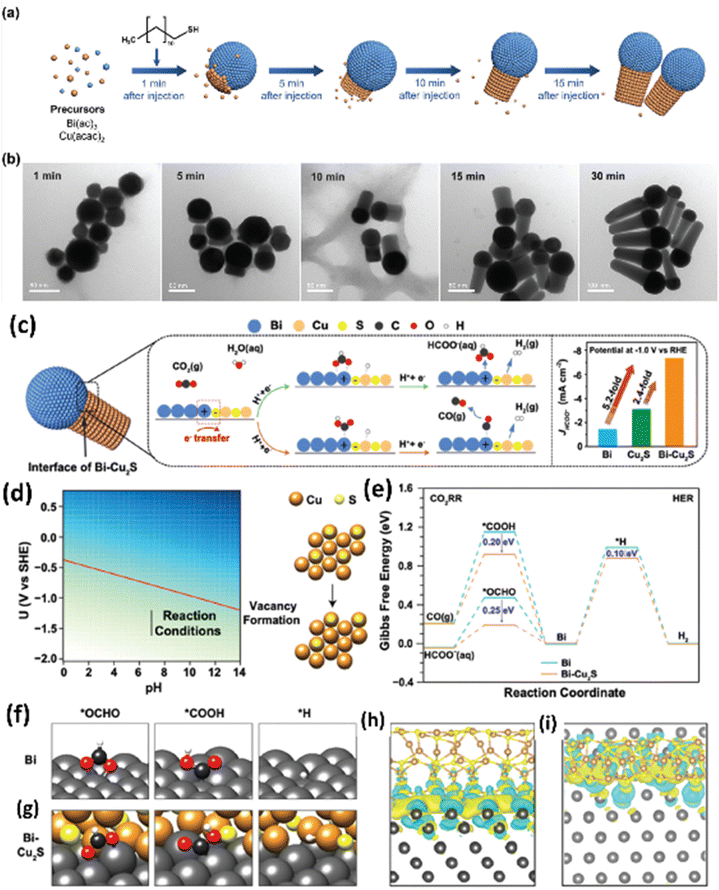 | ||
| Fig. 21 (a) Schematic illustration of the nucleation and growth of the heterostructured Bi–Cu2S nanocrystals. (b) TEM images of Bi–Cu2S at different time intervals after introducing 1-dodecanethiol at 220 1C. (c) Proposed CO2 reduction mechanism on the Bi–Cu2S interfacial system. (d) Pourbaix diagram showing the sulfur vacancy formation. (e) Free energy diagrams of the ECO2R and HER on Bi(001) and Bi–Cu2S model systems. Optimized geometry structures of key intermediates (*OCHO, *COOH, and *H) on Bi(001) and Bi–Cu2S systems are shown in (f) and (g), respectively (dark grey, brown, yellow, black, red, and white spheres denote Bi, Cu, S, C, O, and H atoms, respectively). (h) and (i) are the top and front views of the charge density difference for the Bi–Cu2S interfacial surface, respectively. Cyan corresponds to an isosurface of −0.001 e Bohr−3 and yellow corresponds to an isosurface of +0.001 e Bohr−3 (reproduced with permission from ref. 160, copyright RSC 2022). | ||
Prasanna et al.161 designed a rationally created novel heterostructure of CuS decorated NH4+ ion incorporated stable 1T-WS2/WO3 using a simple hydrothermal method followed by a reflux technique (Fig. 22a). The nanohybrid favoured reaction mechanism through the *OCHO route, which reduces (by obtaining H+ + e−) to HCOO− (HCOO− pathway) as the main product. Thus, CuS@1T-N-WS2/WO3 nanohybrids yielded 55.6% ± 0.5 at −1.3 VRHE and a jgeo of −125.05 mA cm−2. Interestingly, from their analysis, the intercalation of NH4+ ions with effective surfaces that donate WO3 and accept protons from WS2 stabilized the metallic 1T phase. Besides, an effective hydrogen spillover mechanism in the stable heterointerface of 1T-N-WS2/WO3 may provide kinetic support for CuS active centres, leading to notable improvements in product efficiency and selectivity (Fig. 22b).
 | ||
| Fig. 22 (a) Schematic illustration of the synthetic process of 1T-N-WS2/WO3 and CuS@1T-N-WS2/WO3 nanohybrids using a simple solvothermal process and reflux method. (b) Proposed schematic illustration of the ECO2R in an H-type cell (reproduced with permission from ref. 161, copyright Wiley-VCH GmbH 2023). (c) Schematic diagram of the synthesis procedures and formation of the pristine CuS and TMS-decorated CuS microflower-like structures; SEM images of the MoS2–CuS catalysts (d) at a low magnification and (e) at a high magnification (reproduced with permission from ref. 162, copyright Springer Nature 2024). | ||
Guo et al.162 synthesized transition metal sulfide (TMS)-supported CuS catalysts with microflower-shaped frameworks via a facile hydrothermal approach for ECO2R to CO (Fig. 22c). They investigated the effect of doping TMSs (i.e., ZnS, Bi2S3, and MoS2) with CuS on ECO2R performance. All the heterostructured catalysts, i.e., ZnS–CuS, Bi2S3–CuS and MoS2–CuS, exhibited flower-shaped morphology, with the doped TMSs (i.e., ZnS, Bi2S3, and MoS2) attached as microcrystals on the surface of petals. On the other hand, strong Cd2+ and Cu2+ interactions with S2− caused CdS and CuS to aggregate and expand disorderly, forming incomplete sphere-like cage-shaped microstructures. Electrochemical experiments revealed that TMS-supported CuS catalysts outperformed pristine CuS in terms of ECO2R activity, but their CO2 conversion rate was reduced. Decorating CuS with MoS2 resulted in a flower-shaped nanomorphology (Fig. 22d and e), enhancing the catalyst's CO selectivity from 72.67% to 83.20% at −0.6 VRHE. With the increase in applied potential from −0.2 to −1.0 VRHE, CO selectivity initially improved and then decreased, but the CO2 conversion rate increased considerably from 0.2% to 21.9%. It has been observed that, during a 300-minute electrolysis at −0.6 VRHE, the target MoS2–CuS catalyst exhibited constant ECO2R activity, with CO selectivity being improved over a limited range of 73.6–88.5% and jCO remaining stable at about 3.6 mA cm−2. The authors attributed the increased selective CO formation linked to the synergistic effect of the generated GBs, undercoordinated S-vacancies, and edge-exposed Mo sites in boosting CO2 activation, stabilizing *COOH adsorption, facilitating *CO desorption, and reducing the energy barrier of the potential-limiting step.
3.6 Heteroatom doping effect for improving electrocatalytic performance
Doping is widely regarded as an effective method for modulating the electronic structure of a catalyst through charge redistribution.163,164 Doping different elements into the lattice of the pristine material can effectively modulate the electronic and physiochemical properties to fine-tune its electrocatalytic activity.165,166 Also, heteroatom doping has little impact on crystal structure disruption, so the catalyst's main composition remains unchanged. It has been observed that the doping approach can increase the adsorption/desorption energy of the reaction intermediates.167,168 The doping strategies are divided into metal and nonmetal doping, as described below.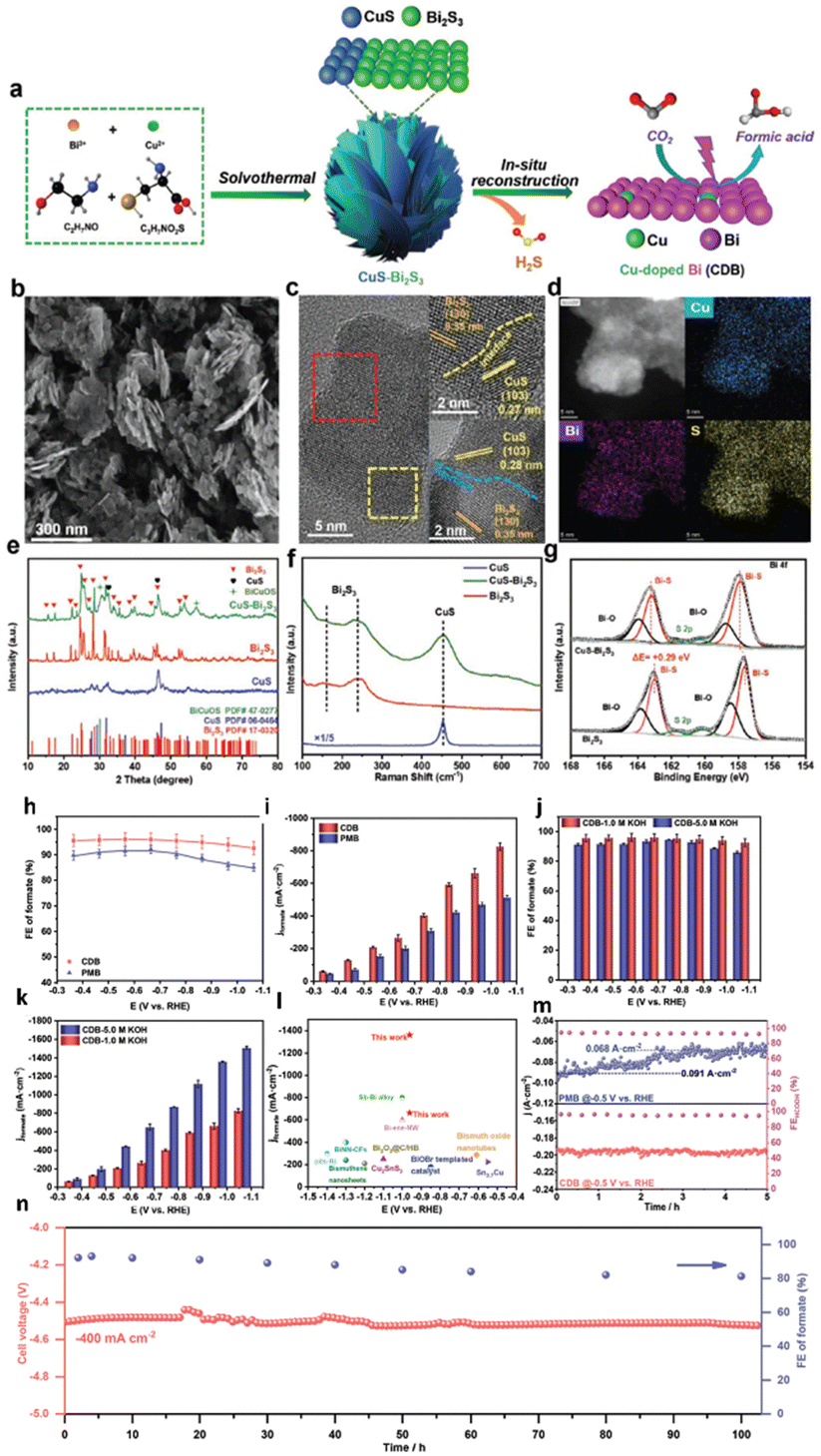 | ||
| Fig. 23 (a) Synthetic strategy of a CuS–Bi2S3 nano-heterojunction precursor. (b) SEM images of a CuS–Bi2S3 nano-heterojunction precursor. (c) HR-TEM images of a CuS–Bi2S3 nano-heterojunction precursor. (d) Corresponding elemental mapping images of S, Cu, and Bi elements. (e) X-ray diffraction (XRD) patterns of CuS, Bi2S3, and CuS–Bi2S3. (f) Raman spectrum of CuS, Bi2S3, and CuS–Bi2S3. (g) The high-resolution XPS spectra of Bi 4f for Bi2S3 and CuS–Bi2S3. ECO2R measurements in the lab-made flow cell. (h) FE of formate and (i) formate partial current densities at different electrolytic potentials in 1 M KOH electrolyte with CDB and PMB electrocatalysts. (j) FE of formate and (k) formate partial current densities of the CDB electrocatalyst at different electrolytic potentials in 1 M and 5 M KOH electrolytes. (l) Performance comparison of the CDB electrocatalyst with the reported electrocatalysts towards ECO2R to formate in the flow cell. (m) Stability test of PMB and CDB electrocatalysts at the potential of −0.5 VRHE for 5 h. (n) Continuous 100 h electrolysis in a 5 cm2 MEA electrolyzer under a constant total current density of −400 mA cm−2. The error bars in (f)–(h) represented the standard deviations of three independent measurements (reproduced with permission from ref. 170, copyright Wiley-VCH GmbH 2022). | ||
 | ||
| Fig. 24 (a) Illustration of the key hypothesis: the rate of C–C coupling by CO dimerization can be manipulated using structurally asymmetric metalδ1+–metalδ2+ pair sites (0 < δ1+ ≠ δ2+). A well-ordered CuSx surface facet with surface Cu atoms adsorbing CO2 molecules is shown. Dopant atoms introduce electronic asymmetries in Cu site pairs by adjusting electron-withdrawing/donating properties. Asymmetric CO chemisorption energies favor CO dimerization and hence ECO2R to C2 products. By contrast, sites with even electronic distributions feature strong dipole–dipole repulsion forces during CO2 activation, which seriously hinders CO dimerization (reproduced with permission from ref. 171, copyright Wiley-VCH GmbH 2022). (b) Schematic showing the synthetic process of SNC@Cu1.96S. Characterization of Cu vacancies in SNC@Cu1.96S. (c) SEM image, (d) TEM image, (e) HR-TEM image, and (f) the elemental mapping images of SNC@Cu1.96S. (g) XRD patterns of SNC@Cu1.96S, SNC@Cu2S, and SNC. (h) EPR spectra and (i) Cu 2p XPS spectra of SNC@Cu1.96S and SNC@Cu2S (reproduced with permission from ref. 172, copyright ACS 2022). | ||
In another work, Li et al.172 demonstrated the effective modulation of ECO2R pathways by designing and synthesizing three kinds of copper sulfides (Fig. 24b). Among all, they observed that SNC@Cu2S (Cu2S coated sulfur, nitrogen-co-doped carbon) without Cu vacancies displays a high FE for formate production. In contrast, the other two catalysts, SNC@CuxS and SNC@Cu1.96S, with Cu vacancies, generated CO as the primary product (Fig. 24c–i). Firstly, the Cu vacancies present in Cu1.96S modify the S sites' electronic structures and significantly increase the energy barrier of H* formation. At the same time, the Cu vacancies generate the appropriate binding energy for the *COOH intermediate while attenuating the adsorbate–metal interaction. These advantageous characteristics, when combined, result in the favoured formation of CO over formate. Furthermore, the Cu vacancies lower charge transfer resistance, enrich the electronic structure of active sites and increase CO2 adsorption capacity, enhancing the ECO2R activity of SNC@Cu1.96S.
Huang and his colleagues173 synthesized Cu2O-derived Cu catalysts doped with sulfur by immersing the Cu substrates in ammonium polysulfide solutions. Regarding cost, availability, longevity, and catalytic efficacy, the catalyst outperforms alternative materials for ECO2R to formate. Their XPS, ToF-SIMS, and μXRF analyses revealed a positive correlation between catalyst sulfur content and formate production. According to their findings, among all the prepared electrocatalysts, Cu-5000S with 2.7% atomic sulfur has satisfactory catalytic activity with a faradaic efficiency of 75% and a current density of −13.9 mA cm−2 at −0.9 VRHE for formate selectivity. Moreover, Cu-5000S exhibits outstanding stability for prolonged CO2 reduction, as evidenced by its formate FE retained at approximately 75% for 12 hours. When sulfur doping was increased, formate production outpaced HER activity, as demonstrated by comparing the SRF-normalized jformate and jH2. According to investigations of mechanisms, sulfur-doped Cu reduces the binding energy of *COOH intermediates for CO generation, facilitating the synthesis of HCOOH. This work demonstrates that sulfur doping is an effective strategy for enhancing the catalytic selectivity of Cu towards formate and expands the material choice for producing this commercially valuable fuel and chemical.
Li et al.174 examined the influence of the doping strategy on the ECO2R to ethylene using S-doped spherical coral-like CuO catalysts. With a current density of 15.5 mA cm−2, the as-synthesized 5% S–CuO observed a good FEC2H4 of 48.4% at −1.3 VRHE compared to pure CuO. The 5% S–CuO also retained a consistent FE for C2H4 upon long-term stability testing, with no appreciable drop in current density. The superior performance of 5% S–CuO was derived from the improved dynamic barriers of *CO intermediate dimerization at the Cu active site caused by S-doping, according to the experimental analysis and contact angle measurements. In addition, the hydrophobicity of the 5% S–CuO surface prevented accessible H2O molecules from interacting, thereby preventing competitive HER.
Wang et al.175 discovered various new morphologies of Cu2S-X materials reduced to S-Cu2O-X during ECO2R in 2022. The researchers found that the optimized electronic structure, aided by the S dopant and microstructure reconstruction, resulted in a large surface area, critical in improving the ECO2R performance and formate selectivity of the S-Cu2O-X catalysts. In addition, as compared to Cu(OH)2 and desulfurized Cu2O materials, S-Cu2O-X (X = 6, 10, and 14) catalysts showed significantly higher catalytic activity in the formation of formate (60–70%). The S-Cu2O-14 catalyst demonstrated a partial current density of 16 mA cm−2 at 1.0 VRHE and a FE of 67.2% for formate selectivity with a stability of 20 h among all evaluated electrocatalysts. DFT studies demonstrated that S-doped Cu(111) and S-vacancy (Vs) species promoted COOH* or OCHO* intermediates, which accelerated selectivity towards HCOOH pathways.
Recently, Zhang et al.176 showed that S-doped Cu derived from hierarchical hollow-liked CuS polyhedron (CuS-HP) nanostructures synthesised from a MOF significantly improves ECO2R performance in neutral pH water environments (Fig. 25a–j). According to their findings, during electrolysis, the CuS-HP was gradually transformed into an S-doped Cu as the reduction process went on. The in situ formed electrocatalyst exhibited a stability of 36 hours at a jformate of 16 mA cm−2 at −0.6 VRHE with a FE of >90%. The DFT study indicates that the Cu(111) facet in S-doped Cu lowers formate activation energy barriers while inhibiting the HER and the reaction intermediates remain unchanged. This work comprehensively explains the mechanisms for improving formate selectivity using CuS electrode materials. Deng et al.177 have experimentally and theoretically demonstrated that sulfur-doped Cu catalyzes the ECO2R to formate. They synthesized an active CuSx (AC-CuSx) catalyst, which exhibited a high FE of 75% and a partial current density of 9 mA cm−2 at −0.9 VRHE toward formate production (Fig. 25k–m). It was discovered that sulfur dopants were the key to the increased formate production on the AC-CuSx surfaces. Operando Raman spectroscopy found that S dopants on the catalyst inhibited the CO intermediate formation during ECO2R, with a lower FE for other products (i.e., CO, hydrocarbons, and alcohols). DFT calculations validated the Raman band assignments. They observed that the adsorption strengths of adsorbed HCOO* were modified by the presence of sulfur on the copper surface, which supported the formation of formate while inhibiting the formation of *COOH, the CO intermediate.
 | ||
| Fig. 25 Structural characterization of CuS-HP. (a) Schematic illustration of the synthetic process. (b) XRD pattern. (c)–(f) TEM images. (g)–(j) HAADF-STEM image and the corresponding EDS elemental mapping images (reproduced with permission from ref. 176, copyright Chinese Chemical Society 2021). (k) SEM images, (l) TEM images, and (m) XPS spectra of (i) fresh CuSx, (ii) AC-CuSx, and (iii) AC-CuSx samples after 40 min of ECO2R in 0.1 M KHCO3 at −0.85 VRHE and (iv) the DS-CuSx sample (reproduced with permission from ref. 177, copyright ACS 2018). | ||
3.7 Catalyst support materials for improving electrocatalytic performance
The supporting materials significantly impact the catalytic performance of electrocatalytic materials. In particular, TMD NSs show promise as supporting materials.178,179 Composite catalysts have high catalytic performance due to the synergistic interaction of TMDs, supporting materials, and electrocatalysts.180,181 Kahsay et al.182 successfully deposited copper sulfide nanoparticles onto thermally synthesized copper oxide using a simple and facile SILAR method and examined its catalytic activity for ECO2R (Scheme 1 in Fig. 26). The modified nanocomposites exhibited high selectivity for formate formation at low overpotential. Remarkably, a maximum faradaic efficiency of 84% and an enhanced partial current density of −20 mA cm−2 were obtained at an overpotential of −0.7 VRHE. It was observed that copper sulfides undergo phase changes during ECO2R, which can contribute to the enhanced electrocatalytic activity. As a result, together with copper oxides, a catalytic synergy is created in the composite, and more favourable adsorption sites are generated to facilitate the ECO2R. This study paves the way for controlling the composition–selectivity relationship using a facile and scalable catalyst synthesis approach. Also, Li et al.183 produced extremely porous Cu2O/CuS nanocomposites, as evidenced by X-ray diffraction (XRD), Raman spectroscopy and scanning electron microscopy (SEM) (Fig. 26a–i), showing a better formate FE of 67.6% at a jHCOOH value of 15.3 mA cm−2 at −0.5 VRHE in ECO2R. More significantly, with the same applied potential, the current density remained unchanged for at least 30 hours with an average FE of 62.9%. DFT modelling showed that CuS(110) facets favour HCOOH over CO, with substitutional surface OS or vacancy VS species expected to result in lower onset potentials and increased catalytic activity, with both COOH*- and OCHO*-mediated ECO2R routes predicted to be involved. As a result, this increases the yield of HCOOH with a better current density than the Cu, CuS, and Cu2O electrocatalysts. | ||
| Fig. 26 Scheme 1: illustration of thermal oxidation and SILAR growth: (i) copper mesh before thermal oxidation, (ii) Cu2O/CuO grown on copper mesh, (iii) adsorption of Cu2+ and NO3− and the formation of an electrical double layer, (iv) rinsing (I) removes excess unabsorbed Cu2+ and NO3−, (v) reaction of S2− with pre-adsorbed Cu2+ ions to form CuS, and (vi) rinsing (II) to remove excess and unreacted species and form the solid solution CuS on surface of the Cu mesh/Cu2O/CuO (reproduced with permission from ref. 182, copyright Springer 2019). (a) Schematic illustration of the synthesis of Cu2O/CuS nanocomposites. (b) and (c) SEM images of Cu2O/CuS nanocomposites grown on a copper gauze collected at different magnifications. (d) HR-TEM image collected from a representative Cu2O/CuS particle. (e) The corresponding FFT image of (d). Blue and yellow dashed lines highlight the diffraction rings of polycrystalline Cu2O and CuS particles, respectively. (f)–(i) HAADF-TEM image of Cu2O/CuS nanocomposites and the corresponding mapping images of elements O, Cu, and S (reproduced with permission from ref. 183, copyright ACS 2021). | ||
4. Theoretical studies of Cu/S-based ECO2R Catalysts
As knowledge of electrocatalysis grows, scientists are no longer satisfied with investigating macroscopic phenomena in experimentation; instead, they are exploring the complex microscopic world to gain insight into the mechanism underlying electrocatalytic reactions. This effort is significant for the design and evaluation of highly efficient electrocatalysts. The rapid development of supercomputers has resulted in significantly greater computational speed and performance, enhancing theoretical calculations. In addition, modelling and simulated environments are now closely consistent with the experimental surroundings, resulting in accurate results.184–186 Nowadays, computational models are used not only to shed light on experimental phenomena but also to direct research strategies and the development of efficient electrocatalysts.184–186Inspired by this, Fonseca et al. discussed theoretically how the tuning ratio of Cu/S on 2D CuSx nanomaterials influenced the ECO2R performance.187 Their findings demonstrate that the adsorption modes and strength of the ECO2R intermediate on CuSx monolayers vary considerably based on the Cu/S ratio, resulting in different catalytic activities. For instance, CuS0.5 catalytic systems showed high onset potential values even though the system was slightly more favourable for reduction toward HCOOH than CO. The CuS system's minimal onset potential for CO or HCOOH suggests that the system could potentially act as a catalyst to form 2e− products. In comparison, CuS1.5 revealed minimal onset potential values for the generation of CO, HCOOH, CH4, and CH3OH, which are, respectively, 0.19, 0.19, 0.53, and 0.53 (VRHE) less than the values determined for Cu(111). The simulation also revealed that, on CuS1.5, the reaction toward CH4/CH3OH was limited by the *CO → *COH formation, while on both the CuS1.5 and CuS surfaces, the CO2 → *COOH generation was the potential determining step for CO and HCOOH formation. According to the calculation, the facile *CO desorption on CuS surfaces could restrict the formation of CH4/CH3OH. Therefore, methods for breaking the *CO and *COOH or *CHO/*COH interactions' linear scaling could be investigated as well in order to enhance most of these systems, while the performance of CuS as a catalyst for the production of CH4 and CH3OH could be improved by simply increasing the CO adsorption strength. In summary, their DFT calculation discovered that increasing the sulfur content in 2D copper sulfide materials promotes CO and HCOOH formation at lower applied potentials and facilitates methane and methanol production. Although these outcomes seem promising from a thermodynamic standpoint, more research into the kinetic behaviour of processes using the most promising systems, considering defect creation under operating conditions, and more research examining additional reaction pathways are essential for strengthening the concept of using these systems as potential electrocatalysts.
Another group proposed experimentally supported theoretical simulation to investigate the mechanism of ECO2R on S-modified Cu electrocatalysts for HCOOH selectivity.188 They discovered that the surrounding environment and symmetry of the remaining sulfur atoms greatly influence their stability. Most sulfur exists in somewhat unstable forms that satisfy the strong CO* surface-enhanced infrared absorption spectroscopy signal and the experimentally confirmed negative XPS shift. From the ECO2R energy graphics, it was revealed that these types of S atoms cannot immediately promote formic acid generation; instead, they produce a highly dominant CO* and have a large CO* adsorption capacity. However, the study discovered that the sulfur atoms' strong CO* adsorption improves the CO* coverage to an almost four times higher level than on a bare Cu. With such a dense CO* coverage, specific surface reactive sites are restricted, resulting in a solution-phase CO2 reduction pathway leading to the formation of highly selective HCOOH. Also, it has been found that CO* can considerably stabilize residual S, and the adsorption and associated electronic structure modulation studies have revealed the source of CO* adsorption improvement. Lastly, the group proposed a synergy between residual S and CO* dominating the HCOOH formation in ECO2R using experimental findings from published research and their DFT calculations.118,177 These findings offer novel insights into the fundamental role of atomic sulfur in the selective formation of ECO2R products and intermediates on metallic Cu electrodes. Advanced characterization techniques, i.e., in situ/operando studies189 and computational approaches,190,191 must be correlated to better understand the structure–stability–efficiency connections of electrocatalysts. This connection is also essential because of the high selectivity of these catalysts regarding particular catalytic processes and product generation.
5. Perspectives on Cu/S-based ECO2R catalysts
The catalyst's nanostructure, porosity, morphology, stability, surface area, and particle size influence ECO2R by affecting the adsorption and desorption of CO2 molecules. These changes in adsorption and desorption processes lead to different products. In addition to the several modification strategies, the performance of Cu/S-based nanomaterials towards ECO2R is strongly connected with several other factors. This section highlighted the novelty aspects related to Cu/S-based nanomaterials.Controlled morphology is critical for understanding the structure–activity relationships of Cu/S-based nanomaterials in ECO2R catalysis. For example, Cu/S-based nanomaterials have been developed with various nanoscale morphologies, including nanoarrays,111 cubes,104 ultrathin nanosheets,150 hollow polyhedrons,176 and hierarchical structures.150 Also, it is to be noted that the morphology and ECO2R performance vary among Cu/S-based nanomaterials due to the precursor effect.121 The morphology-related characterization studies showed that different sulfur source precursor materials have different solubilities and rates of release during hydrothermal synthesis, resulting in diverse surface morphologies at the end.121 Several of these catalysts also have highly porous frameworks,108 aiding electrolyte permeability. Moreover, a greater surface area might offer more multi-active sites, which is also advantageous for producing an optimal nanostructure for improved ECO2R activity.151,153
Notably, the phase engineering approach also has a vital role in ECO2R. For example, phase changes in Cu/S-based nanomaterials are observed due to S leaching during ECO2R. For instance, during the reduction process, XPS and HRTEM analyses showed that thermodynamically unstable CuS undergoes phase changes to metallic Cu, CuO, or Cu2O.117 For example, Shinagawa et al. employed XPS with SAED studies for the post-electrolysis sample. The group observed that during ECO2R, S-modified materials lose excess sulfur irrespective of initial sulfur concentration due to the cathodic environment, resulting in modification of particle size and the co-existence of Cu2S and Cu metal, significantly interfering with the electrocatalyst's activity.128 Zhao's group110 performed XPS and HRTEM to demonstrate that CuS nanosheets undergo partial reduction to metallic Cu, and the metallic Cu phase is partially oxidized to CuO after ECO2R. Zhang et al.176 also did XPS and revealed that CuS-HP synthesized from HKUST-1 was transformed to Cu(S) with a minimal Sδ− (0 ≤ δ ≤ 2) and metallic Cu. This study revealed that although Cu(S) has minimal S concentrations, the polyhedron structures offer high activity and stability during the ECO2R. As a result, in situ reconstruction of CuS in ECO2R promotes the ECO2R activity and product selectivity.
Utilizing highly conducting substrates to load copper sulfides, such as copper oxide,182 carbon-based materials,119 and highly porous 3D foams,111 improves electron conduction and generates more electrocatalytic active sites. Notably, forming copper sulfides–conducting substrate bonds improves catalyst stability, resulting in a longer lifetime.119
Other techniques for increasing catalytic activity include the compositional changes of bi-metallic sulfides in comparison to mono-metallic sulfides to generate a heterostructure capable of providing synergistic coupling,160 interfacial phenomena,161 and defect-rich structures142 to maximize the benefits of metals and sulfides. The number of exposed active sites can be increased, and ECO2R performance can be improved through heterostructure engineering strategies by doping highly functional single-metal nanoparticles169 and non-metals171 on metal sulfides. Also, constructing heterostructures can improve ECO2R efficiency by adjusting the interfaces and electrical states and offering more active sites. So, lower onset potential, increased cathodic current, higher TOF, and lower Tafel slope values can be obtained during the ECO2R process.187
Furthermore, the ECO2R performance varies across Cu/S-based nanomaterials based on their S content or S-vacancy concentration. Vacancy creation through defect engineering techniques, i.e., S-vacancy162 and Cu-vacancy,172 can shift the product selectivity of ECO2R to C2+ products by modifying the electronic structure of S and Cu sites in Cu/S-based nanomaterials.
6. Comparative analysis of Cu/S-based ECO2R catalysts with other best-performing Cu-based and metal–sulfide-based ECO2R catalysts
The performance data and recent research of earlier studies on different Cu/S-based nanomaterials are summarized in Table 1. Tables 2 and 3 present the best-performing Cu-based and metal–sulfide-based nanomaterials (MS-based nanomaterials) for ECO2R (other than copper–sulfide-based) to compare the catalytic efficacy of Cu/S-based nanomaterials (summarized in Table 1) with an emphasis on partial current density and faradaic efficiency. Table 1 indicates that many Cu/S-based nanomaterials have demonstrated acceptable levels of stability during the ECO2R as well as good partial current density and faradaic efficiency for mainly formate/HCOOH production. However, for C1 and C2+ products, the performance of Cu/S-based nanomaterials is unsatisfactory in terms of faradaic efficiency and product selectivity. Other than HCOOH/formate, several Cu/S-based nanomaterials such as N-doped Cu2S thin layers,171 polycrystalline Cu (Cu-s),100 S-doped spherical coral-like CuO,174 CSVE-Cu,142 DSV-engineered CuS,143 S-HKUST-1,154 CeO2-modified CuS nanoplates,158 and Cu2S nanocrystal-decorated Cu nanosheets,155 have yielded C2+ products. However, their selectivities are much lower compared to those of best-performing Cu-based catalysts, i.e., dCu2O/Ag2.3%,192 Ag–Cu2O,193 Cu3N-derived Cu nanowires,194 CuO-derived Cu,195 Cu(salophen)-coated GDE,196 Cu@Cu2(OH)3NO,197 Cu2O films,198 oxygen-bearing copper,199 and nano-defective Cu nanosheets.200 In conclusion, combining Cu/S-based nanomaterials with other nanomaterials, i.e., metal oxides, single-atom metals, metal selenides and metal phosphides, through various engineering/modifications strategies could be effective for higher C2+ product selectivity. In addition, cadmium-based sulfide, molybdenum-based sulfide and zinc-based sulfide, as summarized in Table 3, electrocatalysts exhibited better current density and faradaic efficiency performance than Cu/S-based and Cu-based catalysts for CO production. Furthermore, for CH4 production, the CuS/Ni foam110 electrocatalyst offered better performance than Cu/MoS2201 and Fe4.5Ni4.5S8202 regarding partial current density and faradaic efficiency.| Products | Electrocatalysts | Method of synthesis | Potential (VRHE) | Electrolyte used | Cell | Partial current density (mA cm−2) | Faradaic efficiency (%) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| HCOOH | Sulfur-modified Cu2O | Wet chemistry approach | −0.9 | 0.1 M KHCO3 | H-type cell | 260 ± 16 | ≈90 | 80 | 99 |
| C2H4 | Cu-s | Electrochemical reduction | −1.2 | 0.1 M KHCO3 | H-Type cell | 40.8 | 68.6 | 8 | 100 |
| HCOOH | PTFE-coated CuS/BM | One-step chemical bath process | −0.7 | 0.5 mol L−1 KHCO3 | H-type cell | 50 | 70 | 1.5 | 101 |
| CO | Hollow-shaped CuS microcubes | Galvanic replacement technique | −0.16 | 0.5 M KHCO3 | H-type cell | — | 32.7 | 10 | 104 |
| HCOOH | Hollow ordered porous copper sulfide cuboctahedra | Hard templating strategy | −1.1 | 0.1 M KHCO3 | H-type cell | 7.8 | 70.3 | 26 | 108 |
| C2H4 | Copper sulfide film consisting of nanoparticles | Thermal decomposition | −0.13 | 1 M KHCO3 | H-type cell | ≈0.08 (total) | — | — | 109 |
| HCOOH | Cu2S over copper foam | Electrochemical deposition | −2.0 V (vs. Ag/Ag+) | 0.5 mol L−1 BmimBF4 | H-type cell | 5.3 | 85 | 6 | 111 |
| CH4 | CuS/Ni foam | Hydrothermal method | −1.1 | 0.1 M KHCO3 | H-type cell | 7.32 (total) | 73.5 ± 5 | 60 | 110 |
| HCOOH | CuS nanoflowers | Ethylene glycol solvothermal method | −0.8 to ∼−1.0 | 0.1 M KHCO3 | H-type cell | ≈6.5 | ∼52 | 2 | 117 |
| HCOOH | Cu1.81S@MWCNT-600-OD | Two-step coupling approach | −0.67 | 0.5 M KHCO3 | H-type cell | 3.75 (total) | 82 | 20 | 119 |
| HCOOH | Cu2S@C | Liquid phase sulfidation | −0.78 ± 0.02 | 0.5 M KCl + 0.5 M KHCO3 | H-type cell | 1.5 (total) | 12 | 5 | 120 |
| CO | CuS catalyst thiourea precursors | Hydrothermal synthesis | −0.51 | 0.1 M KHCO3 | H-type cell | — | 72.67 | 5 | 121 |
| HCOOH | Cu2S nanocrystals | Electrochemically driven cation exchange method (ED-CE) | −0.9 | 0.1 M NaHCO3 | H-type cell | 19 | 87.3 | 9 | 122 |
| HCOOH | Cu2S | ED-CE | −0.2 | 0.05 M K2CO3 | H-type cell | — | ≈70 | 16 | 123 |
| CO | S-derived Cu–Sb | Heat-up colloidal nanoparticle route | −1.0 | 1 M KHCO3 | Flow cell | 37.6 | 80.5 | 24 | 124 |
| HCOOH | S-CNWs | Chemical oxidation followed by solvothermal reaction | −0.59 | 0.25 M KHCO3 | H-type cell | ≈10 | ∼60 | 2 | 125 |
| HCOOH | Submicron-sized Cu–S | Solvothermal approach | −0.8 | 0.1 M KHCO3 | H-type Cell | ≈10 (total) | 80 | 12 | 128 |
| HCOOH | CuSx nanoparticles | Dipping | −0.6 | 0.1 M KHCO3 | H-type cell | 2.5 | 72 | 72 | 129 |
| HCOOH | S-Cu2O/Cu | Hydrothermal reduction followed by in situ reconstruction | −1.0 | 0.1 M KHCO3 | H-type cell | 5 | 70 | 6 | 134 |
| HCOOH | CuS 811 derived S-doped Cu2O | Surfactant-free double-replacement reaction | 0.1 M KHCO3 | Flow cell | 321 | 80 | 33 | 135 | |
| Alcohols (ethanol + propanol) | CSVE-Cu | Solution-phase growth | –0.95 | 1.0 M KOH | Flow cell | 126 | 31.6 | 16 | 142 |
| n-Propanol | DSV-engineered CuSx | Lithium tuning approach | −1.05 | 0.1 M KHCO3 | H-type cell | 9.9 | 15.4 ± 1 | 10 | 143 |
| HCOOH | SnO2@C onto snowflake-like Cu2S | Hydrothermal method | −1.0 | 0.1 M KHCO3 | H-type cell | 15.6 | 88 | 50 | 148 |
| HCOOH | CuS/SnO2-S derived Cu/Sn/Cu6.26Sn5 catalysts | Redox reaction | −0.8 | 0.5 M KHCO3 | H-type cell | ≈18.8 | 84.9 | 10 | 149 |
| CO | SnO2 confined on CuS nanosheets | One-pot precipitation synthesis | −1.0 | 0.1 M KHCO3 | H-type cell | 15.24 | >85 | 24 | 150 |
| HCOOH | Cu2SnS3 nanoplates | Step-growth approach | −1.2 | 1 M KHCO3 | Flow cell | 181 | 72.4 | 3 | 151 |
| CO | CuInS2 hollow nanostructures | Template-free approach | −0.7 & −1.0 | 0.1 M KHCO3 | H-type cell | ≈13 (total) | 82.3 | 4 | 152 |
| HCOOH | Graphdiyne/copper sulfide | Hydrothermal followed by in situ growth | −0.9 | 0.1 M KHCO3 | H-type cell | 65.6 (total) | 70 | 4 | 153 |
| C2H4 | S-HKUST-1 | Local sulfur doping strategy | −1.94 | 1.0 M KOH | Flow cell | 228 | 57.2 | 8 | 154 |
| Ethanol | Cu2S nanocrystal-decorated Cu nanosheets | Hydrothermal followed by self-assembly | −1.2 | 0.1 M KHCO3 | H-type cell | jC2H5OH = 20.7 | 46 | 20 | 155 |
| CH4 | CuxZnyS nanoparticles | Seed-mediated growth method | −0.98 | 0.1 M KHCO3 | H-type cell | jmethane = 4.58 | 76 | 5 | 156 |
| Ethanol | SD-CuCd2 | Co-Precipitation | −0.8 | 0.1 M KHCO3 | H-type cell | 0.6 | 32 | 6 | 157 |
| C2+ products | CeO2-modified CuS nanoplates | Self-assembly method | −0.9 | 1 M KOH | Flow cell | JC2+ = 75.3 | 75 | 180 | 158 |
| HCOOH | Bi/CeO2/CuS | Solvothermal process | −0.9 | 0.5 M KHCO3 | H-type cell | 17 (total) | 88 | 6 | 159 |
| HCOOH | Heterostructure Bi–Cu2S nanocrystals | One-pot solution-phase approach | −1.0 | 0.1 M KHCO3 | H-type cell | ∼17 | >98 | 10 | 160 |
| HCOOH | CuS@1T-N-WS2/WO3 | A facile solvothermal process followed by a reflux strategy | −1.3 | 0.5 M KHCO3 | H-type cell | 125.05 | ∼55.6 | 2 | 161 |
| CO | MoS2–CuS | Hydrothermal approach | −0.6 | 0.1 M KHCO3 | H-type cell | 3.6 | 83.2 | 5.55 | 162 |
| Ethanol | Mo4+-doped CuS nanosheet-assembled hollow spheres | One-step solution-phase approach | −0.6 | 1.0 M KOH | Flow cell | Jethanol = 186.5 | 67.5 | 14 | 169 |
| HCOOH | Cu-doped Bi derived from CuS–Bi2S3 | Solvothermal followed by in situ reconstruction | −0.86 | 1 M KOH | MEA cell | 1132 | 90 | 100 | 170 |
| C2H4 | N-doped Cu2S thin layers | Annealing process | −0.98 | 0.1 M KHCO3 | H-type cell | ∼2.6 | 14.72 | 0.25 | 171 |
| CO | SNC@Cu1.96S | Surface coating and calcination | −0.84 | 0.5 M KHCO3 | H-type cell | 37.2 | 85.2 | 13 | 172 |
| HCOOH | Cu-5000S | Electrochemical deposition | −0.8 | 0.1 M KHCO3 | H-type cell | 13.9 | ≈75 | 12 | 173 |
| C2H4 | Sulfur-doped spherical coral-like CuO | Local sulfur doping strategy | −1.3 | 0.1 M KHCO3 | H-type cell | 15 | 48.4 | 20 | 174 |
| HCOOH | S-Cu2O-14 | Hydrothermal sulfurization method followed by an electrochemical reduction process | −1.0 | 0.1 M KHCO3 | H-type cell | 16.3 | 67.2 | 20 | 175 |
| HCOOH | CuS hollow polyhedron derived from a MOF | MOF self-sacrificial template method | −0.6 | 0.5 M K2SO4 aqueous solution | H-type cell | ≈16 | >90 | 36 | 176 |
| HCOOH | AC-CuSx | Pulsed electrochemical deposition | −0.9 | 0.1 M KHCO3 | H-type cell | 9 | 75 | 3 | 177 |
| HCOOH | CuS deposited onto thermally prepared Cu2O/CuO heterostructure | Thermal oxidation followed by SILAR | −0.7 | 0.1 M KHCO3 | H-type cell | 20 | 84 | 2.5 | 182 |
| HCOOH | Cu2S/Cu2O nanocomposites | Hydrothermal approach and electrochemical treatment | −0.9 | 0.1 M KHCO3 | H-type cell | 12.3 | 67.6 | 30 | 183 |
| Electrocatalysts | Main products | Potential (VRHE) | Electrolyte used | Cell | Current density (mA cm−2) | Faradaic efficiency (%) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Single Cu atom | Acetone | −0.76 | 0.1 M KHCO3 | H-type cell | 1.0 | 36.7 | 5 | 203 |
| TS-Cu | Ethylene | 3.5 | 1 M KOH | Flow cell | 800 | 72 | 200 | 204 |
| dCu2O/Ag2.3% | Ethanol | −0.87 | 1 M KOH | H-type cell | 326.4 | 40.8 | 12 | 192 |
| Cu-NC | Ethanol | −1.01 | 0.1 M KHCO3 | H-type cell | — | 18.4 | 8 | 205 |
| Cu2O/NCS | Ethylene | −1.3 | 0.1 M KHCO3 | H-type cell | — | 24.7 | 4 | 206 |
| CuN4 | Ethanol | −1.2 | 0.1 M KHCO3 | Flow cell | 16.2 | 55 | 1 | 207 |
| Cu1Ni2@N-MWCNT | Formate | −0.53 | 0.1 M KHCO3 | H-type cell | 3.0 | 90 | 10 | 208 |
| Cu-doped carbon xerogel | Methane | 0.1 M KHCO3 | H-type cell | — | 5 | 209 | ||
| Cu-nanoparticles | Formate | −1.0 | 0.1 M KHCO3 | H-type cell | 6.53 | 54 | 12 | 210 |
| GO-VB6-Cu-2 | Ethanol | −0.250 | 0.1 M KHCO3 | H-type cell | 7.452 | 56.3 | 24 | 211 |
| Cu–N–C | Carbon mono oxide | −0.7 | 0.1 M KHCO3 | H-type cell | 3.61 | 92 | 60 | 212 |
| Cu2O@Cu-MOF | Methane | −1.71 | 0.1 M KHCO3 | H-type cell | 8.4 | 79.4 | 1 | 213 |
| PcCu–Cu–O | Ethylene | −1.2 | 0.1 M KHCO3 | H-type cell | 7.3 | 50 | 4 | 214 |
| Cu90In10/C | Carbon mono oxide | −0.75 | 0.1 M KHCO3 | H-type cell | 9.85 | 85 | 4 | 215 |
| CuO/CuTCPP | Formic acid | −1.65 | 0.5 M EMIMBF4 | H-type cell | 4.5 | 85.2 | 5 | 216 |
| Cu(111)@Cu-THQ | Ethylene | −1.4 | 0.1 M KHCO3 | H-type cell | 14.3 | 42 | 8 | 217 |
| CuZn | C2+ | −1.0 | 0.1 M KHCO3 | H-type cell | 1.5 | 25 | 7 | 218 |
| Cu@CuxO | C2+ | −1.78 | 0.1 M KHCO3 | H-type cell | 150 | 70 | 20 | 219 |
| Cu–Sn | Carbon monoxide | −0.7 | 0.1 M KHCO3 | H-type cell | 4.5 | 90 | 12 | 220 |
| Cu3N-derived Cu nanowires | C2 | −1.0 | 0.1 M KHCO3 | H-type cell | −50.6 | 86 | 28 | 194 |
| CuO-derived Cu | C2+ | −0.48 | KH2PO4 | H-type cell | −210 | 9.6 | 1 | 195 |
| Cuv–Cu2O | Ethylene | −0.76 | 0.1 M KHCO3 | H-type cell | 15.7 | 51 | 10 | 221 |
| Cu–Zn | Carbon monoxide | −1.1 | 0.1 M KHCO3 | H-type cell | 4.3 | 74 | 4 | 222 |
| Cu@Cu2(OH)3NO | Ethylene | −1.2 | 0.1 M KHCO3 | H-type cell | 80 | 31.8 | 20 | 197 |
| Cu2O-derived Cu/PdCl2 | Ethylene | −1.0 | 0.1 M KHCO3 | Flow cell | — | 30.1 | 8 | 223 |
| Cu2O films | C2 | −0.99 | 0.1 M KHCO3 | H-type cell | −52 | 39 | 1 | 198 |
| Cu2O-derived Cu particles | Ethylene | −0.98 | 0.1 M KHCO3 | H-type cell | −13.3 | 43 | 1 | 224 |
| Branched CuO nanoparticles | Ethylene | −1.0 | 0.1 M KHCO3 | H-type cell | −22.0 | 53 | 12 | 225 |
| Cu–Pd alloy | Carbon monoxide | −0.9 | 0.1 M KHCO3 | H-type cell | — | 87 | 5 | 226 |
| Cu–Pt alloy | Methane | −1.6 | 0.1 M KHCO3 | H-type cell | 0.598 | 21 | 0.5 | 227 |
| CuMgAl LDH | Acetic acid | −0.4 | 0.1 M KHCO3 | Flow cell | — | 24 | 228 | |
| PD-CuOx/C | Ethylene | −1.4 | 0.1 M KHCO3 | MEA cell | 26.0 | 45 | 48 | 229 |
| CuCo2Se4 | Acetate | −0.25 | 0.3 M NaHCO3 | H-type Cell | 26 | 100 | 100 | 230 |
| Cu(salophen)-coated GDE | C2+ | −1.2 | 1 M KOH | H-type cell | 121 | 37 | 2 | 196 |
| Ag–Cu2O | Acetic acid | −0.9 | 1 M KOH | H-type cell | 310 | 70 | 20 | 193 |
| Hydroxo-bridged phenanthroline Cu(II) molecule | Ethylene | −1.25 | 0.1 M CsHCO3 | H-type cell | −5.5 | 42 | 15 | 231 |
| Cu NC spheres | Ethylene | −1.1 | 0.1 M KHCO3 | H-type cell | −9 | 41 | 1 | 232 |
| Copper electrodes | Methane | −1.2 | 0.1 M KHCO3 | H-type cell | 5 | 45 | 8 | 233 |
| Oxygen-bearing copper | Ethylene | −0.95 | 0.3 M NaHCO3 | H-type cell | 44.7 | 45 | 1 | 199 |
| Nano-defective Cu nanosheets | Ethylene | −1.0 | 0.1 M K2SO4 | Flow cell | 66.5 | 83.2 | 1 | 200 |
| Metal–sulfides | Electrocatalysts | Potential (VRHE) | Electrolyte used | Main products | Cell | Partial current density (mA cm−2) | Faradaic efficiency (%) | Stability (h) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Cadmium-based sulfide | Ag–CdS1−x | −1.1 | 1 M KHCO3 | CO | H-type cell | 53.7 | 87.1 | — | 234 |
| P–Cd|S | −0.8 | 0.5 M KHCO3 | CO | H-type cell | 89.8 | 88 | 10 | 235 | |
| Nanoneedle-shaped CdS | −1.2 | 0.1 M KHCO3 | CO | Flow-cell | 212 | 95.5 ± 4.0 | 24 | 236 | |
| CdS nanorods | −1.2 | 0.1 M KHCO3 | CO | H-type cell | ∼22 | 81 | 10 | 237 | |
| CdS with S-vacancy | −0.8 to −1.1 | 0.5 M KHCO3 | CO | H-type cell | ∼1.3 | 95 | 40 | 238 | |
| S vacancy engineered CdS nanorods | −1.1 | 0.5 M KHCO3 | CO | H-type cell | 20.5 | 100 ± 0.5 | 10 | 239 | |
| CdS supported CNTs | −1.2 | 0.1 M KHCO3 | CO | H-type cell | ∼13 (total) | ∼95 | 10 | 240 | |
| CdS/MXene | −1.0 | 0.1 M KHCO3 | CO | H-type cell | ∼6 | 94 | 8 | 241 | |
| Ag2S/CdS | −1.1 | 0.5 M KHCO3 | CO | Flow-cell | 10.6 | 95 | 20 | 242 | |
| Tin-based sulfide | Hybridized composite of defective SnS2 nanosheets and Ag nanowires | −0.1 | 0.5 M KHCO3 | HCOOH | H-type cell | 23.3 | 60 | 10 | 243 |
| 5% Ni-doped SnS2 nanosheets | −0.9 | 0.1 M KHCO3 | CO & HCOOH | H-type cell | 19.6 (total) | 93 | 8 | 244 | |
| Sn(S)/Au | −0.75 | 0.1 M KHCO3 | HCOOH | H-type cell | 51 | 93.3 | 40 | 245 | |
| Stannous sulfide modified with amino-functionalized carbon | −0.9 | 0.5 M KHCO3 | HCOOH | H-type cell | 41.1 | 92.6 | 15 | 246 | |
| SnS2 nanosheets supported on rGO | −1.4 | 0.5 M NaHCO3 | HCOOH | H-type cell | 11.75 | 84.5 | 2 | 247 | |
| Tin(IV) sulfide monolayers | −0.8 | 0.1 M KHCO3 | HCOOH | H-type cell | ∼45 (total) | 94 ± 5 | 80 | 248 | |
| N–Sn(S) nanosheets | −0.7 | 0.1 M KHCO3 | HCOOH | Flow-cell | 25 (total) | 93.3 | 20 | 249 | |
| Semimetal phased 1H-SnS2 | −0.8 | 0.1 M KHCO3 | CO | H-type cell | 10.9 | 98.2 | 15 | 250 | |
| Bismuth-based sulfide | S doped-Bi2O3-CNT | −0.9 | 0.5 M KHCO3 | HCOOH | H-type cell | 48.64 | 97.06 | 10 | 251 |
| S-derived Bi | −0.75 | 0.5 M NaHCO3 | HCOOH | H-type cell | 5 (total) | 84 | 24 | 252 | |
| Bi2S3–Bi2O3/rGO | −0.9 | 0.1 M KHCO3 | HCOOH | H-type cell | ∼3.8 | 90 | 24 | 253 | |
| Bi–Bi2S3 | −1.0 | 0.1 M KHCO3 | HCOOH | H-type cell | ∼14 | 85 | 12 | 254 | |
| S-doped two-dimensional (2D) bismuth subcarbonate (S-BiOC) | −0.9 | 0.5 M KHCO3 | HCOOH | H-type cell | 29 | >90 | 20 | 255 | |
| Bi2S3–PPy | −0.9 | 0.5 M KHCO3 | HCOOH | H-type cell | 56.95 (total) | 91.18 | >110 | 256 | |
| 2D defective Bi2S3 NSs | −0.93 | 0.5 M KHCO3 | HCOOH | H-type cell | ∼30 | >90% | 200 | 257 | |
| Molybdenum-based sulfide | Bulk MoS2 | −0.764 | 4 mol% EMIM-BF4 solution | CO | H-type cell | 65 | 98 | 10 | 258 |
| rGO–PEI–MoSx | −0.65 | 0.5 M NaHCO3 | CO | H-type cell | 55 (total) | 85.1 | — | 259 | |
| N-MoS2@NCDs-180 | −0.9 | 6 mol% EMIM-BF4 solution | CO | H-type cell | 36.2 | 90.2 | 10 | 260 | |
| Cu-g-C3N4/MoS2 | −0.67 | 0.5 M KHCO3 | CH3OH | H-type cell | 78 | 19.7 | 30 | 261 | |
| CuO–ZnO–MoS2 | −0.6 & −0.9 | 0.5 M KHCO3 | Methanol & ethanol | H-type cell | 121 (total) | 24.6 & 11.1 | 3 | 262 | |
| NCMSH | −0.7 | 4 mol% EMIM-BF4 solution | CO | H-type cell | 34.31 | 92.68 | 24 | 263 | |
| Cu/MoS2 | −1.4 VSCE | 0.1 M KHCO3 | CH4 | H-type cell | ∼3.2 | 17.08 | 48 | 201 | |
| VA-Mo0.95Nb0.05S2 | −0.8 | 50 vol% EMIM–BF4 | CO | H-type cell | 237 (total) | 80 | — | 264 | |
| Cu2O@MoS2 nanosheets | −1.3 and −1.1 | 0.5 M KHCO3 | Methanol & ethanol | H-type cell | 113 (total) | 12.3 & 7.9 | 2 | 265 | |
| H-E-MoS2 with decoration of fluorosilane | −0.9 | 6 mol% EMIM-BF4 solution | CO | H-type cell | — | 81 | 10 | 266 | |
| Monolayers of MoSeS alloys | −0.15 | 4 mol% EMIM-BF4 solution | CO | H-type cell | 43 | 45.2 | 10 | 267 | |
| Indium-based sulfide | In nanoparticles on In2S3 nanosheets | −1.0 | 1 M KHCO3 | HCOOH | H-type cell | 40.3 | 76 | 8 | 268 |
| Mn–In2S3 | −0.9 | 0.1 M KHCO3 | HCOOH | H-type cell | 20.1 (total) | 90 | 8 | 269 | |
| ZnIn2S4 | −1.18 | 1 M KHCO3 | HCOOH | Flow cell | ∼298 | 99.3 | 60 | 270 | |
| Flower-shaped In2S3 | −2.3 | BmimPF6/MeCN–H2O solution | HCOOH & CO | H-type cell | 25.6 | 86 | 10 | 271 | |
| S–In | −0.98 | 0.1 M KHCO3 | HCOOH | H-type cell | 84 (total) | 93 | 10 | 272 | |
| ZnO NW/CIGS/InS | −0.24 | 0.1 M KHCO3 | HCOOH | H-type cell | 0.35 | 77.2 | 10 | 273 | |
| ZnO NF/CIGS/InS | −0.24 | 0.1 M KHCO3 | HCOOH | H-type cell | — | 68.1 | — | 273 | |
| Lead-based sulfide | Wafer-structured sulfur-derived Pb | −1.08 | 0.1 M KHCO3 | HCOOH | H-type cell | 12 | 88 | — | 274 |
| PbS nanocrystals | −1.2 | 0.1 M KHCO3 | HCOOH | H-type cell | — | 97.6 ± 5.3 | 10 | 275 | |
| Zinc-based sulfide | ZnS@Zn | −2.4 VFc/Fc+ | Propylene carbonate/(CH3CH2CH2CH2)4N(ClO4) | CO | H-type cell | 6.4 | 92 | 4 | 276 |
| S–Zn–S nanosheets | −0.8 | 0.1 M KHCO3 | CO | H-type cell | ∼11 | 94.2 | 15 | 277 | |
| ZnS@ZnO | −0.56 | 1 M KOH | CO | H-type cell | ∼109 | 97.2 ± 0.5 | 40 | 278 | |
| TA-ZnS | −1.9 VAg|AgCl | 0.1 M KHCO3 | CO | H-type cell | ∼7 | ∼83 | 20 | 279 | |
| Titanium-based sulfide | Semi-metallic titanium disulfide (TiS2) | −0.5 | 0.1 M NBu4–PF6 | CO | H-type cell | 5 | 83 | 16 | 280 |
| Other bimetallic-sulfide | Ni2FeS4 nanosheets | −0.7 | 0.1 M KHCO3 | CO | H-type cell | — | 5.9 | 1 | 281 |
| FeS2/NiS nanocomposite | −0.6 | 0.5 M KHCO3 | CH3OH | H-type cell | 3.1 (total) | 64 | 4 | 282 | |
| Bulk Fe/Ni sulfides (Fe4.5Ni4.5S8 pentlandite) | −1.80 VNHE | 0.1 M TBAPF6 | CO & methane | H-type cell | 3 (total) | 87 & 13 | 15 | 202 | |
| Fe3Ni6S8 | −1.80 VNHE | 0.1 M TBAPF6/acetonitrile solution | CO | H-type cell | 15 (total) | 3.6 | 8 | 283 | |
| Fe4.5Ni4.5S4Se4 | −1.80 VNHE | Acetonitrile | CO | H-type cell | 11 | 84 | 2 | 284 |
7. Practical applications of Cu/S-based ECO2R catalysts
The execution of ECO2R on Cu/S-based catalysts on a practical level is still in its initial stages due to several difficulties. The primary challenges to the commercialization of Cu/S-based ECO2R catalysts are (1) considerable CO2 extraction and purification costs, as ECO2R needs highly pure (99.999%) CO2, (2) high energy consumption, (3) low yield of energy-dense C2+ products and (3) a restricted marketplace that is less attractive to investors.285,286 In addition, there are multiple technical obstacles related to ECO2R, such as low catalytic performance, poor product selectivity, unsatisfactory catalytic stability, and non-optimization of cell design for use in practice.287,288 While Cu/S-based materials yielded several products during ECO2R, producing formate/HCOOH was observed as the main product. However some catalysts can produce hydrocarbons, but their selectivities are much lower than those of copper nanoparticles or oxide-derived copper. This section discusses the possibilities of scaling up ECO2R technology to the industrial level, focusing on the practical applications of formate/HCOOH production.Indeed, scaling up an ECO2R cell for formate/HCOOH production is complex because various factors and constraints must be investigated and carefully evaluated to maintain stable cell efficiency.289 A study revealed that the cell must maintain consistent operation for at least 8000 hours for commercial applications. Additionally, it should exhibit an overpotential below 1.0 VRHE, a partial current density between 200 and 1000 mA cm−2, and a formate/HCOOH faradaic efficiency of above 90% during stable operation.290 Another research stated that the electrolyzer should be stable for a minimum of 20000 hours to be financially viable.291 In this context, the proton exchange membrane and electrode framework also play a vital role in stabilizing the cell. Among the various membranes reported, bipolar membranes have received much attention for large-scale use because they can maintain pH gradient, decrease liquid crossover, and promote water separation at the membrane electrolyte interface.292,293 Operating variables such as electrolyte, flow rate, pH, and CO2 feed type can all be optimized to improve cell efficiency.294 Nowadays, there are only a few huge-scale CO2 electrolyzers for methane, CO, C1, C2+, and formic acid generation.295,296 Few academic institutions executed large-scale manufacture of formate/formic acid using ECO2R. For example, in 2008, a pilot plant was constructed with a maximum output of 146 kg CO2 per day, leading to approximately 110 kg formic acid per day at an operating pressure of around 10 bar.297 A different approach to the current scale-up investigation revealed a pre-pilot facility output of 55 kg CO2 per day, resulting in approximately 12 kg of formate per day at ambient pressure.298
While the aforementioned studies demonstrated the practicality of this approach for scaling up, several obstacles must be addressed to maximize plant functionality. One of the primary challenging tasks is maintaining a higher constant current density (recommended >200 mA cm−2) during the ECO2R process. The higher current density value denotes a high electrochemical reaction rate, which raises the rate at which different products are produced. Notably, the electrode's properties and structure, as well as that of both the catalyst and the support material, significantly impact the current density. The lower energy efficiency presents another difficulty. The system demonstrated 28% and 33% energy efficiency in a lab-scale three-compartment set-up over a 1000-hour operation at current densities >100 mA cm−2.299 But, during 450 minutes of operation on a pilot scale, the energy efficiency exceeds 50%.297 However, data on the energy efficiency of various products formed on a large/pilot scale are scarce. Notably, the FE should be higher and the overpotential lower to maintain high cell energy efficiency. The support material on the working electrode, electrocatalytic properties, and anode materials primarily determine these parameters. In addition, the choice of the membrane is crucial because it may substantially decrease the IR drop between the compartments, i.e., cathodic and anodic, lowering the cell overpotential and increasing energy efficiency. It is advised that the potential is between 2.5 and 3 V and that the FE is higher than 90% for industrial-scale implementation.300–302
The cell construction for ECO2R to formate/formic acid remains to be developed for industrial use. Every design has pros and cons of its own. However, the PEM cell layout appears to be a viable strategy for scaling up.303 The PEM design demonstrated a reasonably higher FE for formic acid generation for Cu/S-based electrocatalysts with a stable cell procedure of more than 100 hours.170 However, multiple challenges restrict the commercial use of PEMs for ECO2R to formate/formic acid and other products (i.e., C1 and C2+ products), including declination of membrane performance over time and GDE flooding. When the cell is operated at high pressure, the mechanical strength of the membrane becomes a significant issue, leading to a higher crossover rate of formic acid and membrane damage. Also, further research must be carried out on enhancing cell stability and efficiency. Although the PEM cell demonstrated a FE of 90% and steady performance for 100 hours,170 industrial applications necessitate cell performance for a minimum of 8000 hours with a FE greater than 90%.290 Design aspects and operational variables should be considered when designing a cell assembly that can sustain long-term operation, encounter commercial operation demands, and be cost-effective. However, membranes are costly and need periodic replacement. According to researchers, membrane-less ECO2R cells require more care to prevent frequent maintenance or membrane damage. Another significant problem is the overflow of GDE with electrolytes, which could harm the electrode structure and lower CO2 conversion. Gas-phase ECO2R cells may avoid such issues, so more research into these cells is needed to develop efficient, stable, and long-lasting ECO2R cells.
8. Summary and outlook
This review discusses several strategies for modifying Cu/S-based nanomaterials, which are recognized as highly promising electrocatalysts for ECO2R. These strategies include adjustments to morphologies, structures, nanosize effects, and heterointerfaces, all of which contribute to triggering electronic modulation effects within the nanomaterials. This modulation leads to the generation of multiple active sites and facilitates charge redistribution, ultimately boosting the adsorption energy of intermediates and enhancing electrocatalytic activity. Additionally, recent advancements in engineered Cu/S-based nanomaterials for ECO2R are summarized, shedding light on the interplay between engineering strategies, reaction mechanisms, and electrocatalytic performance. This article provides insights into the formation mechanisms, synthetic strategies, and the diverse morphological and compositional variations observed in Cu/S-based nanomaterials.Despite the progress achieved for the engineered Cu/S-based nanomaterials, there are still some challenges and opportunities that should be addressed based on the following aspects:
(1) Regarding the design and development of Cu/S-based nanomaterials, novel synthesis procedures are required to develop copper sulfide materials with various unique morphologies (i.e., nano-tips, nanoparticles, two-dimensional layered structures, and so on) and crystal and phase structures (i.e., Cu1.97S, Cu1.80S, and so on) to increase their performance. Also, combining different phases and components of low-cost Cu2−xS templates to create versatile and multifunctional nanocomposite heterostructures promotes ECO2R catalysis with multi-active sites and synergistic effects.
(2) The actual role of phase changing CuS in the electrolysis process without converting it to CuO, Cu2O and metallic Cu should be investigated. It is also conceivable to examine the approach of stabilizing S vacancies from a different angle. Therefore, a long-term stability analysis is advised to evaluate the poisoning or degradation of the catalyst, which is vital for the catalyst's design and development.
(3) C2+ product selectivity and stability of Cu/S-based nanomaterials for CO2 reduction should be improved. Most Cu/S-based nanomaterials have relatively high ECO2R catalytic selectivity and high faradaic efficiency towards CO and HCOOH (as summarised in Table 1), and other C1 products and high-energy-dense C2 products are challenging to generate. Thus, obtaining high-energy-dense C2 products is crucial for commercial electrochemical CO2 conversion.
(4) Sophisticated characterization technologies should be implemented. For heterogenous electrocatalysis, the catalyst's surface is an essential active site for the catalytic reaction. The interaction between the surface-active sites and the reaction intermediates is the primary factor influencing the catalytic activity. As a result, more cutting-edge in situ characterization techniques, such as in situ Raman, Fourier transform infrared spectroscopy (FTIR), and X-ray absorption spectroscopy (XAS), must be used to identify the nature of active sites and binding/adsorption energies between active sites and intermediates during ECO2R on Cu/S-based electrocatalysts.
(5) Execution of comprehensive and advanced theoretical techniques Theoretical modelling and simulation are mandatory to understand the reaction process and establish the structure–activity relationship. Several studies have shown that DFT is a practical approach for determining the energy values of chemical intermediates. Also, many reported theoretically expected processes may differ between DFT models. Also, several computational outcomes may be theoretically possible but not experimentally viable. Thus, DFT analysis is closely linked with experimental outcomes, especially in situ technique results, confirming its viability for understanding reaction processes.
Closing the gap between lab-scale research and industrial applications is crucial. Momentous improvements have been observed in the study and in the lab-scale use of Cu/S-based nanomaterials for ECO2R. However, implementing Cu/S-based electrocatalysts on a large scale for real-world applications remains a significant challenge. Smaller batch preparation and controlled laboratory conditions make academic research much more straightforward, while industrial manufacturing requires additional factors like instrument stability, process consistency, and scaling up experiments. Although Cu/S-based electrocatalysts have advanced significantly in terms of practical uses, there is still more work to be done before vast commercial applications. Therefore, reducing the gap between lab-scale research and industrial applications is crucial for Cu/S-based electrocatalysts.
Author contributions
A. M. conducted the literature search, wrote the final manuscript, and created figures and tables. MA, SSM, and BCR supervised and revised the manuscript. All authors were involved in the discussion and critical revision, and the final version of the manuscript was approved.Data availability
No primary research results, software or codes have been included and no new data have been generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge the Birla Institute of Technology, Mesra, for providing the institute research fellowship and Indian Science Technology and Engineering facilities Map (I-STEM/catalytic grant/acad_18/2022-2023) for financial support.References
- N. Patel and D. Mehta, Int. J. Thermofluids, 2023, 20, 100397 CrossRef CAS.
- B. Doğan, M. Shahbaz, M. F. Bashir, S. Abbas and S. Ghosh, Renewable Sustainable Energy Rev., 2023, 184, 113551 CrossRef.
- Y. Huang, Z. Kuldasheva, S. Bobojanov, B. Djalilov, R. Salahodjaev and S. Abbas, Environ. Sci. Pollut. Res., 2023, 30, 10854–10866 CrossRef CAS PubMed.
- L. J. R. Nunes, Environ., 2023, 10, 66 Search PubMed.
- E. Liu, X. Lu and D. Wang, Energies, 2023, 16, 2865 CrossRef CAS.
- B. Dziejarski, R. Krzyżyńska and K. Andersson, Fuel, 2023, 342, 127776 CrossRef CAS.
- N. Yusuf, F. Almomani and H. Qiblawey, Fuel, 2023, 345, 128178 CrossRef CAS.
- W. Chung, W. Jeong, J. Lee, J. Kim, K. Roh and J. H. Lee, Comput. Chem. Eng., 2023, 170, 108106 CrossRef CAS.
- J.-Y. Chen, M. Li and R.-Z. Liao, Inorg. Chem., 2023, 62, 9400–9417 CrossRef CAS PubMed.
- M. Li, K. Yang, M. Abdinejad, C. Zhao and T. Burdyny, Nanoscale, 2022, 14, 11892–11908 RSC.
- G. Bhattacharya, R. Manna, P. Sardar, S. Rahut and A. N. Samanta, Ind. Eng. Chem. Res., 2024, 63(28), 12361–12372 CrossRef CAS.
- Y. Fang, Y. Liu, L. Qi, Y. Xue and Y. Li, Chem. Soc. Rev., 2022, 51, 2681–2709 RSC.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS PubMed.
- S. Wei, W. Liu, C. Yang, P. Bai, X. Kong, W. Sun and L. Xu, Mater. Chem. Front., 2023, 7, 4723–4743 RSC.
- M. Abdinejad, A. Seifitokaldani, C. Dao, E. H. Sargent, X. A. Zhang and H. B. Kraatz, ACS Appl. Energy Mater., 2019, 2, 1330–1335 CrossRef CAS.
- S. Kaur, M. Kumar, D. Gupta, P. P. Mohanty, T. Das, S. Chakraborty, R. Ahuja and T. C. Nagaiah, Nano Energy, 2023, 109, 108242 CrossRef CAS.
- M. Abdinejad, M. N. Hossain and H. B. Kraatz, RSC Adv., 2020, 10, 38013–38023 RSC.
- C. He, Y. Gong, S. Li, J. Wu, Z. Lu, Q. Li, L. Wang, S. Wu and J. Zhang, Adv. Mater., 2024, 2311628 CrossRef CAS PubMed.
- M. Abdinejad, T. Yuan, K. Tang, S. Duangdangchote, A. Farzi, H. P. Iglesias van Montfort, M. Li, J. Middelkoop, M. Wolff, A. Seifitokaldani, O. Voznyy and T. Burdyny, Chem. – Eur. J., 2023, 29, e202203977 CrossRef CAS PubMed.
- I. Masood ul Hasan, L. Peng, J. Mao, R. He, Y. Wang, J. Fu, N. Xu and J. Qiao, Carbon Energy, 2021, 3, 24–49 CrossRef CAS.
- M. Abdinejad, I. Santos da Silva and H. B. Kraatz, J. Mater. Chem. A, 2021, 9, 9791–9797 RSC.
- M. Abdinejad, E. Irtem, A. Farzi, M. Sassenburg, S. Subramanian, H. P. Iglesias Van Montfort, D. Ripepi, M. Li, J. Middelkoop, A. Seifitokaldani and T. Burdyny, ACS Catal., 2022, 12, 7862–7876 CrossRef CAS PubMed.
- Z. Masood and Q. Ge, Catal. Today, 2023, 409, 53–62 CrossRef CAS.
- Q. Cheng, M. Huang, L. Xiao, S. Mou, X. Zhao, Y. Xie, G. Jiang, X. Jiang and F. Dong, ACS Catal., 2023, 13, 4021–4029 CrossRef CAS.
- A. Mukherjee, M. Abdinejad, S. S. Mahapatra and B. C. Ruidas, J. Mater. Chem. A, 2023, 11, 9300–9332 RSC.
- M. Isegawa, Chem. Phys., 2023, 565, 111758 CrossRef CAS.
- K. Kim, P. Wagner, K. Wagner and A. J. Mozer, Molecules, 2023, 28, 5179 CrossRef CAS PubMed.
- X. Song, L. Xu, X. Sun and B. Han, Sci. China: Chem., 2023, 66, 315–323 CrossRef CAS.
- M. Todoroki, K. Hara, A. Kudo and T. Sakata, J. Electroanal. Chem., 1995, 394, 199–203 CrossRef.
- J. W. Jang, S. Cho, G. Magesh, Y. J. Jang, J. Y. Kim, W. Y. Kim, J. K. Seo, S. Kim, K. H. Lee and J. S. Lee, Angew. Chem., Int. Ed., 2014, 53, 5852–5857 CrossRef CAS PubMed.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2016, 6, 6265–6274 CrossRef.
- B. Ávila-Bolívar, L. García-Cruz, V. Montiel and J. Solla-Gullón, Molecules, 2019, 24, 2032 CrossRef PubMed.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J. J. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- S. Santra, V. Streibel, L. I. Wagner, N. Cheng, P. Ding, G. Zhou, E. Sirotti, R. Kisslinger, T. Rieth, S. Zhang and I. D. Sharp, ChemSusChem, 2024, 17, e202301452 CrossRef CAS PubMed.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- W. Zhu, Y. J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- K. Sun, L. Wu, W. Qin, J. Zhou, Y. Hu, Z. Jiang, B. Shen and Z. Wang, J. Mater. Chem. A, 2016, 4, 12616–12623 RSC.
- R. Wang, H. Haspel, A. Pustovarenko, A. Dikhtiarenko, A. Russkikh, G. Shterk, D. Osadchii, S. Ould-Chikh, M. Ma, W. A. Smith, K. Takanabe, F. Kapteijn and J. Gascon, ACS Energy Lett., 2019, 4, 2024–2031 CrossRef CAS.
- D. H. Won, H. Shin, J. Koh, J. Chung, H. S. Lee, H. Kim and S. I. Woo, Angew. Chemie, 2016, 128, 9443–9446 CrossRef.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- A. Klinkova, P. De Luna, C. T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- J. Zeng, W. Zhang, Y. Yang, D. Li, X. Yu and Q. Gao, ACS Appl. Mater. Interfaces, 2019, 11, 33074–33081 CrossRef CAS PubMed.
- F. A. Hanc-Scherer, M. A. Montiel, V. Montiel, E. Herrero and C. M. Sánchez-Sánchez, Phys. Chem. Chem. Phys., 2015, 17, 23909–23916 RSC.
- M. Umeda, Y. Niitsuma, T. Horikawa, S. Matsuda and M. Osawa, ACS Appl. Energy Mater., 2020, 3, 1119–1127 CrossRef CAS.
- J. Yin, J. Jin, Z. Yin, L. Zhu, X. Du, Y. Peng, P. Xi, C. H. Yan and S. Sun, Nat. Commun., 2023, 14, 1–10 Search PubMed.
- S. Pérez-Rodríguez, G. García, L. Calvillo, V. Celorrio, E. Pastor and M. J. Lázaro, Int. J. Electrochem., 2011, 2011, 1–13 CrossRef.
- H. Bin Yang, S. F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H. Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef.
- J. Han, B. Tu, P. An, J. Zhang, Z. Yan, X. Zhang, C. Long, Y. Zhu, Y. Yuan, X. Qiu, Z. Yang, X. Huang, S. Yan and Z. Tang, Adv. Mater., 2024, 36, 2313926 CrossRef CAS PubMed.
- R. Chen, X. Zu, J. Zhu, Y. Zhao, Y. Li, Z. Hu, S. Wang, M. Fan, S. Zhu, H. Zhang, B. Ye, Y. Sun and Y. Xie, Adv. Mater., 2024, 36, 2314209 CrossRef CAS PubMed.
- Y. Hori, Mod. Asp. Electrochem., 2008, 89–189 CAS.
- C. Kong, G. Jiang, Y. Sheng, Y. H. Liu, F. Gao, F. Liu and X. Duan, Chem. Eng. J., 2023, 460, 141803 CrossRef CAS.
- M. Li and J. N. Zhang, Sci. China: Chem., 2023, 66, 1288–1317 CrossRef CAS.
- J. Yan, H. Ma, J. Ni, J. Ma, J. Xu, J. Qi, S. Zhu and L. Lu, J. Colloid Interface Sci., 2023, 648, 558–566 CrossRef CAS PubMed.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143(16), 6152–6164 CrossRef CAS PubMed.
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1–11 Search PubMed.
- M. Ding, Z. Chen, C. Liu, Y. Wang, C. Li, X. Li, T. Zheng, Q. Jiang and C. Xia, Mater. Rep. Energy, 2023, 3, 100175 CAS.
- D. Song, Y. Lian, M. Wang, Y. Su, F. Lyu, Z. Deng and Y. Peng, eScience, 2023, 3, 100097 CrossRef.
- M. Abdinejad, S. Subramanian, K. Motlagh, M. Noroozifar, S. Duangdangchote, I. Neporozhnii, D. Ripepi, D. Pinto, M. Li, K. Tang, J. Middelkoop, A. Urakawa, O. Voznyy, H.-B. Kraatz, T. Burdyny, M. Abdinejad, S. Subramanian, D. Ripepi, D. Pinto, M. Li, J. Middelkoop, A. Urakawa, T. Burdyny, M. K. Motlagh, M. Noroozifar, S. Duangdangchote, I. Neporozhnii, K. Tang, O. Voznyy and H.-B. Kraatz, Adv. Energy Mater., 2023, 13, 2300402 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- M. Li, Y. Hu, T. Wu, A. Sumboja and D. Geng, Mater. Today, 2023, 67, 320–343 CrossRef CAS.
- K. Yang, Y. Sun, S. Chen, M. Li, M. Zheng, L. Ma, W. Fan, Y. Zheng, Q. Li and J. Duan, Small, 2023, 19, 2301536 CrossRef CAS PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Surf. Sci., 1995, 335, 258–263 CrossRef CAS.
- Y. Hori, I. Takahashi, O. Koga and N. Hoshi, J. Mol. Catal. A: Chem., 2003, 199, 39–47 CrossRef CAS.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- J. Chen, S. K. Iyemperumal, T. Fenton, A. Carl, R. Grimm, G. Li and N. A. Deskins, ACS Catal., 2018, 8, 10464–10478 CrossRef CAS.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2011, 14, 76–81 RSC.
- Z. Lyu, S. Zhu, M. Xie, Y. Zhang, Z. Chen, R. Chen, M. Tian, M. Chi, M. Shao and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 1909–1915 CrossRef CAS PubMed.
- Y. Wang, C. Niu, Y. Zhu, D. He and W. Huang, ACS Appl. Energy Mater., 2020, 3, 9841–9847 CrossRef CAS.
- T. T. H. Hoang, S. Ma, J. I. Gold, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2017, 7, 3313–3321 CrossRef CAS.
- A. S. Malkani, M. Dunwell and B. Xu, ACS Catal., 2019, 9(1), 474–478 CrossRef CAS.
- R. M. Arán-Ais, R. Rizo, P. Grosse, G. Algara-Siller, K. Dembélé, M. Plodinec, T. Lunkenbein, S. W. Chee and B. Roldan Cuenya, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- J. Shen, L. Wang, X. He, S. Wang, J. Chen, J. Wang and H. Jin, ChemSusChem, 2022, 15, e202201350 CrossRef CAS PubMed.
- L. Wan, X. Zhang, J. Cheng, R. Chen, L. Wu, J. Shi and J. Luo, ACS Catal., 2022, 12, 2741–2748 CrossRef CAS.
- D. Majumdar, J. Electroanal. Chem., 2021, 880, 114825 CrossRef CAS.
- K. Samdhyan, P. Chand, H. Anand and S. Saini, J. Energy Storage, 2022, 46, 103886 CrossRef.
- Y. Xie, A. Riedinger, M. Prato, A. Casu, A. Genovese, P. Guardia, S. Sottini, C. Sangregorio, K. Miszta, S. Ghosh, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2013, 135, 17630–17637 CrossRef CAS PubMed.
- R. J. Goble, Can. Mineral., 1985, 23, 6–76 Search PubMed.
- G. Kalimuldina, A. Nurpeissova, A. Adylkhanova, D. Adair, I. Taniguchi and Z. Bakenov, ACS Appl. Energy Mater., 2020, 3, 11480–11499 CrossRef CAS.
- P. Roy and S. K. Srivastava, CrystEngComm, 2015, 17, 7801–7815 RSC.
- F. di Benedetto, M. Borgheresi, A. Caneschi, G. Chastanet, C. Cipriani, D. Gatteschi, G. Pratesi, M. Romanelli and R. Sessoli, Eur. J. Mineral., 2006, 18, 283–287 CrossRef.
- R. Zeinodin, F. Jamali-Sheini and M. Cheraghizade, Mater. Sci. Semicond. Process., 2021, 123, 105501 CrossRef CAS.
- Z. Song, Y. Liu, B. Zhang, S. Song, Z. Zhou, Y. Huang and Z. Zhao, New J. Chem., 2023, 47, 2286–2295 RSC.
- J. Tirado, C. Roldán-Carmona, F. A. Muñoz-Guerrero, G. Bonilla-Arboleda, M. Ralaiarisoa, G. Grancini, V. I. E. Queloz, N. Koch, M. K. Nazeeruddin and F. Jaramillo, Appl. Surf. Sci., 2019, 478, 607–614 CrossRef CAS.
- P. Tetyana, N. Mphuthi, A. N. Jijana, N. Moloto, P. M. Shumbula, A. Skepu, L. S. Vilakazi and L. Sikhwivhilu, Nanomaterials, 2023, 13, 481 CrossRef CAS PubMed.
- E. M. Mkawi, M. W. Iqbal, Y. Al-Hadeethi, H. Hassan, B. Arkook, F. G. ALmehmadi and J. O. Dennis, J. Energy Storage, 2023, 67, 107656 CrossRef.
- J. He, K. Ramachandraiah, T. Huang, T. Yuan, X. Liu, H. Zhang and F. Ke, Biochem. Biophys. Res. Commun., 2023, 638, 51–57 CrossRef CAS PubMed.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- O. C. Pore, A. V. Fulari, C. D. Chavare, D. S. Sawant, S. S. Patil, R. V. Shejwal, V. J. Fulari and G. M. Lohar, Chem. Phys. Lett., 2023, 824, 140551 CrossRef CAS.
- S. K. Godlaveeti, A. R. Somala and R. R. Nagireddy, Appl. Phys. A: Mater. Sci. Process., 2023, 129, 1–12 CrossRef.
- N. Badar, H. M. Yusoff, K. Elong and N. Kamarulzaman, Adv. Powder Technol., 2023, 34, 104102 CrossRef CAS.
- H. Bian, R. Wang, K. Zhang, H. Zheng, M. Wen, Z. Li, Z. Li, G. Wang, G. Xie, X. Liu and L. Jiang, Surf. Coatings Technol., 2023, 459, 129407 CrossRef CAS.
- E. Aboobakri, T. Heidari and M. Jahani, Carbon Lett., 2023, 33, 1629–1638 CrossRef.
- M. H. Patel, T. K. Chaudhuri and V. K. Patel, J. Mater. Sci.: Mater. Electron., 2023, 34, 1–9 CrossRef.
- E. Gribov, E. Koshevoy, I. Chikunova and V. Parmon, Catalysts, 2023, 13, 168 CrossRef CAS.
- T. Singh and V. Mutreja, AIP Conf. Proc., 2023, 2535(1), 060002 CrossRef CAS.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef.
- Y. Tian, B. Li, J. Wang, Y. Ge, W. Gao, L. Yu, L. Ma, Y. Li, L. Wang, Z. Liu and J. Chen, Chem. Eng. J., 2024, 490, 151704 CrossRef CAS.
- C. Ye, B. Liu, Q. Li, M. Yu, Y. Liu, Z. Tai, Z. Pan and Y. Qiu, Small, 2024, 20, 2309856 CrossRef CAS PubMed.
- X. Ma, Y. Zhang, T. Fan, D. Wei, Z. Huang, Z. Zhang, Z. Zhang, Y. Dong, Q. Hong, Z. Chen and X. Yi, Adv. Funct. Mater., 2023, 33, 2213145 CrossRef CAS.
- C. He, D. Duan, J. Low, Y. Bai, Y. Jiang, X. Wang, S. Chen, R. Long, L. Song and Y. Xiong, Nano Res., 2023, 16, 4494–4498 CrossRef CAS.
- T. Dou, Y. Qin, F. Zhang and X. Lei, ACS Appl. Energy Mater., 2021, 4, 4376–4384 CrossRef CAS.
- G. Janani, S. Surendran, D. K. Lee, S. Shanmugapriya, H. Lee, Y. Subramanian and U. Sim, Aggregate, 2024, 5, e430 CrossRef CAS.
- C. Zhou, Y. Wu, X. Zhang, C. Ye, G. Peng and W. Yuan, J. Nanoparticle Res., 2024, 26, 1–11 CrossRef.
- P. Shao, S. Ci, L. Yi, P. Cai, P. Huang, C. Cao and Z. Wen, ChemElectroChem, 2017, 4, 2593–2598 CrossRef CAS.
- L. Zhang, X. Shi, A. Xu, W. Zhong, J. Zhang and S. Shen, Nano Res., 2024, 17, 3693–3699 CrossRef CAS.
- M. Ravipati and S. Badhulika, ACS Appl. Nano Mater., 2024, 7, 7277–7288 CrossRef CAS.
- L. Meng, C. W. Kao, Z. Wang, J. Ma, P. Huang, N. Zhao, X. Zheng, M. Peng, Y. R. Lu and Y. Tan, Nat. Commun., 2024, 15, 1–10 Search PubMed.
- W. Zhu, L. Fan, Q. Geng, C. Wang, X. Fan, Y. Zhang and C. Li, Chem. Eng. J., 2024, 489, 151316 CrossRef CAS.
- A. Yabuki, Y. Iwamura, T. Tachibana and J. H. Lee, New J. Chem., 2022, 46, 19633–19637 RSC.
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, J. Mater. Chem. A, 2017, 5, 20239–20243 RSC.
- Q. G. Zhu, X. F. Sun, X. C. Kang, J. Ma, Q. L. Qian and B. X. Han, Wuli Huaxue Xuebao, 2016, 32, 261–266 CAS.
- Z. Kou, X. Li, L. Zhang, W. Zang, X. Gao and J. Wang, Small Sci., 2021, 1, 2100011 CrossRef CAS.
- C. Yan, J. Huang, C. Wu, Y. Li, Y. Tan, L. Zhang, Y. Sun, X. Huang and J. Xiong, J. Mater. Sci. Technol., 2020, 42, 10–16 CrossRef CAS.
- C. Kim, S. H. Kim, S. Lee, I. Kwon, S. Kim, C. Seok, Y. S. Park and Y. Kim, J. Energy Chem., 2022, 64, 364–371 CrossRef CAS.
- K. Fan, H. Zou, Y. Lu, H. Chen, F. Li, J. Liu, L. Sun, L. Tong, M. F. Toney, M. Sui and J. Yu, ACS Nano, 2018, 12, 12369–12379 CrossRef CAS PubMed.
- T. X. Nguyen, Y. H. Su, C. C. Lin and J. M. Ting, Adv. Funct. Mater., 2021, 31, 2106229 CrossRef CAS.
- J. Chen, Y. Tu, Y. Zou, X. Li and J. Jiang, Mater. Lett., 2021, 284, 128919 CrossRef CAS.
- K. R. Phillips, Y. Katayama, J. Hwang and Y. Shao-Horn, J. Phys. Chem. Lett., 2018, 9, 4407–4412 CrossRef CAS PubMed.
- B. Zhang, M. Wang, J. Ding, Y. Li, G. Cao, M. T. Bernards, Y. He and Y. Shi, J. CO2 Util., 2020, 39, 101169 CrossRef CAS.
- C. H. M. van Oversteeg, M. Tapia Rosales, K. H. Helfferich, M. Ghiasi, J. D. Meeldijk, N. J. Firet, P. Ngene, C. de Mello Donegá and P. E. de Jongh, Catal. Today, 2021, 377, 157–165 CrossRef CAS.
- Y. Gao, Y. Guo, Y. Zou, W. Liu, Y. Luo, B. Liu and C. Zhao, ACS Appl. Energy Mater., 2023, 6, 1340–1354 CrossRef CAS.
- W. He, I. Liberman, I. Rozenberg, R. Ifraemov and I. Hod, Angew. Chem., Int. Ed., 2020, 59, 8262–8269 CrossRef CAS PubMed.
- J. Li, J. Li, C. Dun, W. Chen, D. Zhang, J. Gu, J. J. Urban and J. W. Ager, RSC Adv., 2021, 11, 23948–23959 RSC.
- D. Y. Y. Goh, K. M. Yam, L. Rekhi, A. D. Handoko, Y. C. Tan, Y. Wang, J. M. R. Tan, T. S. Choksi, Y. Lum and L. H. Wong, J. Mater. Chem. A, 2024, 12, 1840–1851 RSC.
- E. Cui, W. Zhang, X. Zhou, L. Lv, W. Chen, L. Zhou and L. Mai, ACS Appl. Nano Mater., 2023, 6, 9361–9368 CrossRef CAS.
- X. Zhang, Y. Zhang, X. Wei, C. Wei and Y. Song, Nanoscale Adv., 2021, 3, 5777–5784 RSC.
- T. Wu, M. Y. Han and Z. J. Xu, ACS Nano, 2022, 16, 8531–8539 CrossRef CAS PubMed.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 CrossRef CAS.
- J. W. Lim, W. J. Dong, J. Y. Park, D. M. Hong and J. L. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 22891–22900 CrossRef CAS PubMed.
- H. K. Park, H. Ahn, T. H. Lee, J. Y. Lee, M. G. Lee, S. A. Lee, J. W. Yang, S. J. Kim, S. H. Ahn, S. Y. Kim, C. H. Lee, E. S. Park and H. W. Jang, Small Methods, 2021, 5, 2000755 CrossRef CAS PubMed.
- X. Zhong, E. Yuan, F. Yang, Y. Liu, H. Lu, J. Yang, F. Gao, Y. Zhou, J. Pan, J. Zhu, C. Yu, C. Zhu, A. Yuan and E. H. Ang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2306673120 CrossRef CAS PubMed.
- X. Xu, Y. Zhong, M. Wajrak, T. Bhatelia, S. P. Jiang and Z. Shao, InfoMat, 2024, e12608 CrossRef CAS.
- R. Zhang, L. Wang, Y. H. Ma, L. Pan, R. Gao, K. Li, X. Zhang and J. J. Zou, J. Mater. Chem. A, 2019, 7, 10010–10018 RSC.
- R. Yang, X. Zheng, H. Fu, X. Cao, Y. Hu and Y. Huang, ChemSusChem, 2024, 17, e202301771 CrossRef CAS PubMed.
- Y. Wang, H. Xu, Y. Liu, J. Jang, X. Qiu, E. P. Delmo, Q. Zhao, P. Gao and M. Shao, Angew. Chem., Int. Ed., 2024, 63, e202313858 CrossRef CAS PubMed.
- K. Wu, C. Lyu, J. Cheng, W. Ding, J. Wu, Q. Wang, W. M. Lau and J. Zheng, Carbon Energy, 2024, 6, e485 CrossRef CAS.
- Y. Wang, Z. Lu, S. Wu, Z. Zou, X. Zhang and Y. Wang, J. Environ. Chem. Eng., 2024, 12, 112839 CrossRef CAS.
- Y. Liu, Z. Yang, Y. Zou, S. Wang and J. He, Energy Environ. Mater., 2024, 7, e12576 CrossRef CAS.
- J. Liang, S. Li, F. Li, L. Zhang, Y. Jiang, H. Ma, K. Cheng and L. Qing, J. Colloid Interface Sci., 2024, 655, 296–306 CrossRef CAS PubMed.
- L. Hong, B. Li, C. Jing, Z. Zhuang, Y. Zhang, H. Huang, Q. Jiang and J. Tang, J. Environ. Chem. Eng., 2024, 12, 111946 CrossRef CAS.
- Z. Guo, M. Bi, H. He, Z. Liu, Y. Duan and W. Cao, J. Colloid Interface Sci., 2024, 654, 785–794 CrossRef CAS PubMed.
- T. T. Zhuang, Z. Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P. N. Chen, X. L. Zheng, H. Liang, W. N. Ge, B. J. Ye, D. Sinton, S. H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- R. He, X. Huang and L. Feng, Energy Fuels, 2022, 36, 6675–6694 CrossRef CAS.
- M. Li and L. Feng, Chin. J. Struct. Chem., 2022, 41, 2201019–2201024 CAS.
- H. Liu, D. Zhao, P. Hu, K. Chen, X. Wu and D. Xue, Mater. Today Phys., 2020, 13, 100197 CrossRef.
- S. Zhang, C. Tan, R. Yan, X. Zou, F. L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202302795 CrossRef CAS PubMed.
- H. Tao, T. Jia, L. Zhang, X. Li, P. Li, Y. Zhou and C. Zhai, J. Colloid Interface Sci., 2024, 655, 909–919 CrossRef CAS PubMed.
- T. Dou, J. He, S. Diao, Y. Wang, X. Zhao, F. Zhang and X. Lei, J. Energy Chem., 2023, 82, 497–506 CrossRef CAS.
- X. Wang, J. Lv, J. Zhang, X. L. Wang, C. Xue, G. Bian, D. Li, Y. Wang and T. Wu, Nanoscale, 2020, 12, 772–784 RSC.
- Y. Liu, Z. Jiang, C. Huang, S. Jeong, A. L. Coughlin, S. Zhang, Y. Liu and X. Ye, Nano Lett., 2023, 23, 5911–5918 CrossRef CAS PubMed.
- C. He, S. Chen, R. Long, L. Song and Y. Xiong, Sci. China: Chem., 2020, 63, 1721–1726 CrossRef CAS.
- S. Cao, Y. Xue, X. Chen, C. Zhang, Y. Gao and Y. Li, Mater. Chem. Front., 2023, 7, 2620–2627 RSC.
- C. F. Wen, M. Zhou, P. F. Liu, Y. Liu, X. Wu, F. Mao, S. Dai, B. Xu, X. L. Wang, Z. Jiang, P. Hu, S. Yang, H. F. Wang and H. G. Yang, Angew. Chemie, 2022, 134, e202111700 CrossRef.
- Y. Li, Y. Chen, T. Chen, G. Shi, L. Zhu, Y. Sun and M. Yu, ACS Appl. Mater. Interfaces, 2023, 15, 18857–18866 CrossRef CAS PubMed.
- V. S. S. Mosali, G. Puxty, M. D. Horne, A. M. Bond and J. Zhang, Electrochim. Acta, 2024, 475, 143628 CrossRef CAS.
- V. S. S. Mosali, X. Zhang, Y. Liang, L. Li, G. Puxty, M. D. Horne, A. Brajter-Toth, A. M. Bond and J. Zhang, ChemSusChem, 2021, 14, 2924–2934 CrossRef CAS PubMed.
- Z. Yang, D. Ji, Z. Li, Z. He, Y. Hu, J. Yin, Y. Hou, P. Xi and C. H. Yan, Small, 2023, 19, 2303099 CrossRef CAS PubMed.
- Q. Wang, T. Bao, X. Zhao, Y. Cao, J. Cao, Q. Li and W. Si, Molecules, 2024, 29, 2948 CrossRef CAS PubMed.
- X. Han, T. Mou, S. Liu, M. Ji, Q. Gao, Q. He, H. Xin and H. Zhu, Nanoscale Horiz., 2022, 7, 508–514 RSC.
- M. Prasanna, N. Logeshwaran, S. Ramakrishnan and D. J. Yoo, Small, 2024, 20, 2306165 CrossRef CAS PubMed.
- Y. Guo, Y. Gao, B. Guo, Y. Luo, G. Zhao, J. Sun, W. Li, R. Wang and C. Zhao, Carbon Neutrality, 2024, 3, 1–20 CrossRef.
- Y. Wang, Y. Wang, J. Zhao, T. Tang, B. Lv, Y. Chang, T. Hu, J. Zhang, E. Luo and J. Jia, ACS Sustainable Chem. Eng., 2024, 12, 6982–6989 CrossRef CAS.
- Y. Zhang, Y. Dong, X. Yan, H. Peng, S. Xu, M. Zhu, Z. Jin, L. Han and J. Zhang, J. Energy Storage, 2024, 84, 111023 CrossRef.
- X. Cheng, D. Wu, H. Xu and W. Zhang, J. Phys. Chem. C, 2024, 128, 12101–12108 CrossRef CAS.
- W. Cheng, H. Yang, T. Wang, X. He, L. Tian and Z. Li, Chem. Rec., 2024, 24, e202300088 CrossRef CAS PubMed.
- Y. Jia, Y. Zhang, H. Xu, J. Li, M. Gao and X. Yang, ACS Catal., 2024, 14, 4601–4637 CrossRef CAS.
- P. Xu, Z. Bao, Y. Zhao, L. Zheng, Z. Lv, X. Shi, H. E. Wang, X. Fang and H. Zheng, Adv. Energy Mater., 2024, 14, 2303557 CrossRef CAS.
- Y. Hu, J. Zhu, X. Wang, X. Zheng, X. Zhang, C. Wu, J. Zhang, C. Fu, T. Sheng and Z. Wu, Inorg. Chem., 2024, 63, 9983–9991 CrossRef CAS PubMed.
- H. Shen, Y. Zhao, L. Zhang, Y. He, S. Yang, T. Wang, Y. Cao, Y. Guo, Q. Zhang and H. Zhang, Adv. Energy Mater., 2023, 13, 2202818 CrossRef CAS.
- L. Liang, L. Yang, T. Heine, A. Arinchtein, X. Wang, J. Hübner, J. Schmidt, A. Thomas and P. Strasser, Adv. Energy Mater., 2024, 14, 2304224 CrossRef CAS.
- S. Li, H. Duan, J. Yu, C. Qiu, R. Yu, Y. Chen, Y. Fang, X. Cai and S. Yang, ACS Catal., 2022, 12, 9074–9082 CrossRef CAS.
- Y. Huang, Y. Deng, A. D. Handoko, G. K. L. Goh and B. S. Yeo, ChemSusChem, 2018, 11, 320–326 CrossRef CAS PubMed.
- T. Jia, L. Wang, L. Zhang, Z. Zhu, K. Zhang, B. Zhu, X. Li, C. Zhai, S. Li, Y. Zhou and H. Tao, Surf. Interfaces, 2023, 38, 102841 CrossRef CAS.
- H. Yuan, Z. Liu, S. Sang and X. Wang, Appl. Surf. Sci., 2023, 613, 156130 CrossRef CAS.
- X. Zhang, R. Sa, F. Zhou, Y. Rui, R. Liu, Z. Wen and R. Wang, CCS Chem., 2021, 3, 199–207 CrossRef CAS.
- Y. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, ACS Appl. Mater. Interfaces, 2018, 10, 28572–28581 CrossRef CAS PubMed.
- E. Meza, R. E. Diaz and C. W. Li, ACS Nano, 2020, 14, 2238–2247 CrossRef CAS PubMed.
- P. Vancsó, Z. I. Popov, J. Petö, T. Ollár, G. Dobrik, J. S. Pap, C. Hwang, P. B. Sorokin and L. Tapasztó, ACS Energy Lett., 2019, 4, 1947–1953 CrossRef PubMed.
- Y. Liu, Y. Guo, Y. Liu, Z. Wei, K. Wang and Z. Shi, Energy Fuels, 2023, 37, 2608–2630 CrossRef CAS.
- H. Wang, T. Yang, J. Wang, Z. Zhou, Z. Pei and S. Zhao, Chem, 2024, 10, 48–85 CAS.
- A. W. Kahsay, K. B. Ibrahim, M. C. Tsai, M. K. Birhanu, S. A. Chala, W. N. Su and B. J. Hwang, Catal. Lett., 2019, 149, 860–869 CrossRef CAS.
- S. Wang, T. Kou, J. B. Varley, S. A. Akhade, S. E. Weitzner, S. E. Baker, E. B. Duoss and Y. Li, ACS Mater. Lett., 2021, 3, 100–109 CrossRef CAS.
- J. Wang, M. G. Sandoval, M. Couillard, E. A. González, P. V. Jasen, A. Juan, A. Weck and E. A. Baranova, ACS Sustainable Chem. Eng., 2024, 12(29), 11044–11055 CrossRef CAS.
- N. K. Singh, P. Kumar, A. Yadav and V. C. Srivastava, J. Colloid Interface Sci., 2024, 654, 895–905 CrossRef CAS PubMed.
- X. Li, L. Jiang, Y. Zhou and Q. Yu, Langmuir, 2024, 40(30), 15580–15587 CAS.
- H. A. B. Fonseca, L. G. Verga and J. L. F. Da Silva, J. Phys. Chem. C, 2023, 127, 24118–24128 CrossRef CAS.
- D. Liu, Y. Liu and M. Li, J. Phys. Chem. C, 2020, 124, 6145–6153 CrossRef CAS.
- C. L. Dong and L. Vayssieres, Chem. – Eur. J., 2018, 24, 18356–18373 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nat., 2018, 559, 547–555 CrossRef CAS PubMed.
- A. Chandrasekaran, D. Kamal, R. Batra, C. Kim, L. Chen and R. Ramprasad, npj Comput. Mater., 2019, 5, 1–7 CrossRef.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 1–11 CAS.
- R. Dorakhan, I. Grigioni, B. H. Lee, P. Ou, J. Abed, C. O’Brien, A. Sedighian Rasouli, M. Plodinec, R. K. Miao, E. Shirzadi, J. Wicks, S. Park, G. Lee, J. Zhang, D. Sinton and E. H. Sargent, Nat. Synth., 2023, 2, 448–457 CrossRef.
- Y. Mi, S. Shen, X. Peng, H. Bao, X. Liu and J. Luo, ChemElectroChem, 2019, 6, 2393–2397 CrossRef CAS.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. P. Veenstra, S. T.-J. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López, J. Pérez-Ramírez and B. S. Yeo, Angew. Chemie, 2020, 132, 21258–21265 CrossRef.
- L. J. Zhu, D. H. Si, F. X. Ma, M. J. Sun, T. Zhang and R. Cao, ACS Catal., 2023, 13, 5114–5121 CrossRef CAS.
- M. Wang, Q. Zhang, Q. Xie, L. Wan, Y. Zhao, X. Zhang and J. Luo, Nanoscale, 2020, 12, 17013–17019 RSC.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef CAS.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS.
- G. Shi, L. Yu, X. Ba, X. Zhang, J. Zhou and Y. Yu, Dalton Trans., 2017, 46, 10569–10577 RSC.
- S. Piontek, K. Junge Puring, D. Siegmund, M. Smialkowski, I. Sinev, D. Tetzlaff, B. Roldan Cuenya and U. P. Apfel, Chem. Sci., 2019, 10, 1075–1081 RSC.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 1–10 Search PubMed.
- W. Fang, R. Lu, F. M. Li, C. He, D. Wu, K. Yue, Y. Mao, W. Guo, B. You, F. Song, T. Yao, Z. Wang and B. Y. Xia, Angew. Chem., Int. Ed., 2024, 63, e202319936 CrossRef CAS PubMed.
- Y. S. Cheng, X. P. Chu, M. Ling, N. Li, K. L. Wu, F. H. Wu, H. Li, G. Yuan and X. W. Wei, Catal. Sci. Technol., 2019, 9, 5668–5675 RSC.
- H. Ning, X. Wang, W. Wang, Q. Mao, Z. Yang, Q. Zhao, Y. Song and M. Wu, Carbon N. Y., 2019, 146, 218–223 CrossRef CAS.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- M. Wang, Z. Cai, B. Zhang, K. Yang, T. Shou, M. T. Bernards, P. Xie, Y. He and Y. Shi, Energy Fuels, 2022, 36, 5833–5842 CrossRef CAS.
- A. F. Pérez-Cadenas, C. H. Ros, S. Morales-Torres, M. Pérez-Cadenas, P. J. Kooyman, C. Moreno-Castilla and F. Kapteijn, Carbon N. Y., 2013, 56, 324–331 CrossRef.
- S. Dongare, N. Singh and H. Bhunia, J. CO2 Util., 2021, 44, 101382 CrossRef CAS.
- J. Yuan, W. Y. Zhi, L. Liu, M. P. Yang, H. Wang and J. X. Lu, Electrochim. Acta, 2018, 282, 694–701 CrossRef CAS.
- F. Yang, X. Mao, M. Ma, C. Jiang, P. Zhang, J. Wang, Q. Deng, Z. Zeng and S. Deng, Carbon, 2020, 168, 528–535 CrossRef CAS.
- X. Tan, C. Yu, C. Zhao, H. Huang, X. Yao, X. Han, W. Guo, S. Cui, H. Huang and J. Qiu, ACS Appl. Mater. Interfaces, 2019, 11, 9904–9910 CrossRef CAS PubMed.
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- X. Ma, J. Tian, M. Wang, X. Jin, M. Shen and L. Zhang, Catal. Sci. Technol., 2021, 11, 6096–6102 RSC.
- J. X. Wu, S. Z. Hou, X. Da Zhang, M. Xu, H. F. Yang, P. S. Cao and Z. Y. Gu, Chem. Sci., 2019, 10, 2199–2205 RSC.
- Z. H. Zhao, K. Zheng, N. Y. Huang, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, Chem. Commun., 2021, 57, 12764–12767 RSC.
- S. Juntrapirom, J. Santatiwongchai, A. Watwiangkham, S. Suthirakun, T. Butburee, K. Faungnawakij, P. Chakthranont, P. Hirunsit and B. Rungtaweevoranit, Catal. Sci. Technol., 2021, 11, 8065–8078 RSC.
- K. Yao, Y. Xia, J. Li, N. Wang, J. Han, C. Gao, M. Han, G. Shen, Y. Liu, A. Seifitokaldani, X. Sun and H. Liang, J. Mater. Chem. A, 2020, 8, 11117–11123 RSC.
- Y. Zhao, C. Wang and G. G. Wallace, J. Mater. Chem. A, 2016, 4, 10710–10718 RSC.
- X. Ren, X. Zhang, X. Cao and Q. Wang, J. CO2 Util., 2020, 38, 125–131 CrossRef CAS.
- J. Zeng, T. Rino, K. Bejtka, M. Castellino, A. Sacco, M. A. Farkhondehfal, A. Chiodoni, F. Drago and C. F. Pirri, ChemSusChem, 2020, 13, 4128–4139 CrossRef CAS PubMed.
- C. S. Chen, J. H. Wan and B. S. Yeo, J. Phys. Chem. C, 2015, 119, 26875–26882 CrossRef CAS.
- A. D. Handoko, C. W. Ong, Y. Huang, Z. G. Lee, L. Lin, G. B. Panetti and B. S. Yeo, J. Phys. Chem. C, 2016, 120, 20058–20067 CrossRef CAS.
- J. Kim, W. Choi, J. W. Park, C. Kim, M. Kim and H. Song, J. Am. Chem. Soc., 2019, 141, 6986–6994 CrossRef CAS PubMed.
- Y. Mun, S. Lee, A. Cho, S. Kim, J. W. Han and J. Lee, Appl. Catal., B, 2019, 246, 82–88 CrossRef CAS.
- X. Guo, Y. Zhang, C. Deng, X. Li, Y. Xue, Y. M. Yan and K. Sun, Chem. Commun., 2014, 51, 1345–1348 RSC.
- M. Serafini, F. Mariani, A. Fasolini, E. T. Brandi, E. Scavetta, F. Basile and D. Tonelli, Adv. Funct. Mater., 2023, 33, 2300345 CrossRef CAS.
- T. Gao, Y. Gu, M. Wu, Y. Liu, L. Yang, T. Han, S. Zhang, S. Li, W. Wei, W. Chen and X. Dong, Energy Fuels, 2023, 37, 19053–19062 CrossRef CAS.
- A. Saxena, S. Kapila, J. E. Medvedeva and M. Nath, ACS Appl. Mater. Interfaces, 2023, 15(11), 14433–14446 CAS.
- N. Liu, S. Bartling, A. Springer, C. Kubis, O. S. Bokareva, E. Salaya, J. Sun, Z. Zhang, S. Wohlrab, A. M. Abdel-Mageed, H. Q. Liang and R. Francke, Adv. Mater., 2024, 36, 2309526 CrossRef CAS PubMed.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- C. Dong, L. Cui, Y. Kong, C. Chen, H. Liu, Y. Zhang, W. Zhu and R. He, J. Phys. Chem. C, 2022, 126, 102–109 CrossRef CAS.
- Y. Wu, P. Zhai, S. Cao, Z. Li, B. Zhang, Y. Zhang, X. Nie, L. Sun and J. Hou, Adv. Energy Mater., 2020, 10, 2002499 CrossRef CAS.
- F. Gao, S. Hu, X. Zhang, Y. Zheng, H. Wang, Z. Niu, P. Yang, R. Bao, T. Ma, Z. Dang, Y. Guan, X. Zheng, X. Zheng, J. Zhu, M. Gao and S. Yu, Angew. Chemie, 2020, 132, 8784–8790 CrossRef.
- R. He, A. Zhang, Y. Ding, T. Kong, Q. Xiao, H. Li, Y. Liu and J. Zeng, Adv. Mater., 2018, 30, 1705872 CrossRef PubMed.
- Y. H. Li, L. Cheng, P. F. Liu, L. Zhang, M. Y. Zu, C. W. Wang, Y. H. Jin, X. M. Cao, H. G. Yang and C. Li, ChemSusChem, 2018, 11, 1421–1425 CrossRef CAS PubMed.
- L. Cheng, Y. Li, A. Chen, Y. Zhu and C. Li, Chem. Commun., 2020, 56, 563–566 RSC.
- B. Qin, Y. Li, H. Wang, G. Yang, Y. Cao, H. Yu, Q. Zhang, H. Liang and F. Peng, Nano Energy, 2019, 60, 43–51 CrossRef CAS.
- Y. Wang, R. Du, Z. Li, H. Song, Z. Chao, D. Zu, D. Chong, N. Gao and C. Li, Ceram. Int., 2021, 47, 28321–28327 CrossRef CAS.
- L. Cheng, Y. Wang, Y. Li, Y. Shen, Y. Zhen, Z. Xing, L. Lin, A. Chen, Y. Zhu and C. Li, ChemCatChem, 2021, 13, 1161–1164 CrossRef CAS.
- R. He, X. Yuan, P. Shao, T. Duan and W. Zhu, Small, 2019, 15, 1904882 CrossRef CAS PubMed.
- A. Zhang, R. He, H. Li, Y. Chen, T. Kong, K. Li, H. Ju, J. Zhu, W. Zhu and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 10954–10958 CrossRef CAS PubMed.
- X. Zheng, P. De Luna, F. Pelayo García De Arquer, X. Du, P. Yang, E. H. Sargent, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler and Y. Cui, Joule, 2017, 1, 794–805 CrossRef CAS.
- Z. Chen, X. Zhang, M. Jiao, K. Mou, X. Zhang and L. Liu, Adv. Energy Mater., 2020, 10, 1903664 CrossRef CAS.
- F. Li, L. Chen, M. Xue, T. Williams, Y. Zhang, D. R. MacFarlane and J. Zhang, Nano Energy, 2017, 31, 270–277 CrossRef CAS.
- J. He, X. Liu, H. Liu, Z. Zhao, Y. Ding and J. Luo, J. Catal., 2018, 364, 125–130 CrossRef CAS.
- H. Cheng, S. Liu, J. Zhang, T. Zhou, N. Zhang, X. S. Zheng, W. Chu, Z. Hu, C. Wu and Y. Xie, Nano Lett., 2020, 20, 6097–6103 CrossRef CAS PubMed.
- J. Xu, S. Lai, M. Hu, S. Ge, R. Xie, F. Li, D. Hua, H. Xu, H. Zhou, R. Wu, J. Fu, Y. Qiu, J. He, C. Li, H. Liu, Y. Liu, J. Sun, X. Liu and J. Luo, Small Methods, 2020, 4, 2000567 CrossRef CAS.
- S. Q. Liu, M. R. Gao, R. F. Feng, L. Gong, H. Zeng and J. L. Luo, ACS Catal., 2021, 11, 7604–7612 CrossRef CAS.
- Y. Zhang, F. Li, X. Zhang, T. Williams, C. D. Easton, A. M. Bond and J. Zhang, J. Mater. Chem. A, 2018, 6, 4714–4720 RSC.
- X. Yang, P. Deng, D. Liu, S. Zhao, D. Li, H. Wu, Y. Ma, B. Y. Xia, M. Li, C. Xiao and S. Ding, J. Mater. Chem. A, 2020, 8, 2472–2480 RSC.
- X. Shao and Y. Liu, J. Electrochem. Soc., 2022, 169, 026505 CrossRef.
- J. Wang, J. Mao, X. Zheng, Y. Zhou and Q. Xu, Appl. Surf. Sci., 2021, 562, 150197 CrossRef CAS.
- C. Li, Z. Liu, X. Zhou, L. Zhang, Z. Fu, Y. Wu, X. M. Lv, G. Zheng and H. Chen, Energy Environ. Sci., 2023, 16, 3885–3898 RSC.
- Y. Li, J. Chen, S. Chen, T. Lu, X. Liao, T. Zhao, F. Cheng and H. Wang, Appl. Catal., B, 2024, 349, 123874 CrossRef CAS.
- M. Asadi, B. Kumar, A. Behranginia, B. A. Rosen, A. Baskin, N. Repnin, D. Pisasale, P. Phillips, W. Zhu, R. Haasch, R. F. Klie, P. Král, J. Abiade and A. Salehi-Khojin, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- F. Li, S. F. Zhao, L. Chen, A. Khan, D. R. MacFarlane and J. Zhang, Energy Environ. Sci., 2016, 9, 216–223 RSC.
- K. Lv, W. Suo, M. Shao, Y. Zhu, X. Wang, J. Feng and M. Fang, Nano Energy, 2019, 63, 103834 CrossRef CAS.
- N. Hussain, M. A. Abdelkareem, H. Alawadhi, K. Elsaid and A. G. Olabi, Chem. Eng. Sci., 2022, 258, 117757 CrossRef CAS.
- N. Hussain, M. A. Abdelkareem, H. Alawadhi, S. Begum, K. Elsaid and A. G. Olabi, J. Power Sources, 2022, 549, 232128 CrossRef CAS.
- H. Li, X. Liu, S. Chen, D. Yang, Q. Zhang, L. Song, H. Xiao, Q. Zhang, L. Gu and X. Wang, Adv. Energy Mater., 2019, 9, 1900072 CrossRef.
- P. Abbasi, M. Asadi, C. Liu, S. Sharifi-Asl, B. Sayahpour, A. Behranginia, P. Zapol, R. Shahbazian-Yassar, L. A. Curtiss and A. Salehi-Khojin, ACS Nano, 2017, 11, 453–460 CrossRef CAS PubMed.
- N. Hussain, M. A. Abdelkareem, H. Alawadhi, A. H. Alami and K. Elsaid, Appl. Phys. A: Mater. Sci. Process., 2022, 128, 1–11 CrossRef.
- K. Lv, C. Teng, M. Shi, Y. Yuan, Y. Zhu, J. Wang, Z. Kong, X. Lu and Y. Zhu, Adv. Funct. Mater., 2018, 28, 1802339 CrossRef.
- J. Xu, X. Li, W. Liu, Y. Sun, Z. Ju, T. Yao, C. Wang, H. Ju, J. Zhu, S. Wei and Y. Xie, Angew. Chem., Int. Ed., 2017, 56, 9121–9125 CrossRef CAS PubMed.
- X. Yuan, Y. Luo, B. Zhang, C. Dong, J. Lei, F. Yi, T. Duan, W. Zhu and R. He, Chem. Commun., 2020, 56, 4212–4215 RSC.
- A. Zhang, Y. Liang, H. Li, X. Zhao, Y. Chen, B. Zhang, W. Zhu and J. Zeng, Nano Lett., 2019, 19, 6547–6553 CrossRef CAS PubMed.
- L. P. Chi, Z. Z. Niu, X. L. Zhang, P. P. Yang, J. Liao, F. Y. Gao, Z. Z. Wu, K. Bin Tang and M. R. Gao, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- J. Feng, H. Gao, J. Feng, L. Liu, S. Zeng, H. Dong, Y. Bai, L. Liu and X. Zhang, ChemCatChem, 2020, 12, 926–931 CrossRef.
- W. Ma, S. Xie, X. G. Zhang, F. Sun, J. Kang, Z. Jiang, Q. Zhang, D. Y. Wu and Y. Wang, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- C. T. Altaf, T. O. Colak, E. Karagoz, J. Wang, Y. Liu, Y. Chen, M. Liu, U. Unal, N. D. Sankir and M. Sankir, ACS Omega, 2024, 9(17), 19209–19218 CrossRef CAS PubMed.
- J. E. Pander, J. W. J. Lum and B. S. Yeo, J. Mater. Chem. A, 2019, 7, 4093–4101 RSC.
- Z. Zhang, C. Liu, J. T. Brosnahan, H. Zhou, W. Xu and S. Zhang, J. Mater. Chem. A, 2019, 7, 23775–23780 RSC.
- J. Z. Zhen, J. X. Liu, T. Y. Chen, F. Shi, Y. N. Dai, B. Yang, Y. F. Li, X. Wang, T. G. Nong, Y. Q. Hu and J. Shi, J. Alloys Compd., 2019, 771, 994–999 CrossRef CAS.
- C. Li, G. Shen, R. Zhang, D. Wu, C. Zou, T. Ling, H. Liu, C. Dong and X. W. Du, J. Mater. Chem. A, 2019, 7, 1418–1423 RSC.
- Y. Song, Y. Wang, J. Shao, K. Ye, Q. Wang and G. Wang, ACS Appl. Mater. Interfaces, 2022, 14(18), 20368–20374 CrossRef CAS PubMed.
- H. il Nam, K. Ryeol Park, Y. W. Choi, H. Ji Sim, K. Yong Sohn and D. H. Lim, Appl. Surf. Sci., 2023, 612, 155646 CrossRef.
- A. Aljabour, H. Coskun, X. Zheng, M. G. Kibria, M. Strobel, S. Hild, M. Kehrer, D. Stifter, E. H. Sargent and P. Stadler, ACS Catal., 2020, 10, 66–72 CrossRef CAS.
- C. Simon, J. Zander, T. Kottakkat, M. Weiss, J. Timm, C. Roth and R. Marschall, ACS Appl. Energy Mater., 2021, 4, 8702–8708 CrossRef CAS.
- S. Zhao, S. Guo, C. Zhu, J. Gao, H. Li, H. Huang, Y. Liu and Z. Kang, RSC Adv., 2017, 7, 1376–1381 RSC.
- D. Tetzlaff, K. Pellumbi, K. Junge Puring, D. Siegmund, W. S. K. Polet, M. P. Checinski and U. P. Apfel, ChemElectroChem, 2021, 8, 3161–3167 CrossRef CAS.
- K. Pellumbi, M. Smialkowski, D. Siegmund and U. P. Apfel, Chem. – Eur. J., 2020, 26, 9938–9944 CrossRef CAS PubMed.
- T. J. Wang, W. S. Fang, Y. M. Liu, F. M. Li, P. Chen and Y. Chen, J. Energy Chem., 2022, 70, 407–413 CrossRef CAS.
- C. Xia, X. Wang, C. He, R. Qi, D. Zhu, R. Lu, F. M. Li, Y. Chen, S. Chen, B. You, T. Yao, W. Guo, F. Song, Z. Wang and B. Y. Xia, J. Am. Chem. Soc., 2024, 146(29), 20530–20538 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2013, 43, 631–675 RSC.
- D. Du, R. Lan, J. Humphreys and S. Tao, J. Appl. Electrochem., 2017, 47, 661–678 CrossRef CAS.
- B.-Q. Miao, W.-S. Fang, B. Sun, F.-M. Li, X.-C. Wang, B.-Y. Xia and Y. Chen, Chin. J. Struct. Chem., 2023, 42, 100095 CrossRef.
- S. Verma, B. Kim, H. R. M. Jhong, S. Ma and P. J. A. Kenis, ChemSusChem, 2016, 9, 1972–1979 CrossRef CAS PubMed.
- R. Lin, J. Guo, X. Li, P. Patel and A. Seifitokaldani, Catal., 2020, 10, 473 CrossRef CAS.
- Y. C. Li, D. Zhou, Z. Yan, R. H. Gonçalves, D. A. Salvatore, C. P. Berlinguette and T. E. Mallouk, ACS Energy Lett., 2016, 1, 1149–1153 CrossRef CAS.
- S. A. Al-Tamreh, M. H. Ibrahim, M. H. El-Naas, J. Vaes, D. Pant, A. Benamor and A. Amhamed, ChemElectroChem, 2021, 8, 3207–3220 CrossRef CAS.
- M. Zeng, W. Fang, Y. Cen, X. Zhang, Y. Hu and B. Y. Xia, Angew. Chem., 2024, 136, e202404574 CrossRef.
- H. Veldhuizen, M. Abdinejad, P. J. Gilissen, J. Albertsma, T. Burdyny, F. D. Tichelaar, S. van der Zwaag and M. A. van der Veen, ACS Appl. Mater. Interfaces, 2024, 16, 34010–34019 CrossRef CAS PubMed.
- O. G. Sánchez, Y. Y. Birdja, M. Bulut, J. Vaes, T. Breugelmans and D. Pant, Curr. Opin. Green Sustainable Chem., 2019, 16, 47–56 CrossRef.
- C. Oloman and H. Li, US Pat., App. 12/090,052, 2008.
- D. Ewis, M. Arsalan, M. Khaled, D. Pant, M. M. Ba-Abbad, A. Amhamed and M. H. El-Naas, Sep. Purif. Technol., 2023, 316, 123811 CrossRef CAS.
- H. Yang, J. J. Kaczur, S. D. Sajjad and R. I. Masel, J. CO2 Util., 2020, 42, 101349 CrossRef CAS.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- A. J. Martín, G. O. Larrazábal and J. Pérez-Ramírez, Green Chem., 2015, 17, 5114–5130 RSC.
- F. Proietto, U. Patel, A. Galia and O. Scialdone, Electrochim. Acta, 2021, 389, 138753 CrossRef CAS.
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He, C. Xia, H. Niu, S. Wang, Y. Liu, Y. Mao, C. Zhang, B. You, Y. Pang, L. Duan, X. Yang, F. Song, T. Zhai, G. Wang, X. Guo, B. Tan, T. Yao, Z. Wang and B. Y. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |